

Abstract
Preclinical drug development studies currently rely on costly and time-consuming animal testing because existing cell culture models fail to recapitulate complex, organ-level, disease processes in humans. Here, we provide the proof-of-principle for using a biomimetic microdevice that reconstitutes organ-level lung functions to create a human disease model-on-a-chip that mimics pulmonary edema. The microfluidic device, which reconstitutes the alveolar-capillary interface of the human lung, consists of channels lined by closely apposed layers of human pulmonary epithelial and endothelial cells that experience air and fluid flow, as well as cyclic mechanical strain to mimic normal breathing motions. In the present study, this device was used to reproduce drug toxicity-induced pulmonary edema observed in human cancer patients treated with interleukin-2 (IL-2) at similar doses and over the same time frame. Studies using this on-chip disease model revealed that mechanical forces associated with physiological breathing motions play a crucial role in the development of increased vascular leakage that leads to pulmonary edema, and that circulating immune cells are not required for the development of this disease. These studies also led to identification of potential new therapeutics, including angiopoietin-1 (Ang-1) and a new transient receptor potential vanilloid 4 (TRPV4) ion channel inhibitor (GSK2193874), which might prevent this life-threatening toxicity of IL-2 in the future.
One-sentence summary:
An in vitro model of human pulmonary edema predicts drug toxicity and efficacy previously observed in humans.
Introduction
Development of safe and effective drugs is currently hampered by the poor predictive power of existing preclinical animal models that often lead to failure of drug compounds late in their development after they enter human clinical trials. Given the tremendous cost of drug development and the long timelines involved, major pharmaceutical companies and government funding agencies, including the U.S. Food and Drug Administration (FDA), National Institutes of Health (NIH), Defense Advanced Research Projects Agency (DARPA), and Defense Threat and Reduction Agency (DTRA) are now beginning to recognize a crucial need for new technologies that can quickly and reliably predict drug safety and efficacy in humans in preclinical studies.
One approach to meeting this challenge is the development of three-dimensional (3D) cell cultures in which cells are grown within extracellular matrix (ECM) gels to induce expression of more tissue-specific functions (1). These models are currently being used for testing drug efficacy and toxicities; however, they fail to provide organ-level functionalities (molecular transport across tissue-tissue interfaces, contributions of vascular and air flow, etc.) that are required for the development of meaningful disease models. A potential solution to this problem is the development of human ‘organs-on-chips’ in which microscale engineering technologies are combined with cultured living human cells to create microfluidic devices that recapitulate the physiological and mechanical microenvironment of whole living organs (2–10). These organomimetic microdevices enable the study of complex human physiology in an organ-specific context and, more importantly, they offer the potential opportunity to develop specialized in vitro human disease models that could revolutionize drug development (2, 11–15). However, human organ-on-chip models that effectively mimic complex disease processes and drug responses have not yet been developed.
We recently microengineered a “lung-on-a-chip” that reconstituted the alveolar-capillary interface of the human lung and exposed it to physiological mechanical deformation and flow; in other words, it breathed rhythmically much like a living lung (4). Although this microengineered system recapitulated organ-level physiological functions, including pulmonary vascular barrier integrity and inflammatory responses to pathogens, it has not yet been shown to mimic complex human disease processes or to predict human responses to pharmaceutical agents at clinically relevant doses. Thus, this state-of-the-art system failed to provide the proof-of-principle required by the pharmaceutical industry and regulatory agencies to prove that organs-on-chips can be used to create human-relevant disease models that have potential to replace preclinical animal experiments.
In the present study, we explored whether we can use the human lung-on-a-chip microdevice to develop a microengineered disease model of pulmonary edema in human lungs, which is a life-threatening condition characterized by abnormal accumulation of intravascular fluid in the alveolar air spaces and interstitial tissues of the lung due to impaired homeostatic fluid balance mechanisms that lead to vascular leakage across the alveolar-capillary barrier (16–18). This disease can result from increased capillary hydrostatic pressure or microvascular permeability produced by various cardiovascular, respiratory, renal, and neurological diseases (16–20), or by dose-limiting drug toxicities. For example, systemic administration of interleukin-2 (IL-2) for the treatment of malignant melanoma and metastatic renal cell carcinoma is limited by toxicities that induce microvascular inflammation and injury leading to extravasation and accumulation of fluid in the alveolar air spaces (21–24), as well as activation of the coagulation cascade leading to intra-alveolar fibrin deposition (25, 26) (Fig. 1A). In this study, we focused on establishing a human lung disease model on-chip that reconstitutes these toxic side effects of IL-2 and resultant pulmonary edema in patients. We also investigated the possibility that mechanical breathing motions might contribute to pulmonary toxicity of IL-2, and applied this in vitro disease model to test whether therapeutics such as angiopoietin-1 (Ang-1) and a new inhibitor of transient receptor potential vanilloid 4 (TRPV4), GSK2193874 (GlaxoSmithKline), can suppress pulmonary vascular leakage in this in vitro human disease model, and hence have the potential to treat human patients.
Fig. 1.
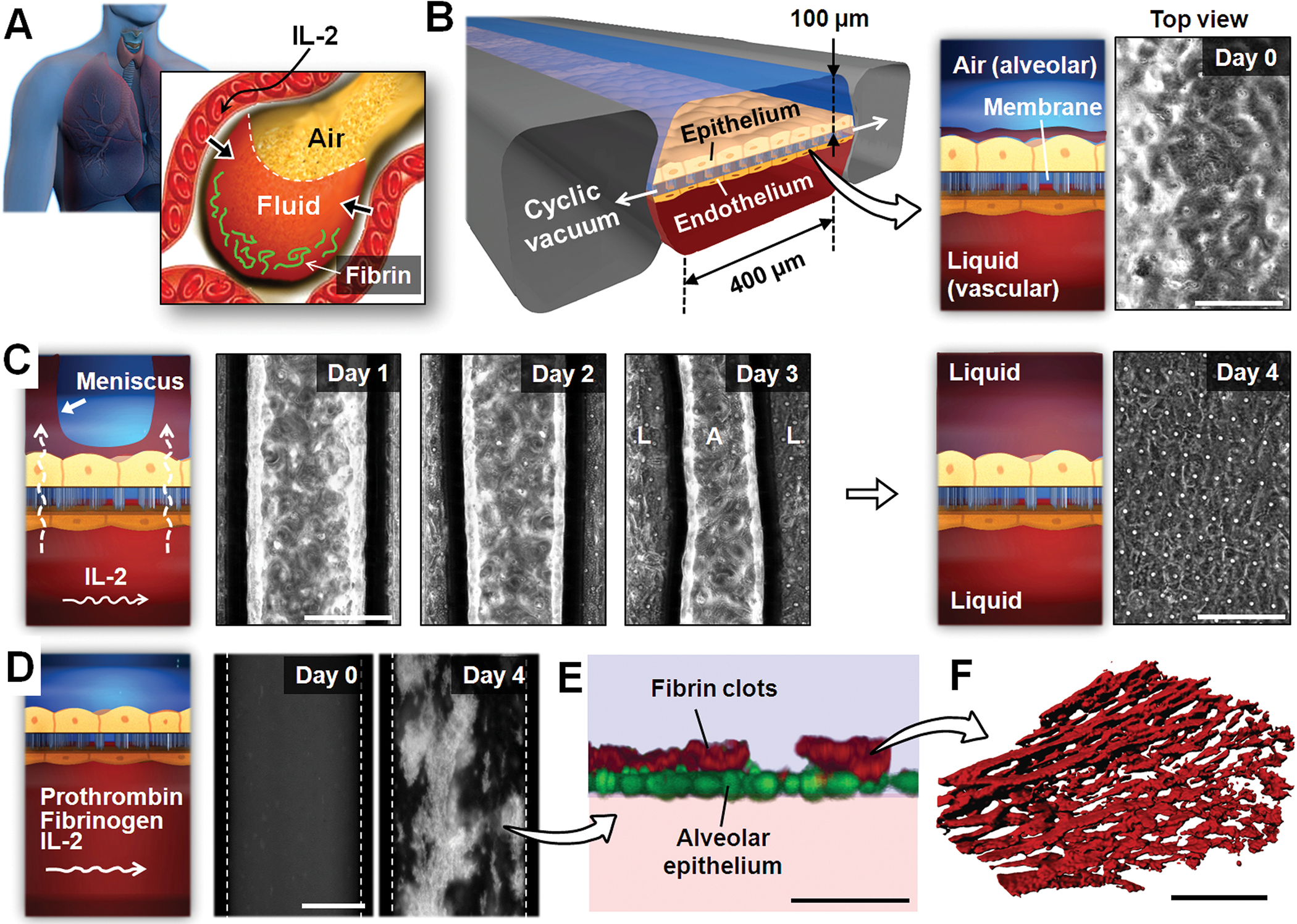
A microengineered model of human pulmonary edema. (A) IL-2 therapy is associated with vascular leakage that causes excessive fluid accumulation (edema) and fibrin deposition in the alveolar air spaces. (B) IL-2–induced pulmonary edema is modeled in a microengineered lung-on-a-chip that reproduces the lung micro-architecture and breathing-induced cyclic mechanical distortion of the alveolar-capillary interface. The top “air” portion is the alveolar channel; the bottom “liquid” portion is the vascular channel. The phase-contrast image shows a top-down view of the apical surface of the alveolar epithelium maintained at an air-liquid interface in the upper microchannel. Scale bar, 200 μm. (C) Endothelial exposure to IL-2 (1000 U/ml) causes liquid in the lower microvascular channel to leak into the alveolar chamber (days 1 to 3) and eventually fill the entire air space (day 4). The meniscus between air (A) and liquid (L) appears as dark bands in the phase contrast images. Scale bars, 200 μm. (D) During IL-2 treatment, prothrombin (100 μg/ml) and fluorescently labeled fibrinogen (2 mg/mL) introduced into the microvascular channel form fluorescent fibrin clots (white) over the course of 4 days. Dotted lines represent channel walls. Scale bar, 200 μm. (E) A fluorescence confocal microscopic image shows that the fibrin deposits (red) in (D) are found on the upper surface of the alveolar epithelium (green). Scale bar, 50 μm. (F) The clots in (D and E) are highly fibrous networks, as visualized at high magnification by confocal fluorescence microscopy. Images are representative of three independent experiments. Scale bar, 5 μm.
Results
Mimicking pulmonary edema in the human lung-on-a-chip
To explore whether we could mimic the dose-limiting toxicity of IL-2 in vitro, we used a lung-on-a-chip microfluidic device made of an optically transparent silicone elastomer that contains closely apposed layers of human alveolar epithelial and pulmonary microvascular endothelial cells cultured in two parallel microchannels separated by a thin (10 μm), porous, flexible membrane coated with ECM (4) (Fig. 1B). In the upper channel, the alveolar epithelium was exposed to air to mimic the alveolar air space; culture medium was flowed through the microvascular channel; and cyclic vacuum was applied to hollow side chambers to cyclically stretch the tissue layers (10% cyclic strain at 0.2 Hz) and thus reproduce physiological breathing movements (Fig. 1B).
To develop a drug toxicity–induced human pulmonary edema model on-chip, we perfused a clinically relevant dose of IL-2 (1000 U/ml) through the microvascular channel of this device. Phase contrast microscopic imaging revealed that clear liquid began to leak across the endothelium lining the microvascular channel and into the previously air-filled alveolar compartment, initially accumulating preferentially in a meniscus at the sides of the channel (Fig. 1C). This leakage occurred continuously over a period of 4 days, causing a progressive reduction in the volume of the air space until the entire alveolar microchannel became completely flooded (Fig. 1C). Moreover, when we introduced IL-2 and physiological concentrations of the human blood plasma proteins, prothrombin and fluorescently labeled fibrinogen, into the microvascular channel, extensive formation of fluorescent fibrin clots was observed on the apical surface of the alveolar epithelium (Fig. 1, D and E; movie S1). These clots also were organized as porous, fibrous, network structures (Fig. 1F), confirming that they mimic natural fibrin clots. Clot formation is likely due to activation of Factor X to Xa by tissue factor expressed or released by inflamed cells during IL-2-induced leakage of plasma proteins into the alveolar space, which then cleaves prothrombin to thrombin that acts as a pro-coagulant to convert fibrinogen to fibrin (27).
Pathological changes in barrier integrity
We then quantitatively examined these pathological changes in lung fluid transport by measuring the permeability of the microengineered alveolar-capillary barrier to fluorescein isothiocynate (FITC)-conjugated inulin that had been injected into the microvascular channel (Fig. 2A). Fluorescence intensity of fluids collected from the alveolar compartment was monitored during endothelial exposure to IL-2. In the absence of cyclic mechanical strain, a gradual increase in fluorescence was detected over time that was small but significant (Fig. 2B). However, when the tissue layers were subjected to mechanical deformations that mimicked physiological breathing movements, the same dose of IL-2 severely compromised pulmonary barrier function and further increased tissue permeability by more than 3-fold within 8 hours (Fig. 2B). We have previously demonstrated that mechanical stretch alone does not have any detrimental effects on barrier integrity in this system, regardless of its duration (4).
Fig. 2.
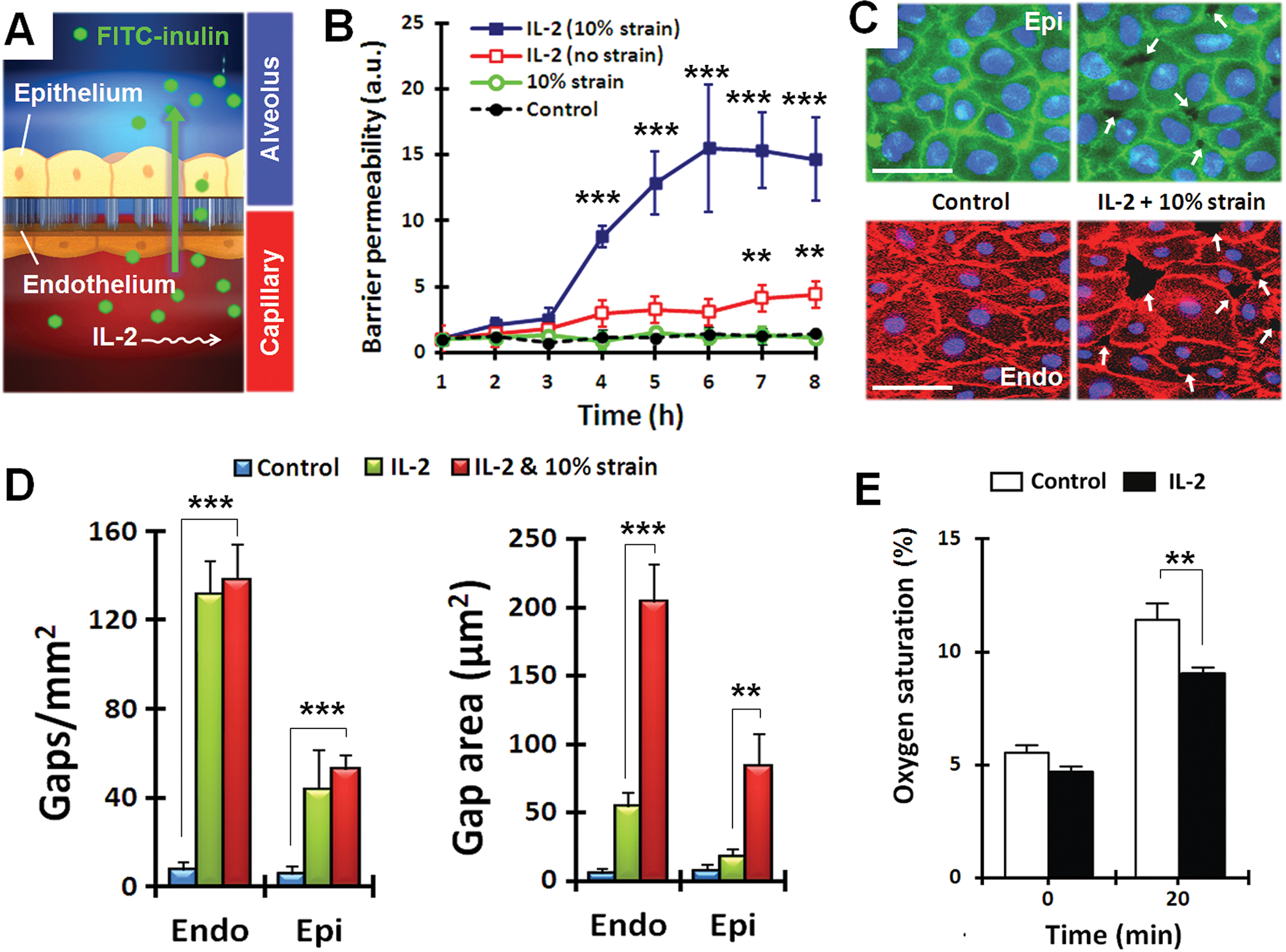
Quantitative analysis of pulmonary edema progression on-chip. (A) Pathological alterations of barrier function were quantified by measuring alveolar-capillary permeability to FITC-inulin (green) introduced into the microvascular channel containing IL-2. (B) Barrier permeability in response to IL-2, with and without cyclical strain. Data are means ± SEM (n = 3) and were normalized to the mean at time 0 (C) Immunostaining of epithelial occludin (green) and endothelial VE-cadherin (red) after 3 days of cyclic stretch with 10% strain without IL-2 (control) or with IL-2. White arrows indicate intercellular gaps; blue, nuclear staining. Scale bars, 30 μm. (D) Compromised barrier integrity due to IL-2 and mechanical strain assessed by quantifying the number and size of intercellular gaps. Data are means ± SEM (n = 3). (E) Quantification of O2 uptake from air in the epithelial channel by deoxygenated medium flowing through vascular channel in control and IL-2-treated lung chips. Data are means ± SEM (n = 3). For (B, D, and E), **P < 0.01, ***P < 0.001, ANOVA followed by post hoc Tukey’s multiple comparison test.
Immunostaining of intercellular junctions revealed that administration of IL-2 in conjunction with physiological mechanical strain disrupted cell-cell junctions and resulted in the formation of multiple paracellular gaps in both the epithelial and endothelial monolayers relative to controls (Fig. 2, C and D). Interestingly, when compared to the effects of IL-2 alone, application of physiological cyclic mechanical strain (10% at 0.2 Hz) during IL-2 treatment did not affect the number of the gaps, but significantly increased their size (area) by ~4-fold (Fig. 2D). Thus, these results reveal an important new contributor to the development of vascular leakage and pulmonary edema during IL-2 therapy that has not been identified in previous studies (31, 32): mechanical forces generated by breathing motions act in synergy with IL-2 to enhance opening of cell-cell junctions in both the alveolar epithelium and capillary endothelium.
Effects on gas transport across the alveolar-capillary barrier
Next we explored whether the leakage of fluid from the vascular channel to the air space induced by IL-2 treatment compromised oxygen transport in our microdevice, as occurs in humans with pulmonary edema. To investigate these effects, we perfused the vascular channel with deoxygenated medium (by bubbling 95% N2 and 5% CO2 through the medium reservoir), exposed the air space to room air (~20% O2), and measured the O2 concentration in the medium using an in-line fluorescent sensor. Perfusion of the deoxygenated medium through control (untreated) lung chips for 20 minutes under these conditions resulted in more than a 2-fold increase in oxygenation of the medium, whereas the lung chip treated with IL-2 for 4 days significantly compromised O2 uptake by the flowing medium (Fig 2E). Thus, this on-chip human disease model also mimics an important physiological consequence of pulmonary edema related to impaired gas exchange in the lung.
Drug testing on-chip
Next, we explored whether this human pulmonary edema-on-a-chip model can be used to identify pharmacological agents that might prevent vascular leakage induced by IL-2. We first tested angiopoietin-1 (Ang-1), which has been shown to stabilize endothelial intercellular junctions (33), and found that co-administration of Ang-1 (100 ng/ml) with IL-2 in the microvascular channel completely inhibited IL-2–induced vascular leakage (Fig. 3A). Ang-1 also prevented paracellular gap formation, even in the presence of 10% cyclic mechanical strain (Fig. 3, B and C). The Ang-1 antagonist, angiopoietin-2 (Ang-2), has been shown to destabilize the capillary barrier (34) and promote pulmonary edema during IL-2 therapy or sepsis (31, 32). Consistent with these observations, we found that Ang-2 (100 ng/ml) also induced pulmonary vascular leakage in the lung-on-a-chip when it was infused into the microvascular channel, and that co-administration of Ang-1 blocked this effect (Fig. 3A).
Fig. 3.
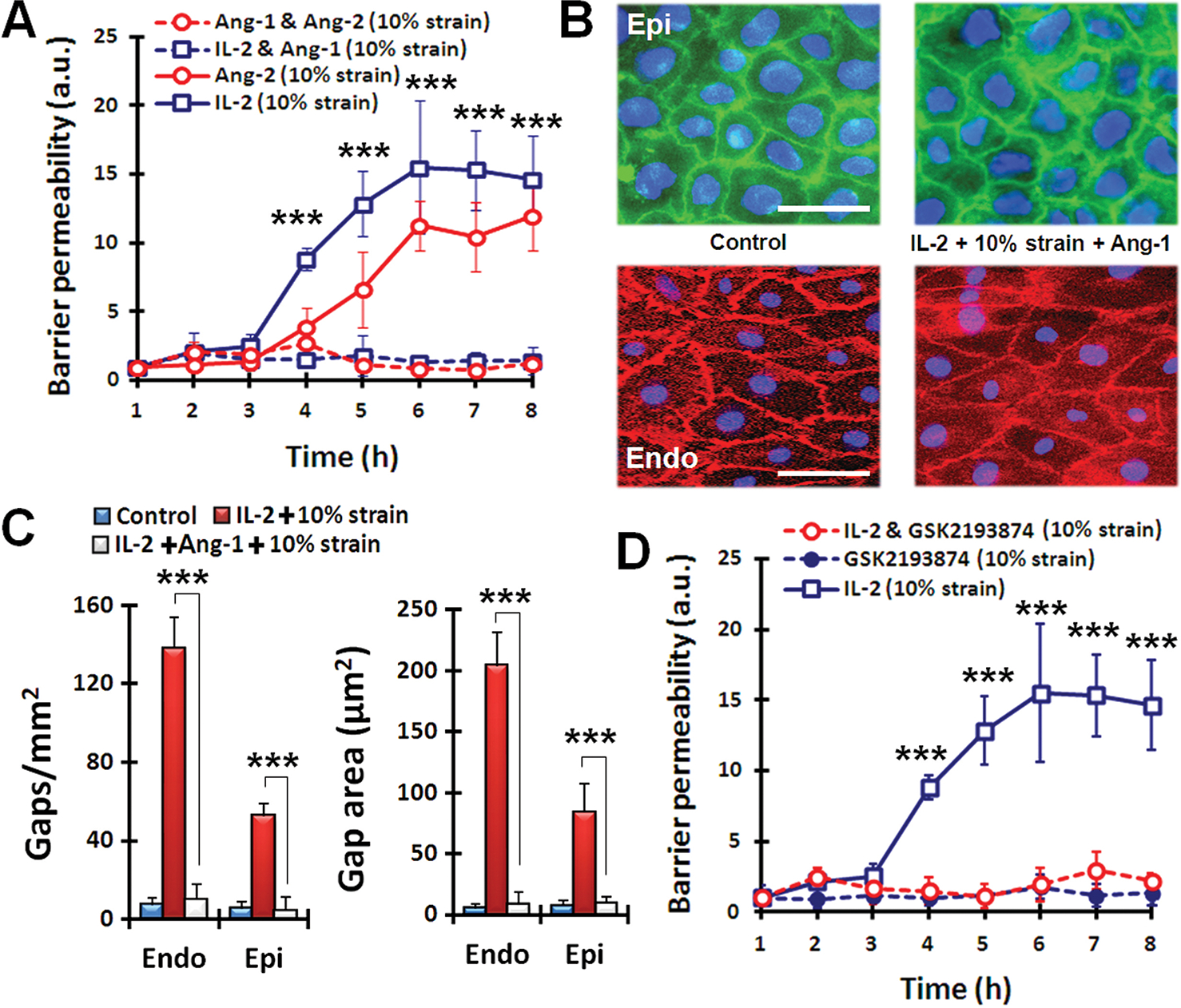
Pharmacological modulation in lung-on-a-chip pulmonary edema model. (A) Administration of Ang-1 (100 ng/ml) prevents fluid leakage caused by IL-2 (1000 U/ml) and Ang-2 (100 ng/ml) applied in conjunction with mechanical stretch. Statistical significance determined for comparison of IL-2 (10% strain) and IL-2 and Ang-1 (10% strain). (B) Immunostaining of epithelial occludin (green) and endothelial VE-cadherin (red) in cell-cell junctions after exposure to cyclic mechanical strain (10% at 0.2 Hz) without IL-2 (control) or with co-administration of IL-2 and Ang-1 for 3 days. Scale bars, 30 μm. (C) Restoration of barrier integrity by Ang-1 treatment assessed by changes in the number and size of intercellular gaps. ***P < 0.001. (D) The effect of TRPV4 inhibitor GSK2193874 (100 nM) on barrier permeability alone and in the presence of IL-2 and 10% strain. Statistical significance determined for comparison of IL-2 alone and IL-2 with GSK inhibitor. For (A, C, and E), data are means ± SEM (n = 3). For (A, C, and E), ***P < 0.001, ANOVA followed by post hoc Tukey’s multiple comparison test.
Mechanical strain can activate TRPV4 ion channels, and stimulation of these channels has been shown to increase alveolar-capillary permeability and cause vascular leakage in the lung (35–37). Therefore, we reasoned that pharmaceutical inhibition of TRPV4 channels might possibly prevent the exacerbation of IL-2-induced permeability owing to physiological cyclic mechanical strain. To test this, we examined the effects of a newly developed TRPV4 channel blocker GSK2193874 (GlaxoSmithKline) (20) on barrier permeability in our disease model. When administered intravascularly (through the microvascular channel, Fig 1B), this compound completely inhibited the increase in vascular permeability induced by IL-2 in combination with 10% cyclic mechanical strain (Fig. 3D). Importantly, these results closely match those obtained using the same TRPV4 inhibitor (GSK2193874) in rodent and canine models of pulmonary edema induced by heart failure, and the same study confirmed that cells of the human lung express TRPV4 channels (20).
Comparison of pulmonary edema-on-a-chip to whole mouse lung
To further investigate the physiological relevance of these results, we administered FITC-inulin and IL-2 intravenously to mice and analyzed alveolar-capillary barrier function in a whole-lung ex vivo ventilation-perfusion model (Fig. 4A). Fluorescence analysis of bronchoalveolar lavage samples collected from the whole lung confirmed that application of mechanical ventilation greatly accentuated the deleterious effect of IL-2 and increased barrier permeability by ~3-fold as compared with IL-2 administration without ventilation (Fig. 4B). These strain-induced changes were strikingly similar to the increases in permeability caused by physiological cyclic strain in our human pulmonary edema-on-a-chip (Fig. 4B). We also verified the ability of Ang-1 to prevent IL-2–induced fluid leakage into the lung in vivo, corroborating the predictions made by our on-chip pulmonary edema model (Fig. 4B).
Fig. 4.
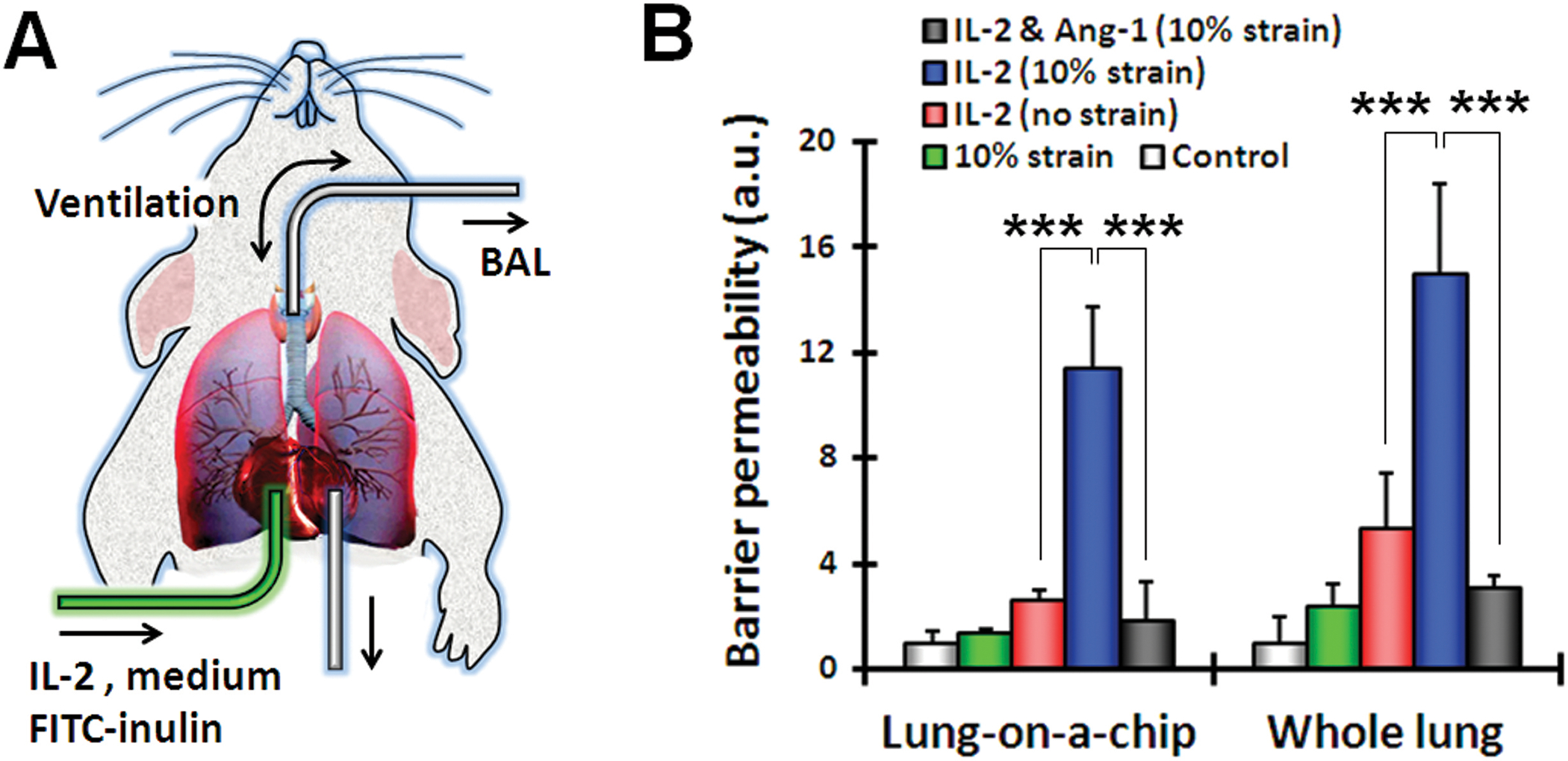
Barrier permeability in the lung-on-a-chip compared to whole mouse lung ex vivo. (A) An excised whole mouse lung was mechanically ventilated ex vivo, and the pulmonary microvasculature was perfused with culture medium containing IL-2 (1000 U/ml) and FITC-inulin (1 mg/ml). After IL-2 treatment, bronchoalveolar lavage (BAL) fluid was collected for fluorescence measurement. (B) Barrier permeability was measured in the lung-on-a-chip and in the whole animal lung model. Control represents no strain and no IL-2. Data are means ± SEM (n = 3). ***P < 0.001, ANOVA followed by post hoc Tukey’s multiple comparison test.
Discussion
These data demonstrate the ability to leverage the unique capabilities of the human lung-on-a-chip microdevice to create a clinically relevant human disease model in vitro, ultimately to reliably predict drug toxicity and efficacy in humans. This on-chip pulmonary edema model effectively reproduces the intra-alveolar fluid accumulation, fibrin deposition, and impaired gas exchange that have been observed in living edematous lungs following 2 to 8 days of IL-2 therapy in humans (23). Using real-time, high-resolution imaging and quantitative analysis of barrier function, we discovered that physiological mechanical forces combined with IL-2 treatment is detrimental to both epithelial and endothelial tissues. These studies provide new insight into the mechanism of IL-2–induced pulmonary edema by revealing that mechanical breathing motions induce increases in the size and number of gaps between cell-cell junctions in both the human pulmonary epithelium and endothelium in the presence of IL-2—a result that could not be readily examined in the animal models, nor can it be quantified easily in human biopsy specimens. IL-2–induced injury of the alveolar-capillary barrier in vivo is caused by apoptosis in both alveolar epithelial and pulmonary microvascular endothelial cells (28, 29). Combined application of IL-2 and mechanical strain in the lung-on-a-chip might similarly trigger apoptosis in cultured cells, as this has been previously shown to destabilize cell-cell junctions and induce formation of intercellular gaps (30).
Recognition of the importance of this mechanical contribution also led to the identification of a potential new therapeutic agent that might be used to prevent IL-2–induced pulmonary toxicities. More specifically, the TRPV4 inhibitor GSK2193874 prevented pulmonary edema induced by IL-2 in the lung-on-a-chip model, closely mimicking in vivo results obtained using the same inhibitor in rodent and canine models of pulmonary edema induced by heart failure (20). This preclinical study by Thornloe et al. serves to validate our findings and confirm the physiological relevance of the lung-on-a-chip model. Moreover, these studies combined suggest that TRPV4 (and hence mechanical signaling) contributes to the development of cardiogenic (20) as well as non-cardiogenic (this study) forms of pulmonary edema. Thus, use of the biomimetic microdevice offers many unique capabilities that provide added value beyond what is available in current preclinical animal models.
Importantly, our studies also revealed that the pulmonary vascular leakage response elicited by IL-2 in this microsystem, which closely mimics the response seen in humans, does not require circulating immune cells. This finding is in contrast to previous in vitro and in vivo studies showing that blood-borne immune cells, such as lymphocytes and neutrophils, activated by IL-2 play a central role in the induction of pulmonary vascular leakage (21, 38, 39). Our results suggest that direct toxic effects of IL-2 on endothelial and epithelial cells, amplified by physiological force, can mediate the onset and progression of IL-2–induced vascular injury and resultant pulmonary edema in the absence of immune cells. It is likely that the mechanically induced IL-2 toxicity might be further aggravated by recruitment of lymphocytes and neutrophils; however, future studies will be required to clarify whether and how particular immune cells contribute to this response. The human lung-on-a-chip system provides a robust means to address this question because it enables an investigator to introduce particular types of immune cells (one-at-a-time or in combination) to determine their contributions. This is not currently possible using genetically engineered or irradiated animal models that are widely employed to study the role of the immune system in pulmonary vascular leakage (38, 39).
The finding that mechanical breathing motions exacerbate IL-2 toxicity and increase the likelihood of the development of pulmonary edema in this model system implies that reducing tidal volumes during mechanical ventilation, which is now standard of clinical care (40, 41), also might be beneficial for minimizing pulmonary edema in patients on ventilators who receive IL-2 or exhibit symptoms of increased vascular leakage (for example, secondary to heart failure or sepsis). However, this requires further studies in animals and humans as we did not examine how different pressure waves or ventilation frequencies influence this response.
Recognition of the importance of mechanical forces for IL-2-induced pulmonary toxicity also led us to identify the TRV4 inhibitor GSK2193874 and Ang-1 as antagonists of intercellular gap formation and pulmonary vascular leakage caused by IL-2. These data suggest that these agents could represent potential new therapeutic agents for prevention of this life-threatening drug toxicity. These insights also might encourage the pharmaceutical industry to develop and screen for IL-2 replacements that produce their anticancer effects, without inducing these mechanosensitive toxic responses (and hence have fewer side effects). From a basic research perspective, it is also important to note that while the qualitative effects of IL-2, Ang-1, and Ang-2 on monolayer permeability can also be demonstrated under static culture conditions, the quantitative changes in vascular permeability induced by these agents in response to physiological breathing motions that we measured in vitro on-chip (and confirmed in vivo in whole mouse lung) could not be discovered using static culture conditions.
One potential limitation in these studies is that the pulmonary alveolar epithelial (NCI H441) cells that we used were originally isolated from a lung tumor, and so they might not fully recapitulate critical barrier functions of the human alveolar epithelium in vivo. Addressing this question directly would require reproducing these experiments with primary human alveolar epithelial cells, but long-term culture of these primary human cells with maintenance of physiological barrier function and proliferative capacity in vitro has not yet been possible. The more important question, however, is whether our system recapitulates the critical pathological responses that are characteristic of pulmonary edema. The fact that our model mimics IL-2-induced pulmonary vascular leakage and clot formation at similar doses and time courses to those observed in humans clearly demonstrates that these transformed epithelial cells exhibit the critical subset of properties required to study key disease processes and drug responses in vitro. We also previously showed that these cells reestablish pulmonary barrier functions that closely mimic those displayed in vivo when cultured in our lung chip device, whereas they do not in standard Transwell culture (4). Moreover, established cell lines, such as the one used in our studies, that have the ability to mimic in vivo phenotypes are much more robust than primary cells, which often can show unwanted batch-to-batch variability; this robustness makes them even more advantageous for technology translation and product development.
There are, however, still important physiological processes such as reabsorption and clearance of fluid in the alveolar air space (42, 43) that could be further studied in our microengineered system to determine their relative contribution to development of pulmonary edema in this model. For drug screening, the effects of serum-containing medium on test compounds also need to be explored in greater depth to ensure optimal evaluation and prediction of drug efficacy and toxicity in this model system.
In summary, analysis of this unique on-chip human disease model revealed three major new insights into the mechanism of IL-2 induced pulmonary edema: 1) vascular leakage results from intercellular gap formation in both the epithelium and endothelium, 2) mechanical breathing motions are a major contributor to IL-2–induced edema, and 3) the onset and progression of pulmonary edema during IL-2 therapy do not require circulating immune cells. Importantly, these findings suggest that a microengineered in vitro human disease model can potentially replace preclinical animal models of pulmonary edema currently used by the pharmaceutical industry to evaluate the effects of drugs on vascular leakage syndrome in the lung. Use of this microengineering approach also allows one to develop the simplest model possible that retains physiological relevance, and then to add complexity to the system as necessary, which is not possible in animal models.
This in vitro platform might be applied to model diseases in other organs and to predict other drug efficacies and toxicities in humans. In this manner, human organ-on-chip disease models could provide a potential new approach to enhance our fundamental understanding of complex disease processes and to enable more rapid, accurate, cost-effective, and clinically relevant testing of drugs, as well as cosmetics, chemicals, and environmental toxins. On-chip human disease models also have the potential to facilitate the translation of basic discoveries into effective new treatment strategies.
Materials and Methods
Device fabrication
Microfluidic devices used in this work were produced by the fabrication methods slightly modified from those that we reported previously (4). Briefly, the upper and lower layers of the microfluidic device were produced by casting PDMS prepolymer against a photolithographilcally prepared master that contained a positive relief of parallel microchannels made of photoresist (SU8–50, MicroChem). The weight ratio of PDMS base to curing agent was 15:1. The cross-sectional size of the microchannels is 400 μm (width) × 100 μm (height) for the central culture channels and 200 μm (width) × 100 μm (height) for the side channels. Thin microporous PDMS membranes were generated by spin-coating PDMS prepolymer (15:1) at 2500 rpm for 10 minutes on a silanized PDMS (10:1) substrate and bringing the spin-coated PDMS layer in conformal contact with a photolithographically prepared master that has an array of 50 μm-tall circular posts. Pressure was applied constantly to the master during curing of PDMS to ensure intimate contact and penetration of posts through the spin-coated layer. This process produced 10 μm-thick PDMS membranes with circular through-holes. After curing at 65°C overnight, the membrane surface was briefly treated with corona plasma generated by a hand-held corona treater (Electro-Technic Products) and bonded to the upper PDMS substrate to achieve irreversible bonding between the layers. After overnight incubation at 65°C, the bottom surface of the membrane was treated with corona and permanently bonded to the lower PDMS layer after careful manual alignment.
The side chambers in the microfluidic device were created by etching the membrane layers in the side microchannels using tetrabutylammonium fluoride (TBAF) and N-methylpyrrolidinone (NMP) mixed at a volumetric ratio of 1:3 (TBAF: NMP). An etching solution was introduced into the side microchannels through inlet reservoirs by using hydrostatic pressure or vacuum suction at the outlet ports. The flow of etchant was driven at a constant flow rate of ~200 μl/min. The membrane layers containing pentagonal holes in the side channels completely etched away within 2 minutes. Etching continued until the thickness of the PDMS walls between the side chambers from the central culture channels became thinner than 30 μm. After etching, the side chambers were washed thoroughly with NMP for at least 3 minutes to remove any remaining PDMS etchant.
Microfluidic cell culture
Human pulmonary microvascular endothelial cells (Lonza) were cultured in EBM-2 medium supplemented with 5% FBS and growth factors according to the manufacturer’s protocols. Alveolar epithelial cells (NCI-H441; ATCC) were grown in RPMI-1640 supplemented with 10% FBS, L-glutamine (0.292 mg/ml), penicillin (100 U/ml), and streptomycin (100 μg/ml). The cells were maintained at 37°C in a humidified incubator under 5% CO2 in air. Prior to cell seeding, microfluidic devices were sterilized by UV irradiation, and the porous membranes embedded in the central culture channels were coated with fibronectin (5 μg/ml in carbonate buffer). Alveolar epithelial cells were seeded into the upper channel at approximately 2×104 cells/cm2 and allowed to attach to the membrane surface for 2 hours under static conditions. The attached cells were then perfused with culture medium by a syringe pump at a volumetric flow rate of 50 μl/h. After 24 hours, the flow of culture medium was stopped, and the microfluidic device was inverted to seed endothelial cells onto the opposite side of the membrane.
After cell attachment, steady flows of culture media were driven in both the upper and lower channels at 50 μl/h (fluid shear stress ~ 0.2 dyne/cm2). The cells in both microchambers were grown to confluence within 5 days. Once alveolar epithelial cells reached confluence, they were treated with culture medium containing 1 μM of glucocorticoid dexamethasone (Sigma) to promote the formation of tight junctions (44). For air-liquid interface culture, culture medium was gently aspirated from the upper channel on day 5, and a 50:50 mixture of epithelial and endothelial medium was introduced into the lower channel to feed the alveolar epithelial cells on their basolateral side. The epithelial cells were grown at an air-liquid interface for 15 days. Microfluidic culture was maintained at 37°C in a humidified incubator with 5% CO2 in air.
Mechanical stimulation
Prior to mechanical stretching, the outlet holes of the side chambers were blocked, and the inlet ports were connected to a computer-controlled vacuum pump via vacuum-resistant tubing. Cyclic stretching was achieved by applying vacuum to the two side chambers simultaneously in a cyclic fashion. To mimic physiological breathing motion, the alveolar-capillary barriers formed in the microfluidic device were stretched with 10% strain at a frequency of 0.2 Hz (sinusoidal waveform). Prior to experiments, cells were stretched for 3 days using the specified regimen.
Immunostaining
To stain for Occludin, alveolar epithelial cells in the upper microchannel were washed with phosphate buffered saline (PBS), fixed with 4% paraformaldehyde (PFA) in PBS for 20 minutes, rinsed with PBS, and permeabilized with 0.5% Triton-TX-100 in PBS for 10 minutes. After washing with PBS and blocking with 2% bovine serum albumin (BSA) in PBS for 1 hour, the cells were incubated with FITC-conjugated mouse anti-human occludin antibody (Invitrogen) for 1 hour and washed with PBS.
For staining of vascular endothelial (VE)-cadherin, microvascular endothelial cells in the microfluidic device were fixed, permeabilized, and blocked using the same methods described above. The cells were then incubated with mouse anti-human VE-cadherin antibody for 1 hour and washed with PBS. Subsequently, Alexa 594-conjugated secondary antibody was added and incubated for 1 hour before fluorescence imaging.
Permeability assay
The permeability of the alveolar-capillary barrier was assessed by measuring the rate of FITC-inulin transport from the lower microvascular channel to the upper alveolar compartment. FITC-inulin (1 mg/ml in endothelial medium) was introduced into the lower channel, and inulin transport across the barrier was determined by serially sampling fluid from the upper channel and measuring its fluorescence intensity, which was used as an index of barrier permeability.
Oxygen transport measurements
Medium oxygenation was measured with an in-line fluorescent sensor off-chip (PreSens) before and after the device incubated under 5% CO2 in air at 37°C. Supplemented medium was deoxygenated by bubbling 95% N2 and 5% CO2 and infused into the device and sensors. Flow was regulated to 500 μL/hr using a syringe pump, and the O2 concentrations measured in the first sensor before introduction into the microdevice were consistently below 2%; O2 saturation levels were also measured after 20 min of perfusion through the lung chip at the outlet sensor. IL-2 was administered for four days to the device to induce fluid leakage into the air channel prior to initiating the experiment.
Ex vivo lung ventilation-perfusion experiments
All experimental animal protocols were approved by the Institutional Animal Care and Use Committee at Children’s Hospital Boston and Harvard Medical School. Eight-week–old male C57BL/6 mice (The Jackson Laboratory) were weighed and then anesthetized with Avertin (200 mg/kg IP). Ex vivo ventilation and perfusion of the mouse lung was facilitated by the IL-1 ex vivo mouse lung ventilation perfusion system (Harvard Apparatus). The trachea was incised via a surgical tracheotomy, and a 22G stainless steel endotracheal tube was secured inside the trachea. The lungs were subsequently ventilated at a rate of 60 breaths/min, with a peak inspiratory pressure (Pip) of 10 cm H2O and a positive end expiratory pressure (Peep) of 3 cm H2O with compressed air using a mouse ventilator (VCM-R, Hugo Sachs Elektroniks).
Following the initiation of mechanical ventilation, the chest was opened via thoracotomy, and heparin (100 IU) was injected into the right ventricle. After 30 seconds, the thoracic aorta and superior vena cava were cut and the animal ex-sanguinated. A suture was placed around the pulmonary artery and aorta. Flare tipped cannulae custom made using PE-90 tubing (0.86 mm ID, 1.7 mm OD, Becton Diskinson) were placed in the pulmonary artery (PA) and left atrium (LA), and lungs were perfused with RPMI-1640 with 4% bovine albumin (ProbuminTM Reagent Grade), 0.7 g NaCl/500 ml, and FITC-inulin (1 mg/ml) via a roller pump (ISM 834C, Ismatec SA) set at a constant flow rate of 0.5 ml/min in a recirculating system with a system volume of 6 ml. For 10 minutes following initiation of perfusion, the lungs were observed for leakage and or obstruction to perfusion or airflow. If leakage or obstruction were observed, the experiment was aborted; if not, the initial 2 ml of perfusate, which contained residual blood cells and plasma, were discarded and not recirculated.
Following the 10-minute observation period, and depending on the experiment, IL-2 (1,000 units/ml of recirculating perfusate) or carrier fluid (serving as a control) were injected into the reservoir containing the recirculating perfusion media. Ang-1 was used at a concentration of 100 ng/ml of recirculating perfusate. The lungs were subsequently ventilated and perfused for 5 hours. Perfusate and lung temperatures were maintained at 37°C by housing the entire ex-vivo ventilation perfusion system inside a standard cell incubator without CO2 (Forma Scientific). Humidity was maintained in the range of 90–95%. Pulmonary arterial and left atrial pressures and airway flow and pressures were recorded with dedicated Type 379 vascular pressure and DLP2.5 flow and MPX Type 399/2 airway pressure transducers and TAM-A amplifiers (Hugo Sachs Elektroniks). Vascular pressures were zeroed at the mid lung level prior to each experiment and recorded using Polyview16 software (Grass Technologies) running on a desktop PC running Windows XP SP2 (Microsoft Corporation). At the end of each experiment, the lungs were lavaged with 1.5 ml (500 μl x 3) of 0.9% normal saline, and the lavage fluid stored at −20°C for later analysis.
Statistical analysis
Data are represented as means ± SEM. Statistical significance was determined by analysis of variance (ANOVA) followed by post hoc Tukey’s multiple comparison test.
Supplementary Material
Movie S1. Fluorescent fibrin clots in the alveolar microchannel of the human lung-on-a-chip.
Acknowledgments:
We thank M. Khan and D. Shea for assistance in device fabrication, T. C. Ferrante and M. Montoya-Zavala for confocal microscopy, and A. Bahinski for helpful discussions.
Funding:
This research was supported by funding from NIH/FDA (U01 NS073474-01) and DARPA (W911NF-12-2-0036), and by the Wyss Institute for Biologically Inspired Engineering at Harvard University.
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability: Dr. McAlexander indicated in the authorship/COI forms that an MTA will be needed to “obtain novel reagents disclosed in the manuscript.” I am assuming this is the GSK inhibitor. Can you please make a statement to the effect here that such an MTA will be required between the requestor and GSK? Also, please indicate patent information here for the “Organ Mimetic Device and Methods of Use and Manufacturing Thereof.”
References and Notes
- 1.Pampaloni F, Reynaud EG, Stelzer EH, The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol 8, 839 (October, 2007). [DOI] [PubMed] [Google Scholar]
- 2.Huh D, Hamilton GA, Ingber DE, From 3D cell culture to organs-on-chips. Trends Cell Biol 21, 745 (December, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jang KJ, Suh KY, A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells. Lab Chip 10, 36 (January 7, 2010). [DOI] [PubMed] [Google Scholar]
- 4.Huh D et al. , Reconstituting organ-level lung functions on a chip. Science 328, 1662 (June 25, 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee PJ, Hung PJ, Lee LP, An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol Bioeng 97, 1340 (August 1, 2007). [DOI] [PubMed] [Google Scholar]
- 6.Sung JH, Shuler ML, A micro cell culture analog (microCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip 9, 1385 (May 21, 2009). [DOI] [PubMed] [Google Scholar]
- 7.Huh D et al. , Acoustically detectable cellular-level lung injury induced by fluid mechanical stresses in microfluidic airway systems. Proc Natl Acad Sci U S A 104, 18886 (November 27, 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grosberg A, Alford PW, McCain ML, Parker KK, Ensembles of engineered cardiac tissues for physiological and pharmacological study: heart on a chip. Lab Chip 11, 4165 (December 21, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HJ, Huh D, Hamilton G, Ingber DE, Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 12, 2165 (June 21, 2012). [DOI] [PubMed] [Google Scholar]
- 10.Domansky K et al. , Perfused multiwell plate for 3D liver tissue engineering. Lab Chip 10, 51 (January 7, 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douville NJ et al. , Combination of fluid and solid mechanical stresses contribute to cell death and detachment in a microfluidic alveolar model. Lab Chip 11, 609 (February 21, 2011). [DOI] [PubMed] [Google Scholar]
- 12.Torisawa YS et al. , Microfluidic platform for chemotaxis in gradients formed by CXCL12 source-sink cells. Integr Biol (Camb) 2, 680 (November, 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung S et al. , Cell migration into scaffolds under co-culture conditions in a microfluidic platform. Lab Chip 9, 269 (January 21, 2009). [DOI] [PubMed] [Google Scholar]
- 14.Sung KE et al. , Transition to invasion in breast cancer: a microfluidic in vitro model enables examination of spatial and temporal effects. Integr Biol (Camb) 3, 439 (April, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song JW et al. , Microfluidic endothelium for studying the intravascular adhesion of metastatic breast cancer cells. PLoS One 4, e5756 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robin ED, Cross CE, Zelis R, Pulmonary edema. 1. N Engl J Med 288, 239 (February 1, 1973). [DOI] [PubMed] [Google Scholar]
- 17.Robin ED, Cross CE, Zelis R, Pulmonary edema. 2. N Engl J Med 288, 292 (February 8, 1973). [DOI] [PubMed] [Google Scholar]
- 18.Hurley JV, Current views on the mechanisms of pulmonary oedema. J Pathol 125, 59 (June, 1978). [DOI] [PubMed] [Google Scholar]
- 19.Ware LB, Matthay MA, Clinical practice. Acute pulmonary edema. N Engl J Med 353, 2788 (December 29, 2005). [DOI] [PubMed] [Google Scholar]
- 20.Thorneloe KS et al. , An Orally Active TRPV4 Channel Blocker Prevents and Resolves Pulmonary Edema Induced by Heart Failure. submitted (2012). [DOI] [PubMed]
- 21.Baluna R, Vitetta ES, Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology 37, 117 (October, 1997). [DOI] [PubMed] [Google Scholar]
- 22.Rosenstein M, Ettinghausen SE, Rosenberg SA, Extravasation of intravascular fluid mediated by the systemic administration of recombinant interleukin 2. J Immunol 137, 1735 (September 1, 1986). [PubMed] [Google Scholar]
- 23.Conant EF, Fox KR, Miller WT, Pulmonary edema as a complication of interleukin-2 therapy. AJR Am J Roentgenol 152, 749 (April, 1989). [DOI] [PubMed] [Google Scholar]
- 24.Briasoulis E, Pavlidis N, Noncardiogenic pulmonary edema: an unusual and serious complication of anticancer therapy. Oncologist 6, 153 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Kradin RL et al. , Tumor-derived interleukin-2-dependent lymphocytes in adoptive immunotherapy of lung cancer. Cancer Immunol Immunother 24, 76 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satchi-Fainaro R et al. , Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell 7, 251 (March, 2005). [DOI] [PubMed] [Google Scholar]
- 27.Bastarache JA et al. , The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax 62, 608 (July, 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guan H, Nagarkatti PS, Nagarkatti M, Blockade of hyaluronan inhibits IL-2-induced vascular leak syndrome and maintains effectiveness of IL-2 treatment for metastatic melanoma. J Immunol 179, 3715 (September 15, 2007). [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Wenthold RJ Jr., Yu ZX, Herman EH, Ferrans VJ, Characterization of the pulmonary lesions induced in rats by human recombinant interleukin-2. Toxicol Pathol 23, 653 (Nov-Dec, 1995). [DOI] [PubMed] [Google Scholar]
- 30.Corfe BM, Dive C, Garrod DR, Changes in intercellular junctions during apoptosis precede nuclear condensation or phosphatidylserine exposure on the cell surface. Cell Death Differ 7, 234 (February, 2000). [DOI] [PubMed] [Google Scholar]
- 31.Parikh SM et al. , Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med 3, e46 (March, 2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallagher DC et al. , Angiopoietin 2 is a potential mediator of high-dose interleukin 2-induced vascular leak. Clin Cancer Res 13, 2115 (April 1, 2007). [DOI] [PubMed] [Google Scholar]
- 33.Peters KG, Vascular endothelial growth factor and the angiopoietins: working together to build a better blood vessel. Circ Res 83, 342 (August 10, 1998). [DOI] [PubMed] [Google Scholar]
- 34.Maisonpierre PC et al. , Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55 (July 4, 1997). [DOI] [PubMed] [Google Scholar]
- 35.Hamanaka K et al. , TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol 293, L923 (October, 2007). [DOI] [PubMed] [Google Scholar]
- 36.Alvarez DF et al. , Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res 99, 988 (October 27, 2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiter B et al. , TRPV4-mediated regulation of epithelial permeability. FASEB J 20, 1802 (September, 2006). [DOI] [PubMed] [Google Scholar]
- 38.Assier E et al. , NK cells and polymorphonuclear neutrophils are both critical for IL-2-induced pulmonary vascular leak syndrome. J Immunol 172, 7661 (June 15, 2004). [DOI] [PubMed] [Google Scholar]
- 39.Melencio L et al. , Role of CD4(+)CD25(+) T regulatory cells in IL-2-induced vascular leak. Int Immunol 18, 1461 (October, 2006). [DOI] [PubMed] [Google Scholar]
- 40.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 342, 1301 (May 4, 2000). [DOI] [PubMed] [Google Scholar]
- 41.Lipes J, Bojmehrani A, Lellouche F, Low Tidal Volume Ventilation in Patients without Acute Respiratory Distress Syndrome: A Paradigm Shift in Mechanical Ventilation. Crit Care Res Pract 2012, 416862 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ware LB, Matthay MA, Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163, 1376 (May, 2001). [DOI] [PubMed] [Google Scholar]
- 43.Matthay MA, Folkesson HG, Clerici C, Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 82, 569 (July, 2002). [DOI] [PubMed] [Google Scholar]
- 44.Hermanns MI, Unger RE, Kehe K, Peters K, Kirkpatrick CJ, Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: development of an alveolo-capillary barrier in vitro. Lab Invest 84, 736 (June, 2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Fluorescent fibrin clots in the alveolar microchannel of the human lung-on-a-chip.