

Abstract
BIM is the master BH3-only BCL-2 family regulator of lymphocyte survival. To understand how long-term loss of BIM affects apoptotic resistance in T cells we studied animals with T cell-specific deletion of Bim. Unlike CD19CRE Bimfl/fl animals, LCKCRE Bimfl/fl mice have pronounced early lymphocytosis followed by normalization of lymphocyte counts over time. This normalization occurred in mature T cells, as thymocyte development and apoptotic sensitivity remained abnormal in LCKCRE Bimfl/fl mice. T cells from aged mice experienced normalization of their absolute cell numbers and responses against various apoptotic stimuli. mRNA expression levels of BCL-2 family proteins in CD4+ and CD8+ T cells from young and old mice revealed upregulation of several BH3-only proteins, including Puma, Noxa, and Bmf. Despite upregulation of various BH3 proteins, there were no differences in anti-apoptotic BCL-2 protein dependency in these cells. However, T cells had continued resistance to direct BIM BH3-induced mitochondrial depolarization. This study further highlights the importance of BIM in cell death maintenance in T cells and provides new insight into the dynamism underlying BH3-only regulation of T cell homeostasis versus induced cell death and suggests that CD4+ and CD8+ T cells compensate differently in response to loss of Bim.
Keywords: BCL-2, T cells, BIM, Apoptosis
Introduction
Apoptosis is critical for organismal development, maintenance of normal immune cell number, and removal of abnormal cells [1]. Apoptosis is triggered by two main pathways, with the extrinsic pathway reliant on external ligands binding to death receptors outside of the cell and the intrinsic pathway mediated at the level of the mitochondrion [2, 3]. The BCL-2 family of proteins is key in regulating both pathways and is divided into three major subgroups: the multidomain anti-apoptotic proteins (BCL-2, BCL-XL, BCL-W, MCL-1, BFL-1/A1), the multidomain pro-apoptotic proteins (BAK, BAX), and the BH3-only proteins (BIM, BID, PUMA, NOXA, BAD, BMF, HRK) [4, 5]. BH3-only proteins act as cellular sentinels that mediate interactions between the two groups of multidomain proteins and initiate cell death via two different mechanisms [6]. Indirectly, BH3-only proteins bind to and sequester the anti-apoptotic proteins leading to the release and oligomerization of BAX and BAK resulting in mitochondrial outer membrane permeabilization (MOMP) [7]. A subset of BH3-only proteins, namely BIM, BID, and PUMA (and possibly NOXA), can also induce cell death through their direct binding and subsequent activation of BAK and BAX [8–10].
Of the BH3-only proteins, BIM plays the most fundamental role in apoptotic regulation of the immune system, particularly with respect to T and B cells [11–17]. Young mice with global Bim deletion have increased white blood cell counts (WBC), abnormal thymopoiesis, and lymphocytes that are resistant to a wide range of apoptotic stimuli [11, 12]. By one year of age, over 50% of Bim null mice become moribund, typically due to B cell-mediated autoimmunity [11]. Targeted loss of BIM within the entire hematopoietic compartment recapitulates this phenotype, emphasizing the importance of BIM in immune stability and pointing to a lymphocyte-intrinsic mechanism by which BIM regulates immune cell ontogeny, homeostasis, and apoptotic resistance [18, 19].
Given the central importance of BIM in immune cell homeostasis, genetic ablation of Bim in specific lymphocyte subsets can provide a mechanistic framework to better understand how BCL-2 family compensation occurs in non-malignant lymphocytes, as what might occur in patients treated with BH3-mimetics [5, 20]. To evaluate if long-term deletion of Bim in T cells recapitulates the early phenotype measured in globally deleted animals we characterized the peripheral lymphoid compartment in aged mice harboring Bim-deficient T or B cells. Unlike CD19CRE Bimfl/fl mice, LCKCRE Bimfl/fl mice experienced early lymphocytosis that normalized over time. We examined several potential mechanisms that may be responsible for this normalization, including global phenotypic changes in the T cell repertoire, apoptotic resistance patterns, and quantitative mRNA expression of the BCL-2 family of proteins. We found a consistent pattern of BH3-only mRNA upregulation, particularly of Bmf, in CD4+ and CD8+ T cells lacking Bim. Despite similarities in mRNA expression patterns, CD4+ and CD8+ T cells had differences in cell death sensitivities to apoptotic stimuli, suggesting divergent compensatory mechanisms. However there were no changes in anti-apoptotic BCL-2 dependence patterns between the two cell types. Our studies shed light on the importance of BH3-only proteins in T cell homeostasis and emphasizes the importance of BIM in T cell apoptotic control. This study also highlights the difference between qualitative versus quantitative BCL-2 family compensatory mechanisms in T cells, which may prove critical to the mechanistic understanding of normal immune cell sensitivities in patients treated with BH3 mimetics [21].
Materials and methods
Animals
CD19CRE Bimfl/fl, LCKCRE Bimfl/fl, and LCKCRE Neg (referred to hereafter as WT) mice were generated as described previously [22–24]. LCKCRE Bimfl/fl mice were kind gifts from Loren Walensky, M.D., Ph.D., Dana-Farber Cancer Institute, Boston, MA and were generated through cross of LCKCRE mice with Bimfl/fl animals [22, 25, 24]. Complete blood counts were obtained from peripheral blood using the ADVIA 120 (Siemens Healthineers) specifically calibrated and used for animal studies. All animal experiments were approved by and performed in accordance with the guidelines and regulations set forth by the Institutional Animal Care and Use Committee of the University of Chicago.
Fluorescence activated cell sorting
Splenocytes and thymocytes were isolated from animals and single cell suspensions were generated by disruption of the spleen and thymus through 100 µm and 70 µm filters respectively as previously described [26]. Individual lymphocyte populations were sorted on a BD FACSAria (University of Chicago Flow Cytometry Core) using CD4-APC, 1:100 (BD) and CD8-FITC, 1:100 (BD). Sorted populations were ≥ 98% pure.
Flow cytometric analysis
Single cell suspensions of splenocytes and thymocytes were prepared as described above. The following antibodies were used for staining at a dilution of 1:100 in PBS (all antibodies were from BD unless otherwise indicated): CD44-PE-Cy7, CD62L-PE, CD25-PerCPCy5.5, CD4-APC, CD8-BUV395 and CD8-FITC. For intracellular FOXP3 staining, cells were fixed and permeabilized with the FOXP3 Fixation/Permeabilization kit (eBioscience) per the manufacturer’s protocol. After fixation and permeabilization, cells were stained with FOXP3-FITC (eBioscience) at 1:50 in permeabilization buffer for 25 min in the dark on ice. Samples were analyzed using a LSRII cytometer (BD) or FACSAria (BD). Data analysis was performed using FlowJo (Tree Star).
Viability assays
CD4+ splenocytes, CD8+ splenocytes, and CD4+CD8+ double positive (DP) thymocytes were plated in 96 well round bottom plates at a density of 50,000 cells/well. Cells were cultured in cDMEM (DMEM with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% l-glutamine, 1% non-essential amino acids, and 10 µM β-mercaptoethanol). Cells were treated with: cDMEM alone (cytokine deprivation), 1 µg/mL ionomycin, 4 ng/mL PMA, or 1 µM etoposide. Cells were incubated for 24 and 48 h, at 37 °C, 5% CO2. At the indicated timepoints, viability was assessed by staining cells with Annexin V-FITC (BD) and propidium iodide (Life Technologies) and analyzed via flow cytometry, with percent viable cells reported as Annexin Vneg PIneg as previously reported [27].
Quantitative real-time PCR (qRT-PCR)
Pure populations of CD4+ splenocytes, CD8+ splenocytes and CD4+CD8+ thymocytes were lysed with Trizol (Life Technologies) and mRNA extracted using the Direct-zol RNA MiniPrep kit (Zymo). mRNA concentration and purity was assessed using a DeNovix DS-11 spectrophotometer and reverse transcription was performed using the Superscript III first strand synthesis reverse transcription kit (Invitrogen) per the manufacturer’s guidelines. qRT-PCR was performed using TaqMan Master Mix and Gene Expression Probes (Applied Biosystems) for each of the BCL-2 family members as follows: Mcl-1: Mm00725832_s1; Bak1: Mm00432045_m1; Bcl2l11 (Bim): Mm00437796_m1; Bcl2a1a (A1): Mm03646861_mH; Bbc3 (Puma): Mm00519268_m1; Pmaip1 (Noxa): Mm00451763_m1; Bad: Mm00432042_m1; Bid: Mm00477631_m1; Bcl-2: Mm00477631_m1; Bcl2l1 (Bcl-xL): Mm00437783_m1; Bcl2l2 (Bcl-w): Mm03053297_s1; Bax: Mm00432051_m1; Bmf: Mm00506773_m1; B2m: Mm00437762_m1. Samples were run on the 7500 Fast Real-Time PCR System (Applied Biosystems). Data was analyzed with the ExpressionSuite software using the ∆∆CT method with B2m as the housekeeping gene and age-matched LCKCRE neg Bimfl/fl mice for the reference samples.
BH3 profiling
BH3 profiling was performed as described previously [28, 29]. The basis of this assay was to determine the mechanism of apoptosis regulation in T cells. BH3 profiling on intact cells uses BH3-only peptides (prepared using solid-phase peptide synthesis) and digitonin, which selectively permeabilizes the cellular plasma membrane and thereby exposes intact intracellular mitochondria to the peptide treatment. Mitochondrial outer membrane permeabilization (MOMP), and loss of the transmembrane mitochondrial potential, is measured through changes in fluorescence intensity of mitochondrial fluorescent probes. Briefly, cells were stained for CD4 and CD8 for 30 minutes, washed, and incubated with 75 µM total peptide (NOXA + BAD comprised 37.5 µM each) with 20 µg/mL oligomycin (Sigma) and 0.002% digitonin for 60 minutes. DMSO and carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (Sigma) were used as negative and positive controls respectively. Cells were stained for an additional 30 minutes with 450 nM rhodamine123 (Sigma) and analyzed via flow cytometry. Titration of BIM BH3 peptide was used to determine overall apoptotic priming in individual cells because of its ability to bind to all multidomain anti- and pro-apoptotic BCL-2 proteins. % Depolarization caused by each BH3-peptide was calculated using the following formula: % Depolarization = (1 − (sample-FCCP)/(DMSO-FCCP)) × 100.
Statistical analysis
A one-way ANOVA test was used to evaluate statistically significant changes in young, middle, and old mice. For all other comparisons, an unpaired Student t test was performed and data are expressed as the means ± SEM. Plots were created using Prism (GraphPad Software). Expression data was depicted by RQ Min/Max with a 95% confidence level using the ExpressionSuite Software (Thermo Fisher/Life Technologies). Statistical significance was defined as p < 0.05.
Results
Loss of BIM in T cells results in lymphocytosis that normalizes over time
Previous work has confirmed BIM as a master BH3-only regulator of T and B cell homeostasis [11, 18]. To explore how loss of BIM affects the lymphocyte compartment during the natural lifespan, we analyzed the consequences of T and B cell-specific deletions of BIM in LCKCRE and CD19CRE Bimfl/fl animals, respectively. Complete blood counts (CBCs) from the peripheral blood of these animals were analyzed in young (6–12 weeks), middle-aged (15–39 weeks) and old-aged (≥ 40 weeks) mice. Loss of BIM in T cells (LCKCRE Bimfl/fl) resulted in a significant elevation of total white blood cells in young mice as previously described [11, 18]. However, this lymphocytosis normalized to levels equivalent to wild type (WT) mice by middle age and was maintained past 40 weeks of life (Fig. 1a). In contrast, CD19CRE Bimfl/fl mice developed a lower level of lymphocytosis when young, as previously reported, that remained relatively unchanged through old age (Fig. 1b) [23]. The changes in peripheral blood lymphocyte counts in LCKCRE Bimfl/fl mice were also reflected in the absolute cell number from spleens and thymi indicating that the changes measured were not isolated to peripherally circulating T cells (Fig. 1c, d). While the absolute number of lymphocytes was increased in young LCKCRE Bimfl/fl mice, the relative percentage of B cells and T cells were similar to those of wild-type animals (Supplemental Fig. 1). These data point to a BIM-independent compensatory mechanism responsible for lymphocyte normalization in LCKCRE Bimfl/fl mice over time.
Fig. 1.
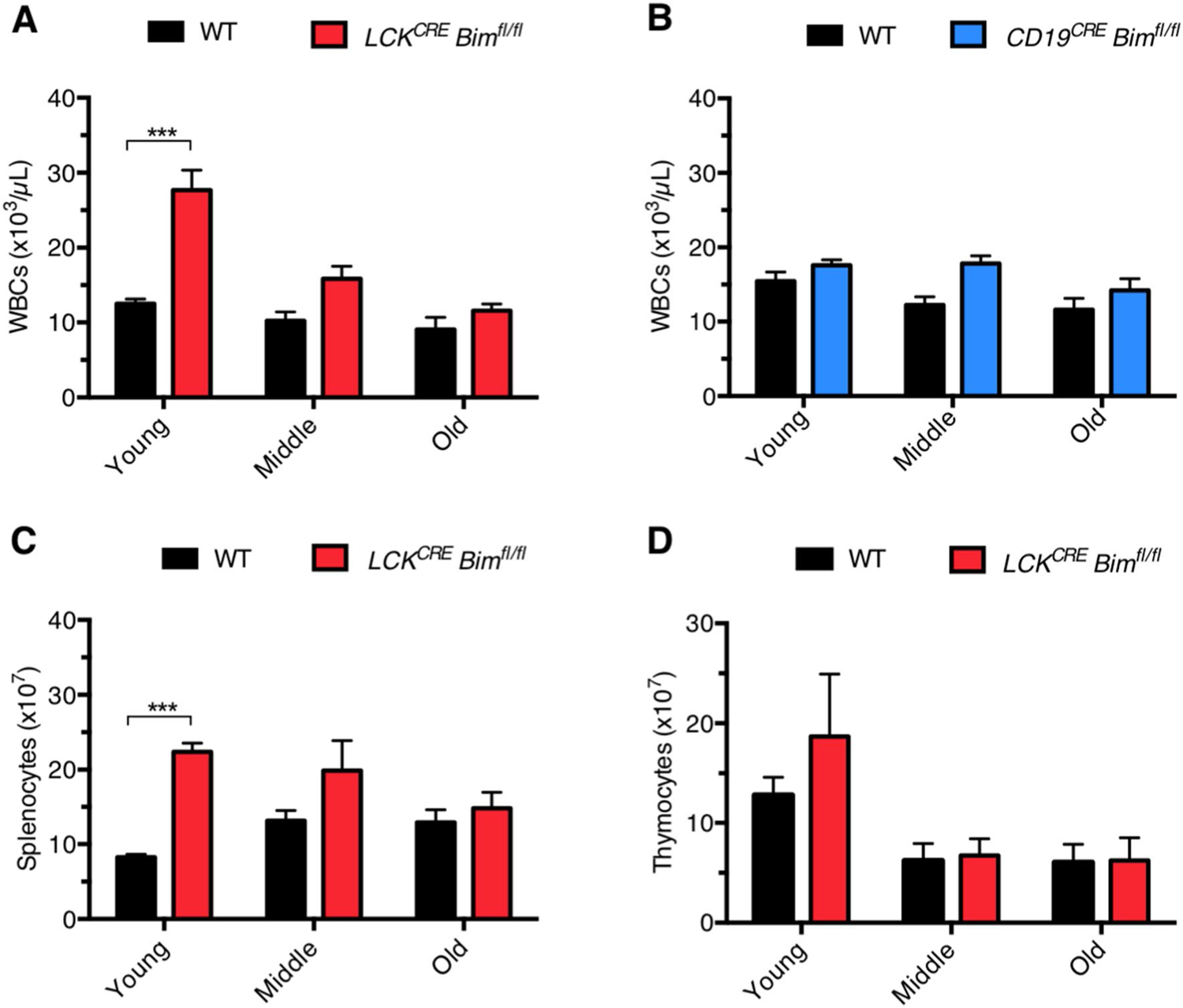
T cell-specific deletion of Bim leads to lymphocytosis early in life that normalizes as mice age. a CBC analysis was performed on peripheral blood from WT and LCKCREBimfl/fl and b CD19CRE Neg and CD19CRE Pos (referred to hereafter as CD19CRE) Bimfl/fl mice at the indicated ages. The peripheral blood lymphocytosis and normalization was also reflected in absolute cell numbers of c splenocytes and d thymocytes isolated from LCKCRE Bimfl/fl animals. Young: 6–12 weeks of age, middle: 15–39 weeks of age, old: ≥ 40 weeks of age. LCKCRE Bimfl/fl n ≥ 14, CD19CRE Bimfl/fl n ≥ 22, and WT n ≥ 5 for each age group. Data represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Thymocyte development remains abnormal in LCKCREBimfl/fl mice as they age
Normal thymocyte development is impaired in young mice with either global or hematopoietic cell-specific Bim deletion [11, 12, 18, 30]. Such animals harbor low numbers of CD4+CD8+ double positive (DP) thymocytes and increased numbers of double negative (DN) and single positive (SP) CD4+ and CD8+ thymocytes [18, 30]. To determine if T cell contraction was due to changes in thymopoiesis in LCKCRE Bimfl/fl animals, CD4 and CD8 expression was analyzed on thymocytes from young, middle-aged, and elderly mice. Thymocyte development remained similarly abnormal in young and aged LCKCRE Bimfl/fl mice, as reflected by reduced DP thymocytes and increased DN and SP cells (Fig. 2a, b). These proportions remained stable through all age groups (Fig. 2c–f). The overall similar decline in absolute thymocyte numbers in both WT and LCKCRE Bimfl/fl mice was likely due to age-dependent thymic involution (Fig. 2g–j) [31]. Our results are consistent with previous data showing that 6–8 week-old native Bim−/− mice have lower absolute number of thymocytes and a higher content of DN and SP CD4+ and CD8+ thymocytes compared to WT animals [11, 32, 30]. Therefore, normalization of thymopoiesis does not appear to be responsible for peripheral WBC normalization in LCKCRE Bimfl/fl mice as animals age.
Fig. 2.
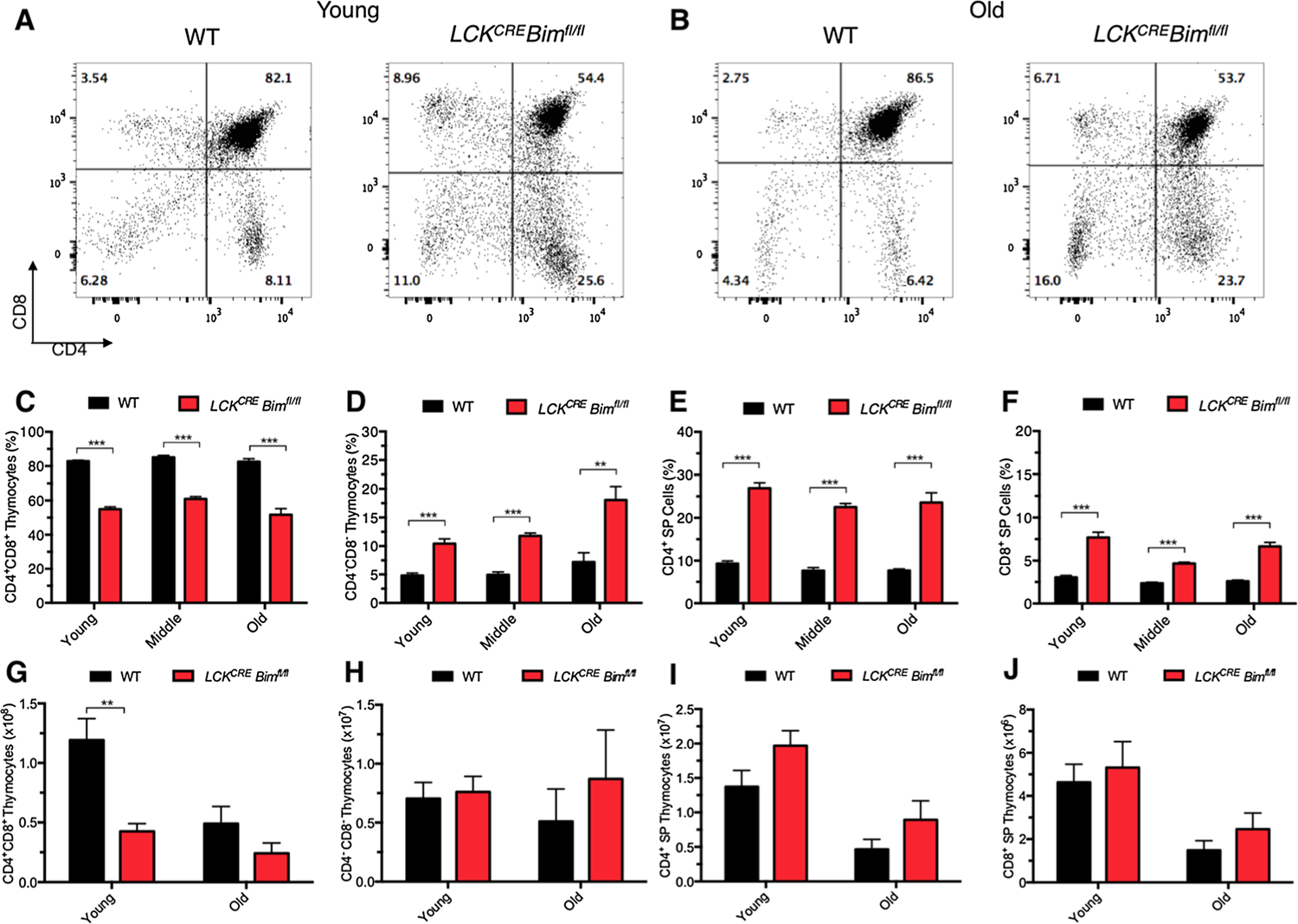
Thymocyte development remains abnormal as LCKCRE Bimfl/fl mice age. Thymocytes were analyzed from WT and LCKCRE Bimfl/fl mice at the indicated age groups. Representative flow plots of a young WT and LCKCRE Bimfl/fl mice, as well as b old WT and LCKCRE Bimfl/fl mice. Although thymus size decreases over time, the percentage of c DP CD4+CD8+, d DN CD4−CD8−, e SP CD4+, and f SP CD8+ thymocytes continues to reflect abnormal thymopoiesis of BIM-deficient cells. Absolute numbers of g DP CD4+CD8+, h DN CD4−CD8−, i SP CD4+, and j SP CD8+ thymocytes are shown below. LCKCRE Bimfl/fl and WT n ≥ 4 for each age group. Data represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Mature T cell numbers normalize in aged LCKCREBimfl/fl mice
Given there were no significant changes in thymopoiesis in young versus aged LCKCRE Bimfl/fl mice, we next turned our analysis to determine if splenic T cell numbers normalized over time instead. The proportions of CD4+ and CD8+ splenic T cells were lower in LCKCRE Bimfl/fl mice compared to age-matched WT controls until they reached old age, at which point the proportions equalized (Fig. 3a–c). Despite lower proportions of T cells in the spleens of young LCKCRE Bimfl/fl animals, there were significantly higher absolute numbers of CD4+ and CD8+ splenic T cells that normalized by middle age (Fig. 3d, e). These findings are consistent with the peripheral blood data from these animals (Fig. 1). Naïve T cells (CD44low CD62Lhigh) decreased over time to a greater extent in LCKCRE Bimfl/fl mice compared to WT mice (Fig. 4a, f). Conversely, LCKCRE Bimfl/fl mice accumulated more CD4+ effector/memory T cells (CD44high CD62Llow) as has been previously described in animals with global Bim deficiency (Fig. 4b,f) [32]. Like naïve CD4+ T cells, naïve CD8+ T cells decreased over time to a greater extent in the spleens of LCKCRE Bimfl/fl mice compared to WT mice (Fig. 4c, f). However unlike CD4+ T cells, effector/memory CD8+ cell percentages and absolute numbers were equivalent in all age groups (Fig. 4d, f). Importantly, spleens from young WT and LCKCRE Bimfl/fl mice had equivalent percentages of both CD4+ and CD8+ naïve and effector memory cells. It was only as mice aged that differences became apparent (Fig. 4a–d). In contrast to conventional T cells, splenic CD4+FOXP3+ T cells increased in proportion in all age groups but more so in LCKCRE Bimfl/fl mice. Interestingly, while Treg absolute numbers were more elevated in LCKCRE Bimfl/fl mice, their absolute numbers remained stable throughout life (Fig. 4e, g). These data support previous studies demonstrating the importance of BIM in Treg ontogeny and the normal accumulation of Tregs as mice age [33–35]. Decreased levels of BIM in old versus young WT Tregs allows them to naturally accumulate and preferentially survive in older animals though chronic stimulation with IL-2 [33]. Taken together, young LCKCRE Bimfl/fl mice had higher overall numbers of CD4+ and CD8+ cells but equivalent proportions of naïve and effector memory cells when compared to WT controls. As the global numbers of CD4+ and CD8+ cells normalized, LCKCRE Bimfl/fl mice accumulated memory CD4+ T cells but not memory CD8+ T cells. Both WT and LCKCRE Bimfl/fl animals developed an increased percentage of CD4+ Tregs throughout their lives but maintained overall constant Treg numbers.
Fig. 3.
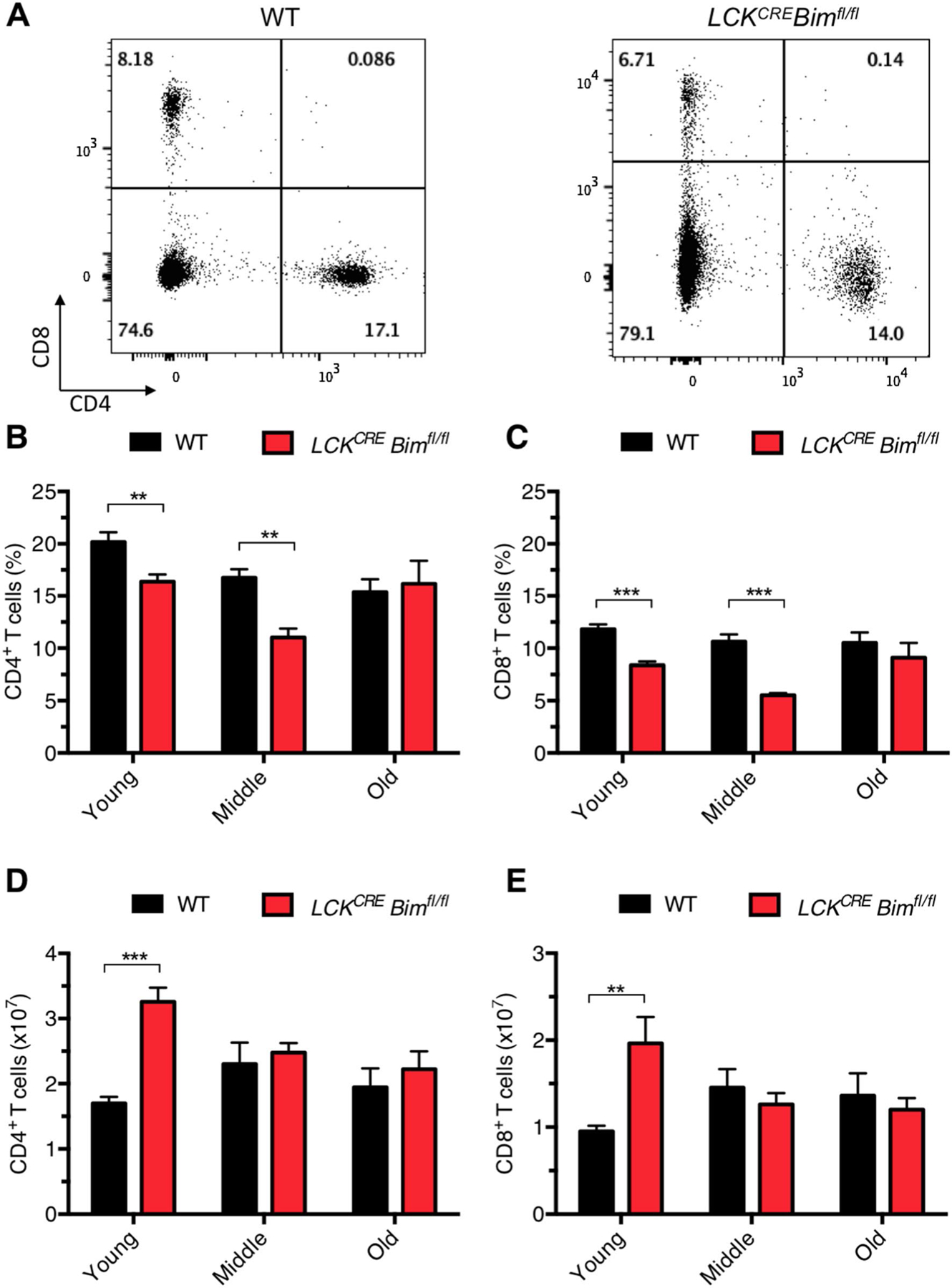
Mature CD4+ and CD8+ T cell numbers normalize over time in LCKCRE Bimfl/fl mice. a Representative flow plots of CD4+ and CD8+ splenic T cells from WT and LCKCRE Bimfl/fl mice. b, c While the ratios of mature splenic CD4+ and CD8+ T cells from WT and LCKCRE Bimfl/fl mice do not change as mice age the d, e absolute numbers of both decrease to WT levels in older aged mice. LCKCRE Bimfl/fl and WT n ≥ 3 for each age group. Data represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Fig. 4.
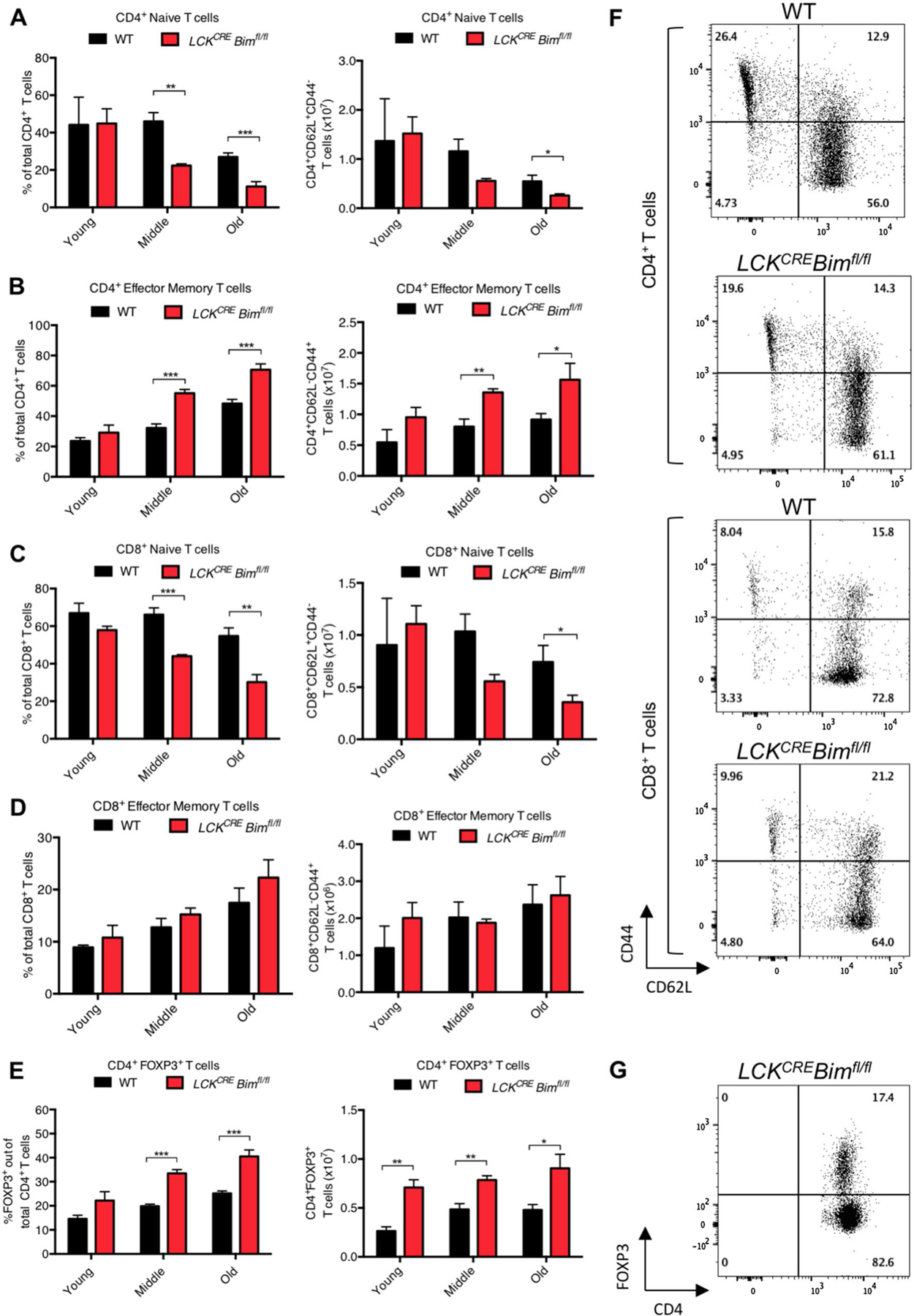
There is a decreased proportion of naïve CD4+ and CD8+ T cells and accumulation of CD4+ effector/memory and regulatory T cells in aged LCKCRE Bimfl/fl mice. Old LCKCRE Bimfl/fl mice have reduced proportions of a, b naïve (CD44lowCD62Lhigh) CD4+ T cells and accumulation of effector memory (CD44highCD62Llow) CD4+ T cells. c, d Old LCKCRE Bimfl/fl mice have reduced proportions of naïve CD8+ T cells but similar numbers of effector memory CD8+ T cells compared to WT controls. e Both WT and LCKCRE Bimfl/fl mice increase their percentage CD4+FOXP3+ regulatory Tregs as they age. Absolute numbers of Tregs are greater in LCKCRE Bimfl/fl animals compared to WT controls but their overall numbers remain relatively constant throughout their lives. Representative flow plots of f CD4+, CD8+ and g FOXP3+ CD4+ Treg populations from young mice are shown to the right of each bar graph. LCKCRE Bimfl/fl and WT n ≥ 4 for each age group. Data represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Mature CD4+ and CD8+ LCKCRE Bimfl/fl T cells have apoptotic sensitivity profiles reflecting partial BCL-2 family compensation
One possibility for the normalization of overall T cell numbers in LCKCRE Bimfl/fl mice is that they were able to compensate for the lack of BIM and normalize their apoptotic resistance tendencies. CD4+ and CD8+ T cells from the spleens of young WT and LCKCRE Bimfl/fl mice were tested to determine if they were increasingly resistant to apoptotic stimuli given their longer, albeit variable, existence out of the thymus. This would allow for detection of BCL-2 family compensation in T cells that have survived longer than the ~ 5 days it takes for a SP T cell to mature and emigrate from the thymus [36, 37]. The young age group was chosen to ensure equal percentages of naïve and effector memory T cells. CD4+ and CD8+ T cells from young LCKCRE Bimfl/fl mice were challenged to undergo cell death following treatment with cytokine deprivation (BIM, PUMA, BAD, HRK reliant death trigger), ionomycin (BIM reliant), phorbol 12-myristate 13-acetate (PMA; PUMA reliant), and etoposide (BID reliant) [38, 18]. BIM deficient CD4+ and CD8+ T cells were equally resistant to cytokine deprivation and etoposide treatment as age-matched controls (Fig. 5a, b). While LCKCRE Bimfl/fl CD4+ T cells were equally sensitive to ionomycin as WT controls, LCKCRE Bimfl/fl CD8+ T cells were more resistant (Fig. 5a, b). The converse was measured against PMA treatment. These data indicate that mature CD4+ and CD8+ T cells lacking BIM were differentially sensitive against cell death triggers. We hypothesized that different BCL-2 family compensation mechanisms may underlie such distinctive responses in these cells. As such, we would expect CD4+CD8+ DP thymocytes to retain their apoptotic resistance patterns over time as thympoiesis remained stably abnormal in LCKCRE Bimfl/fl mice as they aged. As expected, DP thymocytes from LCKCRE Bimfl/fl mice were more resistant to all treatments compared to DP thymocytes from WT control animals (Fig. 5c).
Fig. 5.
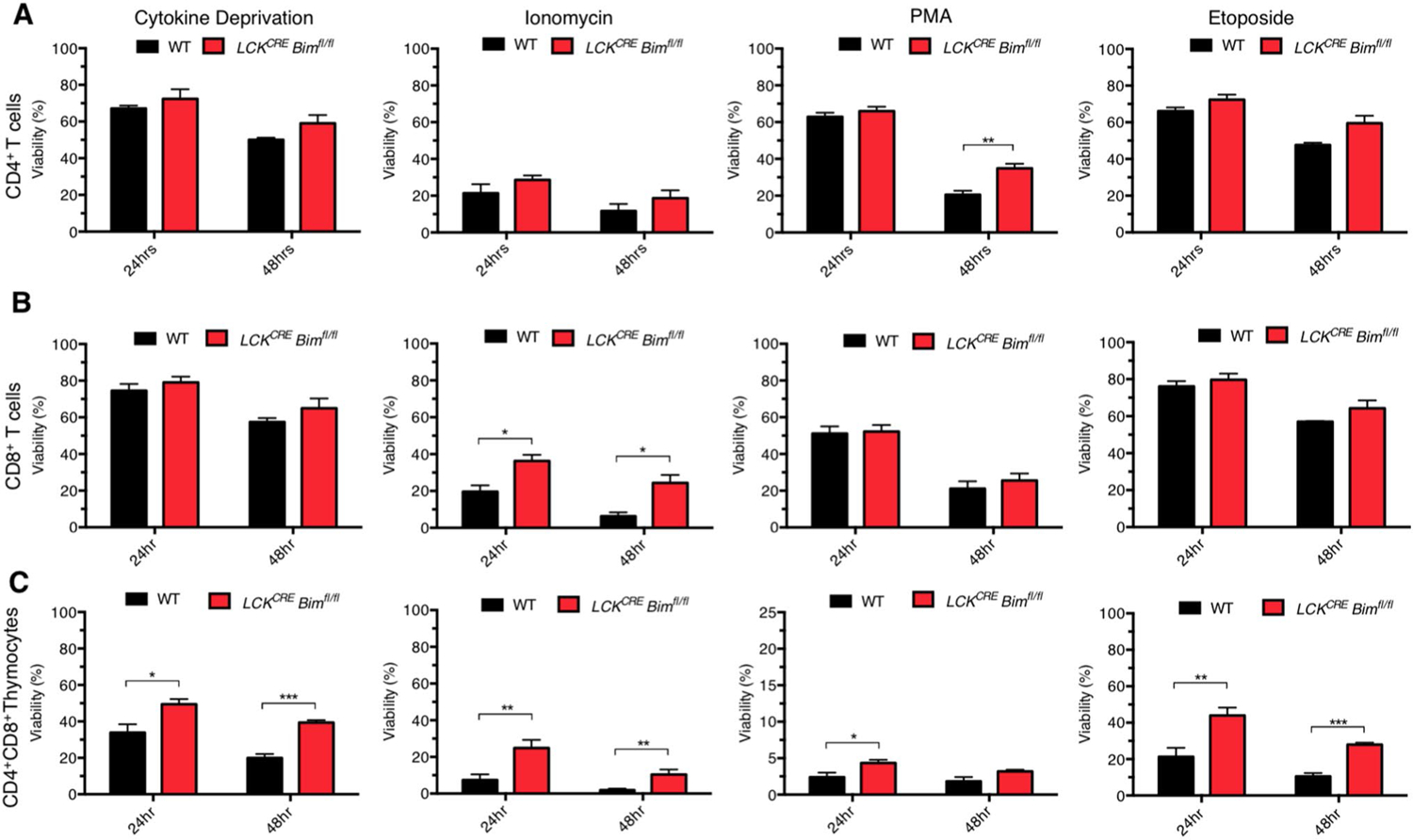
CD4+ and CD8+ T cells from LCKCRE Bimfl/fl mice were differentially sensitive to apoptotic stimuli compared to age-matched WT controls. a CD4+, b CD8+ splenic T cells, and c CD4+CD8+ thymocytes from young mice were cultured for 24 and 48 h in the absence of cytokines, 1 µg/mL ionomycin, 4 ng/mL PMA, or 1 µM etoposide. Viability was assessed by Annexin V/PI staining with Annexin Vneg PIneg cells considered viable. n ≥ 4 for each age group and genotype. Data represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
One possible explanation for our cell death results is that CD4+ and CD8+ T cells may have distinct patterns of BCL-2 family member expression in response to loss of BIM. Compared to age-matched WT controls, CD4+ splenic T cells from young LCKCRE Bimfl/fl mice demonstrated a significant increase in the expression of Bmf (~ 5× fold), A1 (~ 2× fold), Puma (~ 2× fold) and Noxa (~ 1.5× fold) (Fig. 6a). CD8+ splenic T cells from young LCKCRE Bimfl/fl mice displayed a similar pattern of BCL-2 family expression changes apart from lacking upregulation of Noxa. Despite differences in the ratio of naïve:effector cells in older animals, Puma and Noxa transcripts continued to increase in CD4+ and CD8+ splenic T cells from older LCKCRE Bimfl/fl mice (Fig. 6b). Bmf remained elevated in older CD4+ and CD8+ splenic T cells albeit to a lower extent than those from young animals. Notably, the changes in BCL-2 family mRNA expression were largely confined to BH3-only proteins. CD4+ and CD8+ splenic T cells had minimally increased expression (~ 1.5–2× fold) of the anti-apoptotic BCL-2 protein A1 (Fig. 6a, b). Consistent with their apoptotic resistance (Fig. 5c), BCL-2 family expression patterns in CD4+CD8+ DP thymocytes from young and old WT mice were not appreciably different confirming that BH3-protein compensation occurred over time in mature T cells and not in the thymus (Supplementary Fig. 2a). Importantly, thymocytes from young and old LCKCRE Bimfl/fl mice had persistent, and equivalent, deletion of Bim (Supplementary Fig. 2b).
Fig. 6.
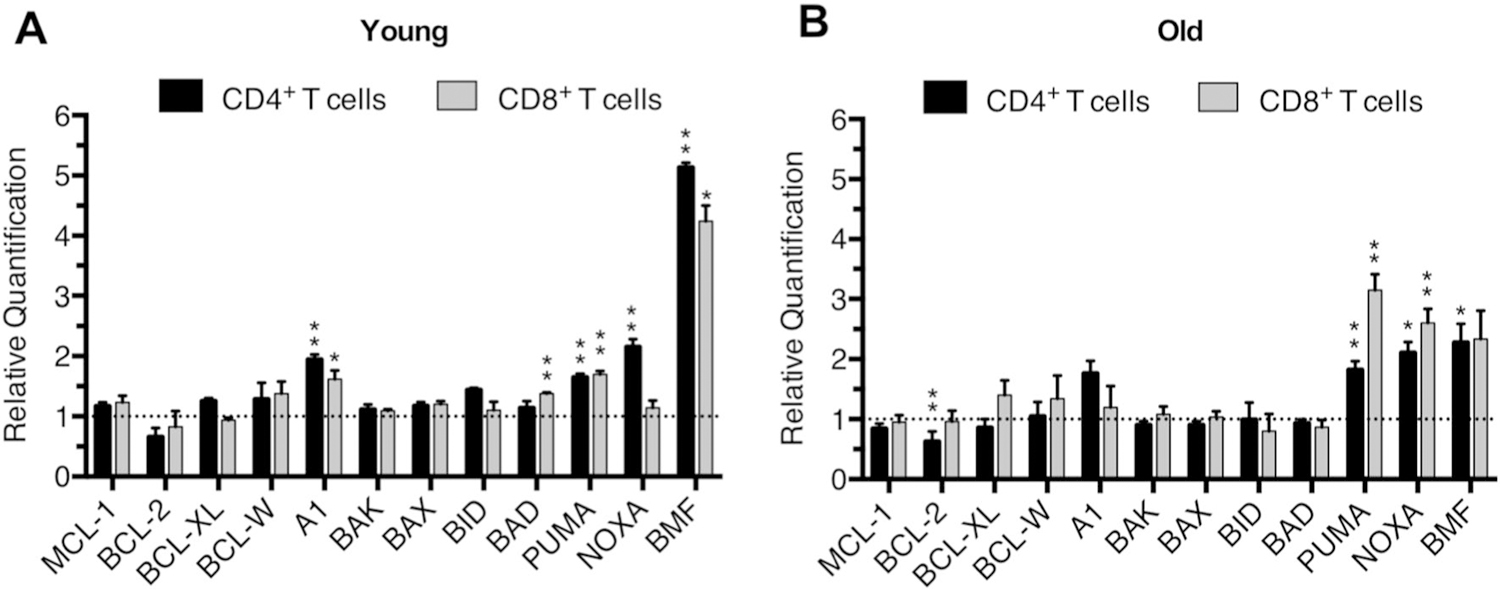
CD4+ and CD8+ T cells from LCKCRE Bimfl/fl mice upregulate expression of other BH3-only proteins in response to loss of BIM. mRNA expression levels of BCL-2 proteins from CD4+ and CD8+ splenic T cells from a young and b old LCKCRE Bimfl/fl mice. Age-matched WT mice were used as a reference sample. Expression was normalized using B2M and analyzed using the ∆∆CT method. n ≥ 3 for young and n ≥ 4 for old age group and genotype. Significance was compared to age-matched reference samples (normalized to 1, dashed line) Data represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 as determined by calculating RQ Min/Max with a 95% confidence level using the ExpressionSuite Software
The loss of BIM in T cells led to global changes in BH3-only mRNA expression but little alteration in anti-apoptotic BCL-2 protein transcript. This would predict that anti-apoptotic dependency patterns were similar in T cells from young LCKCRE Bimfl/fl and WT animals despite clear changes in BH3-only protein profiles and partial normalization in cell death sensitivity (Figs. 5 and 6). In this regard, loss of BIM would have been compensated by other BH3-only proteins to regulate T cell homeostasis as has been measured with respect to functional upregulation of BCL-2 in response to loss of MCL-1 [21]. Such a quantitative model would predict that other BH3-only proteins are able to functionally compensate for the loss of BIM. To test this, we utilized BH3-profiling, a technique in which death domains of select BH3-only proteins with focused BCL-2 anti-apoptotic affinities are intracellularly introduced to determine the anti-apoptotic protein dependency patterns of individual cells (Fig. 7a) [39, 40, 28, 29]. Supporting our gene expression analyses, mitochondrial depolarization measurements from individual CD4+ and CD8+ splenic T cells as well as CD4+CD8+ thymocytes from LCKCRE Bimfl/fl and WT animals showed no difference in BCL-2 family anti-apoptotic protein dependency (Fig. 7b). Despite this result, challenge with apoptotic stimuli found Bim deficient T cells to be variably more resistant to cell death (Fig. 5). To determine if this cell death resistance was also reflected at the level of the mitochondrion, BIM BH3 titration was used to compare apoptotic priming to the intrinsic apoptotic pathway in CD4+and CD8+ splenic T cells and CD4+CD8+ thymocytes from LCKCRE Bimfl/fl and WT animals. Mitochondria within T cells from LCKCRE Bimfl/fl mice were progressively resistant to depolarization following treatment with decreasing doses of BIM BH3 (Fig. 7c). Akin to our cell death data, these results suggest that T cells lacking BIM upregulate other BH3-only members and that these proteins partially compensate for apoptotic sensitivity in mature T cells lacking BIM.
Fig. 7.
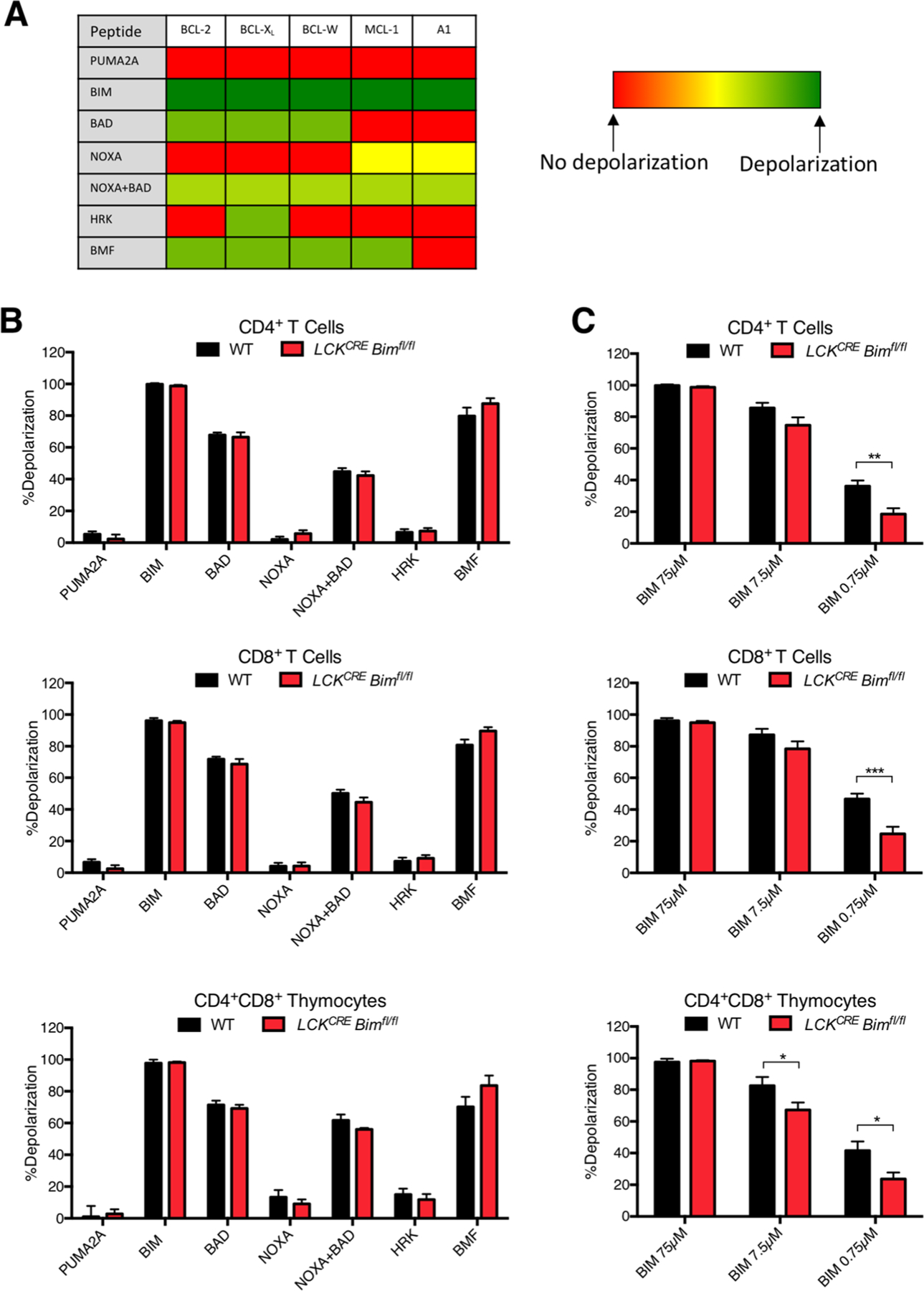
T cells lacking BIM do not shift their anti-apoptotic dependency and remain less primed to die compared to WT T cells. a Table of BH3 peptides used for BH3 profiling and expected mitochondrial depolarization patterns. b There were no significant changes in mitochondrial depolarization as a result of any of the peptide treatments in mature CD4+ and CD8+ splenic T cells, or CD4+CD8+ thymocytes. c To measure the level of apoptotic priming, cells were treated with decreasing concentrations of the BIM BH3 peptide. Cells from LCKCRE Bimfl/fl mice were less primed to die compared to WT controls. n ≥ 4 for each genotype. Data represented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Understanding the roles that BCL-2 family proteins play in the regulation of immune cell viability and function has become increasingly important as the clinical use of highly-specific small molecule and peptide BH3-mimetics increases. The role of BCL-2 anti-apoptotic proteins in regulating death and survival of various lymphocyte populations has been widely studied [41, 21]. Less well characterized however is the role that BH3-only proteins play in long-term immune homeostasis of these same populations. In this report, we have begun to characterize how the long-term loss of Bim in T cells affects total lymphocyte numbers and T cell apoptotic sensitivity over time. We and others confirm that BIM is the master BH3-only regulator of T cell homeostasis, as its deletion leads to abnormal thymocyte development, elevated white blood cell counts, and resistance to apoptotic stimuli [18]. These phenotypes are recapitulated in mice with deletion of Bim restricted to T cells. Importantly, the elevation in peripheral lymphocytes in LCKCRE Bimfl/fl mice normalized as they aged suggesting the existence of T cell-specific BCL-2 family compensatory mechanism(s) as a result of BIM loss. Our study thereby supports the fundamental importance of BIM in T cell apoptotic homeostasis and shows that, unlike BCL-2 anti-apoptotic proteins, BH3-only proteins are unable to fully functionally replace BIM in T cells [42]. Therefore, our work corroborates that of others pointing to the notion that BH3-only proteins, like anti-apoptotic BCL-2 proteins, can contribute at least somewhat quantitatively to the survival of T cells [41, 21].
Global, conditional hematopoietic (VAV-CreER), and T cell-specific (LCKCRE Bimfl/fl, as shown here) deletion of BIM results in abnormal maturation of thymocytes, including an increase of DN thymocytes and increased time in the DN phases [11, 12, 18, 19]. If compensatory mechanisms leading to white count normalization occurred in the thymus, aged LCKCRE Bimfl/fl mice would have been expected to develop an increasingly normal distribution of DN and DP thymocytes. However, thymocyte development remained abnormal in aged animals, pointing to a normalization mechanism that occurred over a longer period of time than that needed for a mature T cell to exit the thymus. To determine this fully, a model system that efficiently and stably deletes BIM in all mature post-thymic T cells would have to be employed. However, to our knowledge, such a model is not currently available.
While CD4+ and CD8+ splenocyte numbers approached those of WT animals over time, there was an increased percentage of CD4+ T cells that were FOXP3+ in both WT and LCKCRE Bimfl/fl animals as they age (Fig. 4e). BIM is a critical regulator of Treg levels and partially mediates their accumulation in aged mice through decreased BIM expression [33, 35]. Treg accumulation may rely predominantly on BIM rather than anti-apoptotics as Tregs from older animals have decreased expression of both BCL-2 and MCL-1, the major partners of BIM in T cells [21, 43]. Tregs from aged mice have increased suppressive capacity compared to Tregs from young mice, suggesting that the increased proportion of Tregs may be a contributing factor to the normalization of lymphocyte counts in aged mice [44]. We found that Treg accumulation was more robust in LCKCRE Bimfl/fl mice but did not increase significantly with age compared to WT animals. Perhaps the accumulation of Tregs in LCKCRE Bimfl/fl animals is partially responsible for the normalization of CD4+ and CD8+ splenic T cells deficient in Bim. However, a presumed natural rise in Tregs was unable to normalize WBC in 20–25 week aged CD19CRE Bimfl/fl or EµCycD1CD19CRE Bimfl/fl animals indicating a T cell-specific phenomenon [23]. Additionally, there were equivalent CD4+ T cells in older mice from both groups (Fig. 3d, e), suggesting that the mechanism for WBC normalization does not depend, at least fully, on Tregs. The direct role that BIM plays in Treg-specific maintenance and function continues to be an active area of investigation.
We observed a consistent transcript upregulation in other BH3-only proteins in response to loss of BIM. This likely played a role in lymphocyte number and cell death sensitivity normalization as there were no significant differences in anti-apoptotic expression or BH3 profiling between splenic T cells from LCKCRE Bimfl/fl and WT animals (Figs. 6 and 7). This would suggest that a qualitative compensation mechanism takes either time or cell death pressure to develop. The exception was CD8+ T cells, which remained significantly resistant to ionomycin suggesting that there are fundamental differences in how CD4+ and CD8+ T cells respond to the loss of BIM. This distinction may reflect previously unresolved discrepancies in cell death sensitivity profiles and BCL-2 family compensation in non-malignant T cells from patients treated with BH3-mimetics which result in disruption of BIM bound to various anti-apoptotic BCL-2 proteins as their primary mechanism of action [40, 45, 46].
This study, and that of others, supports the notion that the full functionality of BIM in lymphocytes is irreplaceable by other BH3-only proteins. There may also exist different levels of BH3 compensation in T cells following the loss of BIM. This is also reflected in work showing that mice expressing BIM protein with altered BH3-defined specificities (BimBad, BimPuma, BimNoxa) show partial, yet incomplete, WBC normalization compared to mice lacking Bim [42]. DP thymocytes from these animals showed partial in vitro normalization to cell death stimuli. Mature CD4+ and CD8+ T cells were not examined nor was BimBmf. The supremacy of BIM for initiation of apoptosis extends to its capacity to directly activate BAX and BAK to a higher degree than other potential direct activators, such as BID and PUMA [47, 10]. According to this model, the BH3 region of these proteins led to “very substantial” apoptosis but “maximal” apoptosis was only manifested by BIM [42]. This idea is supported by our data and mechanistically by the ability of BIM BH3 peptide to fully depolarize mitochondria when used in BH3 profiling, and thus serve as a positive control, independent of a cell’s anti-apoptotic dependency patterns [40, 48, 28].
While there were slightly different levels of BH3-only protein upregulation in CD4+ and CD8+ T cells lacking Bim, the genes involved were identical, perhaps indicating a conserved T cell mechanism of BH3 protein cell death control (Fig. 6). Specifically, there was increased transcription of Puma and Noxa, and most particularly, Bmf. Several studies have suggested BIM and BMF may have evolved from a common ancestry with shared transcriptional control and overlapping functions and regulatory programs [8, 49]. Both proteins are normally sequestered by the cytoskeleton and global deletion of either leads to defects in the immune system, although Bmf deletion leads to a more dramatic effect in B cells [50, 51]. Like loss of BIM, BMF deficiency also leads to lymphocyte accumulation through a cell autonomous mechanism and results in increased B cell accumulation compared to T cells [50]. Bim−/−Bmf−/− double knockout mice develop thymocytes more resistant than those from single knockout mice to a number of agents, including steroids and HDAC inhibition [50, 51]. However, apoptotic resistance in mature SP T cells was completely BIM dependent [51]. Additionally, combined loss of BIM and BMF led to a compounded increase in B cells but T cell numbers were not increased above single Bim−/− animals, suggesting a redundant mechanism of apoptotic control in B cells and not T cells. These studies support our observation that normal T cell death regulation and numbers are predominantly regulated by BIM. Given their functional overlap, it is conceivable that BMF compensates for loss of BIM in aged LCKCRE Bimfl/fl animals. T cell-specific genetic regulation and possible function for BMF has not been previously reported. We also cannot discount a combined cooperative compensation by multiple BH3 proteins in the absence of BIM that are able to control some, but not all, aspects of cell death.
Identification of how BCL-2 proteins compensate for functional or deletional loss of one another may have important clinical implications. This study provides new insight into the redundancy and fluidity of the BH3 subset of the BCL-2 family of proteins in T cells. It will be essential to learn more about how these proteins are able to compensate for one another in response to long-term pressure such as encountered during prolonged treatment with BH3 mimetics.
Supplementary Material
Acknowledgements
This work was supported by research funds from the National Institutes of Health: NCI K08-CA151450 (to J.L.L.) and the Comer Children’s Hospital Development Board, University of Chicago (to J.L.L). We would like to thank Loren Walensky for initial critical review of our manuscript.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10495-020-01593-6) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of Interest The authors declare no conflict of interests.
References
- 1.Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281(5381):1305–1308 [DOI] [PubMed] [Google Scholar]
- 3.Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15(1):49–63. 10.1038/nrm3722 [DOI] [PubMed] [Google Scholar]
- 4.Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116(2):205–219 [DOI] [PubMed] [Google Scholar]
- 5.Ludwig LM, Nassin ML, Hadji A, LaBelle JL (2016) Killing two cells with one stone: pharmacologic BCL-2 family targeting for cancer cell death and immune modulation. Front Pediatr 4:135. 10.3389/fped.2016.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW (2011) BH3-only proteins: orchestrators of apoptosis. Biochim Biophys Acta 1813(4):508–520. 10.1016/j.bbamcr.2010.11.024 [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17(3):393–403. 10.1016/j.molcel.2004.12.030 [DOI] [PubMed] [Google Scholar]
- 8.Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang Y, Ren D, Kim H, Takeda S, Reyna DE, Chan PM, Ganesan YT, Liao CP, Gavathiotis E, Hsieh JJ, Cheng EH (2015) An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol 17(10):1270–1281. 10.1038/ncb3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17(4):525–535. 10.1016/j.molcel.2005.02.003 [DOI] [PubMed] [Google Scholar]
- 10.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2010) BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330(6009):1390–1393. 10.1126/science.1190217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A (1999) Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286(5445):1735–1738 [DOI] [PubMed] [Google Scholar]
- 12.Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A (2002) BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415(6874):922–926. 10.1038/415922a [DOI] [PubMed] [Google Scholar]
- 13.Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, Strasser A (2003) Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J Exp Med 198(7):1119–1126. 10.1084/jem.20030411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernandez JB, Newton RH, Walsh CM (2010) Life and death in the thymus–cell death signaling during T cell development. Curr Opin Cell Biol 22(6):865–871. 10.1016/j.ceb.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renault TT, Chipuk JE (2013) Getting away with murder: how does the BCL-2 family of proteins kill with immunity? Ann N Y Acad Sci 1285:59–79. 10.1111/nyas.12045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sionov RV, Vlahopoulos SA, Granot Z (2015) Regulation of Bim in health and disease. Oncotarget 6(27):23058–23134. 10.18632/oncotarget.5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strasser A (2005) The role of BH3-only proteins in the immune system. Nat Rev Immunol 5(3):189–200. 10.1038/nri1568 [DOI] [PubMed] [Google Scholar]
- 18.Herold MJ, Stuchbery R, Merino D, Willson T, Strasser A, Hildeman D, Bouillet P (2014) Impact of conditional deletion of the pro-apoptotic BCL-2 family member BIM in mice. Cell Death Dis 5:e1446. 10.1038/cddis.2014.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hildeman D, Jorgensen T, Kappler J, Marrack P (2007) Apoptosis and the homeostatic control of immune responses. Curr Opin Immunol 19(5):516–521. 10.1016/j.coi.2007.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shukla S, Saxena S, Singh BK, Kakkar P (2017) BH3-only protein BIM: an emerging target in chemotherapy. Eur J Cell Biol 96(8):728–738. 10.1016/j.ejcb.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 21.Carrington EM, Zhan Y, Brady JL, Zhang JG, Sutherland RM, Anstee NS, Schenk RL, Vikstrom IB, Delconte RB, Segal D, Huntington ND, Bouillet P, Tarlinton DM, Huang DC, Strasser A, Cory S, Herold MJ, Lew AM (2017) Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ 24(5):878–888. 10.1038/cdd.2017.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biswas S, Shi Q, Matise L, Cleveland S, Dave U, Zinkel S (2010) A role for proapoptotic Bax and Bak in T-cell differentiation and transformation. Blood 116(24):5237–5246. 10.1182/blood-2010-04-279687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katz SG, Labelle JL, Meng H, Valeriano RP, Fisher JK, Sun H, Rodig SJ, Kleinstein SH, Walensky LD (2014) Mantle cell lymphoma in cyclin D1 transgenic mice with Bim-deficient B cells. Blood 123(6):884–893. 10.1182/blood-2013-04-499079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ (2005) Essential role of BAX,BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci USA 102(32):11272–11277. 10.1073/pnas.0504783102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ (2003) Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 426(6967):671–676. 10.1038/nature02067 [DOI] [PubMed] [Google Scholar]
- 26.LaBelle JL, Hanke CA, Blazar BR, Truitt RL (2002) Negative effect of CTLA-4 on induction of T-cell immunity in vivo to B7–1+, but not B7–2+, murine myelogenous leukemia. Blood 99(6):2146–2153 [DOI] [PubMed] [Google Scholar]
- 27.Pitter K, Bernal F, Labelle J, Walensky LD (2008) Dissection of the BCL-2 family signaling network with stabilized alpha-helices of BCL-2 domains. Methods Enzymol 446:387–408. 10.1016/S0076-6879(08)01623-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murase K, Kim HT, Bascug OR, Kawano Y, Ryan J, Matsuoka K, Davids MS, Koreth J, Ho VT, Cutler C, Armand P, Alyea EP, Blazar BR, Antin JH, Soiffer RJ, Letai A, Ritz J (2014) Increased mitochondrial apoptotic priming of human regulatory T cells after allogeneic hematopoietic stem cell transplantation. Haematologica 99(9):1499–1508. 10.3324/haematol.2014.104166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan J, Letai A (2013) BH3 profiling in whole cells by fluorimeter or FACS. Methods 61(2):156–164. 10.1016/j.ymeth.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutcheson J, Perlman H (2007) Loss of Bim results in abnormal accumulation of mature CD4-CD8-CD44-CD25-thymocytes. Immunobiology 212(8):629–636. 10.1016/j.imbio.2007.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Hsu HC, Grizzle WE, Stockard CR, Ho KJ, Lott P, Yang PA, Zhang HG, Mountz JD (2003) Cellular mechanism of thymic involution. Scand J Immunol 57(5):410–422 [DOI] [PubMed] [Google Scholar]
- 32.Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, Michalak E, Strasser A, Villunger A (2006) Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med 203(13):2939–2951. 10.1084/jem.20061552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chougnet CA, Tripathi P, Lages CS, Raynor J, Sholl A, Fink P, Plas DR, Hildeman DA (2011) A major role for Bim in regulatory T cell homeostasis. J Immunol 186(1):156–163. 10.4049/jimmunol.1001505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harpaz I, Bhattacharya U, Elyahu Y, Strominger I, Monsonego A (2017) Old mice accumulate activated effector CD4 T cells refractory to regulatory T cell-induced immunosuppression. Front Immunol 8:283. 10.3389/fimmu.2017.00283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tischner D, Gaggl I, Peschel I, Kaufmann M, Tuzlak S, Drach M, Thuille N, Villunger A, Jan Wiegers G (2012) Defective cell death signalling along the Bcl-2 regulated apoptosis pathway compromises Treg cell development and limits their functionality in mice. J Autoimmun 38(1):59–69. 10.1016/j.jaut.2011.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCaughtry TM, Wilken MS, Hogquist KA (2007) Thymic emigration revisited. J Exp Med 204(11):2513–2520. 10.1084/jem.20070601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yates AJ (2014) Theories and quantification of thymic selection. Front Immunol 5:13. 10.3389/fimmu.2014.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Happo L, Strasser A, Cory S (2012) BH3-only proteins in apoptosis at a glance. J Cell Sci 125(Pt 5):1081–1087. 10.1242/jcs.090514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9(5):351–365. 10.1016/j.ccr.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 40.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A (2007) BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12(2):171–185. 10.1016/j.ccr.2007.07.001 [DOI] [PubMed] [Google Scholar]
- 41.Carrington EM, Tarlinton DM, Gray DH, Huntington ND, Zhan Y, Lew AM (2017) The life and death of immune cell types: the role of BCL-2 anti-apoptotic molecules. Immunol Cell Biol 95(10):870–877. 10.1038/icb.2017.72 [DOI] [PubMed] [Google Scholar]
- 42.Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O’Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, Bouillet P (2009) The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 186(3):355–362. 10.1083/jcb.200905153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, Hildeman DA (2007) Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med 204(7):1665–1675. 10.1084/jem.20070618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, Belkaid Y, Chougnet C (2008) Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol 181(3):1835–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, Jin S, Smith M, Xue J, Zhang H, Oleksijew A, Magoc TJ, Vaidya KS, Albert DH, Tarrant JM, La N, Wang L, Tao ZF, Wendt MD, Sampath D, Rosenberg SH, Tse C, Huang DC, Fairbrother WJ, Elmore SW, Souers AJ (2015) Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med 7(279):279ra240. 10.1126/scitranslmed.aaa4642 [DOI] [PubMed] [Google Scholar]
- 46.Renault TT, Elkholi R, Bharti A, Chipuk JE (2014) B cell lymphoma-2 (BCL-2) homology domain 3 (BH3) mimetics demonstrate differential activities dependent upon the functional repertoire of pro-and anti-apoptotic BCL-2 family proteins. J Biol Chem 289(38):26481–26491. 10.1074/jbc.M114.569632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lopez J, Bessou M, Riley JS, Giampazolias E, Todt F, Rochegue T, Oberst A, Green DR, Edlich F, Ichim G, Tait SW (2016) Mitopriming as a method to engineer Bcl-2 addiction. Nat Commun 7:10538. 10.1038/ncomms10538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2(3):183–192 [DOI] [PubMed] [Google Scholar]
- 49.Delgado M, Tesfaigzi Y (2013) BH3-only proteins, Bmf and Bim, in autophagy. Cell Cycle 12(22):3453–3454. 10.4161/cc.26696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Labi V, Erlacher M, Kiessling S, Manzl C, Frenzel A, O’Reilly L, Strasser A, Villunger A (2008) Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. J Exp Med 205(3):641–655. 10.1084/jem.20071658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Labi V, Woess C, Tuzlak S, Erlacher M, Bouillet P, Strasser A, Tzankov A, Villunger A (2014) Deregulated cell death and lymphocyte homeostasis cause premature lethality in mice lacking the BH3-only proteins Bim and Bmf. Blood 123(17):2652–2662. 10.1182/blood-2013-11-537217 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.