

Abstract
Adolescence marks a key developmental window during which emotion dysregulation increases, along with risk for the onset of anxiety and other affect-related pathologies. Although emotion dysregulation and related pathologies normatively decline during the transition into adulthood, this does not occur for a sizable minority of individuals. Finally, sex differences in anxiety emerge during adolescence, with females developing a 2-fold increase in risk relative to males. Unfortunately, a neurobiological model of the mechanisms that cause these changes during adolescence has yet to be proposed. In the present work, we first provide brief reviews of relevant literature. Next, we outline a dual-mechanism model focused on (i) the influence of pubertal testosterone on key emotion-regulation circuitry (i.e., orbitofrontal cortex-amygdala coupling) and (ii) myelination of the fiber bundles connecting such circuitry (i.e., uncinate fasciculus). The proposed model offers a set of specific, testable hypotheses that will hopefully spur much needed cross-disciplinary research.
Keywords: Anxiety, Adolescence, Puberty, Testosterone, Myelination, Sex Differences, Amygdala, Orbitofrontal Cortex, Uncinate Fasciculus
1.0. Introduction
Adolescence marks a unique developmental window wherein individuals learn to navigate complex social landscapes, individuate from caregivers, and cultivate the self-regulatory capacity necessary for long-term goal pursuit. A key mechanism underlying such growth is a unique increase in behavioral and neural plasticity, relative to both children and adults, which allows for a more nuanced adaptation to the environment (Tottenham & Galván, 2016). In fact, the relative duration of adolescence is significantly extended in humans, compared to other primates (D.J. Miller et al., 2012), paralleling the greater intricacy of the adult environments that human adolescents must learn to navigate.
Unfortunately, the mechanisms that allow for such flexible and complex development also leave adolescents with increased vulnerability for a variety of negative health outcomes. Although adolescents are at their peak physical health, overall morbidity and mortality rise 200–300% during this stage (Dahl, 2004). Consequently, this extended period of plasticity can confer resilience to or risk for pathology, depending on the environments encountered and the vulnerabilities carried by the individual into adolescence. For example, enhanced attention to threat may be optimal if an individual is frequently in hazardous contexts but prove maladaptive in the long term if that individual shifts into safer environments.
To obtain insight into the specific mechanisms that confer this risk during adolescence, it is useful to identify the processes shared among the various forms of psychopathology that emerge during this period. Specifically, the peak ages of onset for anxiety, depression, and risk-taking behavior (e.g., substance use, reckless driving, unprotected sex) all occur during adolescence. Central to these pathologies are deficits in the regulation of emotion. For example, difficulty regulating reactivity to a potential threat is a central facet of anxiety disorders (Dillon et al., 2014). Anxiety disorders remain one of the most common forms of mental illness (lifetime prevalence = 28%; Kessler et al., 2005) and are estimated to be the sixth leading cause of disability in both developed and developing countries (Baxter et al., 2014). Similarly, unipolar depression, which is the 2nd leading cause of disability worldwide (Baxter et al., 2014), involves both disturbances in the downregulation of threat-related responses and hypo-responsivity to reward (Dillon et al., 2014). Finally, over 70% of adolescent deaths each year are due to preventable risk-taking behaviors (Steinberg, 2008), and these behaviors are thought to result from deficient down-regulation of reward reactivity (Richards et al., 2012).
Anxiety, depression, and risk taking tend to decrease during the transition into young adulthood, thus evidencing a u-shaped timecourse in their prevalence across adolescence. However, in approximately half of those with adolescent pathology, anxiety becomes chronic and persists well into adulthood (Olino et al., 2010; Pine, 2007). Thus, the developmental period from early adolescence through young adulthood represents a critical time window for understanding (i) why pathology tends to onset during this time and (ii) why symptoms that are present during adolescence remit for some individuals but become chronic for others.
Sex differences in the diagnosis of anxiety and depression also arise during adolescence. In particular, rates are approximately equal pre-adolescence, become significantly higher in females during adolescence, and remain unbalanced throughout adulthood (Altemus et al., 2014). The emergence of these sex differences during adolescence further underscores the unique importance of this developmental window for addressing significant public health concerns.
It is perhaps not surprising that emotion-related pathology emerges during adolescence, given the tremendous maturation that occurs in affect systems during this period. As discussed in more detail in section 3.1, reactivity to both positively and negatively valenced stimuli increases during the transition from childhood into adolescence and then decreases during the transition into young adulthood. Importantly, these changes appear to be due, in part, to differences in the regulation of emotion rather than emotional reactivity alone. Thus, adolescence comprises a unique period of regulatory change, during which broader regulatory abilities show a steady linear improvement from childhood through adolescence and into adulthood, but normative maturation in emotion-regulation capacity evidences a u-shaped curve.
2.0. Current Paper
In the current paper, we attempt to address a set of key questions:
Why does emotion dysregulation tend to increase – both normatively and pathologically – with adolescence and decrease with emerging adulthood?
Why do some individuals develop pathological levels of emotion dysregulation (and related symptoms) during adolescence whereas others do not?
Why do some individuals fail to show a decline in emotion dysregulation (and related symptoms) during emerging adulthood (i.e., pathology becomes chronic)?
Why do sex differences in affect-related pathology emerge during adolescence?
In the following work, we first briefly review relevant literatures, then draw these threads together to propose a novel neuromaturational model to address the questions posed above. In particular, we focus on two potential mechanisms – pubertal hormones and axonal myelination. Note that, although the model proposed in section 4.0 is likely also relevant for depression and risk-taking, for the sake of simplicity and clarity we focus herein on anxiety-related pathology. Similarly, with regard to pubertal hormones, we focus on testosterone, with brief discussions of estradiol and dehydroepiandrosterone (DHEA).
3.0. Brief Literature Reviews
3.1. Developmental Course of Emotion-Regulation Capacity
Unlike most regulatory processes, that maintain a steady (linear) increase in capacity from childhood to young adulthood (Best & Miller, 2010), the capacity to regulate emotion appears to suffer a decrement during the transition into adolescence. In contrast, increases in the capacity to regulate emotion are not typically observed until individuals transition into young adulthood (S.P. Ahmed et al., 2015). In other words, dysregulation in emotion appears to peak during adolescence. Compared to both children and adults, adolescents exhibit increased reactivity to both appetitive and aversive stimuli across a number of behavioral, physiological, and neural measures. For example, mood sampling studies indicate that adolescents show greater emotional responsivity to both positive and negative events than adults (Larson et al., 1998; Larson et al., 2002; Weinstein & Mermelstein, 2007), with negative moods, in particular, increasing in prevalence from early adolescence until a peak at age 15 (Larson et al., 2002). Similarly, adolescents consistently show greater neural activation in nucleus accumbens (NAcc) in response to a variety of rewarding stimuli (for review, see Galvan, 2010). The evidence for increased reactivity to aversive stimuli is more limited but growing. For example, adolescents exhibit greater levels of startle reflex (Quevedo et al., 2009), which is linked to the defensive motivation system (Lang et al., 1990). Similarly, amygdala activation to aversive faces increases longitudinally from childhood into early adolescence (Spielberg et al., 2014).
As mentioned above, evidence suggests that changes in affect reactivity during adolescence are due, at least in part, to deficits in the capacity to regulate emotion. For example, fear extinction is impaired during adolescence in both human and rodents, whereas fear acquisition does not differ (for review, see Baker et al., 2014). A wealth of research has attempted to identify the mechanisms responsible for such regulation differences in adolescence, with limited success. One area of examination has been executive function, with inhibitory control being the most relevant for the present discussion. When examined in non-(explicitly) affective contexts (e.g., traditional go-nogo task), a linear trajectory is observed, with inhibitory capacity increasing steadily from childhood to adulthood (Best & Miller, 2010). Findings have been mixed when the stimuli to be ignored are affective in nature. For example, Tottenham et al. (2011) used an emotional variant of the go/no-go task with happy, fear, sad, and anger facial expressions in a sample with children (n=53, ages 5–12), adolescents (n=24, ages 13–18), and adults (n=23, ages 19–28). Successful regulation was defined as inhibition of a prepotent behavioral response in the context of irrelevant emotional information (i.e., facial expression). Using these criteria, a linear pattern of regulation capacity was observed, with emotion-related interference decreasing across age. However, this study used both (i) a wide age range of within groups and (ii) relatively few age groups, each of which may obscure m0re complex patterns of emotion regulation. For example, given that general inhibitory control contributes to affect-specific regulation (Hofmann et al., 2012), lumping early childhood (with very underdeveloped inhibitory control) with late childhood will bias the mean capacity downward. To address this problem, Cohen-Gilbert and Thomas (2013) used a more fine-grained age distribution across adolescence, with 5 groups (each with n=20): ages 11–12, 13–14, 15–16, 18–19, and 20–25. Results revealed that aversive stimuli were the most distracting (relative to neutral) in the 13–14 age range, compared to both younger and older age groups. A sex by emotion interaction was also observed in the 15–16 age group, wherein girls exhibited worse accuracy in the aversive condition, but boys did not. Thus, there is at least some evidence of a non-linear sex-dependent trajectory, with the highest levels of disinhibition observed in mid-adolescence (see Ahmed et al., 2015, for a more detailed review of this issue).
The use of explicit emotion-regulation strategies has also been examined in adolescence. The most well-studied strategy is reappraisal, whereby the meaning attached to a situation is altered in order to adjust the subsequent affective response (Ochsner & Gross, 2005). Overall, adolescents are less successful in using emotion-regulation strategies compared to both children and adults, whereas levels of emotional reactivity do not appear to differ. For example, Zimmermann and Iwaski (2014) examined the use of emotion regulation strategies in response to sadness, fear, and anger in a large sample (n=1,305) divided into nine age groups (ages 11, 13, 15, 17, 19, 22, 25, 29, and 50). The authors reported that children in mid-adolescence (ages 13–17) were least likely to use adaptive strategies (e.g., reappraisal) and support seeking compared to both earlier (i.e., 11) and later age groups (i.e., 19–50). Gullone et al. (2010) longitudinally examined changes in adaptive strategy use across a 2-year period in a large sample (n=1,128), with participants ranging in age from 9–15 at time 1. A similar non-linear pattern was observed for both within- and between-subject effects of reappraisal use, with use decreasing from late childhood to mid-adolescence. Finally, McRae et al. (2012) examined reported affect intensity in response to aversive stimuli after either no regulation or reappraisal in three age groups: 10–13, 14–17, and 18–23. Reappraisal was found to be least successful in lowering affect intensity in the 14–17 group, relative to both younger and older groups. Importantly, affect intensity in the no-regulation condition did not differ between groups, suggesting that these effects are specific to emotion regulation.
In summary, evidence garnered from a range of methodologies supports the presence of heightened emotional reactivity during adolescence, in comparison to both children and adults. Furthermore, mounting evidence suggests that regulation of emotion decreases as individuals enter adolescence and increases again during the transition into adulthood.
An interesting issue arises from these collective data, given the relative consistency across individuals in the trajectory of affect development (i.e., increased reactivity/decreased control). In particular, this consistency suggests that there is an evolutionary advantage conferred by this pattern of emotion regulation. However, it is not immediately clear why decreased regulatory capacity would be adaptive in adolescence. A full discussion of this issue is beyond the scope of this paper, but one hypothesis is that increased reactivity leads to engagement in the types of encounters that build skills necessary for independence (Casey et al., 2008).
3.2. Developmental Course of Anxiety
The developmental course of anxiety appears to be inversely related to the maturation of emotion-regulation capacity, with anxiety increasing during the transition into adolescence and decreasing during the transition into young adulthood. To explore the timecourse of anxiety disorders, we used data from the National Comorbidity Survey Replication reported in Kessler et al. (2005) to illustrate the middle 5o% age-of-onset distributions for individual anxiety disorders. Specifically, the horizontal bars in Figure 1 begin at the 25th percentile onset age (i.e., 25% of cases will have onset by this age) and end at the 75th percentile, and the background panels reflect the dominant hormonal events at that time: adrenarche (red) and gonadarche (purple) (see section 3.6 below for a discussion of these developmental windows). As illustrated, some disorders are most likely to onset after adolescence (e.g., generalized anxiety disorder), and some largely beforehand (e.g., separation anxiety). However, most anxiety disorders have large periods during adolescence when onset is likely. For example, 50% of individuals with social phobia have an onset between ages 8 and 15. Perhaps most importantly, the peak age-of-onset for any anxiety disorder is age 11.
Figure 1.
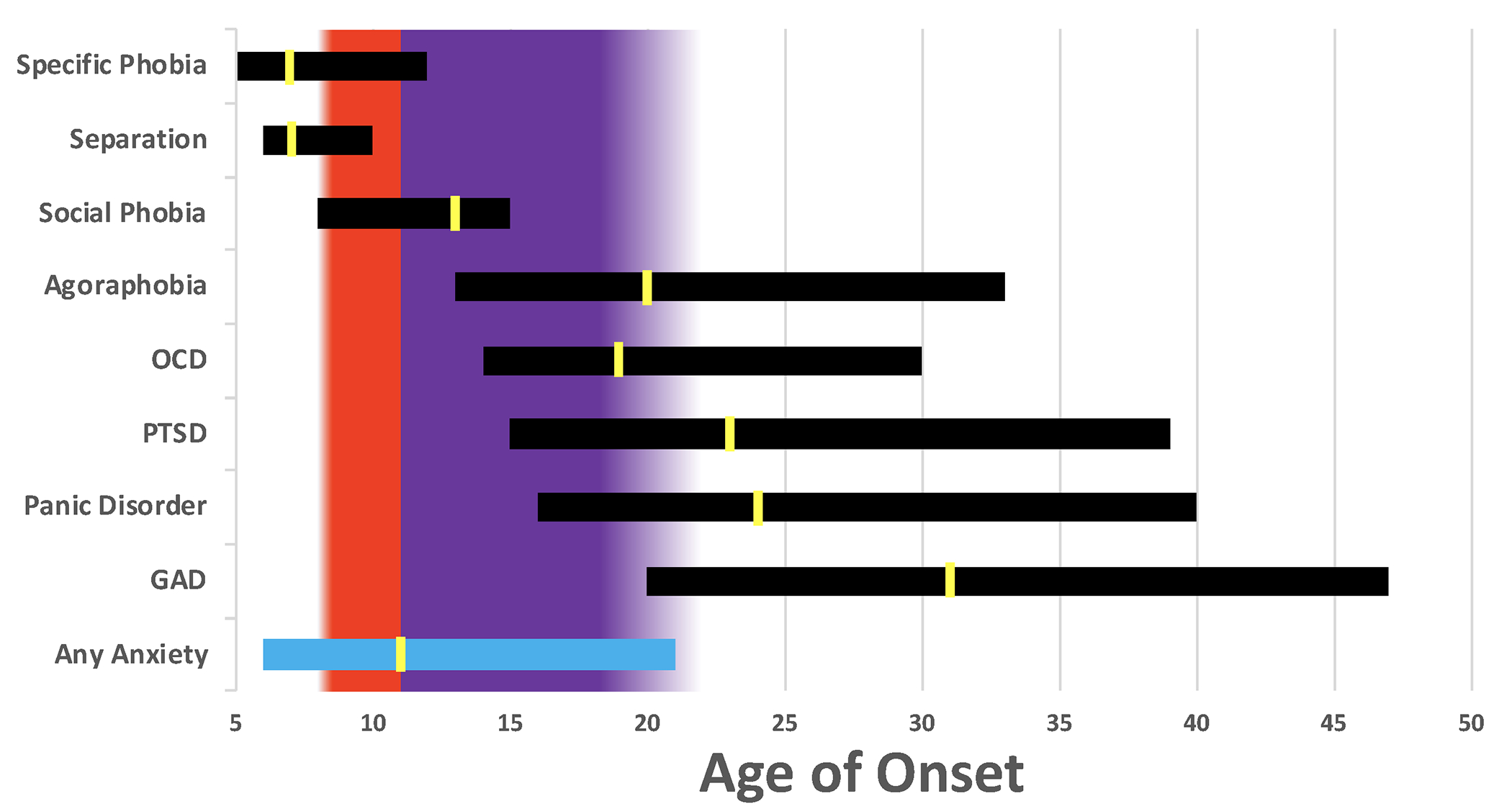
Age of Onset Distributions for Anxiety Disorders
Each horizontal bar represents the middle 50% age of onset distribution for each disorder based on the data presented in Kessler et al. (2005). Specifically, the bars begin at the 25th percentile onset age (i.e., 25% of cases will have onset by this age) and end at the 75th percentile. The yellow marks are located at the 50th percentile age of onset. The background panels reflect the dominant hormonal events at that time: adrenarche (red) and gonadarche (purple). OCD=obsessive compulsive disorder; PTSD=posttraumatic stress disorder; GAD=generalized anxiety disorder.
Although research has been more limited, a similar pattern emerges when anxiety is examined dimensionally. For example, Morin et al. (2011) examined longitudinal change in a general measure of anxiety (i.e., Beck Anxiety Inventory, Beck et al., 1988) in a large sample (n=1,370) across adolescence (ages 12–18) and performed mixture modelling to uncover groups of individuals with distinct timecourses of anxiety. Although the largest group of individuals (74% of the sample) evidenced consistently low anxiety across time, the next largest group (17%) showed increasing anxiety until about age 16, after which levels trended downwards. Though this study did not observe changes in anxiety across time in the majority of the sample, the measure used was designed to capture pathological levels of anxiety (Beck et al., 1988), and thus there may be a floor effect that prevented changes from being observed in a non-clinical population. Conway et al. (2017) examined longitudinal change in anxious arousal, a dimension reflecting physiological manifestations of anxiety (e.g., racing heart), in a large sample (n=604). This study found that anxious arousal is greater in adolescence (ages 16–17) relative to young adulthood (early 20s).
In summary, mounting evidence suggests that measures of anxiety tend to increase during adolescence and decrease during young adulthood, at least among those individuals who experience moderate to high levels of anxiety. Moreover, this idea is supported by solid evidence that, across a number of anxiety disorders, onset tends to occur predominantly during adolescence.
3.3. Sex Differences in the Developmental Course of Anxiety
Although pathological anxiety tends to onset during adolescence, this trend does not occur equally across both sexes. Indeed, a wealth of evidence indicates that rates of anxiety are approximately equal pre-adolescence, become significantly higher in females during adolescence, and remain so into adulthood (Altemus et al., 2014). For example, Costello et al. (2003) examined longitudinal changes in anxiety disorders in a large sample (n=1,420) ages 9–16. Increases in social phobia symptoms emerged over time in girls but not boys, and girls evidenced significantly more anxiety diagnoses than boys during this period. Finally, having one anxiety disorder connoted a greater risk for developing another anxiety disorder in girls but not boys. Similarly, Hale et al. (2008) examined longitudinal changes in symptoms of anxiety disorders in a large sample (n=1,318) ages 12–20. Generalized anxiety disorder (GAD) symptoms were initially similar across sexes and increased over time in girls, whereas boys remained stable. Social phobia symptoms were higher at age 12 in girls than boys and remained so across time. In addition, a close examination of the mean values in girls indicated that social phobia symptoms increased until mid-adolescence and subsequently decreased, whereas they remained flat in boys (although this quadratic interaction did not appear to be tested statistically). Finally, Nelemans et al. (2014) examined longitudinal changes in symptoms of anxiety disorders in a large sample (n=239) ages 11–22. Sex differences were observed in the developmental trajectories of both GAD and social phobia. In particular, girls began with higher GAD symptom levels, and these levels also increased at a faster rate in girls. Social phobia symptoms were similar pre-adolescence and decreased in boys in both mid-adolescence and emerging adulthood. Conversely, girls showed an increase in social phobia symptoms in middle adolescence, followed by a slow decrease in late adolescence, with levels remaining higher in girls than boys into emerging adulthood.
In an attempt to identify the particular developmental factors driving these differences, several studies have co-examined indices of pubertal development. For example, Patton et al. (1996) cross-sectionally examined symptoms of anxiety disorders in a large sample (n=2,525) ages 12–17. Anxiety was found to increase over adolescence, with girls showing higher levels throughout. Importantly, menarchal status was found to be the greatest predictor of anxiety in girls: girls who were >3 years post-menarche had a two-fold increase in risk for anxiety compared to girls <3 years from menarche. Similarly, Kaczkurkin et al. (2016) examined the relationship between pubertal stage and trait and state anxiety (controlling for age) in a large sample (n=875) ages 12–23. Results revealed that state anxiety levels were higher in both post- and mid-pubertal females, relative to males, but this difference was not evident pre-puberty. A similar pattern was observed for trait anxiety, although the sex difference was only significant for post-puberty in this case.
In summary, although there remains some discrepancy in the exact shape of the developmental trajectories, particularly when examining specific disorders, there is clear evidence that levels of anxiety increase in females during adolescence to a greater extent than males. Furthermore, mounting evidence specifically implicates the role of pubertal factors in these differences.
3.4. Emotion-Regulation Neural Circuitry
Given the availability of several excellent reviews of the neural circuitry supporting the production and regulation of emotion (e.g., Frank et al., 2014; Ochsner & Gross, 2005; Öner, 2018) we will briefly review only what is necessary to understand the model presented below in section 4.0. In particular, there is a wealth of evidence for the involvement of amygdala in determining the affective salience of a stimulus (Cunningham & Brosch, 2012), with threat-related stimuli producing reliable amygdala activation across both normative and pathological samples (Cisler & Koster, 2010). Importantly for the model proposed below, amygdala hyperactivation to threat is consistently observed across anxiety disorders (Duval et al., 2015). Furthermore, the magnitude of amygdala activation is correlated with symptom severity, such that hyperactivation actually decreases or even normalizes following successful treatment of anxiety disorders (Duval et al., 2015). Thus, identifying the mechanisms by which amygdala responses are regulated appears to be a particularly crucial step in the effort to understand the development of anxiety.
Orbitofrontal cortex (OFC) and nearby ventral and midline prefrontal structures appear particularly crucial for the regulation of amygdala, including both enhancement and suppression of amygdala responses (Roberts et al., 2007). For example, the magnitude of coupling between OFC and amygdala during emotion regulation predicts both the level of negative affect experienced post-regulation (Banks et al., 2007; Morawetz et al., 2017) and the extent to which physiological correlates of negative affect are observed (Lee et al., 2012). In fact, a recent meta-analysis identified consistent OFC-amygdala coupling during the regulation of emotion (Di et al., 2017). At the molecular level, one avenue by which amygdala is influenced by OFC and related prefrontal structures appears to be via activation of GABAergic (inhibitory) interneurons in amygdala (Quirk et al., 2003; Rosenkranz & Grace, 2002).
Among other functions, OFC maintains information about stimulus value, particularly with respect to current goals (vs. the mean value of a stimulus across time, which is represented in amygdala, basal ganglia, and other structures) (Rolls, 1999; Zald & Kim, 2001). For example, OFC activation to food-related stimuli is proportional to the level of satiation, with higher activation observed when participants are hungry than when satiated (Thomas et al., 2015). Furthermore, evidence indicates that OFC is involved in top-down regulation of amygdala in order to modulate amygdala responses with regard to current goals (for review, see Roesch & Schoenbaum, 2006). For example, patients with OFC lesions exhibit higher baseline amygdala activity and greater reactivity to aversive images (Motzkin et al., 2015). Importantly, functional coupling between amygdala and OFC is decreased in pathological anxiety (Duval et al., 2015), suggesting that OFC fails to down-regulate inappropriate amygdala responses, consistent with clinical descriptions of anxiety (e.g., PTSD flashbacks).
Anatomically, the uncinate fasciculus (UF) is a large white-matter bundle connecting amygdala/hippocampus with OFC/ventromedial prefrontal cortex (PFC) (Olson, Von Der Heide et al., 2015). In fact, individual differences in the maturational profile of UF have been proposed as key determinates in emotion-regulation behavior (e.g., reversal learning) (Olson et al., 2015). Interestingly, UF appears particularly relevant for emotion regulation in anxiety. Greater anatomical size and integrity of UF predicts reduced amygdala reactivity to negatively valenced faces, and reduced amygdala reactivity, in turn, predicts lower internalizing symptoms (e.g., anxiety) (Swartz et al., 2014). Furthermore, the size/integrity of UF is (i) reduced in both adults and adolescents with anxiety disorders (Hettema et al., 2012; Phan et al., 2009; Tromp et al., 2012, 2017, 2019; Tröstl et al., 2011) and (ii) inversely associated with trait anxiety in both rhesus monkeys (Tromp et al., 2017) and healthy adult humans (Baur et al., 2012; Tromp et al., 2017; Kim & Whalen, 2009). Thus, both structural and functional connections between OFC and amygdala appear central to anxiety development.
3.5. Developmental Course of Emotion-Regulation Neural Circuitry
Several excellent reviews of the developmental course of emotion regulation are available (e.g., S.P. Ahmed et al., 2015; Geise et al., 2014; Martin & Ochsner, 2016; Scherf et al., 2013). As such, we present only a brief review necessary for the model outlined below in section 4.o. Generally, development of subcortical structures (e.g., amygdala, striatum) during adolescence precedes maturation of PFC (Ahmed et al., 2015). For example, adolescents exhibit greater subcortical reactivity and similar PFC reactivity relative to children, whereas PFC reactivity in adolescents is weaker when compared to adults (Geise et al., 2014).
Across a number of studies, adolescents have demonstrated increased amygdala activation, compared to both children and adults, when viewing emotional faces and during emotion expression tasks (for review, see Scherf et al., 2013). For example, a longitudinal study found that amygdala activity (in response to emotional faces) increased during the transition from late childhood (age 10) to early adolescence (age 13) (Pfeifer et al., 2011). In addition, higher activation of amygdala in adolescents has been found during fear learning (Lau et al., 2011) and during emotional go/no-go (Hare et al., 2008). Anatomically, amygdala volume appears to follow quadratic pattern over ages 4–18 years, with peak volume occurring in mid-adolescence (Hu et al., 2013). With regard to the regulation of emotion in particular, larger amygdala volumes during adolescence predict worse emotion-regulation success (Pagliaccio et al., 2014).
Heightened amygdala reactivity in adolescence is thought to be due, in part, to less effective top-down regulation by PFC (Heller & Casey, 2016). One mechanism by which this may occur is delayed myelination of UF. Myelination of axon bodies facilitates saltatory conduction of action potentials, thus increasing communication efficiency (Sherman & Brophy, 2005). For example, higher indices of myelination along a tract are correlated with stronger functional connectivity between the endpoints of that tract (Cohen et al., 2008; Damoiseaux & Greicius, 2009), along with higher improved cognitive and social functioning (Fornari et al., 2007). In the central nervous system, myelination is carried out by oligodendrocytes, each of which myelinates several neighboring axons, thus also increasing structural stability by allowing axons to physically support one another (Baumann & Pham-Dinh, 2001).
Myelination occurs throughout development, with circuits myelinating at different times. Connections between PFC and subcortical structures, including UF, undergo a prolonged period of myelination stretching from late adolescence to the early 30s (peak myelination in the early 30s) (Lebel & Beaulieu, 2011). This protracted developmental window allows for a longer period of plasticity in these circuits and, thus, the potential for both finer adaptation to the environment and a longer window of vulnerability (Tottenham & Galván, 2016). Interestingly, protracted PFC-subcortical myelination may be unique to humans, as chimpanzees evidence adult-like levels of myelination in these circuits by sexual maturity (Miller et al., 2012). As mentioned in the previous section, UF integrity is reduced in those with anxiety disorders (Hettema et al., 2012; Phan et al., 2009; Tromp et al., 2012, 2017, 2019; Tröstl et al., 2011), and variation in these general measures of tract integrity (e.g., fractional anisotropy) reflects myelination, along with other factors (Mädler et al., 2008). Thus, there is evidence suggestive of a role for variation in UF myelination in the development of anxiety.
3.6. Pubertal Development
The beginning of adolescence is typically defined by the onset of puberty, or gonadarche to be more specific. Puberty consists of a suite of physiological and psychological changes that can be grouped into three independent sets of endocrine processes: adrenarche, gonadarche, and activation of the axial growth axis (for review, see Blakemore et al., 2010). Adrenarche typically occurs first (onset between 6–9 in girls, 7–10 in males) and involves the activation of the hypothalamic-pituitary-adrenal (HPA) axis. During this period, the adrenal glands dramatically increase the production of several androgens, including dehydroepiandrosterone (DHEA), which binds to the same androgen receptors as testosterone, albeit to a much weaker extent (Gao et al., 2005). Importantly, DHEA also serves as precursors to testosterone and dihydrotestosterone (DHT) and is thus produced at relatively high levels compared to other steroid hormones, although a large proportion of DHEA is converted to testosterone/DHT rather than being released. Increases in adrenal androgens contribute to the development of secondary sex characteristics (e.g., underarm/pubic hair), although adrenarche is not necessary to achieve sexual maturation (Havelock et al., 2004). Interestingly, adrenarche is found only in higher-order primates (i.e., humans, old world monkeys), suggesting that it is a relatively recent evolutionary developmental process (Havelock et al., 2004).
Gonadarche comprises a second set of processes that begins with the activation of the hypothalamic-pituitary-gonadal axis (onset between 8–14 in girls, 9–15 in boys) and ends with sexual maturity, including maturation of the reproductive organs. Development of the ovaries and testes includes dramatic increases in the production of estrogen and testosterone, respectively, which have widespread neural and behavioral effects, discussed in more detail in section 3.6.2 below. Importantly, the onset of gonadarche is associated with reduced behavioral plasticity in some domains (e.g., language) and increased plasticity in others (e.g., affect, motivation) (Piekarski et al., 2017). Finally, a third set of processes involves activation of the axial growth axis by growth hormone, which leads to increases in height and changes in body composition (e.g., increased muscle mass in males), and typically occurs at ages 12 and 14 for girls and boys, respectively.
Although we will not delve into detail here, it is important to briefly mention the different ways in which puberty is measured. Correspondence between measures is moderate (Shirtcliff et al., 2009), and there are likely systematic differences in what is measured by each. Pubertal measures can be grouped into (i) physical examination, (ii) self-report, and (iii) hormonal assay. Physical examination by a trained clinician is considered the gold-standard measure of current pubertal development as a whole (in comparison to specific components of puberty) (Dorn et al., 2006). Such exams assay the Tanner stage of an individual, ranging from stage 1 (no development) to stage 5 (adult level), and are based upon visible secondary sexual characteristics. Self-report measures largely also seek to measure puberty as a whole and are less invasive that physical examination. These measures include the commonly used Pubertal Development Scale (PDS) (Petersen et al., 1988) questionnaire, along with methods in which adolescents view images of each Tanner stage and identify the one closest to their own level of development (Morris & Udry, 1980). Finally, hormonal assays measure in vivo levels of gonadal (e.g., testosterone, estrogens) and other (e.g., DHEA) steroids. Thus, hormonal assays measure specific components of the various processes involved in puberty, in contrast to the measures discussed above which capture a more general picture of development.
3.6.1. Puberty and Anxiety
A number of studies have examined the relationship between anxiety and current global pubertal status (over and above age), with somewhat mixed findings (for review, see Reardon et al., 2009). For example, when examining a sample of female adolescents (n=754), Hayward et al. (1992) found a 2-fold increase in the likelihood of panic attacks for each increase in Tanner stage. Also examining female adolescents (n=971), and Huerta and Brizuela-Gamino (2002) found that pubertal status was linked to higher trait anxiety. In a mixed-sex sample (n=867), Ge et al. (2006) found that pubertal status was linked to higher GAD symptoms in both boys and girls. Finally, Susman and colleagues (1991) found that puberty status was associated with higher anxiety in a sample of boys (n=56). However, using a mixed-sex sample (n=534), Canals et al. (1992) found no links between pubertal status and trait anxiety and a small effect in the opposite direction for state anxiety in girls. Thus, greater overall pubertal status appears to be related to higher anxiety levels, although there is some evidence to the contrary, potentially due to variation in the pubertal and anxiety measures used.
More consistency has been observed in the impact of individual differences in the timecourse of puberty, including both onset and tempo (how quickly puberty occurs) (Ge & Natsuaki, 2009). For example, earlier sexual maturity – due to earlier onset and/or faster tempo – is associated with higher rates of depression (Galvao et al., 2014) and eating disorders (Klump, 2013), particularly in girls. More relevant to the present work, several studies have found that earlier pubertal onset (i.e., ‘precocious puberty’) is related to higher levels of anxiety in girls. For example, Sonis et al. (1985), using a cross-sectional design (n=33), found that early onset was linked to higher levels across several types of anxiety symptoms (e.g., somatic, obsessive). Using a longitudinal design (n=1,463), Hayward et al. (1997) found that early onset was linked to an increase in the risk of developing panic symptoms. In fact, this relationship was more robust than that found for depression or eating disorders. Unfortunately, little attention has been focused on the relationship between pubertal onset and anxiety in boys (for review, see Reardon et al., 2009). In summary, although there is fairly consistent evidence that early pubertal onset is linked to greater levels of anxiety in girls, the exact mechanisms at play remain unknown.
3.6.2. Pubertal Hormones
There has been a recent shift in our understanding of the type of impact that steroid hormones enact in the brain during different developmental windows. In particular, the influential ‘organization/activation’ hypothesis (Phoenix et al., 1959) distinguished between ‘organizational’ and ‘activational’ effects on the brain (for review, see Schulz & Sisk, 2016). Organizational effects were thought to be permanent and occur only during sensitive periods, whereas activational effects are (potentially) transient, do not require a sensitive period, and require organization of the brain circuitry to have occurred first. In the original formation of this hypothesis, the organizational effects of steroid hormones were thought to occur only pre- and perinatally, whereas pubertal exposure was posited to have only activational effects on already ‘organized’ circuits. Soon after this proposal, it was recognized that organizational effects can occur outside of sensitive periods, given that lasting changes can be enacted at later points in the lifespan (Arnold & Breedlove, 1985; McCarthy & Arnold; 2011). However, the idea that adolescent exposure to steroid hormones was only activational remained dominant until animal work by Sisk and others provided methodological advances that allowed for prenatal and pubertal effects of such hormones to be parsed apart. In particular, this and related work has provided evidence that steroid hormones contribute to the organization of affective systems during puberty, and this impact is qualitatively different than the effect of the same hormones post-puberty (Schulz & Sisk, 2016). Moreover, there is mounting evidence that sensitivity to the organizational impact of such hormones declines with age, such that exposure will have a larger effect the earlier in adolescence that it occurs. For example, early developing boys show better visuospatial abilities than late developers, and early developing girls show better verbal ability (Beltz & Berenbaum, 2013).
Testosterone is one of the primary androgens produced by the testes and was originally named for its male generating effects on the body. Sex hormone-binding globulin (SHBG) is a carrier protein in the blood that allows testosterone produced in the testes to reach its requisite androgen receptor in various target tissues, including the brain. Once released, the “free” testosterone can activate the androgen receptor to induce a number of important rapid and genomic effects on the cellular target. Testosterone can be converted to dihydrotestosterone (DHT), which also activates the androgen receptor and, similar to testosterone, has potent biological effects on the brain and male reproduction. Testosterone can also be produced by the adrenal glands, though at much lower levels than what is produced by the testes. In contrast, the majority of DHEA, which can also act as a weak androgen, is produced in the adrenal glands, with a small percentage produced via the gonads.
Although typically thought of as a “sex hormone”, testosterone is not male-specific. Females also produce androgens, including testosterone, throughout puberty and adulthood, though to a much lesser extent than males (Burger, 2002; Liening et al., 2010). In fact, ovaries produce testosterone as a precursor to estrogens, but those androgens are typically converted to estrogens by the presence of aromatizing enzymes in the ovaries. Interestingly, testosterone can also be converted to estrogen in other tissues that contain the enzyme aromatase, like the brain, where testosterone can have masculinizing effects but via its actions at the estrogen receptor (McCarthy, 2008).
Androgen receptors are found throughout the brain, in many regions necessary for modulating sex-specific behaviors, social behaviors, reward and fear, as well as executive functions (see Moraga-Amaro et al., 2018, for a review on novel techniques used to image steroid-hormone receptors in the brain). As a result, testosterone can alter the development of many neural structures via its actions at the androgen receptors during puberty. As mentioned above, Sisk and colleagues provided many of the findings that allowed for prenatal and pubertal effects of testosterone on the developing brain to be dissociated. They found that, similar to the first (prenatal) increase in testosterone, pubertal increases in testosterone can also organize neural structures and facilitate the development of sex-typical behaviors. Based on their findings, they proposed that testosterone-mediated organization of neural circuits during adolescence is a “refinement” of the neural circuits that were initially differentiated prenatally. For example, during puberty, testosterone can further modulate the proliferation and survival of cells that were sexually differentiated prenatally (i.e., the sexually dimorphic nucleus of the preoptic area and the anteroventral periventricular nucleus). Although many of these structures are important for sex behavior in rodents and humans, testosterone can also influence the development of more general brain regions during adolescence. For example, the posterodorsal medial amygdala (MePD) is significantly larger in male than in female rodents as the result of increased proliferation of new cells, specifically astrocytes, in this region of amygdala during puberty (E.I. Ahmed et al., 2008; Cooke et al., 2007; Johnson et al., 2012). MePD also projects to the bed nucleus of the stria terminalis (BNST), where there is a robust sex difference in size between males and females in both rodents and primates. The size of BNST increases throughout adolescence in males, but not females, thus producing a male-biased sexual dimorphism in BNST size (Chung et al., 2002). Interestingly, preadolescent treatment with testosterone increases the volume of the MePD and BNST to adult-typical size, though this same treatment is ineffective at inducing earlier male sexual behaviors (Schulz et al., 2009). Thus, the adolescent brain is uniquely sensitive to the organizational effects of pubertal testosterone, even at earlier ages. Taken together, these data suggest that testosterone can have important effects on the development of the adolescent brain, not only for the maturation of structures important for sex behaviors, but also regions crucial for emotion regulation.
3.6.3. Pubertal Hormones and Anxiety in Humans
Very few studies have examined the relationship between pubertal hormones and anxiety in humans which, along with methodological concerns, renders this small body of work relatively uninformative. Susman et al. (1991) examined a sample of boys (ages 10–14, n=56) and girls (ages 9–14, n=52). In boys, testosterone and estradiol were correlated positively with anxiety, whereas DHEA showed a negative relationship, and no relationships were found for girls. The observed relationships in boys were no longer significant when age and pubertal stage were covaried, which suggests, at first glance, that there is no link between T and anxiety. However, when examining a sample such as this, with at least moderate variation in age, there will necessarily be a high correlation between testosterone and age and pubertal status. Thus, partialling the variance associated with these variables removes a large portion of the meaningful variance in testosterone changes, rendering uninformative any tests using this partialled testosterone variable (see G.A. Miller & Chapman, 2001, for a general discussion of these issues). As discussed elsewhere (e.g., Spielberg et al., 2015), specific study designs must be employed in order to properly dissociate the effects of age and change in pubertal variables (e.g., hormones). In particular, age (or change in age) must be held relatively constant across participants, and participants must be sampled at the ages when there will be maximum variance in pubertal status (i.e., 11–12 in girls, 12–13 in boys). Thus, in longitudinal work with two assessments, 11–12/12–13 would be the ideal first assessment, and a subsequent assessment would occur when the majority of participants have reached full puberty (note that having greater than two assessment times is preferable because this allows for more complex designs that are beyond the scope of this discussion).
The other study in this area (Olweus et al., 1980, n=58) found no significant relationship between testosterone and anxiety in a sample of boys ages 15–17. Although this design held age relatively constant, participants would already have reached full puberty, and thus this study is also not informative regarding the specific relationship between anxiety and adolescent increases in testosterone level. Finally, Granger et al. (2003) examined a mixed sex sample (ages 11–16, n=213) and found that basal testosterone level was negatively related to a mixed measure of anxiety and depression. Although this study did not covary age, the lack of a pure anxiety measure also renders the findings this study fairly uninformative. Thus, there remains an overall dearth of appropriately designed research on the specific relationships between pubertal hormones and anxiety, and more work in this area is sorely needed.
Some insights may be gained via studies of women with polycystic ovary syndrome (PCOS), which is associated with androgen excess and related physical consequences (e.g., male-pattern hair growth; Norman et al., 2007). Meta-analytic evidence has demonstrated higher levels of anxiety in women with PCOS (Barry et al., 2011; Dokras et al., 2012), and this increase was evident even when matching for body mass index (Jedel et al., 2009). Although interesting, these relationships remain confounded by the physical consequences of PCOS (e.g., excess hair growth) that could reasonably lead to increases in anxiety in these patients.
3.6.4. Pubertal Hormones and Anxiety in Animals
Similar to research in humans, there is an increase in behaviors thought to index anxiety in male rodents that onsets during adolescence. Prior to adolescence and puberty, juvenile male rats will eagerly interact (even play) with one another in a novel environment. After puberty, however, adult male rats show significantly reduced social interaction in novel environments, which is thought to reflect anxiety due to the novelty of the environment. This response to novel environments is normative and is not modulated by testosterone in adulthood, but rather by testosterone during puberty (Primus & Kellogg, 1990). In particular, if a male rat is castrated prior to puberty, this behavior never develops. If testosterone is replaced during puberty in the castrated male rat, “anxiety” behavior develops as normal, but if testosterone is replaced in adulthood, it does not reinstate the reduction in social interaction in novel environments (Primus & Kellogg, 1990). Other anxiety-like behaviors also develop in rodents during puberty in a similar manner. Prepubertal castration of male rats increases the time spent in the open arms of an elevated plus maze and the time spent in the bright side of a light-dark box, indicating that, in the absence of testosterone, these typical anxiety-like behaviors never develop (Brown et al., 2015; McDermott et al., 2012). Castration prior to puberty also results in deficits in contextual (hippocampal-dependent) fear memory, with no effect on cued-fear memory. In a separate study, castration prior to adolescence resulted in less anxiety-like behavior in four different tasks (some social, some not) and reduced startle compared to sham operated controls, despite the fact that testosterone was replaced in adulthood in the castrated rats to the same levels measured in the sham operated controls (Zuloaga et al., 2011). Together, these studies suggest that, in rodents, testosterone organizes the typical emergence of anxiety-like behaviors into adulthood.
Several non-human primate studies also suggest that testosterone can impact emotion regulation and vigilance or alertness to negative social stimuli. Lacreuse and colleagues found that testosterone treatment of young male rhesus monkeys results in enhanced attention to videos of negative social stimuli (Lacreuse et al., 2009, 2010). Furthermore, testosterone treatment also decreased interactions with negative objects in an approach/avoidance task (Lacreuse et al., 2012).
In sum, studies in animals suggest that, similar to humans, anxiety-like behaviors increase naturally throughout adolescence as a result of the onset of puberty. These data also indicate that testosterone has an important role in the development of these “naturally occurring” anxiety-like behaviors, and thus testosterone may also have an important role in the development of pathological anxiety via similar mechanisms.
3.7. Impact of Testosterone on Affect-Relevant Circuitry
As discussed in section 3.5 above, the OFC-amygdala circuit appears critically important for the regulation of affect, and both regions contain high concentrations of androgen receptors (Finley & Kritzer, 1999; Pelletier, 2000). Accordingly, this circuit appears to be a prime candidate for the mechanism by which testosterone (and other hormones) impact emotion regulation. In fact, a growing body of research suggests that testosterone does impact these neural structures and their interaction (Bos et al., 2012). For example, using a double-blind placebo-controlled design in sample of young women (ages 18–28, mean=23, n=12), Hermans et al. (2008) found that testosterone administration led to increased activation to threat-related faces in both amygdala and OFC. When comparing younger (ages 19–30, mean=23, n=17) and middle-aged (ages 37–50, mean=42, n=25) women, van Wingen et al. (2009) found that younger women had both higher testosterone levels and higher amygdala reactivity to threat-related faces. After double-blind placebo-controlled testosterone administration, amygdala reactivity increased in the middle-aged group, such that levels were no longer different from young women. Finally, longitudinal work in our lab in a mixed-sex sample found that increases in circulating (endogenous) pubertal testosterone over a two-year period (time 1 mean ages = 11/12 for females/males, time 2 mean ages = 13/14 for females/males, n=38) were linked to increases in amygdala activation to threat-related faces (Spielberg et al., 2014). Interestingly, increases in amygdala activation predicted increases in anxiety symptoms, but only in adolescents without a concurrent increase in nucleus accumbens (NAcc) activation (to threat-related faces). In contrast, amygdala increases predicted sensation seeking in those with a concurrent increase in NAcc activation. Given that both high anxiety and sensation seeking are linked to emotion dysregulation (Amstadter, 2008; Joseph et al., 2009), these findings suggest that testosterone-related increases in amygdala activation may lead to such dysregulation, but the behavioral impact of this dysregulation depends on other factors.
Beyond activation in individual regions, testosterone also appears to influence connectivity between amygdala and OFC. The first study in this area found that double-blind placebo-controlled administration of testosterone in a sample of middle-aged women (ages 37–50, mean=42, n=25) lead to decreases in the magnitude of functional coupling between these regions while participants viewed emotion-related faces (van Wingen et al., 2010). Using a double-blind placebo-controlled design in a sample of younger women (mean age=21, n=16), Bos et al. (2012) found that testosterone administration decreased the magnitude of connectivity between amygdala and OFC during judgments of unfamiliar faces. Furthermore, testosterone administration increased amygdala responses to faces rated as untrustworthy. Finally, connectivity between amygdala and brain stem was not affected by testosterone administration, supporting the specificity of the impact of testosterone on the OFC-amygdala link. A third study using a double-blind placebo-controlled design, in a sample of younger women (ages 18–37, mean=21, n=22), found that testosterone administration lead to increased activation in OFC to escapable threat (vs. inescapable threat) (Heany et al., 2018) and decreases in the magnitude of coupling between this OFC region and amygdala.
The impact of endogenous testosterone on OFC-amygdala coupling has also been investigated. For example, endogenous testosterone was negatively related to OFC-amygdala coupling magnitude during resting state in a male sample (ages 12–26, mean=16, n=87), but not in a comparable female sample (n=86) (Peters et al., 2015). This study also found that OFC-amygdala coupling predicted alcohol use in males, but when the sample was stratified by age, this effect was only significant for mid-adolescents (ages 14–17). Longitudinal work in our lab, using an overlapping data set as Spielberg et al. (2014), found that increases in pubertal testosterone over two years were linked to decreases in the magnitude of OFC-amygdala coupling while participants (n=41) viewed threat-related faces (Spielberg et al., 2015). This effect was significant in both boys and girls (see Figure 2), even though the range of ng/dl testosterone change was considerably smaller in girls, suggesting a greater sensitivity in girls to the impact of testosterone in this circuit. Finally, decreases in coupling magnitude were associated with increased levels of withdrawal temperament, a normative anxiety-related dimension.
Figure 2.
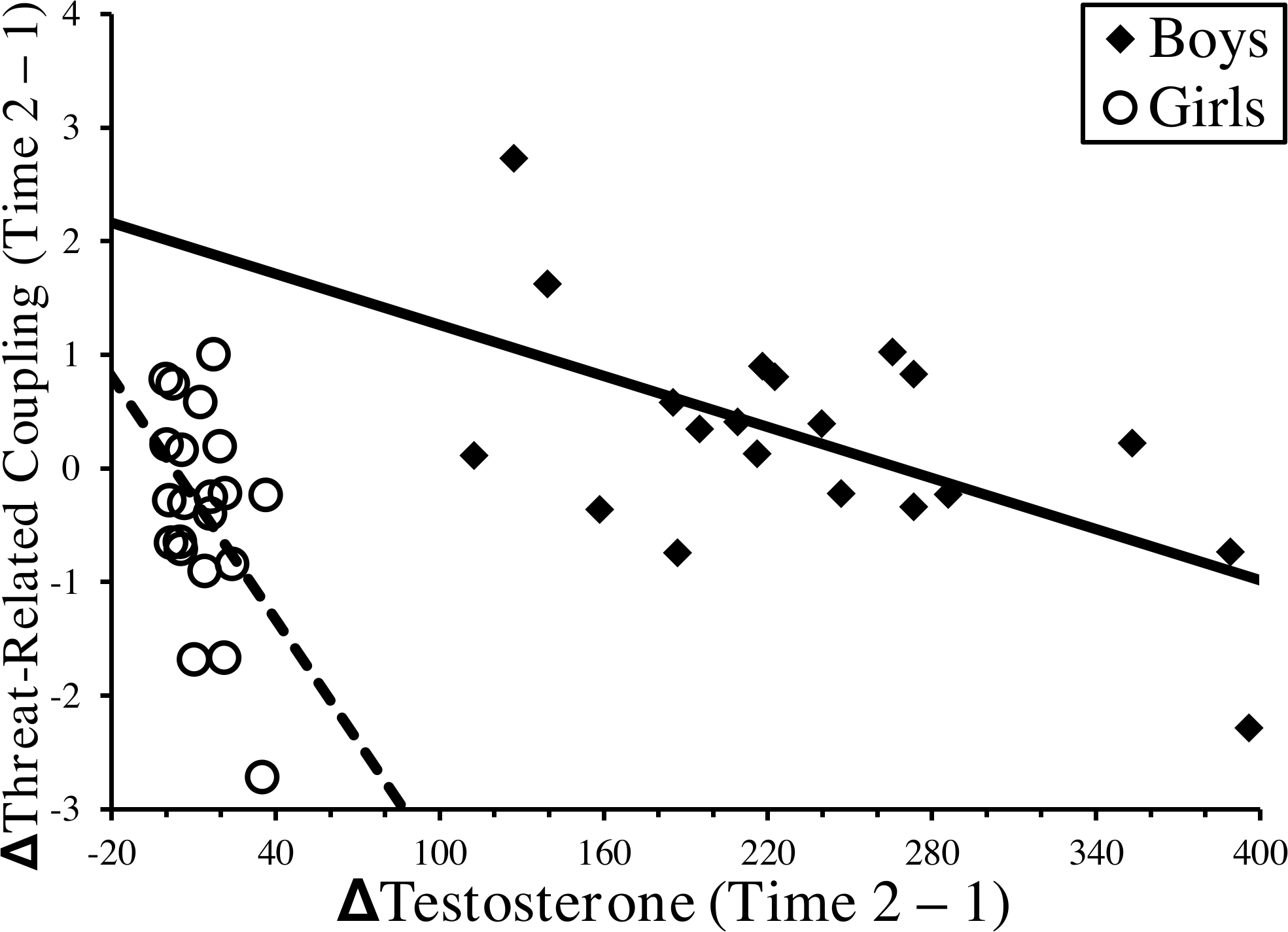
Sex-Specific Relationships Between Change in Pubertal Testosterone and Change in Threat-Related Coupling between Orbitofrontal Cortex and Amygdala
The scatterplot illustrates the relationship between longitudinal change over 2 years in pubertal testosterone (i.e., x-axis, ΔTestosterone (Time 2–1)) and longitudinal change over the same period in coupling between amygdala and OFC (i.e., y-axis, ΔThreat-Related Coupling (Time 2–1)). Relationships are depicted separately for girls (open circles, dotted line) and boys (filled diamonds, solid line). Based on the data presented in Spielberg et al. (2015).
The molecular mechanisms by which testosterone influences OFC-amygdala coupling are less clear. Testosterone does not appear to have an overall downregulating effect on OFC (e.g., testosterone leads greater threat activity in OFC; Hermans et al., 2008), and thus it is likely that the mechanism must occur be via interruption of the communication (or reception of that communication) itself. For example, testosterone may disrupt the reception of OFC signals in amygdala, and there is some evidence that this may be mediated by dopaminergic function. In particular, activation of dopamine receptors in basolateral amygdala attenuates the (inhibitory) action of GABAergic interneurons, leading to a simultaneous decrease in prefrontal influence on amygdala and an increase in the influence from other (e.g., sensory-related) inputs to amygdala (Grace & Rosenkranz, 2002; Marowsky et al., 2005). The mechanism by which testosterone impacts dopaminergic function in amygdala remains unclear, although there is evidence that it does so in other structures. For example, administration of testosterone to male rats leads to increased dopamine levels in the striatum (de Souza Silva et al., 2009). Thus, there is emerging evidence suggesting a dopaminergic-mediated mechanism by which testosterone influences OFC-amygdala coupling.
In summary, evidence supports the influence of testosterone on amygdala, OFC, and their intercommunication. Furthermore, mounting evidence suggests that the influence of testosterone on this circuitry leads to difficulties with emotion regulation (e.g., alcohol use, anxiety). Unfortunately, although the majority of this research was done in adults, two studies provide preliminary evidence that this is also true for pubertal testosterone.
It is important to stress that we cannot know that the effects discussed above, wherein testosterone was measured or administered outside the brain, are directly due to testosterone, given that androgens also function as prohormones (e.g., for estradiol). The studies employing a double-blind placebo design are highly suggestive that testosterone is part of the mechanistic chain at some point, but the extent to which testosterone has a more proximal or distal impact on neural function remains unknown.
3.8. OFC-Amygdala Connectivity Meta-Analysis
Given that OFC represents a reasonably large and diverse piece of the cortical mantle, it is possible that the connectivity studies discussed in the previous section reflect disparate areas of OFC and do not reflect anatomically consistent effects. Thus, we conducted an Activation-Likelihood Estimation (ALE) meta-analysis using GingerALE v 2.3.6 (Eickhoff et al., 2012; Turkeltaub et al., 2012) and the relevant OFC coordinates published in the five studies discussed above (9 foci used; 279 total participants). Given differences in whether connectivity was investigated for right, left, or combined amygdala, we initially collapsed across hemisphere. Finally, we used the recommended settings for cluster-level inference (cluster-forming threshold p=.001, 1,000 threshold permutations, cluster-level p=.05). This analysis resulted in two significant clusters, which were located in middle OFC as illustrated in Figure 3. The first cluster (center xyz=±29,51,−10, 1,920mm3) was contributed to by foci from Bos et al. (2012), Heany et al. (2018), Peters et al. (2015), and Spielberg et al. (2015), and the second cluster (center xyz=±19,38,−14, 1,208mm3) was contributed to by foci from Peters et al. (2015) and van Wingen et al. (2010). We repeated this analysis with the same parameters and input, but without collapsing across hemisphere. This resulted in only one significant cluster (center xyz=31,50,−14, 944mm3), overlapping cluster 1 in the unilateral analysis. Interestingly, this cluster was driven by foci from the two studies involving adolescent samples (Peters et al., 2015; Spielberg et al., 2015).
Figure 3.
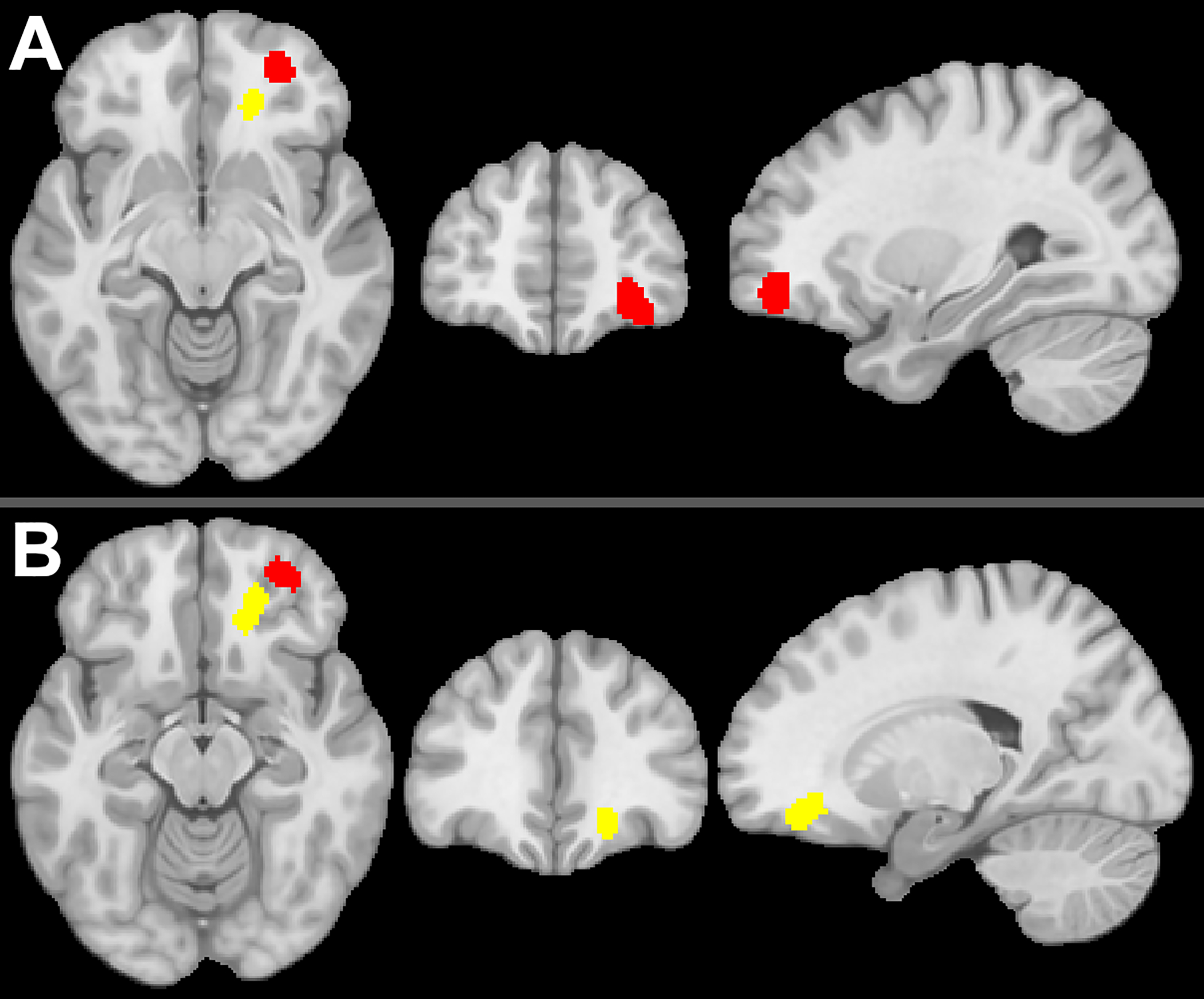
Meta-Analysis of Orbitofrontal Cortex Regions Showing Testosterone-Dependent Coupling with Amygdala
Two clusters in orbitofrontal cortex that emerged from an ALE meta-analysis of studies of testosterone-dependent coupling with amygdala. Panel A: The location of the first (red) cluster, with slices at x=29, y=51, z=−10. Panel B: The location of the second (yellow) cluster, with slices at x=19, y=38, z=−14.
Although based on a relatively small set of studies, the findings of this meta-analysis suggest that there is anatomical correspondence between the studies discussed above. This, in turn, provides further evidence of the reliability of testosterone modulation of OFC-amygdala connectivity.
4.0. Proposed Neuromaturational Model
We now return to the four questions posed above and propose a novel neuromaturational model to address these key gaps in the literature.
4.1. Why does emotion dysregulation tend to increase – both normatively and pathologically – with adolescence and decrease with emerging adulthood?
The research summarized above suggests that (i) greater amygdala reactivity contributes to the development of anxiety, (ii) disruption in top-down control of amygdala by OFC contributes to greater amygdala reactivity, and (iii) testosterone disrupts OFC-amygdala coupling. Based on this work, we propose that the large increase in testosterone (blue line in Figure 4a) that occurs during gonadarche (purple background in Figure 4a) disrupts communication between OFC and amygdala (yellow line in Figure 4a). Consequently, OFC will exert weaker top-down control over amygdala responses, resulting in the increased in amygdala reactivity observed during adolescence, and consequent higher levels of affect dysregulation and anxiety (orange line in Figure 4a). This emotion dysregulation contrasts with broader (‘cognitive’) regulatory capacity, which shows a linear increase across adolescence (gray line in Figure 4a). Given that DHEA and other adrenal androgens, which increase earlier in development than testosterone (i.e., during adrenarche, red background in Figure 4a), can function like weaker forms of testosterone, it is possible that the process of OFC-amygdala decoupling begins earlier than gonadarche. This may explain, in part, why the onset distributions for some anxiety disorders begin before what is typically considered adolescence (see Figure 1).
Figure 4.
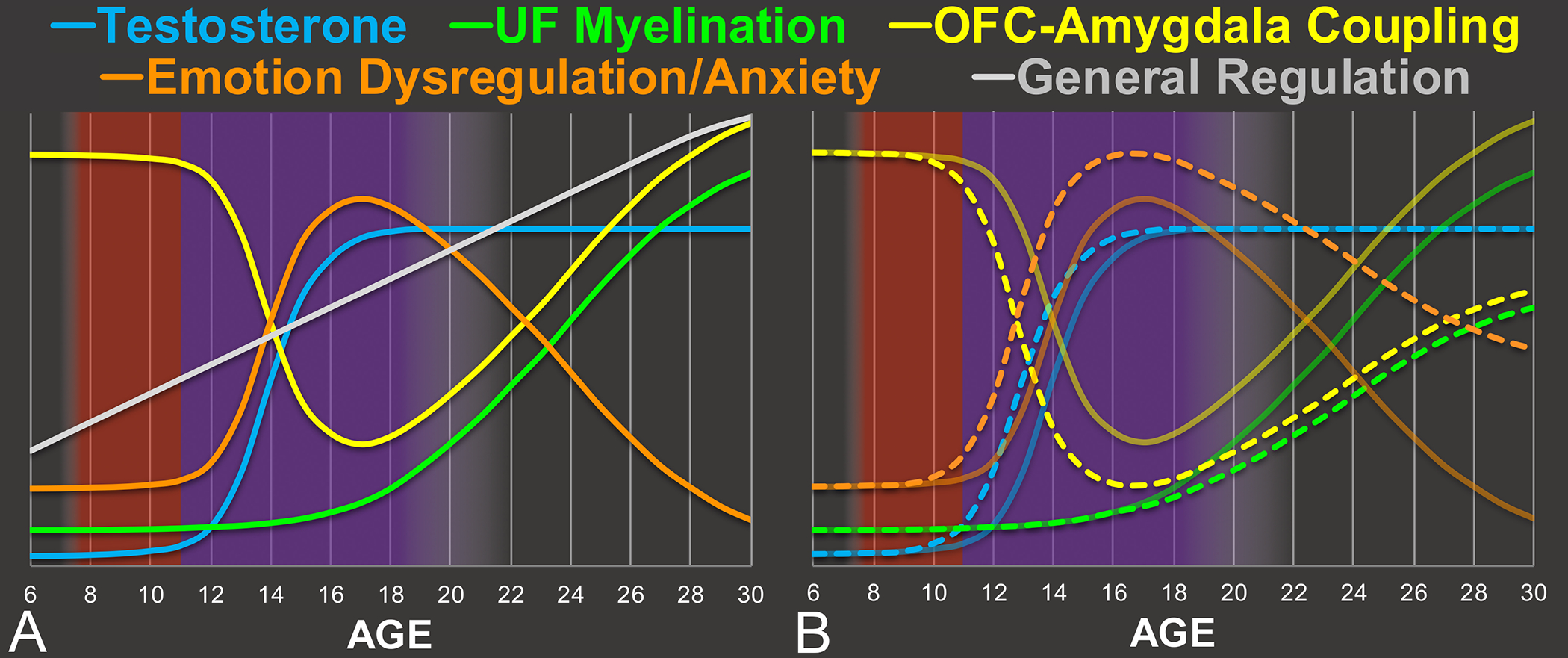
Proposed Normative and Pathological Maturational Timecourses
Panel A: An example illustration of the proposed normative timecourses of development across age (x-axis). Panel B: Illustration of the manner in which pathology might be associated with divergence from the normative timecourses. In particular, the normative timecourses (i.e., those in Panel A) are depicted as transparent solid lines, whereas departures from these timecourses are depicted as dashed lines. For example, the dashed blue line reflects an earlier onset of puberty, leading testosterone to increase earlier. The dashed green line reflects weaker myelination of uncinate fasciculus (UF). Coupling between orbitofrontal cortex (OFC) and amygdala is proposed to depend on both testosterone and UF myelination, and thus the dashed yellow line both decreases earlier (due to earlier increase in testosterone) and stays lower over time (due to both weaker myelination and the organizational effect of earlier testosterone) than the solid yellow line. Emotion dysregulation/anxiety is proposed to be inversely related to OFC-amygdala coupling, and thus the dashed orange line increases earlier and stays higher over time, relative to the solid orange line.
We also propose that UF myelination (green line in Figure 4a), which begins in earnest in late adolescence, increases the efficacy of communication between OFC and amygdala. Over time, this counteracts the impact of testosterone, resulting in a restoration of OFC-amygdala coupling and a consequent decrease in affect dysregulation and anxiety. These transitional processes are also illustrated in Figure 5.
Figure 5.
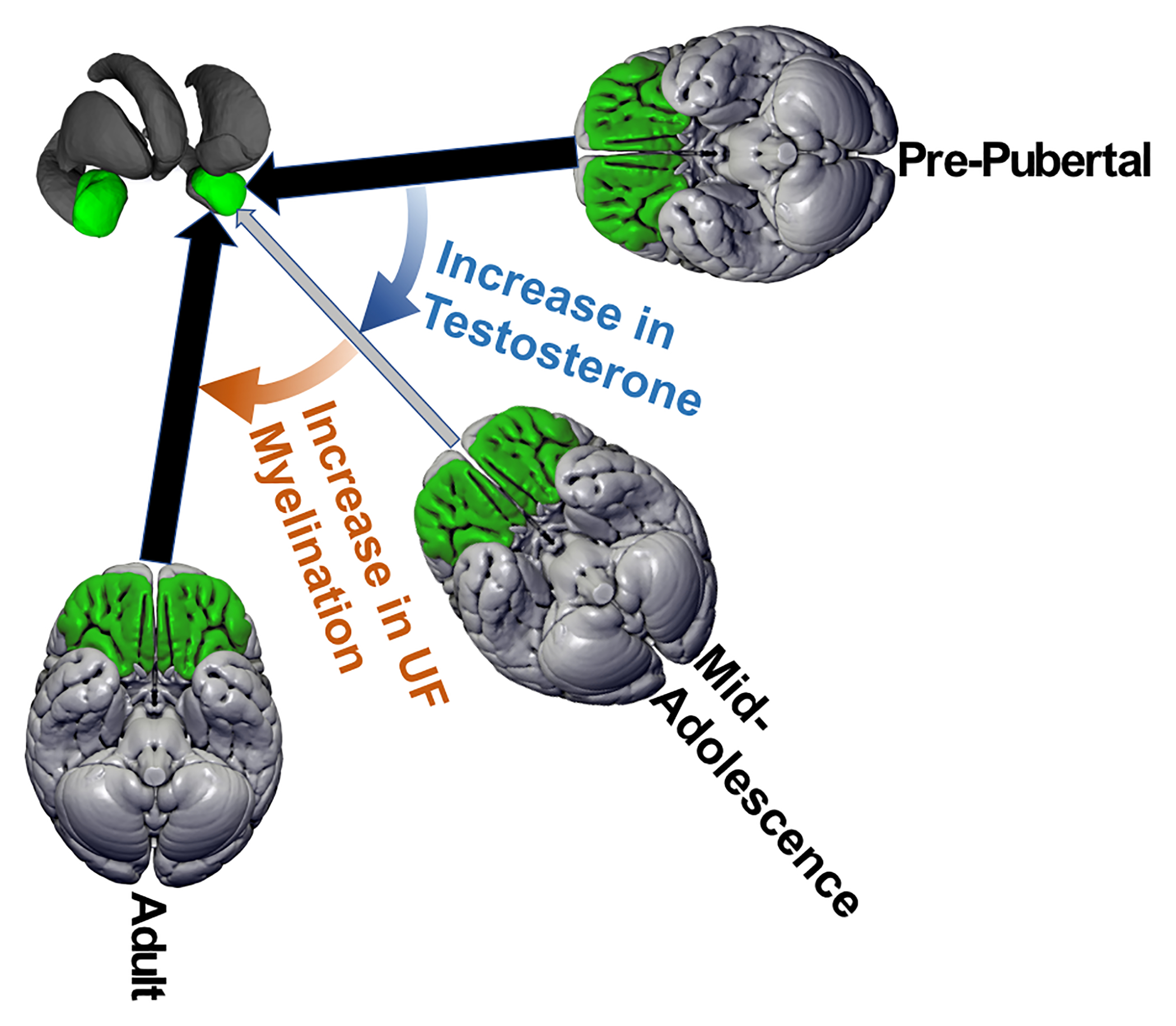
Graphical Representation of the Proposed Model
Illustration of the changes over development in coupling between amygdala (green structures in top-left) and orbitofrontal cortex (green cortex in the three brains). Each brain represents a different period of development, and the size and color of the arrow between that brain and amygdala is proportional to the strength of coupling (i.e., smaller arrow, lighter color represents weaker coupling). The blue and orange colored arrows represent the processes that influence coupling
4.2. Why do some individuals develop pathological levels of emotion dysregulation (and related symptoms) during adolescence whereas others do not?
We propose that individual differences in the maturational timecourse of testosterone contributes to the development of pathological anxiety during adolescence. In particular, earlier pubertal onset, faster pubertal tempo, and/or larger mean (due to baseline and/or ending) testosterone levels could all lead to greater disruption in OFC-amygdala coupling and consequent emotion-regulation capacity. Although there is a dearth of research in this area, rendering unclear which (if any) of these possibilities occurs, there are two reasons to believe that earlier onset is the most likely option: First, earlier pubertal onset has already been associated with greater levels of pathology. Second, given that the magnitude of the organizational neural changes induced by testosterone appears to decrease across adolescence, early onset of puberty would lead to higher testosterone levels at precisely the time during which this would have the greatest impact. In other words, higher testosterone levels during adolescence are not created equal, and thus the option that allows for higher testosterone to occur earlier in development appears more likely. This idea has been supported by the work of others (Beltz & Berenbaum, 2013) who have similarly postulated that, if the “organizational” effects of testosterone on the brain decreases across adolescence, then the age at which adolescence undergo puberty could significantly impact cognitive development. The impact of earlier onset puberty is depicted in Figure 4b, wherein the increase in testosterone occurs earlier (dotted blue line), which induces OFC-amygdala decoupling (dotted yellow line) to occur both earlier and to a greater extent. Similarly, the increase in emotion-dysregulation and anxiety (dotted orange line) would also occur earlier and to a greater extent.
We also propose that lower levels of pre-adolescence UF myelination serve as a vulnerability for the development of emotion dysregulation and anxiety. Namely, if communication in this circuit is already weakened at the time of adolescence, the impact of testosterone on this pathway may be more disruptive. For simplicity’s sake, we have not illustrated the impact of lower UF myelination in Figure 4b, as this is not the primary mechanism proposed herein.
4.3. Why do some individuals fail to show a decline in dysregulation (and related symptoms) during emerging adulthood (i.e., pathology becomes chronic)?
We propose that individual differences in the timecourse of UF myelination contribute to the resolution or maintenance of pathological anxiety. In particular, if UF myelination is less effective in increasing top-down regulation of amygdala by OFC, this would result in amygdala remaining more reactive, which would lead to the maintenance of pathological anxiety. Similar to testosterone, there could be differences in the onset, tempo, and overall effect size of UF myelination. For example, a later onset or slower tempo would result in a longer period of amygdala reactivity, and consequent higher anxiety levels. Lower endpoint UF myelination would have the greatest effect, as depicted in Figure 4b (dotted green line), in that amygdala reactivity and related pathology would be chronically higher. Given the dearth of work in this area, it is unclear which of these options is most likely, and further research is sorely needed.
4.4. Why do sex differences in affect-related pathology emerge during adolescence?
Given that males evidence a much greater increase in pubertal testosterone, the central role of this hormone in the proposed model appears inconsistent with evidence that females experience significantly higher rates of anxiety, particularly during/after puberty. Thus, it is necessary to refine the proposed model to account for this discrepancy, and work by De Vries and colleagues (for review, see De Vries, 2004, or McCarthy & Konkle, 2005) provides key insights to do so. They proposed that sex differences in the brain subserve two purposes, the first being the prototypical view that such divergence produces needed differences, for example in overt behavior (e.g., mating behavior). Counterintuitively, the second purpose is to prevent differences when that would be maladaptive. In other words, when the development of a behavior (e.g., attention to threat), for example, is influenced by physiology that differs across sexes (e.g., testosterone levels), but there is an evolutionary cost associated with that behavior differing across sexes (e.g., both sexes must be able to attend to threat), a compensatory mechanism is needed with which to equalize behavior. Returning to our model, recall that we (Spielberg et al., 2015) found that both boys and girls achieved the same levels of OFC-amygdala decoupling (see Figure 2), but girls did so with a much smaller increase in testosterone. Thus, it appears that the biological effect of a unit change in testosterone was greater for girls than boys. Consequently, we propose that testosterone has a greater organizational effect on OFC-amygdala connectivity in women, which thus equalizes normative decoupling across sexes.
This proposal explains why male anxiety levels are not greater than females. However, it does not explain why female anxiety levels are actually typically higher. One solution to this problem lies in noting that testosterone levels are not static within (e.g., testosterone levels vary with menstrual cycle in women; Al-Dujaili & Sharp, 2012) or between individuals. Thus, variations in testosterone levels of a similar magnitude would ultimately have a much larger impact on females than males. Consequently, early pubertal onset, including an earlier increase in testosterone, could have a stronger impact on girls than boys. More broadly, individual differences in testosterone level may have a stronger impact in females due to the proposed increase in sensitivity.
In summary, we propose that the organizational impact of testosterone on OFC-amygdala coupling is larger in girls, leading to increases in the influence of within-person fluctuations and/or individual differences in the timecourse or level of testosterone. Thus, we propose that a mechanism that normatively ensures equal levels of coupling across sexes also increases vulnerability in a subset of girls.
4.5. Vulnerability vs. Pathology
An astute reader may have noticed that the model, as described up to this point, fails to account for why a large proportion of anxiety disorders tend to onset after adolescence, particularly certain disorders (e.g., GAD). In other words, the model outlined above provides potential mechanisms for why pathology onsets during adolescence and why such pathology becomes chronic into adulthood, but at first glance it does address why pathology would onset during adulthood. Of course, there are likely to be a number of confluent causes, and we do not pretend that the proposed model encompasses all such mechanisms. Thus, should the proposed model turn out to be accurate, there may still be a separate causal pathway responsible for adult onset anxiety. Moreover, individuals with adult onset anxiety disorders may have significant subthreshold anxiety beforehand due to the mechanisms described above, and some other factor may push them over the diagnostic threshold at a later date.
More broadly, however, it is perhaps most accurate to frame the neural and hormonal mechanisms proposed above as creating a vulnerability for pathological anxiety and other pathology related to emotion dysregulation. In particular, it is important to reiterate that the impact of pubertal testosterone on the brain can be organizational in nature rather than solely activational. In other words, exposure to testosterone may create lasting changes in brain organization that result in weaker emotion regulation and greater threat reactivity. Thus, neural reorganization due to testosterone may not result in pathological levels of anxiety immediately, but rather serves as a diathesis that can interact with environmental and other biological and psychological processes to create pathology, particularly when combined with weaker UF myelination. This may help to explain why social phobia tends to onset earlier than most anxiety disorders: given that peer interaction is particularly salient during adolescence, social stressors are likely particularly potent during this time and may interact with the vulnerabilities described above to create pathology. Conversely, the specific stressors that lead to other disorders may be more likely to occur later, and thus the diathesis described above does not manifest until that time.
4.6. Estradiol
The model proposed herein focuses on testosterone, and to a lesser extent DHEA, but it is almost certain that estradiol and other estrogens have a critical impact on emotion regulation and anxiety. A full account of this work is beyond the scope of this paper, but we will briefly mention several key findings. For example, estrogen receptors (ER) are relatively densely distributed in the same regions as androgen receptors, with both ER-α and ER-β found in amygdala and ER-β in OFC (Shugrue et al., 1997). In addition, there is mounting research indicating that estradiol is linked to both anxiety and depressive symptoms (Walf & Frye, 2006). Finally, testosterone may also be aromatized into estradiol. Given that this can occur within presynaptic terminals (Balthazart & Ball, 2006), there may be no observable changes in testosterone plasma levels. Thus, the role for testosterone in the proposed model may, wholly or in part, be only as a precursor for estradiol. Further research is needed to parse these contributions.
4.7. The Role of Societal and Temporal Context
Unfortunately, the majority of the work discussed above, upon which our model is based, has been conducted recently in a Western and 1st-world context. Thus, we do not know the impact of factors associated with this context (e.g., nutrition, modern healthcare) on the proposed model. Although this is true of most psychology/neuroscience research, there are reasons to believe that this issue may be particularly salient for neuroendocrine development. For example, the declining age of menarche over time (Demerath et al., 2004) and differences in age of menarche across countries, with more developed countries having earlier menarche (Morabia et al., 1998). Thus, although there is no evidence to suggest that the proposed model would not hold across countries or in earlier eras, we also make no claims of universality. It will be paramount to expand the field to conduct such research across a more diverse array of populations.
5.0. Conclusions
In the present work, we have proposed a dual-mechanism biological model of the factors promoting the onset of anxiety during adolescence, the development of chronic adult anxiety, and the emergence of sex differences in anxiety. Given that the proposed model is based on data from a range of disciplines (e.g., human and animal neuroscience, psychology), it has the potential to generate a diverse array of research.
Funding
This work was supported by University of Delaware Research Foundation grant 17a00947 to Spielberg, National Institute of Mental Health L30 MH117662 to Spielberg, and National Institute of Mental Health R01 MH106553 to Schwarz.
References
- Ahmed EI, Zehr JL, Schulz KM, Lorenz BH, DonCarlos LL, & Sisk CL (2008). Pubertal hormones modulate the addition of new cells to sexually dimorphic brain regions. Nature Neuroscience, 11(9), 995–7. 10.1038/nn.2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SP, Bittencourt-Hewitt A, & Sebastian CL (2015). Neurocognitive bases of emotion regulation development in adolescence. Developmental Cognitive Neuroscience, 15, 11–25. 10.1016/j.dcn.2015.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dujaili EAS, & Sharp MA (2012). Female salivary testosterone: Measurement, challenges and applications. Steroids. 10.5772/53648 [DOI] [Google Scholar]
- Altemus M, Sarvaiya N, & Neill Epperson C (2014). Sex differences in anxiety and depression clinical perspectives. Frontiers in Neuroendocrinology, 35(3), 320–30. 10.1016/j.yfrne.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstader A (2008). Emotion regulation and anxiety disorders. Journal of Anxiety Disorders, 22, 211–221. 10.1007/s11920-012-0262-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold AP, & Breedlove SM (1985). Organizational and activational effects of sex steroids on brain and behavior: A reanalysis. Hormones and Behavior, 19(4), 469–498. 10.1016/0018-506X(85)90042-X [DOI] [PubMed] [Google Scholar]
- Baker KD, Den ML, Graham BM, & Richardson R (2014). A window of vulnerability: Impaired fear extinction in adolescence. Neurobiology of Learning and Memory, 113, 90–100. 10.1016/j.nlm.2013.10.009 [DOI] [PubMed] [Google Scholar]
- Balthazart J, & Ball GF (2006). Is brain estradiol a hormone or a neurotransmitter? Trends in Neurosciences, 29(5), 241–249. 10.1016/j.tins.2006.03.004 [DOI] [PubMed] [Google Scholar]
- Banks SJ, Eddy KT, Angstadt M, Nathan PJ, & Luan Phan K (2007). Amygdala-frontal connectivity during emotion regulation. Social Cognitive and Affective Neuroscience, 2(4), 303–312. 10.1093/scan/nsm029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry JA, Kuczmierczyk AR, & Hardiman PJ (2011). Anxiety and depression in polycystic ovary syndrome: a systematic review and meta-analysis. Human Reproduction, 26(9), 2442–2451. 10.1093/humrep/der197 [DOI] [PubMed] [Google Scholar]
- Baumann N, & Pham-Dinh D (2001). Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiological Reviews, 81(2), 871–927. 10.1152/physrev.2001.81.2.871 [DOI] [PubMed] [Google Scholar]
- Baur V, Hänggi J, & Jäncke L (2012). Volumetric associations between uncinate fasciculus, amygdala, and trait anxiety. BMC Neuroscience, 13(4). 10.1186/1471-2202-13-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter AJ, Vos T, Scott KM, Ferrari AJ, & Whiteford HA (2014). The global burden of anxiety disorders in 2010. Psychological Medicine, 44(11), 2363–2374. 10.1017/S0033291713003243 [DOI] [PubMed] [Google Scholar]
- Beck AT, Epstein N, Brown G, & Steer RA (1988). An inventory for measuring clinical anxiety: Psychometric properties. Journal of Consulting and Clinical Psychology, 56(6), 893–7. 10.1037/0022-006X.56.6.893 [DOI] [PubMed] [Google Scholar]
- Beltz AM, & Berenbaum SA (2013). Cognitive effects of variations in pubertal timing: Is puberty a period of brain organization for human sex-typed cognition? Hormones and Behavior, 63, 823–828. 10.1016/j.yhbeh.2013.04.002 [DOI] [PubMed] [Google Scholar]
- Best JR, & Miller PH (2010). A developmental perspective on executive function. Child Development, 81(6), 1641–60. 10.1111/j.1467-8624.2010.01499.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore SJ, Burnett S, & Dahl RE (2010). The role of puberty in the developing adolescent brain. Human Brain Mapping, 31(6), 926–933. 10.1002/hbm.21052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PA, Panksepp J, Bluthé RM, & Honk J. van. (2012). Acute effects of steroid hormones and neuropeptides on human social-emotional behavior: A review of single administration studies. Frontiers in Neuroendocrinology, 33, 17–35. 10.1016/j.yfrne.2011.01.002 [DOI] [PubMed] [Google Scholar]
- Brown GR, Kulbarsh KD, Spencer KA, & Duval C (2015). Peri-pubertal exposure to testicular hormones organizes response to novel environments and social behaviour in adult male rats. Hormones and Behavior, 73, 135–141. 10.1016/j.yhbeh.2015.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger HG (2002). Androgen production in women. Fertility and Sterility, 77(4), 3–5. 10.1016/S0015-0282(02)02985-0 [DOI] [PubMed] [Google Scholar]
- Canals J, Martí-Henneberg C, Fernández-Ballart J, Clivillé R, & Domènech E (1992). Scores on the State-Trait Anxiety Inventory for children in a longitudinal study of pubertal Spanish youth. Psychological Reports, 71, 503–512. 10.2466/pr0.1992.71.2.503 [DOI] [PubMed] [Google Scholar]
- Casey BJ, Getz S, & Galvan A (2008). The adolescent brain. Developmental Review, 28(1), 62–77. https://doi.org/doi:10.1016/j.dr.2007.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WCJ, De Vries GJ, & Swaab DF (2002). Sexual differentiation of the bed nucleus of the stria terminalis in humans may extend into adulthood. The Journal of Neuroscience, 22(3), 1027–33. 10.1523/JNEUROSCI.22-03-01027.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisler JM, & Koster EHW (2010). Mechanisms of attentional biases towards threat in anxiety disorders: An integrative review. Clinical Psychology Review, 30(2), 203–16. 10.1016/j.cpr.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Gilbert JE, & Thomas KM (2013). Inhibitory control during emotional distraction across adolescence and early adulthood. Child Development, 84(6), 1954–66. 10.1111/cdev.12085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MX, Elger CE, & Weber B (2008). Amygdala tractography predicts functional connectivity and learning during feedback-guided decision-making. NeuroImage, 39(3), 1396–1407. 10.1016/j.neuroimage.2007.10.004 [DOI] [PubMed] [Google Scholar]
- Conway CC, Zinbarg RE, Mineka S, & Craske MG (2017). Core dimensions of anxiety and depression change independently during adolescence. Journal of Abnormal Psychology, 126(2), 160–172. 10.1037/abn0000222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke BM, Stokas MR, & Woolley CS (2007). Morphological sex differences and laterality in the prepubertal medial amygdala. Journal of Comparative Neurology, 501(6), 904–915. 10.1002/cne.21281 [DOI] [PubMed] [Google Scholar]
- Costello EJ, Mustillo S, Erkanli A, Keeler G, A. A. (2003). Prevalence and Development of Psychiatric Disorders in Childhood and Adolescence. Archives of General Psychiatry, 60(8), 837–844. 10.1001/archpsyc.60.8.837 [DOI] [PubMed] [Google Scholar]
- Cunningham WA, & Brosch T (2012). Motivational salience: Amygdala tuning from traits, needs, values, and goals. Current Directions in Psychological Science, 21(1), 54–59. 10.1177/0963721411430832u [DOI] [Google Scholar]
- Dahl RE (2004). Adolescent brain development: A period of vulnerabilities and opportunities. Annals of the New York Academy of Sciences, 1021, 1–22. 10.1196/annals.1308.001 [DOI] [PubMed] [Google Scholar]
- Damoiseaux JS, & Greicius MD (2009). Greater than the sum of its parts: A review of studies combining structural connectivity and resting-state functional connectivity. Brain Structure and Function, 213, 525–533. 10.1007/s00429-009-0208-6 [DOI] [PubMed] [Google Scholar]
- de Souza Silva MA, Mattern C, Topic B, Buddenberg TE, & Huston JP (2009). Dopaminergic and serotonergic activity in neostriatum and nucleus accumbens enhanced by intranasal administration of testosterone. European Neuropsychopharmacology, 19(1), 53–63. 10.1016/j.euroneuro.2008.08.003 [DOI] [PubMed] [Google Scholar]
- De Vries GJ (2004, March 1). Minireview: Sex differences in adult and developing brains: compensation, compensation, compensation. Endocrinology. Oxford University Press. 10.1210/en.2003-1504 [DOI] [PubMed] [Google Scholar]
- Demerath EW, Towne B, Chumlea WC, Sun SS, Czerwinski SA, Remsberg KE, & Siervogel RM (2004). Recent decline in age at menarche: the Fels Longitudinal Study. American Journal of Human Biology, 16(4), 453–457. 10.1002/ajhb.20039 [DOI] [PubMed] [Google Scholar]
- Di X, Huang J, & Biswal BB (2017). Task modulated brain connectivity of the amygdala: A meta-analysis of psychophysiological interactions. Brain Structure and Function, 222(1), 619–634. 10.1007/s00429-016-1239-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon DG, Rosso IM, Pechtel P, Killgore WDS, Rauch SL, & Pizzagalli DA (2014). Peril and pleasure: An RDoC-inspired examination of threat responses and reward processing in anxiety and depression. Depression and Anxiety, 31, 233–249. 10.1002/da.22202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokras A, Clifton S, Futterweit W, & Wild R (2012). Increased prevalence of anxiety symptoms in women with polycystic ovary syndrome: systematic review and meta-analysis. Fertility and Sterility, 97(1), 225–230. 10.1016/j.fertnstert.2011.10.022 [DOI] [PubMed] [Google Scholar]
- Dorn LD, Dahl RE, Woodward HR, & Biro F (2006). Defining the boundaries of early adolescence: A user’ s guide to assessing pubertal status and pubertal timing in research with adolescents. Applied Developmental Science, 10(1), 30–56. 10.1207/s1532480xads1001 [DOI] [Google Scholar]
- Duval ER, Javanbakht A, & Liberzon I (2015). Neural circuits in anxiety and stress disorders: A focused review. Therapeutics and Clinical Risk Management, 11, 115–26. 10.2147/TCRM.S48528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff SB, Bzdok D, Laird AR, Kurth F, & Fox PT (2012). Activation likelihood estimation meta-analysis revisited. NeuroImage, 59(3), 2349–2361. 10.1016/j.neuroimage.2011.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley SK, & Kritzer MF (1999). Immunoreactivity for intracellular androgen receptors in identified subpopulations of neurons, astrocytes and oligodendrocytes in primate prefrontal cortex. Journal of Neurobiology, 40, 446–457. [DOI] [PubMed] [Google Scholar]
- Fornari E, Knyazeva MG, Meuli R, & Maeder P (2007). Myelination shapes functional activity in the developing brain. NeuroImage, 38, 511–518. 10.1016/j.neuroimage.2007.07.010 [DOI] [PubMed] [Google Scholar]
- Frank DW, Dewitt M, Hudgens-Haney M, Schaeffer DJ, Ball BH, Schwarz NF, … Sabatinelli D (2014). Emotion regulation: Quantitative meta-analysis of functional activation and deactivation. Neuroscience and Biobehavioral Reviews, 45, 202–211. 10.1016/j.neubiorev.2014.06.010 [DOI] [PubMed] [Google Scholar]
- Galvan A (2010). Adolescent development of the reward system. Frontiers in Human Neuroscience, (February 2010). 10.3389/neuro.09.006.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvao TF, Silva MT, Zimmermann IR, Souza KM, Martins SS, & Pereira MG (2014). Pubertal timing in girls and depression: A systematic review. Journal of Affective Disorders, 155, 13–19. 10.1016/j.jad.2013.10.034 [DOI] [PubMed] [Google Scholar]
- Gao W, Bohl CE, & Dalton JT (2005). Chemistry and structural biology of androgen receptor. Chemical Reviews, 105(9), 3352–70. 10.1021/cr020456u [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Brody GH, Conger RD, & Simons RL (2006). Pubertal maturation and African American children’s internalizing and externalizing symptoms. Journal of Youth and Adolescence, 35(4), 531–540. 10.1007/s10964-006-9046-5 [DOI] [Google Scholar]
- Ge X, & Natsuaki MN (2009). Current directions in psychological science in Search of explanations for early pubertal timing effects on developmental psychopathology. Current Directions in Psychological Science, 18(6), 327–331. 10.1111/j.1467-8721.2009.01661.x [DOI] [Google Scholar]
- Geise C, Barzman D, & Strakowski S (2014). Pediatric emotion dysregulation: Biological and developmental evidence for a dimensional approach. Psychiatric Quarterly, 85(3), 383–389. 10.1007/s11126-014-9300-z [DOI] [PubMed] [Google Scholar]
- Granger DA, Shirtcliff EA, Zahn-Waxler C, Usher B, Klimes-Dougan B, & Hastings P (2003). Salivary testosterone diurnal variation and psychopathology in adolescent males and females: Individual differences and developmental effects. Development and Psychopathology, 15, 431–449. 10.1017/S0954579403000233 [DOI] [PubMed] [Google Scholar]
- Gullone E, Hughes EK, King NJ, & Tonge B (2010). The normative development of emotion regulation strategy use in children and adolescents: A 2-year follow-up study. Journal of Child Psychology and Psychiatry, 51(5), 567–574. 10.1111/j.1469-7610.2009.02183.x [DOI] [PubMed] [Google Scholar]
- Hale WW, Raaijmakers Q, Muris P, Hoof A. Van, & Meeus W. im. (2008). Developmental trajectories of adolescent anxiety disorder symptoms: A 5-year prospective community study. Journal of the American Academy of Child & Adolescent Psychiatry, 47(5), 556–564. 10.1097/CHI.0b013e3181676583 [DOI] [PubMed] [Google Scholar]
- Hare TA, Tottenham N, Galvan A, Voss HU, Glover GH, & Casey BJ (2008). Biological substrates of emotional reactivity and regulation in adolescence during an emotional go-nogo task. Biological Psychiatry, 63(10), 927–934. 10.1016/j.biopsych.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havelock JC, Auchus RJ, & Rainey WE (2004). The rise in adrenal androgen biosynthesis: Adrenarche. Seminars in Reproductive Medicine, 22(4), 337–347. 10.1055/s-2004-861550 [DOI] [PubMed] [Google Scholar]
- Hayward C, Killen JD, Hammer LD, Litt IF, Wilson DM, Simmonds B, & Taylor CB (1992). Pubertal stage and panic attack history in sixth- and seventh-grade girls. American Journal of Psychiatry, 149(9), 1239–1243. 10.1176/ajp.149.9.1239 [DOI] [PubMed] [Google Scholar]
- Hayward C, Killen JD, Wilson DM, Hammer LD, Litt IF, Kraemer HC, … Taylor CB (1997). Psychiatric risk associated with early puberty in adolescent girls. Journal of the American Academy of Child and Adolescent Psychiatry, 36(2), 255–62. 10.1097/00004583-199702000-00016 [DOI] [PubMed] [Google Scholar]
- Heany SJ, Bethlehem RAI, van Honk J, Bos PA, Stein DJ, & Terburg D (2018). Effects of testosterone administration on threat and escape anticipation in the orbitofrontal cortex. Psychoneuroendocrinology, 96, 42–51. 10.1016/j.psyneuen.2018.05.038 [DOI] [PubMed] [Google Scholar]
- Heller AS, & Casey BJ (2016). The neurodynamics of emotion: Delineating typical and atypical emotional processes during adolescence. Developmental Science, 19(1), 3–18. 10.1111/desc.12373 [DOI] [PubMed] [Google Scholar]
- Hermans EJ, Ramsey NF, & van Honk J (2008). Exogenous testosterone enhances responsiveness to social threat in the neural circuitry of social aggression in humans. Biological Psychiatry, 63(3), 263–270. 10.1016/j.biopsych.2007.05.013 [DOI] [PubMed] [Google Scholar]
- Hettema JM, Kettenmann B, Ahluwalia V, McCarthy C, Kates WR, Schmitt JE, … Fatouros P (2012). Pilot multimodal twin imaging study of generalized anxiety disorder. Depression and Anxiety, 29, 202–209. 10.1002/da.20901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann W, Schmeichel BJ, & Baddeley AD (2012). Executive functions and self-regulation. Trends in Cognitive Sciences, 16(3), 174–80. 10.1016/j.tics.2012.01.006 [DOI] [PubMed] [Google Scholar]
- Hu S, Pruessner JC, Coupé P, & Collins DL (2013). Volumetric analysis of medial temporal lobe structures in brain development from childhood to adolescence. NeuroImage, 74, 276–287. 10.1016/j.neuroimage.2013.02.032 [DOI] [PubMed] [Google Scholar]
- Huerta R, & Brizuela-Gamiño OL (2002). Interaction of pubertal status, mood and self-esteem in adolescent girls. The Journal of Reproductive Medicine, 47(3), 217–25. [PubMed] [Google Scholar]
- Jedel E, Waern M, Gustafson D, Landen M, Eriksson E, Holm G, Nilsson L, Lind AK, Janson PO & Stener-Victorin E (2009). Anxiety and depression symptoms in women with polycystic ovary syndrome compared with controls matched for body mass index. Human Reproduction, 25(2), pp.450–456. 10.1093/humrep/dep471 [DOI] [PubMed] [Google Scholar]
- Johnson RT, Schneider A, DonCarlos LL, Breedlove SM, & Jordan CL (2012). Astrocytes in the rat medial amygdala are responsive to adult androgens. Journal of Comparative Neurology, 520(11), 2531–44. 10.1002/cne.23061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JE, Liu X, Jiang Y, Lynam D, & Kelly TH (2009). Neural correlates of emotional reactivity in sensation seeking. Psychological Science, 20(2), 215–223. 10.1111/j.1467-9280.2009.02283.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczkurkin AN, Moore TM, Ruparel K, Ciric R, Calkins ME, Shinohara RT, … Satterthwaite TD (2016). Elevated amygdala perfusion mediates developmental sex differences in trait anxiety. Biological Psychiatry, 80(10), 775–785. 10.1016/j.biopsych.2016.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, & Walters EE (2005). Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry, 62(6), 593. 10.1001/archpsyc.62.6.593 [DOI] [PubMed] [Google Scholar]
- Kim MJ, & Whalen PJ (2009). The structural integrity of an amygdala-prefrontal pathway predicts trait anxiety. Journal of Neuroscience, 29(37), 11614–11618. 10.1523/JNEUROSCI.2335-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klump KL (2013). Puberty as a critical risk period for eating disorders: A review of human and animal studies. Hormones and Behavior, 64, 399–410. 10.1016/j.yhbeh.2013.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacreuse A, Chiavetta MR, Shirai AAC, Meyer JS, & Grow DR (2009). Effects of testosterone on cognition in young adult male rhesus monkeys. Physiology and Behavior, 98(5), 524–531. 10.1016/j.physbeh.2009.08.007 [DOI] [PubMed] [Google Scholar]
- Lacreuse A, King HM, Kurdziel LB, Partan SR, Caldwell KM, Chiavetta MR, … Grow DR (2010). Testosterone may increase selective attention to threat in young male macaques. Hormones and Behavior, 58(5), 854–863. 10.1016/j.yhbeh.2010.08.010 [DOI] [PubMed] [Google Scholar]
- Lang PJ, Bradley MM, & Cuthbert BN (1990). Emotion, attention, and the startle reflex. Psychological Review, 97(3), 377–95. 10.1037/0033-295X.97.3.377 [DOI] [PubMed] [Google Scholar]
- Larson RW, Moneta G, Richards MH, & Wilson S (2002). Continuity, stability, and change in daily emotional experience across adolescence. Child Development, 73(4), 1151–1165. 10.1111/1467-8624.00464 [DOI] [PubMed] [Google Scholar]
- Larson RW, & Richards M (1998). Waiting for the weekend: Friday and Saturday night as the emotional climax of the week. New Directions for Child and Adolescent Development, (82), 37–52. 10.1002/cd.23219988204 [DOI] [PubMed] [Google Scholar]
- Lau JY, Britton JC, Nelson EE, Angold A, Ernst M, Goldwin M, … Pine DS (2011). Distinct neural signatures of threat learning in adolescents and adults. Proceedings of the National Academy of Sciences, 108(11), 4500–4505. 10.1073/pnas.1005494108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, & Beaulieu C (2011). Longitudinal development of human brain wiring continues from childhood into adulthood. Journal of Neuroscience, 31(30), 10937–10947. 10.1523/JNEUROSCI.5302-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Heller AS, van Reekum CM, Nelson B, & Davidson RJ (2012). Amygdala-prefrontal coupling underlies individual differences in emotion regulation. NeuroImage, 62(3), 1575–81. 10.1016/j.neuroimage.2012.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liening SH, Stanton SJ, Saini EK, & Schultheiss OC (2010). Salivary testosterone, cortisol, and progesterone: Two-week stability, interhormone correlations, and effects of time of day, menstrual cycle, and oral contraceptive use on steroid hormone levels. Physiology and Behavior, 99(1), 8–16. 10.1016/j.physbeh.2009.10.001 [DOI] [PubMed] [Google Scholar]
- Mädler B, Drabycz SA, Kolind SH, Whittall KP, & MacKay AL (2008). Is diffusion anisotropy an accurate monitor of myelination? Correlation of multicomponent T2 relaxation and diffusion tensor anisotropy in human brain. Magnetic Resonance Imaging, 26, 874–888. 10.1016/j.mri.2008.01.047 [DOI] [PubMed] [Google Scholar]
- Marowsky A, Yanagawa Y, Obata K, & Vogt KE (2005). A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron, 48(6), 1025–1037. 10.1016/j.neuron.2005.10.029 [DOI] [PubMed] [Google Scholar]
- Martin RE, & Ochsner KN (2016). The neuroscience of emotion regulation development: Implications for education. Current Opinion in Behavioral Sciences, 10, 142–148. 10.1016/j.cobeha.2016.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM (2008). Estradiol and the developing brain. Physiological Reviews, 88(1), 91–124. 10.1152/physrev.00010.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, & Arnold AP (2011). Reframing sexual differentiation of the brain. Nature Neuroscience, 14(6), 677–83. 10.1038/nn.2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, & Konkle ATM (2005). When is a sex difference not a sex difference? Frontiers in Neuroendocrinology, 26(2), 85–102. 10.1016/j.yfrne.2005.06.001 [DOI] [PubMed] [Google Scholar]
- McDermott CM, Liu D, & Schrader LA (2012). Role of gonadal hormones in anxiety and fear memory formation and inhibition in male mice. Physiology and Behavior, 105(5), 1168–1174. 10.1016/j.physbeh.2011.12.016 [DOI] [PubMed] [Google Scholar]
- McRae K, Gross JJ, Weber J, Robertson ER, Sokol-Hessner P, Ray RD, … Ochsner KN (2012). The development of emotion regulation: An fMRI study of cognitive reappraisal in children, adolescents and young adults. Social Cognitive and Affective Neuroscience, 7(1), 11–22. 10.1093/scan/nsr093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DJ, Duka T, Stimpson CD, Schapiro SJ, Baze WB, McArthur MJ, … Sherwood CC (2012). Prolonged myelination in human neocortical evolution. Proceedings of the National Academy of Sciences, 109(41), 863–869. 10.1073/pnas.1117943109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GA, & Chapman JP (2001). Misunderstanding analysis of covariance. Journal of Abnormal Psychology, 110(1), 40–48. 10.1037/0021-843X.110.1.40 [DOI] [PubMed] [Google Scholar]
- Moraga-Amaro R, van Waarde A, Doorduin J, & de Vries EFJ (2018). Sex steroid hormones and brain function: PET imaging as a tool for research. Journal of Neuroendocrinology, 30(2), e12565. 10.1111/jne.12565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawetz C, Bode S, Baudewig J, & Heekeren HR (2017). Effective amygdala-prefrontal connectivity predicts individual differences in successful emotion regulation. Social Cognitive and Affective Neuroscience, 12(4), 569–585. 10.1093/scan/nsw169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin AJS, Maïano C, Nagengast B, Marsh HW, Morizot J, & Janosz M (2011). General growth mixture analysis of adolescents’ developmental trajectories of anxiety: The impact of untested invariance assumptions on substantive interpretations. Structural Equation Modeling, 18(4), 613–648. 10.1080/10705511.2011.607714 [DOI] [Google Scholar]
- Morris NM, & Udry JR (1980). Validation of a self-administered instrument to assess stage of adolescent development. Journal of Youth and Adolescence, 9(3), 271–280. 10.1007/BF02088471 [DOI] [PubMed] [Google Scholar]
- Morabia A, Costanza MC, & World Health Organization Collaborative Study of Neoplasia and Steroid Contraceptives. (1998). International variability in ages at menarche, first livebirth, and menopause. American Journal of Epidemiology, 148(12), 1195–1205. 10.1093/oxfordjournals.aje.a009609 [DOI] [PubMed] [Google Scholar]
- Motzkin JC, Philippi CL, Wolf RC, Baskaya MK, & Koenigs M (2015). Ventromedial prefrontal cortex is critical for the regulation of amygdala activity in humans. Biological Psychiatry, 77(3), 276–284. 10.1016/j.biopsych.2014.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelemans SA III, Branje WWH, Raaijmakers SJT, Frijns QAW, om T, Lier P. A. C. Van, & Meeus WHJ (2014). Heterogeneity in development of adolescent anxiety disorder symptoms in an 8-year longitudinal community study. Development and Psychopathology, 26, 181–202. 10.1017/S0954579413000503 [DOI] [PubMed] [Google Scholar]
- Norman RJ, Dewailly D, Legro RS, & Hickey TE (2007). Polycystic ovary syndrome. The Lancet, 370(9588), 685–697. 10.1016/S0140-6736(07)61345-2 [DOI] [PubMed] [Google Scholar]
- Ochsner KN, & Gross JJ (2005). The cognitive control of emotion. Trends in Cognitive Sciences, 9(5), 242–9. 10.1016/j.tics.2005.03.010 [DOI] [PubMed] [Google Scholar]
- Olino TM, Klein DN, Lewinsohn PM, Rohde P, & Seeley JR (2010). Latent trajectory classes of depressive and anxiety disorders from adolescence to adulthood: Descriptions of classes and associations with risk factors. Comprehensive Psychiatry, 51(3), 224–35. 10.1016/j.comppsych.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson IR, Von Der Heide RJ, Alm KH, & Vyas G (2015). Development of the uncinate fasciculus: Implications for theory and developmental disorders. Developmental Cognitive Neuroscience, 14, 50–61. 10.1016/j.dcn.2015.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olweus D, Mattsson Å, Schalling D, & Low H (1980). Testosterone, aggression, physical, and personality dimensions in normal adolescent males. Psychosomatic Medicine, 42(2), 253–269. 10.1097/00006842-198003000-00003 [DOI] [PubMed] [Google Scholar]
- Öner S (2018). Neural substrates of cognitive emotion regulation: A brief review. Psychiatry and Clinical Psychopharmacology, 28(1), 91–96. 10.1080/24750573.2017.1407563 [DOI] [Google Scholar]
- Pagliaccio D, Luby JL, Luking KR, Belden AC, & Barch DM (2014). Brain-behavior relationships in the experience and regulation of negative emotion in healthy children: Implications for risk for childhood depression. Development and Psychopathology, 26(4pt2), 1289–1303. 10.1017/S0954579414001035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton GC, Hibbert ME, Carlin J, Shao Q, Rosier M, Caust J, & Bowes G (1996). Menarche and the onset of depression and anxiety in Victoria, Australia. Journal of Epidemiology and Community Health, 50, 661–666. 10.1136/jech.50.6.661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier G (2000). Localization of androgen and estrogen receptors in rat and primate tissues. Histology and Histopathology, 15, 1261–70. 10.14670/HH-15.1261 [DOI] [PubMed] [Google Scholar]
- Peters S, Jolles DJ, Duijvenvoorde ACKV, Crone EA, & Peper JS (2015). The link between testosterone and amygdala-orbitofrontal cortex connectivity in adolescent alcohol use. Psychoneuroendocrinology, 53, 117–126. 10.1016/j.psyneuen.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Pfeifer JH, Masten CL, Moore WE, Oswald TM, Mazziotta JC, Iacoboni M, & Dapretto M (2011). Entering adolescence: Resistance to peer influence, risky behavior, and neural changes in emotion reactivity. Neuron, 69(5), 1029–1036. 10.1016/j.neuron.2011.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan KL, Orlichenko A, Boyd E, Angstadt M, Coccaro EF, Liberzon I, & Arfanakis K (2009). Preliminary evidence of white matter abnormality in the uncinate fasciculus in Generalized Social Anxiety Disorder. Biological Psychiatry, 66(7), 691–694. 10.1016/j.biopsych.2009.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoenix CH, Goy RW, Gerall AA, & Young WC (1959). Organizing action of prenatally administerd testosterone proprionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology, 65, 369–382. 10.1210/endo-65-3-369 [DOI] [PubMed] [Google Scholar]
- Piekarski DJ, Johnson CM, Boivin JR, Thomas AW, Lin WC, Delevich K, … Wilbrecht L (2017). Does puberty mark a transition in sensitive periods for plasticity in the associative neocortex? Brain Research, 1654, 123–144. 10.1016/j.brainres.2016.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pine DS (2007). Research review: A neuroscience framework for pediatric anxiety disorders. Journal of Child Psychology and Psychiatry, 48(7), 631–648. 10.1111/j.1469-7610.2007.01751.x [DOI] [PubMed] [Google Scholar]
- Primus RJ, & Kellogg CK (1990). Gonadal hormones during puberty organize environment-related social interaction in the male rat. Hormones and Behavior, 24(3), 311–323. 10.1016/0018-506X(90)90012-M [DOI] [PubMed] [Google Scholar]
- Quevedo KM, Benning SD, Gunnar MR, & Dahl RE (2009). The onset of puberty: Effects on the psychophysiology of defensive and appetitive motivation. Development and Psychopathology, 21(1), 27–45. 10.1017/S0954579409000030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, & Paré D (2003). Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. Journal of Neuroscience, 23(25), 8800–8807. 10.1523/JNEUROSCI.23-25-08800.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon LE, Leen-Feldner EW, & Hayward C (2009). A critical review of the empirical literature on the relation between anxiety and puberty. Clinical Psychology Review, 29, 1–23. 10.1016/j.cpr.2008.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JM, Plate RC, & Ernst M (2012). Neural systems underlying motivated behavior in adolescence: Implications for preventive medicine. Preventive Medicine, 55, S7–S16. 10.1016/j.ypmed.2011.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AC, Reekie Y, & Braesicke K (2007). Synergistic and regulatory effects of orbitofrontal cortex on amygdala-dependent appetitive behavior. Annals of the New York Academy of Sciences, 1121(1), 297–319. 10.1196/annals.1401.019 [DOI] [PubMed] [Google Scholar]
- Roesch M, & Schoenbaum G (2006). From associations to expectancies: Orbitofrontal cortex as gateway between the limbic system and representational memory. The Orbitofrontal Cortex. 10.1093/acprof:oso/9780198565741.003.0008 [DOI] [Google Scholar]
- Rolls ET (1999). The functions of the orbitofrontal cortex. Neurocase, 5(4), 301–312. 10.1080/13554799908411984 [DOI] [Google Scholar]
- Rosenkranz JA, & Grace AA (2002). Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. Journal of Neuroscience, 22(1), 324–337. 10.1523/JNEUROSCI.22-01-00324.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherf KS, Smyth JM, & Delgado MR (2013). The amygdala: An agent of change in adolescent neural networks. Hormones and Behavior, 64(2), 298–313. 10.1016/j.yhbeh.2013.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz KM, & Sisk CL (2016). The organizing actions of adolescent gonadal steroid hormones on brain and behavioral development. Neuroscience and Biobehavioral Reviews, 70, 148–158. 10.1016/j.neubiorev.2016.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz KM, Zehr JL, Salas-Ramirez KY, & Sisk CL (2009). Testosterone programs adult social behavior before and during, but not after, adolescence. Endocrinology, 150(8), 3690–8. 10.1210/en.2008-1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman DL, & Brophy PJ (2005). Mechanisms of axon ensheathment and myelin growth. Nature Reviews Neuroscience, 6(9), 683–690. 10.1038/nrn1743 [DOI] [PubMed] [Google Scholar]
- Shirtcliff EA, Dahl RE, & Pollak SD (2009). Pubertal development: Correspondence between hormonal and physical development. Child Development, 80(2), 327–337. 10.1111/j.1467-8624.2009.01263.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, & Merchenthaler I (1997). Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. Journal of Comparative Neurology, 388, 507–525. [DOI] [PubMed] [Google Scholar]
- Sonis WA, Comite F, Blue J, Pescovitz OH, Rahn CW, Hench KD, … Klein RP (1985). Behavior problems and social competence in girls with true precocious puberty. The Journal of Pediatrics, 106(1), 156–60. 10.1016/S0022-3476(85)80489-3 [DOI] [PubMed] [Google Scholar]
- Spielberg JM, Forbes EE, Ladouceur CD, Worthman CM, Olino TM, Ryan ND, & Dahl RE (2015). Pubertal testosterone influences threat-related amygdala-orbitofrontal cortex coupling. Social Cognitive and Affective Neuroscience, 10, 408–415. 10.1093/scan/nsu062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielberg JM, Olino TM, Forbes EE, & Dahl RE (2014). Exciting fear in adolescence: Does pubertal development alter threat processing? Developmental Cognitive Neuroscience, 8, 86–95. 10.1016/j.dcn.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg L (2008). A Social Neuroscience Perspective on Adolescent Risk-Taking. Developmental Review, 28(1), 78–106. 10.1016/j.dr.2007.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susman EJ, Dorn LD, & Chrousos GP (1991). Negative affect and hormone levels in young adolescents: Concurrent and predictive perspectives. Journal of Youth and Adolescence, 20(2), 167–190. 10.1007/BF01537607 [DOI] [PubMed] [Google Scholar]
- Swartz JR, Carrasco M, Wiggins JL, Thomason ME, & Monk CS (2014). Age-related changes in the structure and function of prefrontal cortex-amygdala circuitry in children and adolescents: A multi-modal imaging approach. NeuroImage, 86, 212–220. 10.1016/j.neuroimage.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JM, Higgs S, Dourish CT, Hansen PC, Harmer CJ, & McCabe C (2015). Satiation attenuates BOLD activity in brain regions involved in reward and increases activity in dorsolateral prefrontal cortex: An fMRI study in healthy volunteers. The American Journal of Clinical Nutrition, 101, 697–704. 10.3945/ajcn.114.097543 [DOI] [PubMed] [Google Scholar]
- Tottenham N, & Galván A (2016). Stress and the adolescent brain: Amygdala-prefrontal cortex circuitry and ventral striatum as developmental targets. Neuroscience and Biobehavioral Reviews, 70, 217–227. 10.1016/j.neubiorev.2016.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottenham N, Hare TA, & Casey BJ (2011). Behavioral assessment of emotion discrimination, emotion regulation, and cognitive control in childhood, adolescence, and adulthood. Frontiers in Psychology, 2, 1–9. 10.3389/fpsyg.2011.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromp DPM, Grupe DW, Oathes DJ, McFarlin DR, Hernandez PJ, Kral TRA, … Nitschke JB (2012). Reduced structural connectivity of a major frontolimbic pathway in Generalized Anxiety Disorder. Archives of General Psychiatry, 69(9), 925. 10.1001/archgenpsychiatry.2011.2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromp DPM, Williams LE, Fox AS, Oler JA, Roseboom PH, Rogers GM, … Kalin NH (2019). Altered uncinate fasciculus microstructure in childhood anxiety disorders in boys but not girls. American Journal of Psychiatry. 10.1176/appi.ajp.2018.18040425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tromp DPM, Williams LE, Oler JA, Rogers GM, Benson BE, Fox AS, … Kalin NH (2017). Males, but not females, demonstrate altered uncinate fasciculus microstructure in anxiety disorders. Symposium conducted at the Society of Biological Psychiatry 72nd Annual Scientific Convention, San Diego, CA. [Google Scholar]
- Tröstl J, Sladky R, Hummer A, Kraus C, Moser E, Kasper S, … Windischberger C (2011). Reduced connectivity in the uncinate fiber tract between the frontal cortex and limbic subcortical areas in social phobia. European Psychiatry, 26, 182. 10.1016/S0924-9338(11)71893-4 [DOI] [Google Scholar]
- Turkeltaub PE, Eickhoff SB, Laird AR, Fox M, Wiener M, & Fox P (2012). Minimizing within-experiment and within-group effects in activation likelihood estimation meta-analyses. Human Brain Mapping, 33, 1–13. 10.1002/hbm.21186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wingen G, Mattern C, Verkes RJ, Buitelaar J, & Fernández G (2010). Testosterone reduces amygdala-orbitofrontal cortex coupling. Psychoneuroendocrinology, 35, 105–113. 10.1016/j.psyneuen.2009.09.007 [DOI] [PubMed] [Google Scholar]
- van Wingen G, Zylicz SA, Pieters S, Mattern C, Verkes RJ, Buitelaar JK, & Fernández G (2009). Testosterone increases amygdala reactivity in middle-aged women to a young adulthood level. Neuropsychopharmacology, 34, 539–547. 10.1038/npp.2008.2 [DOI] [PubMed] [Google Scholar]
- Walf AA, & Frye CA (2006). A review and update of mechanisms of estrogen in the hippocampus and amygdala for anxiety and depression behavior. Neuropsychopharmacology, 31(6), 1097–1111. 10.1038/sj.npp.1301067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein SM, & Mermelstein R (2007). Relations between daily activities and adolescent mood: The role of autonomy. Journal of Clinical Child and Adolescent Psychology, 36(2), 182–94. 10.1080/15374410701274967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zald DH, & Kim SW (2001). The orbitofrontal cortex. In The frontal lobes and neuropsychiatric illness (pp. 33–69). Arlington, VA, US: American Psychiatric Publishing, Inc. [Google Scholar]
- Zimmermann P, & Iwanski A (2014). Emotion regulation from early adolescence to emerging adulthood and middle adulthood: Age differences, gender differences, and emotion-specific developmental variations. International Journal of Behavioral Development, 38(2), 182–194. 10.1177/0165025413515405 [DOI] [Google Scholar]
- Zuloaga DG, Jordan CL, & Breedlove SM (2011). The organizational role of testicular hormones and the androgen receptor in anxiety-related behaviors and sensorimotor gating in rats. Endocrinology, 152(4), 1572–81. 10.1210/en.2010-1016 [DOI] [PMC free article] [PubMed] [Google Scholar]