

Introduction
Stress and the stress response are fundamentally central features of any disease. Stress and the stress response are also clearly central features in the pathophysiology of tauopathy and the biology of AD. Chapters 16, 23, 28 and 29 in this book address how stress modifies the phosphorylation of tau, the role of stress in the biology of the endoplasmic reticulum and the translational stress response, and the effects of behavioral stress and glucocorticoids on tau. The stress response is undoubtedly necessary to cope with the harm arising from chronic exposure to β-amyloid (Aβ), oligomeric tau and age or disease related reductions in cerebral blood flow. However, this chapter will address the paradoxical concept that particular elements of the stress response mediated by RNA binding proteins actually accelerate tau oligomerization and thereby accelerate disease progression in tauopathy. Conversely, we will also show how RNA binding proteins are therefore key targets for therapeutic intervention in tauopathies, including Alzheimer’s disease.
Dysfunction of RNA Binding Proteins in Neurodegenerative Disorders
Reviewing the basic biology of RBPs, stress granules and the translational stress response is necessary in order to understand how and why tau would interact with RBPs. An increasing number of genetic studies show that RNA binding proteins (RBPs) are central to the pathophysiology of multiple neurological disorders. Mutations in genes that encode RBPs cause motor neuron diseases such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), multisystem proteinopathy (MSP), and frontotemporal lobar degeneration (FTLD) [40]. ALS is the most common motor neuron disorder that leads to progressive loss of upper and lower motor neurons, muscle weakness, atrophy and ultimately death [78].
TAR DNA-binding proteins 43 (TDP-43) is the major constituent of pathological ubiquitinated inclusions present in ALS and frontotemporal dementias defined by progranulin haploinsufficiency (FTD-TDP-43 or FTD-U) [69]. Mutations in TDP-43 also cause rare forms of familial ALS, which demonstrates that dysfunction of TDP-43 is sufficient to cause disease [83]. TDP-43 is not the only RBP to exhibit mutations linked to ALS, though. Multiple RBPs exhibit mutations that are genetically linked to ALS: heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP A2/B1), fused in sarcoma/translocates in liposarcoma (FUS/TLS), Ewing’s sarcoma breakpoint region 1 (EWSR1), TATA-box binding protein associated factor 15 (TAF15), Matrin3 (MATR3), and TIA1 cytotoxic associated granule binding protein (TIA1) [40, 53] (Table 26.1). In addition, polyglutamine (polyQ) expansions (27–33 Qs) in Ataxin-2 (ATXN-2) are an important genetic risk for ALS [19]. RBPs are generally defined by the presence of a homologous RNA binding domain, the RNA recognition motif (RRM); many RBPs also share homologous low complexity Gly-rich domains (LCDs) [31, 53]. FUS, EWSR1, and TAF15, which comprises the FET family, also share common zinc finger domains [31]. The RRMs allow the binding of these proteins to DNA and RNA in a sequence-specific manner, while the LCD is a prion-like domain that plays a critical role in the formation and the dynamic biophysical state of stress granules (SGs) [30]. These RBPs are multifunctional RNA processing proteins that predominantly reside in the nucleus and are generally expressed in many different types of cells and tissues [40].
Table 26.1.
Physiological and pathophysiological functions of RBPs and non-RBPs implicated in neurodegenerative diseases
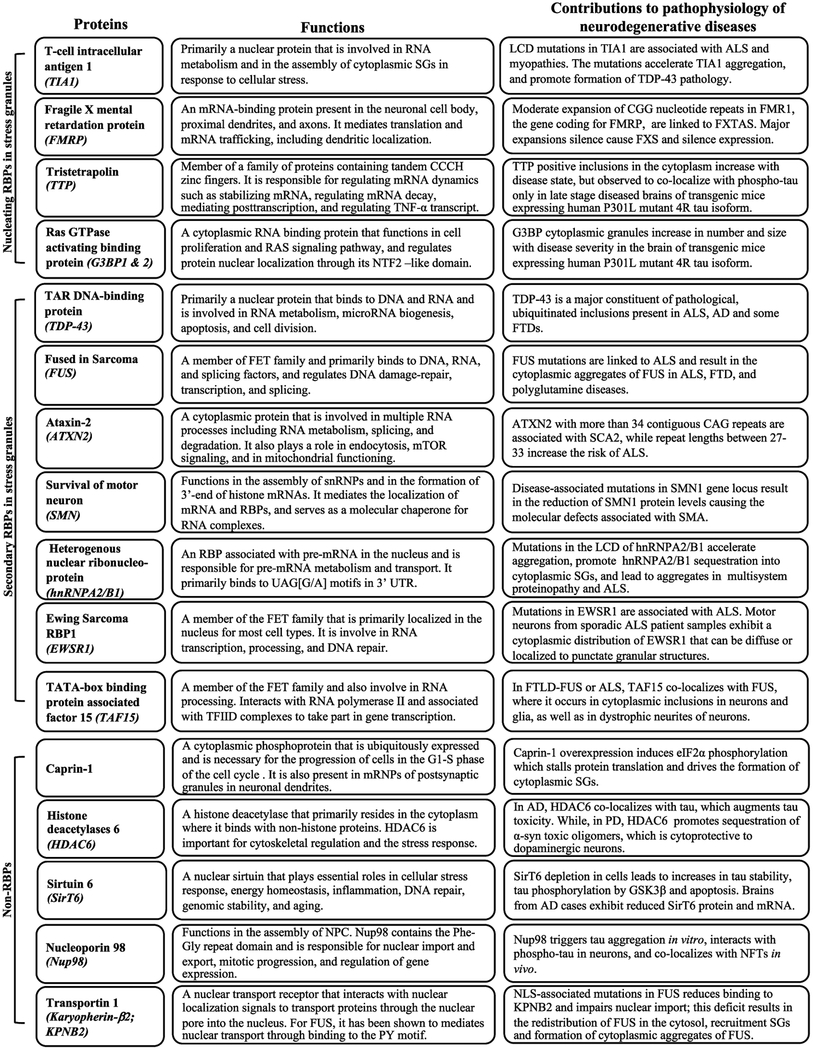
|
ALS: Amyotrophic Lateral Sclerosis; fALS: familial ALS; AD: Alzheimer’s disease; FXTAS: Fragile X-associated Tremor/Ataxia syndrome; FXS: fragile X syndrome; TNF-α: Tumor Necrosis Factor Alpha; NTF2-like domain: N-terminal Nuclear Transport Factor 2-like domain; FTD: Frontotemporal Dementia; SCA2: Spinocerebellar Ataxia Type 2; NPCs: Nuclear Pore Complex; NLS: Nuclear Localization Signal; mRNP: Ribonuceloprotein; RBPs: RNA binding proteins; snRNPs: Small Nuclear Ribonucleoproteins; SMA: Spinal Muscular Atrophy.
RNA Binding Proteins Mediate Disease Through Stress Granules
The identification of disease-linked mutations in the genes that encode these RBPs, particularly TDP-43 and FUS, introduced a paradigm shift in the study of ribostasis and proteostasis in disease. Under physiological conditions, TDP-43 and FUS localize in the nucleus. But, in the presence of a cellular stress, they redistribute from the nucleus to the cytoplasm where they associate with SGs (Fig. 26.1). These granules serve as a cytoprotective mechanism against stress as it temporarily inhibits the translation of non-essential mRNA and promote the translation of transcripts necessary for cell survival [41, 49]. However, mutations in TDP-43 and FUS increase the propensity to aggregate, leading to the accumulation of persistent cytoplasmic SGs, and the formation of pathological inclusions of these proteins in the human brain [9, 16, 48]. Support for the hypothesis that the pathological inclusions in brain derive from SGs comes from observed colocalization with SG markers such as eIF3, eIF4G, TIA1 and PABP [48, 16].
Fig. 26.1.
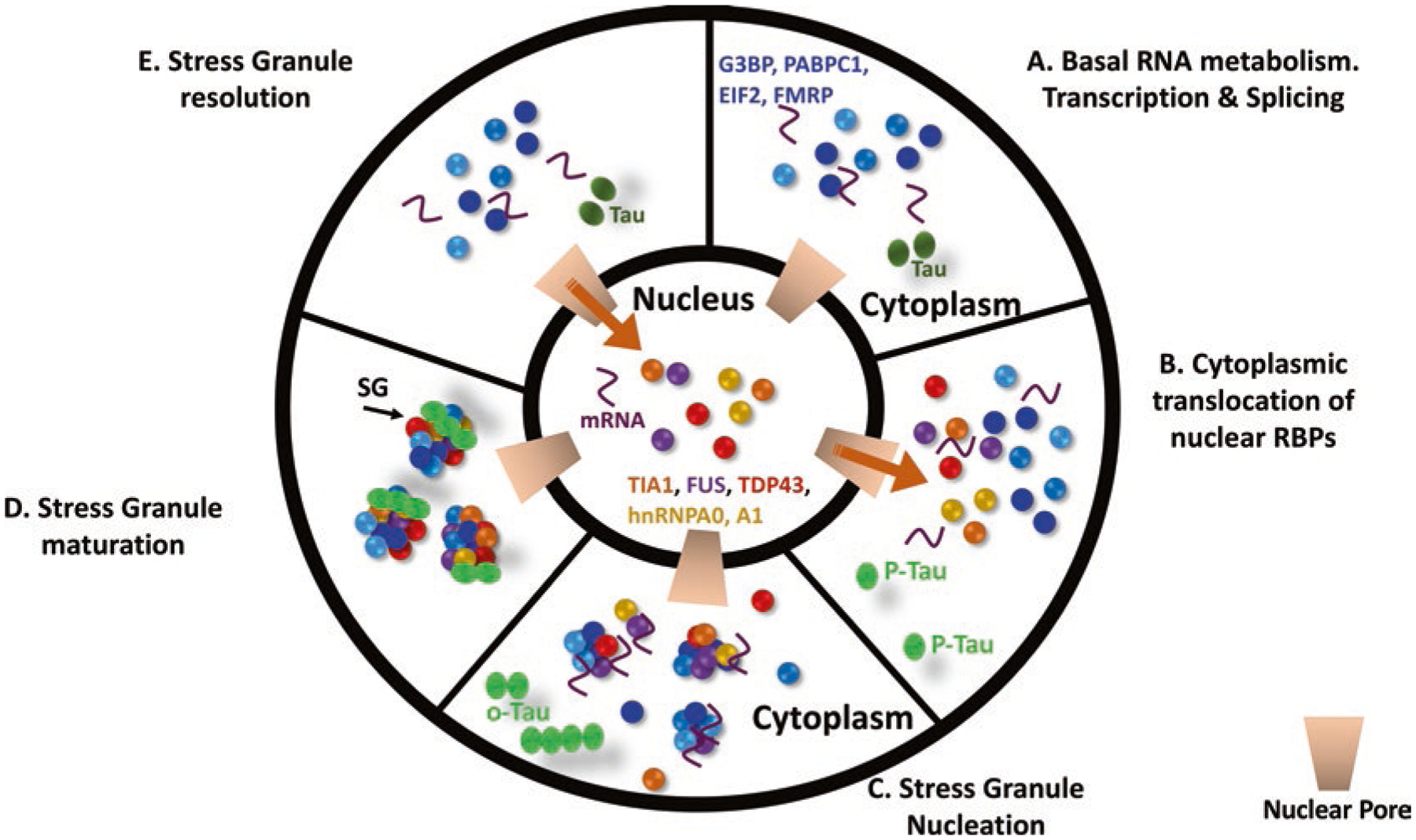
The stress granule cycle. (a) Under basal conditions most RNA binding proteins are nuclear, while some are predominantly cytoplasmic. (b) With stress, nuclear RNA binding proteins exit the nucleus through the nuclear pore (arrow) and enter the cytoplasm. (c) The SGs begin to nucleate. Proteins such as TIA1, TIAR, TTP, G3BP and FMRP are primary nucleators of SGs. (d) The SGs mature over time, consolidating and beginning to incorporate multiple secondary proteins that include RNA binding proteins as well as proteins with functions independent of RNA (e.g., autophagy, apoptosis). (e) Upon resolution of the stress, the SGs dissolve and nuclear RNA binding proteins return to the nucleus
This chapter will focus on SGs because the relationship to disease is clear, SGs provide a linear pathway between chronic stress and disease, and the disease-associated RBPs examined to date have been shown to co-localize with SGs. However, RBPs form many types of RNA granules to mediate many different functions, including splicing, transcription, ribosome genesis, RNA transport, RNA degradation, RNA translation, viral defense and many other functions. The pathophysiological principles linking SGs to disease presented below might also apply to these other types of RBPs, particularly for familial RBPs that exhibit mutations rendering the RBPs more aggregation prone. However, differing types of RNA granules likely differ in their propensity to precipitate the irreversible protein aggregation that causes disease because of differences in the types of proteins associated with each RNA granule type and/or differences in post-translational modifications, as discussed below. SGs currently appear to represent the RNA granule exhibiting the greatest propensity to elicit the irreversible protein aggregation associated with neurodegenerative diseases.
Many of the mutations in FUS are thought to increase cytoplasmic localization by disrupting the nuclear localization signal, which prevents the transportation of these proteins to the nucleus [16, 71, 94]. The disease associated mutations in TDP-43 occur predominantly in the LCD domain, increase the tendency to aggregate, and formation of cytoplasmic SGs [48, 38]. In most cases, the increased tendency to aggregate shifts the biophysical state of these granules, that is thought to extend their persistence, resulting in the formation of persistent pathological SGs which can then consolidate to form the classic pathologic structures that are associated with disease [55]. Finally, it is important to note that cleavage of TDP-43 can produce fragments with a strong tendency to aggregate through a mechanism that is not strongly linked to the translational stress response [71, 94].
The Biology of Stress Granules and the Translational Stress Response
SGs are cytoplasmic complexes that form in concert with inhibition of RNA translation; stimulate of SG include a wide variety of stresses that include nutritional stress, heat or osmotic shock, DNA damage or proteostatic dysfunction [14, 25, 41, 42, 56, 57, 64]. SGs are classically made up of mRNAs, RNA-binding proteins, small 40S ribosome subunits, translation initiation factors, and broadly non-RNA binding proteins [12, 64, 76]. SG formation occurs in stages, with core nucleating RBPs initiating SG formation, followed by secondary association of a wide variety of proteins. The complexity of SGs varies with the type of stress, the type of cell and the duration of the stress [54].
The initial changes in RNA metabolism induced by stress result in polysome stalling and nucleation of SGs by a set of core nucleating RBPs, which include TIA1, G3BP1 and 2, FMRP, TTP and TIAR; proteomic experiments point to a comprehensive list of core RBPs (Fig. 26.1) [5, 54]. Nucleation of these RBPs is controlled by PTMs, which are described below, and by location. TIA1 for instance, translocates from the nucleus to the cytoplasm during stress [41]. These core nucleating components associate with the mRNA transcripts and protein components of the stalled initiation complexes including eIF3, eIF4F (consist of eIF4E, eIF4A and eIF4G), eIF4B, small ribosomal subunits and Poly A Binding protein 1 (Table 26.1) [41, 42]. A wide variety of secondary proteins associate with SGs. These proteins include many different RBPs, including those associated with ALS, such as TDP-43, FUS and hnRNPA0 [41, 42]. However, secondary proteins that associate with SGs also include scaffolding proteins, such as caprin, signaling proteins such as HDAC6 and SirT6, nuclear pore proteins such as nup98 [17], disaggregases such as karyopherin-b2 (Transportin-1, Kapnb2) (Table 26.1) [16, 27, 28, 32], and proteins linked to cell death pathways, such as TRAF and FAST (Table 26.1) [11, 23, 44].
Phase Transition and the Role of Protein Aggregation in the Biology of SGs
The formation and consolidation of SGs (and other RNA granules) appears to depend on the biophysical processes of liquid-liquid phase separation (LLPS) and protein aggregation. The process of LLPS is described in detail in Chaps. 24 and 25 (Kosik & Han, Wegmann), but a brief overview will be given in this chapter because of its fundamental importance for understanding the biology of SGs. The roles of LLPS and protein aggregation are the essential features that make SGs so important for the pathophysiology of neurodegenerative diseases generally, and tauopathies specifically. Under transient stress conditions SG components assemble and disassemble quickly, forming the highly dynamic structures that are governed by the biophysics of phase separation [5, 46]. The dynamic nature of these phase separated proteins enables transitions between multiple protein conformations. A fundamental weakness of this biology gives rise to neurodegenerative diseases. With extended time, such as might occur with chronic stress, some SG proteins transit into highly stable amyloid like states, similar to that observed in the protein aggregates that form in neurodegenerative diseases [65, 74]; mutations that are associated with familial disease frequently increase the rate of amyloidogenic transition [65, 74]. Secondary nucleation also allows the association of SGs with other proteins, such as tau, that exhibit a high propensity to aggregate into stable amyloids [88].
Three different biophysical considerations explain the biology of LLPS. (1) The physical chemistry of liquids creates the conditions for LLPS. Chemicals that are in liquid form and exhibit strong physical differences, such as oil and water, will phase separate to minimize the free energy of the mixture, reduce unfavorable interactions and promote weak bonding. The aqueous nature of proteins thus provides the conditions that allow for phase separation. The phase separation is promoted by weak bonding of low complexity protein sequences that consist primarily of alanine, glycine, glutamine and proline, with some extra complexity arising from interspersed arginine and asparagine [34, 75]. (Note that the phase separation of proteins does not produce the extreme concentrations (e.g., 55 M) occurring when oil and water phase separate because the weak interactions present in proteins are only moderately favored over interactions between proteins and water in the aqueous solution) (Fig. 26.1a). The low affinity binding of multiple short regions of low complexity domains produces the dynamic phase separation that characterizes RNA granules [91, 92]. (2) The low complexity regions that promote LLPS occur in intrinsically disordered regions (referred to as IDRs in the literature). The lack of order enables the “sticky” sequences in these regions to move in a dynamic manner forming the multiple weak associations that drive the LLPS. (3) The final consideration is polymer chemistry. RNA greatly facilitates protein based LLPS by forming a scaffold that helps to stabilize the phase separating proteins, keeping them generally in the same region [20]. Thus, RBPs bound to RNA phase separate at a lower concentration than is required in absence of RNA [20]. The tendency of RBPs to cluster around RNA combines with the presence of intrinsically disordered regions that can self-associate in a low affinity manner to render LLPS a prominent feature of RBP biology, leading to formation of many types of RNA granules, including SGs.
Tau and Stress Granules
Tau Is Sorted to the Somatodendritic Domain in Stress
The information presented above provide a clear mechanistic pathway through which RBPs, SGs and RNA metabolism contribute to pathological aggregation and the pathophysiology of motoneuron diseases. The section below will explain how this mechanism involves tau, and the profound manner in which RBPs, SGs and the translational stress response contribute to the pathophysiology of tauopathy.
Tau is normally most abundant in the axons of neurons, where its primary function is to stabilize microtubules [4]. In AD, as well as in stress, tau becomes phosphorylated by proline directed kinases, such as glycogen synthase kinase β (GSK3β), cyclin dependent kinase 5 (CDK5) and microtubules affinity-regulating kinases (MARKs); this type of phosphorylation will be referred to as “hyperphosphorylation” [89]; this hyperphosphorylated tau accumulates in the somatodendritic arbor where it eventually forms neurofibrillary tangles. Originally hyperphosphorylated tau was through to translocate from the axon to the cytoplasm, but the translocation model didn’t fit the pathology, which does not show extensive axonal tau phosphorylation. More recent data indicate that accumulation occurs as newly synthesized tau becomes phosphorylated and is prevented from entering the axon [33, 84, 96]. In the presence stress, tau is phosphorylated and localizes in the soma and dendrite where it can interact with RBPs associated with SGs [33, 84]. The accumulating phospho-tau arises from phosphorylation of de novo synthesized tau rather than translocation of phospho-tau from the axon [96]. The physiological logic for which the neuron would change the distribution of tau represents a fundamental question for tau research, and is one that has never been explained. We propose that hyperphosphorylated tau accumulates in the somatodendritic arbor during stress to adapt protein synthesis to address the stress [88].
Tau Regulates Stress Granules
The role of tau in the translational stress response is evident from studies of RBPs in neurons during stress. The relationship of tau to SGs is readily apparent when examining TIA1, an RBP that is one of the core nucleating SG proteins. In cell lines TIA1 is completely nuclear under basal conditions, and translocates into the cytoplasm under stress. Translocation of TIA1 has been demonstrated in response to many different stresses as well as viral infections (for a general review of TIA1, see Anderson et al. [1]), but also includes stresses that are very relevant to disease, such as arsenite, glucocorticoids, Aβ and tau oligomers [37, 82, 87, 88]. In each case, the resulting TIA1 SGs co-localize with hyperphosphorylated tau. However, comparison of SGs associated with TIA1 and G3BP1 demonstrate that SGs are not uniform species [54]. The neuronal SY5Y cell line exhibits a strong stress response after glucocorticoid treatment, exhibiting both TIA1 and G3BP1 positive SGs [82]. Interestingly, hyperphosphorylated tau inclusions strongly colocalize with TIA1-positive SGs, but show little colocalization with G3BP1-positive SGs [82]. The relevance of this point becomes clearer when considering tau pathology in vivo (discussed below), where one sees induction of both TIA1 and G3BP1 pathology with disease, but only TIA1 colocalizes with hyperphosphorylated tau [87]. In neurons under basal conditions TIA1 is also abundant in the nucleus, but also has some presence in the cytoplasm [88]. However, in tau knockout neurons, TIA1 is completely nuclear [88]. In addition, under conditions of stress (e.g., Aβ toxicity), TIA1 exhibits reduced translocation to the cytoplasm in tau KO neurons (Wolozin, personal communication). Conversely, over-expressing tau increases SG formation and the associated repression of protein synthesis [60, 87, 88].
Independent approaches support the intimate link between tau, SGs and translational control. Tau is known to exist in dendrites near dendritic boutons [36]. These small tau granules appear to be linked to translational control/protein synthesis because chemicals that modulate protein synthesis affect the tau granule distribution. Cycloheximide, which inhibits protein synthesis and inhibits SG formation, also prevents clustering of tau into granules in the dendritic arbor. Conversely, puromycin, which also inhibits protein synthesis but stimulates SG formation, enhances clustering of tau into granules in the dendritic arbor [88]. Other SG inhibitors, such as the protein kinase R inhibitor C16 or the PERK inhibitor GSK2606414 also prevent coalescence of tau with SGs [88]. Immunoprecipitating TIA1 identifies tau in the protein interactome network, as well as many other RBPs for which binding to TIA1 requires tau, and immunoprecipitating tau identifies multiple co-associating RBPs [26, 35, 55, 88, 90]. These data demonstrate that the biology of tau is tightly connected with that of SGs and translational control.
Tau exhibits a tendency to phase separate in the presence of RNA in vitro much like RBPs, as discussed in the Chaps. 24 and 25 by Kosik and by Wegmann [18, 97]. The propensity of tau to phase separate might facilitate its interaction with RBPs and the formation of SGs, although this point has yet to be empirically demonstrated. Hyperphosphorylation of tau increases its propensity to form droplets in vitro, which suggests that tau hyperphosphorylation might function to promote the formation of phase separated complexes of tau, RBPs and RNA [18]. However, tau also has a strong tendency to fibrillize, and studies of human tau in neurons show that hyperphosphorylation renders tau prone to irreversible aggregation [18]. Thus, the consolidation of tau into droplets and SGs might increase the tendency of tau to aggregate, thereby enhancing a pathway that leads to neurodegeneration.
Tau Colocalizes with RNA Binding Proteins in Disease
These cell culture studies complement cogent pathological data. The connection between tau and RBPs is evident in pathological tissues from human cases of AD, FTD-tau as well as animal models of tauopathy. Molecular pathology studies show colocalization of pathological tau (hyperphosphorylated or misfolded) with multiple RBPs [2, 3, 55, 82, 87, 88]. The degree of co-localization detected in human tissues, though, likely under-represents reality because detection of RBPs in situ decreases dramatically with fixation time [55]. RBPs are abundantly detectable at <2 h of fixation and remain readily detectable at <24 h of fixation, but become difficult to detect at >48 h of fixation [55]; this sensitivity to fixation duration impacts greatly on staining of human tissues because most human cases have been fixed for much more than 48 h.
The pattern of SG pathology and co-localization with tau differs dramatically based on the RNA binding protein examined. TIA1, colocalizes strongly with pathological tau in human tissues [87]. In contrast, rasGAP-binding protein (G3BP) only shows weak colocalization with phosphorylated tau, despite exhibiting increased accumulation in neurons with increasing disease severity [87]. In animal tissues, where shorter fixation times are possible, tau is observed to co-localize with multiple other RBPs, including DDX6, eIF2α, hnRNPA0, and PABP [55, 82]. Interestingly, the pattern of reactivity appears to differ with the type of pathology. Co-localization of tau with RBPs is strongest with smaller inclusions; mature neurofibrillary tangles exhibit accumulation of RBPs adjacent to the pathological tau tangles, suggesting the hypothesis that the RPBs become excluded as tau consolidates to form the mature tangle [55].
The putative dysfunction of RBPs and RNA metabolism in tauopathy can be tested by examining RNA splicing. If RBPs become sequestered as protein aggregates in persistent pathological SGs, then one might expect to observe effects on RNA metabolism when examined through the lens of RNAseq. Indeed, multiple transcriptome studies show dramatic changes in RNA transcriptomes in tauopathies. Studies from several laboratories, including our own, show that splicing is dramatically altered in animal models of tauopathy as well as in cases of AD [2, 3, 6, 27, 28, 63, 81]. The changes in splicing are far greater than the comparatively modest changes in transcript levels. Since the splicesome is made up of RBPs, the large changes in splicing that occur with disease are consistent with a model in which RBPs become sequestered away from splicesomes in the nucleus leading to dysfunctional splicing.
Tau Oligomers Mediate Interactions with RNA Binding Proteins
Animal models provide insight into the mechanisms underlying the interaction of tau with RBPs, SGs and the translational stress response. Our laboratory recently crossed PS19 P301S tau mice with TIA1−/− mice, and demonstrated that reducing TIA1 in vivo provides striking rescue of the degenerative phenotype associated with the PS19 P301S tau mice [2, 3]. Reducing TIA1 expression by 50% greatly decreased the number and size of cytoplasmic pathological TIA1 granules (which colocalize with SG markers). The TIA1 reduction also yielded striking rescue of behavior, neuronal and synaptic degeneration, cortical thickness, as well as a 26% increase in survival despite the continued five-fold over-expression of tau [2, 3]. TIA1 reduction also decreased the amount of hyperphosphorylated tau evident at 3 months of age [2, 3]. This acute reduction in tau pathology is consistent with cell culture studies showing that TIA1 knockdown also provides neuroprotection and reduces tau pathology [88].
Insights into the mechanism of tau/TIA1 interactions arise from our studies examining the mice at later ages, as well as from a subsequent study comparing the propagation of oligomeric and fibrillar tau. Aging of the P301S tau::TIA1+/+ and +/− mice showed dramatic changes in the aggregation of tau. The P301S tau::TIA1+/− exhibited striking (90%) reduction in the accumulation of oligomeric tau at 9 months, and an equally striking increase (>10-fold) in fibrillar tau at 9 months of age. Analysis by immune-electron microscopy demonstrated that TIA1 binds to phosphorylated tau oligomers but not tau fibrils; the ability of TIA1 to increase tau oligomerization (assessed by ELISA) confirmed this observation [88]. These data suggest the hypothesis that TIA1 interacts selectively with phosphorylated tau oligomers.
The selective interaction between tau oligomers and TIA1 is supported by independent studies of tau propagation. Tau oligomers and fibrils were isolated from 9-month P301S tau mice, and propagated in both WT and P301S tau mice; the results were similar for both but more striking in the mice over-expressing human tau [37]. Both oligomeric and fibrillar tau exhibited robust propagation, which is consistent with Chaps. 30 and 31 and multiple reports in the literature [15, 47, 79]. The experimental design also allowed side by side comparison of toxicity, with the results showing that oligomeric tau produced abundant tau pathology and abundant neurodegeneration while the fibrillar tau produced abundant tau pathology but no degeneration evident after 3 months of propagation [37]. This confirms prior studies suggesting that oligomeric tau is much more toxic than fibrillar tau. [80, 86]. These studies suggest that oligomeric or misfolded forms of tau are toxic, drive cognitive decline, and act through a mechanism that occurs before or independently of the development of NFTs [73, 80, 95].
The propagation studies also demonstrated the strong link between oligomeric tau and TIA1-positive SGs. The oligomeric tau propagated tau pathology that co-localized with cytoplasmic TIA1-positive SGs, as shown by co-localization with TIA1, PABP and eIF3η; this was true in both the ipsilateral and contralateral cortex providing clear demonstration of tau propagation [37]. In contrast, the fibrillar tau propagated nicely, but showed no colocalization with TIA1-positive granules. An additional mechanistic link between TIA1 and tau was evident from similar studies performed in P301S tau::TIA1+/−. These mice showed abundant propagation of fibrillar tau, but very little (if any) propagation of oligomeric tau, which provides support for the hypothesis that TIA1 selectively interacts with oligomeric tau.
A Model for the Interactions of Tau in Stress and with RNA Binding Proteins
The accumulating data presented above suggest a model in which phosphorylated tau accumulates in the somatodendritic arbor where it oligomerizes and then interacts with TIA1 and possibly other RNA binding proteins and/or ribosomal proteins. Binding of tau to these proteins appears to promotes the translational stress response, which reduces synthesis of specialized proteins (such as those related to synaptic function) and increases synthesis of proteins needed for the stress response. The proline directed phosphorylation that is characteristic of the stress response also inhibits binding of tau to microtubules, perhaps allowing for more tau oligomerization and interaction with the translational machinery. Chronic stress, though, leads to the accumulation of oligomeric tau which is toxic, although the mechanism of toxicity is not currently known.
Therapeutic Approaches Based on Modulating SGs and the Translational Stress Response
One of the most important aspects of studying the relationship between tau, RBPs, SGs and the translational stress response is the possibility of innovative disease therapies. The biology of SGs involves multiple biochemical pathways, some of which not been considered previously in the context of neurodegenerative diseases.
The classic SG/translational stress response is regulated by phosphorylation of eukaryotic initiation factor 2 (eIF2α-P); this pathway has been studied by a number of different laboratories. In presence of stress, stress-related kinases, including PERK, PKR, HRI and GCN2, phosphorylate eIF2α trigger the assembly of SGs that inhibit global cellular protein synthesis [93]. Chronic diseases produces a sustained stress response, persistent SGs and continued global translational repression [8, 45, 77, 85]. These observations suggest the hypothesis that the translational stress response is too active, and inhibiting the stress response might be beneficial in neurodegenerative disease. One of the first studies demonstrating the value of inhibiting the translational stress response was performed in a mouse prion model, where Mallucci and colleagues demonstrated that reducing eIF2α phosphorylation (by expressing the phosphatase adapter protein, GADD34) relieves the translational repression caused by eIF2α-P and delays neurodegeneration [66]. Studies in drosophila and in rat primary cortical neurons expressing TDP-43 show that inhibiting the phosphorylation of eIF2α alleviates the toxicity induced by TDP-43 [43]. Similar approaches have now also been shown to apply to inhibition of toxicity associated with tau, as well as with Aβ [51, 52, 59, 88]. These studies point to inhibition of eIF2α phosphorylation as a potential therapeutic intervention to neurodegenerative diseases. However, the clinical value of each of these approaches is limited by toxic liabilities. For example, PERK inhibition leads to pancreatic toxicity, although partial inhibition might be clinically tolerated [29], and inhibition of PKR enables reemergence of retroviruses [68].
Other teams are developing innovative small molecule therapeutics to inhibit the accumulation of persistent pathological SGs. Our group used neuronal PC12 cells inducibly over-expressing TDP-43 to screen a library of brain penetrant small molecules [10]. This screen lead to the identification of 16 hits that reduced the accumulation of TDP-43 inclusions [10]. Some of these compounds were able to improve survival of neurons in a C. elegans model of over-expressing TDP-43, suggesting the potential for in vivo efficacy [10]. Cell lines expressing TDP-43 have also been used to screen a variety of other putative therapeutics, with promising results although these approaches have yet to be tested in vivo [13, 67].
An alternative approach for disease modifying therapy has been direct therapeutic targeting of RBPs for therapy. Studies in this area have focused on delaying neurodegeneration in models of tauopathy, based on tau over-expression, and models of ALS based on TDP-43 over-expression [2, 3, 7]. The discussion of tau, described above, demonstrates how reducing TIA1 in the P301S tau mouse model rescues memory loss, reduces neurodegeneration and improves survival [2, 3]. Protection is also be achieved by TIA1 knockdown with shRNA directed against TIA1 [88]. TIA1 reduction also protects against tauopathy in a model of tau propagation, which indicates that reducing TIA1 provides generalized protection against tau pathology produced within neurons as well as propagated among neurons [37]. These results suggest that reducing TIA1 might provide broad-based neuroprotection in AD and other tauopathies.
The therapeutic potential of reducing RNA binding proteins has also been examined in a model of ALS based on over-expressing TDP-43 [7]. These studies focused on ATXN2, which participates in RNA metabolism, contributing to RNA splicing, and degradation [39, 72]. ATXN2 contains a domain with a small number of CAG trinucleotide repeats (producing glutamines) whose expansion is associated with disease. Disease linked mutations in ATXN2 that expand the CAG domain to 34 or more repeats cause the neurodegenerative disorder spinocerebellar ataxia type 2 (SCA2) [24]. However, disease linked mutations that expand the CAG domain to 27–33 repeats increase the risk of ALS, with associated TDP-43 pathology [19]. The link between ATXN2 and TDP-43 was strengthened with the observation that reducing ATXN2 significantly extends survival in an animal model of ALS based on TDP-43 overexpression [7]. Knockout and antisense-mediated knockdown of ATXN2 in TDP-43 transgenic mice decreased SGs containing TDP-43, reduced the accumulation of phosphorylated TDP-43 spinal cord inclusions, improved the motor performance and increased the median lifespan by 35% [7]. These findings indicate that ATXN2 plays a crucial role in the development of pathological SGs and augmentation of TDP-43 toxicity.
Targeting other regulators of RNA metabolism has also been shown to ameliorate disease. McGurk et al. [58] found that downregulating Tankyrase 1 and 2, a poly (ADB-ribose) polymerase, reduced the formation of cytoplasmic TDP-43 foci without affecting the SG assembly. PAR binds to the PAR-binding motif in the N-terminal region of TDP-43 and is necessary for its sequestration in SGs in mammalian cells and neurons upon exposure to stress [58]. Inhibiting Tankyrase significantly increases nuclear TDP-43 and decreases cytoplasmic TDP-43 without affecting the total TDP-43 expression levels and increases survival percentage of flies expressing TDP-43 in the brain. Inhibiting Tankyrase prevents the stress-induced formation of cytoplasmic TDP-43 foci without altering the dynamics and assembly of SGs. TDP-43 also becomes phosphorylated upon prolonged exposure to stress, but the phosphorylation appears to stimulate aggregation of TDP-43 through a pathway that does not co-localize with SG markers [58]. Since phospho-TDP-43 is associated with disease pathology, this TDP-43 phosphorylation pathway might identify a disease-relevant pathway [70]. Since tankyrase downregulation increases nuclear TDP-43 and decreases cytoplasmic TDP-43, inhibiting tankyrase might reduce the amount of cytoplasmic TDP-43 that is available to be phosphorylated and provide a potential pharmacological intervention for diseases associated with pathological TDP-43 [58].
The most striking success in delay of neurodegenerative disease based on targeting RNA metabolism comes from the field of spinal muscular atrophy (SMA). This disease is caused by a missense mutation that causes exon skipping that produces an inactive form of the gene survival of motor neuron 1 (SMN1) [50]. Teams of investigators from Ionis Pharmaceuticals and Biogen developed antisense oligomers capable of correcting the exon skipping, which increases levels of SMN1. Multiple clinical trials now demonstrate that application of these antisense oligomers to children with SMA produces striking rescue from disease, and prolonged delay of disease progression [21, 22, 61, 62]. The striking ability of these antisense oligomers to delay disease progression and actually improve clinical conditions in patients with SMA now serves as a bench post for future therapies.
Conclusion
The work covered in this chapter presents a cogent paradigm for understanding the pathophysiology of tauopathy. Accumulating data suggest that tau becomes hyperphosphorylated and oligomerize as part of an endogenous mechanism to promote the translational stress response. Tauopathies, though, are chronic diseases. The prolonged stress leads to a persistent stress response, which provides time for oligomeric tau to accumulate, cause toxicity and neurodegeneration. We show that tau-mediated neurodegeneration occurs through a mechanism that is mediated by RNA binding proteins and the translational stress response. Discovery of the role of RNA metabolism in tauopathy opens a wide variety of novel therapeutic approaches. The abundant studies targeting TDP-43-mediated disease demonstrate the large variety of approaches designed to therapeutically modulate RNA metabolism, and indicate strong potential for success in disease modification in models of ALS based on abrogating TDP-43 mediated disease. These studies complement the documented success of targeting RNA metabolism modify disease progression in animal models of tauopathy as well as β-amyloidosis. The success in treating SMA point to a new era for therapy of neurodegenerative diseases, and suggest that the right type of therapy can actually go beyond delay of disease progression to actually halt disease progression and improve clinical outcomes for some neurodegenerative diseases. Taken together, these approaches point to a future in which novel therapeutic approaches might be able to significantly delay disease progression in tauopathy.
Acknowledgements
This work was supported by funding to BW from the NIH (AG050471, NS089544, AG056318), BrightFocus Foundation, Alzheimer Association, Cure Alzheimer’s Fund and the Thome Medical Foundation.
Footnotes
Conflict of Interest BW is co-founder and Chief Scientific Officer of Aquinnah Pharmaceuticals Inc., www.aquinnahpharma.com.
References
- 1.Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33(3):141–50. 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Apicco DJ et al. Dysregulation of RNA splicing in tauopathies, Submitted. 2018 [DOI] [PMC free article] [PubMed]
- 3.Apicco DJ, et al. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci. 2018; 10.1038/s41593-017-0022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–72. 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 5.Banani SF, et al. Compositional control of phase-separated cellular bodies. Cell. 2016;166(3):651–63. 10.1016/j.cell.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbash S, et al. Alzheimer’s brains show inter-related changes in RNA and lipid metabolism. Neurobiol Dis. 2017;106:1–13. 10.1016/j.nbd.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker LA, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–71. 10.1038/nature22038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bentmann E, Haass C, Dormann D. Stress granules in neurodegeneration – lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 2013;280(18):4348–70. 10.1111/febs.12287. [DOI] [PubMed] [Google Scholar]
- 9.Bosco DA, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010; 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyd JD, et al. A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J Biomol Screen. 2014; 10.1177/1087057113501553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brehm MA, et al. Intracellular localization of human Ins(1,3,4,5,6)P5 2-kinase. Biochem J. 2007; 10.1042/BJ20070382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buchan JR, Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol Cell. 2009;36(6):932–41. 10.1016/j.molcel.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang CF, et al. Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J Biomed Sci. 2016;23(1):72. 10.1186/s12929-016-0290-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dang Y, et al. Eukaryotic initiation factor 2alpha-independent pathway of stress granule induction by the natural product pateamine A. J Biol Chem. 2006;281(43):32870–8. 10.1074/jbc.M606149200 [DOI] [PubMed] [Google Scholar]
- 15.de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73(4):685–97. 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dormann D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29(16):2841–57. 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eftekharzadeh B, Daigle JG, et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron. 2018a;99(5):925–940.e7. 10.1016/j.neuron.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eftekharzadeh B, Tepper K, et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018b;37:e98049. 10.15252/embj.201798049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–75. 10.1038/nature09320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feric M, et al. Coexisting liquid phases underlie nucleolar subcompartments. Cell. 2016;165(7):1686–97. 10.1016/j.cell.2016.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finkel RS, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388(10063):3017–26. 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 22.Finkel RS, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–32. 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 23.Gennarelli M, et al. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem Biophys Res Commun. 1995;213(1):342–8. 10.1006/bbrc.1995.2135 [DOI] [PubMed] [Google Scholar]
- 24.Geschwind DH, et al. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet. 1997;60(4):842–50. [PMC free article] [PubMed] [Google Scholar]
- 25.Gilks N, et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15(12):5383–98. 10.1091/mbc.e04-08-0715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunawardana CG, et al. The human tau interactome: binding to the ribonucleoproteome, and impaired binding of the proline-to-leucine mutant at position 301 (P301L) to chaperones and the proteasome. Mol Cell Proteomics. 2015;14(11):3000–14. 10.1074/mcp.M115.050724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo C, et al. Tau activates transposable elements in Alzheimer’s disease. Cell Rep. 2018a;23(10):2874–80. 10.1016/j.celrep.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo L, et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell. 2018b;173(3):677–692.e20. 10.1016/j.cell.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halliday M, et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015;6:e1672. 10.1038/cddis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han TW, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149(4):768–79. 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 31.He Y, Smith R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci. 2009;66(7):1239–56. 10.1007/s00018-008-8532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofweber M, et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell. 2018;173(3):706–719 e13. 10.1016/j.cell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Hoover BR, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68(6):1067–81. 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes MP, et al. Atomic structures of low-complexity protein segments reveal kinked beta sheets that assemble networks. Science. 2018;359(6376):698–701. 10.1126/science.aan6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikezu T, et al. Tau phosphorylation is impacted by rare AKAP9 mutations associated with Alzheimer disease in African Americans. J Neuroimmune Pharmacol. 2018; 10.1007/s11481-018-9781-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ittner LM, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142(3):387–97. 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 37.Jiang L, et al. TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 2018; 10.1007/s00401-018-1937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson BS, et al. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–39. 10.1074/jbc.M109.010264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaehler C, et al. Ataxin-2-like is a regulator of stress granules and processing bodies. PLoS One. 2012; 10.1371/journal.pone.0050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapeli K, Martinez FJ, Yeo GW. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet. 2017;136(9):1193–214. 10.1007/s00439-017-1830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kedersha N, et al. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151(6):1257–68. 10.1083/jcb.151.6.1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kedersha N, et al. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13(1):195–210. 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim HJ, et al. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46(2):152–60. 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, et al. Cell proteins TIA-1 and TIAR interact with the 3 ′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J Virol. 2002;76(23):11989–2000. 10.1128/JVI.76.23.11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li YR, et al. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201(3):361–72. 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin Y, et al. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell. 2015;60(2):208–19. 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu L, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu-Yesucevitz L, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5(10):e13250. 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu-Yesucevitz L, et al. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31(45):16086–93. 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lorson CL, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci. 1999; 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lourenco MV, et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18(6):831–43. 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 52.Ma T, et al. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci. 2013;16(9):1299–305. 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mackenzie IR, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and Alter stress granule dynamics. Neuron. 2017;95(4):808–816.e9. 10.1016/j.neuron.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Markmiller S, et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018;172(3):590–604.e13. 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maziuk BF, et al. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol Commun. 2018;6(1):71. 10.1186/s40478-018-0574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mazroui R, et al. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol Biol Cell. 2006;17(10):4212–9. 10.1091/mbc.e06-04-0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McEwen E, et al. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem. 2005;280(17):16925–33. 10.1074/jbc.M412882200 [DOI] [PubMed] [Google Scholar]
- 58.McGurk L, et al. Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell. 2018;71(5):703–717.e9. 10.1016/j.molcel.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meier S, et al. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer’s disease brain. J Alzheimers Dis. 2015;48(3):687–702. 10.3233/JAD-150298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meier S, et al. Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J Neurosci. 2016; 10.1523/JNEUROSCI.3029-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mendell JR, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–22. 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 62.Mercuri E, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625–35. 10.1056/NEJMoa1710504. [DOI] [PubMed] [Google Scholar]
- 63.Mills JD, et al. RNA-Seq analysis of the parietal cortex in Alzheimer’s disease reveals alternatively spliced isoforms related to lipid metabolism. Neurosci Lett. 2013;536:90–5. 10.1016/j.neulet.2012.12.042. [DOI] [PubMed] [Google Scholar]
- 64.Mokas S, et al. Uncoupling stress granule assembly and translation initiation inhibition. Mol Biol Cell. 2009;20(11):2673–83. doi: E08–10-1061 [pii] 10.1091/mbc.E08-10-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Molliex A, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163(1):123–33. 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moreno JA, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012; 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moujalled D, et al. Kinase inhibitor screening identifies cyclin-dependent kinases and glycogen synthase kinase 3 as potential modulators of TDP-43 cytosolic accumulation during cell stress. PLoS One. 2013;8(6):e67433. 10.1371/journal.pone.0067433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muto NF, et al. Inhibition of replication of reactivated human immunodeficiency virus type 1 (HIV-1) in latently infected U1 cells transduced with an HIV-1 long terminal repeat-driven PKR cDNA construct. J Virol. 1999;73(11):9021–8. Available at: https://www.ncbi.nlm.nih.gov/pubmed/10516008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- 70.Neumann M, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009; 10.1007/s00401-008-0477-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nonaka T, et al. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009; 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- 72.Nonhoff U, et al. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18(4):1385–96. 10.1091/mbc.e06-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oddo S, et al. Reduction of soluble Aβ and tau, but not soluble Aβ alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281(51):39413–23. 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 74.Patel A, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162(5):1066–77. 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 75.Prilusky J, et al. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21(16):3435–8. 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 76.Protter DSW, Parker R. Principles and properties of stress granules. Trends Cell Biol. 2016;26(9):668–79. 10.1016/j.tcb.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154(4):727–36. 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robberecht W, Eykens C. The genetic basis of amyotrophic lateral sclerosis: recent breakthroughs. Adv Genomics Genet. 2015;5:327–45. 10.2147/AGG.S57397. [DOI] [Google Scholar]
- 79.Sanders DW, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82(6):1271–88. 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.SantaCruz K Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–81. 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scheckel C, et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and noncoding RNAs in human brain. Elife. 2016;5 10.7554/eLife.10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Silva JM, et al. Dysregulation of autophagy and stress granule-related proteins in stress-driven Tau pathology. Cell Death Differ. 2018;26(8):1411–27. 10.1038/s41418-018-0217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–72. 10.1126/science.1154584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stamer K, et al. Tau blocks traffic of organelles, neuro-filaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156(6):1051–63. https://10.1083/jcb.200108057jcb.200108057 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thomas M, Alegre-Abarrategui J, Wade-Martins R. RNA dysfunction and aggrephagy at the centre of an amyotrophic lateral sclerosis/frontotemporal dementia disease continuum. Brain. 2013;136(Pt 5):1345–60. 10.1093/brain/awt030. [DOI] [PubMed] [Google Scholar]
- 86.Van der Jeugd A, et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012;123(6):787–805. 10.1007/s00401-012-0987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vanderweyde T, et al. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in Tauopathies. J Neurosci. 2012;32(24):8270–83. 10.1523/JNEUROSCI.1592-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vanderweyde T, et al. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 2016; 10.1016/j.celrep.2016.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17(1):5–21. 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 90.Wang P, et al. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017;133(5):731–49. 10.1007/s00401-016-1663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang J, et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell. 2018;174(3):688–699.e16. 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wei MT, et al. Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nat Chem. 2017;9(11):1118–25. 10.1038/nchem.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wek RC, Jiang H-Y, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006; 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 94.Winton MJ, et al. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008; 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zempel H, Mandelkow E. Lost after translation: mis-sorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014;37(12):721–32. 10.1016/j.tins.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 96.Zempel H, et al. Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32(22):2920–37. 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang X, et al. RNA stores tau reversibly in complex coacervates. PLoS Biol. 2017;15(7):e2002183. 10.1371/journal.pbio.2002183. [DOI] [PMC free article] [PubMed] [Google Scholar]