

Abstract
Objective
Weight loss is associated with clinical progression in Huntington disease (HD), but whether body weight causally affects disease onset or progression is unknown. Therefore, we aimed to assess whether genetically determined variations in body weight are causally related to age at onset in HD.
Methods
Using data from different recent genome-wide association studies, we performed a 2-sample mendelian randomization (MR) analysis to assess whether genetic markers of body mass index (BMI) are causally related to residual age at onset in HD, i.e., the difference between observed and expected age at onset based on mutation size. Our study had a statistical power of 90% to detect a causal effect of ≥3.8 months per BMI unit change at a type I error rate of 0.05.
Results
Inverse-variance weighted MR estimates showed that a higher genetically determined BMI was not causally related to residual age at onset in HD (β = −0.44 years per unit increase in BMI, confidence interval: −1.33 to 0.46, p = 0.34). All other complementary (nonparametric) MR regression methods yielded similar results.
Conclusions
Although maintaining a healthy and stable body weight remains important in patients with HD, promoting weight gain with the aim of delaying disease onset or slowing down disease progression should be discouraged. Our findings point toward the existence of underlying pathologic processes that dictate both the rate of clinical progression and weight loss in HD, which need further elucidation as targeting these pathways, rather than body weight per se, could be of therapeutic value.
Unintended weight loss is a hallmark of Huntington disease (HD), an autosomal dominant inherited neurodegenerative disorder caused by a CAG repeat expansion in the HTT gene, and occurs in both patients and premanifest mutation carriers, as well as many genetic animal models of the disease.1,2 Recently, we found that body weight is a robust predictor of the rate of clinical progression in patients with HD.3 Patients with a higher body mass index (BMI) at their first visit deteriorated more slowly in the motor, cognitive, and functional domains, independent of disease severity at baseline and mutant CAG repeat size.3 However, given that these findings were based on observational data, it remains to be clarified whether a higher body weight slows down disease progression, or alternatively, should be regarded as an epiphenomenon (i.e., a marker of slow disease progression without affecting the underlying pathologic processes). Elucidating this question is of critical importance as, in the first instance, measures to increase body weight would be expected to decrease the rate of deterioration in HD, whereas in the latter instance, search for the mechanisms that underlie weight loss in HD should be continued as targeting these mechanisms, rather than body weight per se, could be of therapeutic relevance. To address this issue, here we applied mendelian randomization (MR), a method to explore causal effects based on genetic instrumental variables, to assess the directionality of effect between BMI and age at onset, which could also be regarded as a proxy for disease progression, in HD.
Methods
Two-Sample Mendelian Randomization
We performed a 2-sample MR study—a statistical method that allows the exploration of the causal effects of an exposure (here BMI) on an outcome (here residual age at onset) by combining summary statistics from genome-wide association studies (GWASs) performed in different populations.4 We specifically assessed whether single nucleotide polymorphisms (SNPs) that have been associated with BMI were also associated with residual age at onset in HD, assuming unconfounded effects of these genetic instruments on the outcome mediated through BMI.4
GWAS Summary Statistics for BMI
As genetic instrumental variables for BMI, we selected SNPs from the combined summary statistic files of the 2 largest genetic association studies of body weight available to date (i.e., the GWAS meta-analysis of the Genetic Investigation of Anthropometric Traits (GIANT) consortium and the UK BioBank study), based on combined data from ∼700,000 individuals.5,6 A clumping procedure was performed to retain the index SNPs that were independently related to BMI at a genome-wide significance level of p < 5 × 10−8. SNPs that were in linkage disequilibrium with the index SNPs (R2 threshold ≤0.001) or within 10,000 kb distance thereof, based on the 1000 Genomes reference panel, were clumped with the index SNPs,7 resulting in an initial set of 521 SNPs as genetic instrumental variables for the exposure.
GWAS Summary Statistics for HD
Genotype data for HD were obtained from the largest GWAS of residual age at onset recently published by the Genetic Modifiers of Huntington Disease (GeM-HD) Consortium.5 This GWAS included data on a total of 9,064 HD patients. The summary effect sizes for the outcome SNPs were based on the results of the continuous analysis as reported by the GeM-HD consortium.5 Residual age at onset was defined as the difference in years between observed and expected age at motor onset based on mutant CAG repeat size.5
MR Analysis
The MR analysis was performed using the TwoSampleMR package (version 0.5.5) in R.7 The SNPs were harmonized between the exposure and outcome data sets by removing SNPs with incompatible alleles as well as palindromic SNPs with intermediate allele frequencies (n = 176), resulting in a final set of 345 SNPs used for MR analysis (accounting for 3.55% of BMI variance). For calculating statistical power, from our previous study, we first estimated the SDs of BMI within 1 year of disease onset and the residual age at onset, which were 4.75 kg/m2 and 8.10 years, respectively.3 With this information, we then estimated that the sample size for this MR analysis had a statistical power of 90% to detect a mean causal effect of BMI on residual age at onset of ≥3.8 months per unit change of BMI (at a type I error rate of 0.05 and assuming no additional confounding after accounting for mutant CAG repeat size).8 We defined the results of the random-effects inverse-variance weighted method as our primary outcome. Horizontal pleiotropy was assessed by testing whether the intercept of the MR-Egger regression line significantly differed from zero, as well as with the MR pleiotropy residual sum and outlier (MR-PRESSO) test.9 In addition, we performed extensive sensitivity analyses, which included: (1) leave-one-out analyses; (2) alternative estimation methods using maximum likelihood, simple and weighted median and MR-Egger; and (3) robust MR methods, including mr-raps (as implemented in the mr-raps package version 0.3.1) and the recently introduced contamination mixture method (as implemented in the MendelianRandomization package version 0.5.0), which yield valid causal estimates even in the presence of some invalid instrumental variables.10 p Values <0.05 were considered statistically significant.
Data Availability
GIANT/UK Biobank (portals.broadinstitute.org/collaboration/giant) and Gem-HD study (cegeme.partners.org/gem.euro.9k.html) summary statistics are freely available to qualified researchers.5,11
Results
The summary of the outcomes of the different MR methods is presented in the Table. The random-effects inverse-variance weighted overall causal effect of BMI on residual age at onset in HD was not statistically significant, i.e., a higher genetically determined BMI was not related to residual age at onset in HD (β = −0.33 years per 1-unit increase in BMI, confidence interval [CI]: −1.22 to 0.56, p = 0.47). All other complementary (nonparametric and robust) MR regression methods yielded comparable results (Table and Figure 1).
Table.
Estimates of the Causal Effects of Genetically Determined BMI on Age at Onset in Huntington Disease
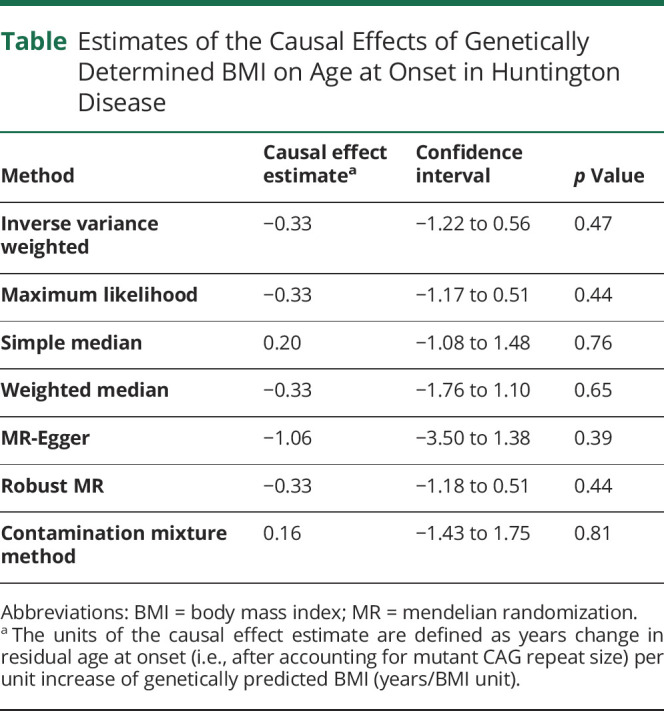
Figure 1. Scatterplot of the Effect of Each Single Genetic Instrument on BMI and Residual Age at Onset.

Each dot represents the combined effect of a single nucleotide polymorphism (SNP) on body mass index (BMI, horizontal axis) and residual age at onset in Huntington disease (vertical axis). The thin lines emanating from each dot represent the associated standard errors. Please note that all SNPs with a negative effect on BMI are shown to be positive with the sign of the effect on residual age at onset flipped. The causal association is represented by the slope of the regression lines for 4 representative mendelian randomization (MR) methods. All MR methods consistently indicated that there was not a causal effect of BMI on residual age at onset in Huntington disease.
An overview of each single SNP's effect on residuals age at onset in HD is presented in eFigure 1 (links.lww.com/NXG/A436). We did not find any indication for horizontal pleiotropy (Egger regression intercept = 0.01 years, CI: −0.03 to 0.05, p = 0.52, Figure 1) or pleiotropic outliers (MR-PRESSO global test p = 0.054). Furthermore, leave-one-out sensitivity analysis confirmed that the causal estimate was not disproportionately influenced by any single SNP (eFigure 2, links.lww.com/NXG/A437).
Discussion
Unintended progressive weight loss is frequently observed in HD and may even start many years before clinical onset.1 We recently found that higher BMI is robustly associated with a slower rate of disease progression in HD.3 Using data from the largest GWAS of age at onset in HD and taking advantage of recent advancements in MR analysis methods, which enable robust causal inference based on evaluation of the effects of genetic instrumental variables in different cohorts, here we demonstrate that BMI is unlikely to be causally related to age at onset in HD, at least not to a clinically relevant degree.
Recently, we showed that about two-thirds of the determinants of age at onset and clinical progression in HD are similar.12 Given this large extent of overlap between age at onset and disease progression determinants in HD,12 our findings thus also suggest that a high body weight can be regarded as a marker rather than a cause of slow disease progression in HD. We did not, however, directly assess the effect of BMI on the rate of disease progression in patients with manifest disease, which therefore remains to be addressed in future studies. Nevertheless, our findings are well in line with a recent study in which it was shown that although transgenic HD mice on a leptin-deficient background displayed increased body weight, this did not result in amelioration of the extensive neuropathology.13 Similarly, a previous small-scale study did not find an effect of a high caloric intervention on disease progression in HD despite stabilization or increase of body weight in the majority of them.14 However, it remains to be elucidated whether other dietary interventions, especially fasting mimicking diets that have shown some efficacy in transgenic mouse models,15 would benefit patients with HD.
Our results are of direct clinical relevance and indicate that the practice of actively promoting weight gain with the aim of delaying disease onset or slowing down disease progression in HD mutation carriers should be discouraged, especially given recent findings that metabolic disturbances like diabetes could in fact hasten disease onset.16 It is important though to stress that maintaining a healthy and stable body weight should remain a priority in HD mutation carriers because patients with HD are at an increased risk of malnutrition and cachexia, which are both associated with an increased risk of morbidity and mortality in general.1 The finding that BMI predicts disease progression but does not causally influence age at disease onset in patients with HD points to the existence of underlying pathologic processes that drive both the rate of clinical progression and weight loss in HD. These mechanisms may involve pathology of both central (hypothalamic/brain stem) and peripheral structures and need further elucidation as targeting these pathways, and not body weight per se, could be of therapeutic value.13,17-19
The MR paradigm has also been applied to assess the effect of BMI in other neurodegenerative diseases. Although BMI was not associated with the risk of developing Alzheimer disease,20 a recent study found that higher genetically determined BMI decreased the risk of developing Parkinson disease.21 However, given that Parkinson disease is commonly a disease of old age, survival bias might, at least partially, have confounded this latter finding.21
The main strength of our study is that it allows assessment of causality based on the application of the MR paradigm. However, despite consistent estimates among different MR tests and a range of sensitivity analyses to ensure compliance with the basic assumptions of MR analysis, we cannot definitely exclude all sources of bias, including weak and pleiotropic instrument bias, which is a challenge inherent to all MR studies.4,7
Acknowledgment
The authors would like to thank all participants and researchers who were involved in the studies whose summary data were used, including the GIANT, UK Biobank, and GeM-HD studies. Furthermore, they are grateful to the GeM-HD Consortium for making the GWAS summary statistics on the genetic determinants of age at onset in HD available for analysis.
Glossary
- BMI
body mass index
- CI
confidence interval
- GeM-HD
Genetic Modifiers of Huntington Disease
- GIANT
Genetic Investigation of Anthropometric Traits
- GWAS
genome-wide association study
- HD
Huntington disease
- MR
mendelian randomization
- MR-PRESSO
MR pleiotropy residual sum and outlier
- SNP
single nucleotide polymorphism
Appendix. Authors

Contributor Information
Jorien M.M. van der Burg, Email: jorienvanderburg@gmail.com.
Patrick Weydt, Email: patrick.weydt@ukbonn.de.
Georg Bernhard Landwehrmeyer, Email: bernhard.landwehrmeyer@uni-ulm.de.
Study Funding
No targeted funding reported.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Aziz NA, Roos RA. Characteristics, pathophysiology and clinical management of weight loss in Huntington's disease. Neurodegener Dis Manag. 2013;3(3):253-266. [Google Scholar]
- 2.Stan TL, Soylu-Kucharz R, Burleigh S, et al. Increased intestinal permeability and gut dysbiosis in the R6/2 mouse model of Huntington's disease. Sci Rep. 2020;10(1):18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Burg JMM, Gardiner SL, Ludolph AC, Landwehrmeyer GB, Roos RAC, Aziz NA. Body weight is a robust predictor of clinical progression in Huntington disease. Ann Neurol. 2017;82(3):479-483. [DOI] [PubMed] [Google Scholar]
- 4.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington's disease onset. Cell. 2019;178(4):887-900.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hemani G, Zheng J, Elsworth B, et al. The MR-base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brion MJA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2012;42(5):1497-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgess S, Foley CN, Allara E, Staley JR, Howson JMM. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun. 2020;11(1):376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yengo L, Sidorenko J, Kemper KE, et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641-3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aziz NA, van der Burg JM, Tabrizi SJ, Landwehrmeyer GB. Overlap between age-at-onset and disease progression determinants in Huntington disease. Neurology. 2018;90(24):e2099-e2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sjogren M, Soylu-Kucharz R, Dandunna U, et al. Leptin deficiency reverses high metabolic state and weight loss without affecting central pathology in the R6/2 mouse model of Huntington's disease. Neurobiol Dis. 2019;132:104560. [DOI] [PubMed] [Google Scholar]
- 14.Trejo A, Boll MC, Alonso ME, Ochoa A, Velasquez L. Use of oral nutritional supplements in patients with Huntington's disease. Nutrition. 2005;21(9):889-894. [DOI] [PubMed] [Google Scholar]
- 15.Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci USA. 2003;100(5):2911-2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogilvie AC, Gonzalez-Alegre P, Schultz JL. Diabetes mellitus is associated with an earlier age of onset of Huntington's disease. Mov Disord. 2021;36(4):1033-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheong RY, Gabery S, Petersén Å. The role of hypothalamic pathology for non-motor features of Huntington's disease. J Huntingtons Dis. 2019;8(4):375-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Wamelen DJ, Aziz NA, Roos RA, Swaab DF. Hypothalamic alterations in Huntington's disease patients: comparison with genetic rodent models. J Neuroendocrinol. 2014;26(11):761-775. [DOI] [PubMed] [Google Scholar]
- 19.van der Burg JM, Bjorkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009;8(8):765-774. [DOI] [PubMed] [Google Scholar]
- 20.Nordestgaard LT, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Body mass index and risk of Alzheimer's disease: a Mendelian randomization study of 399,536 individuals. J Clin Endocrinol Metab. 2017;102(7):2310-2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noyce AJ, Kia DA, Hemani G, et al. Estimating the causal influence of body mass index on risk of Parkinson disease: a Mendelian randomisation study. PLoS Med. 2017;14(6):e1002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
GIANT/UK Biobank (portals.broadinstitute.org/collaboration/giant) and Gem-HD study (cegeme.partners.org/gem.euro.9k.html) summary statistics are freely available to qualified researchers.5,11