

Abstract
Zuranolone (SAGE-217) is a novel, synthetic, clinical stage neuroactive steroid GABAA receptor positive allosteric modulator designed with the pharmacokinetic properties to support oral daily dosing. In vitro, zuranolone enhanced GABAA receptor current at nine unique human recombinant receptor subtypes, including representative receptors for both synaptic (γ subunit-containing) and extrasynaptic (δ subunit-containing) configurations. At a representative synaptic subunit configuration, α1β2γ2, zuranolone potentiated GABA currents synergistically with the benzodiazepine diazepam, consistent with the non-competitive activity and distinct binding sites of the two classes of compounds at synaptic receptors. In a brain slice preparation, zuranolone produced a sustained increase in GABA currents consistent with metabotropic trafficking of GABAA receptors to the cell surface. In vivo, zuranolone exhibited potent activity, indicating its ability to modulate GABAA receptors in the central nervous system after oral dosing by protecting against chemo-convulsant seizures in a mouse model and enhancing electroencephalogram β-frequency power in rats. Together, these data establish zuranolone as a potent and efficacious neuroactive steroid GABAA receptor positive allosteric modulator with drug-like properties and CNS exposure in preclinical models. Recent clinical data support the therapeutic promise of neuroactive steroid GABAA receptor positive modulators for treating mood disorders; brexanolone is the first therapeutic approved specifically for the treatment of postpartum depression. Zuranolone is currently under clinical investigation for the treatment of major depressive episodes in major depressive disorder, postpartum depression, and bipolar depression.
Keywords: Neuroactive steroid, GABAA receptor, Positive allosteric modulator
1. Introduction
GABA is the primary inhibitory neurotransmitter in the central nervous system (CNS) and plays a critical role in maintaining balanced neuronal activity in the brain (Engin et al., 2018). Multiple disorders result from an imbalance between neuronal excitation and inhibition including spasticity, insomnia, anxiety, seizures and epilepsy (Nuss, 2015; Sheean and McGuire, 2009; Treiman, 2001; Winsky-Sommerer, 2009). In the human CNS, ionotropic GABAergic signaling occurs through GABAA receptors, which are a large heterogeneous class of pentameric chloride channels comprised of two α, two β, and one γ, δ, ρ, θ, or ε subunits (Sigel and Steinmann, 2012). GABAA receptors containing the γ subunit are the predominant subtype present at synapses, while δ-containing receptors are present largely extra synaptically (Belelli et al., 2009; Sigel, 2002).
Neuroactive steroids (NASs) can act as positive allosteric modulators (PAMs) of both synaptic and extrasynaptic GABAA receptors with little or no direct activation (Belelli et al., 2009; Majewska et al., 1986). In addition, some NASs can enhance GABAergic signaling through a possible metabotropic mechanism that increases the surface expression of GABAA receptors (Abramian et al., 2014; Modgil et al., 2017; Parakala et al., 2019). The ability of NASs to potentiate both synaptic and extrasynaptic GABAA receptor populations and to enhance extrasynaptic GABAA receptor expression is different from benzodiazepines, which only modulate GABAA receptors that contain γ subunits (Sigel, 2002). This multimodal nature of NAS GABAA receptor PAMs may provide an opportunity for improved therapeutic benefit relative to benzodiazepines.
Due to this differentiated profile, there has been a renewed interest in the therapeutic potential of GABAA receptor modulating NASs (Carver and Reddy, 2013; Martinez-Botella et al., 2017; Zorumski et al., 2000). Brexanolone injection, a proprietary intravenous (IV) formulation of the NAS GABAA receptor PAM allopregnanolone (brexanolone), showed clinical efficacy in patients with postpartum depression (Kanes et al., 2017a; Meltzer-Brody et al., 2018) and essential tremor (Ellenbogen et al., 2016), providing evidence of the clinical utility of this class. In addition, both brexanolone and synthetic NAS GABAA receptor PAMs have demonstrated anticonvulsant, anxiolytic-like, and anti-depressant like activity in preclinical models (Hammond et al., 2017; Hawkins et al., 2017; Melón et al., 2018), further supporting the broad therapeutic potential of this class of compounds.
NAS GABAA receptor PAM drugs like Althesin (Sear, 1996) and brexanolone require intravenous administration, which can limit ease of administration and patient access relative to oral therapies. Therefore, newer synthetic analogs in this class have been engineered to retain their GABAA receptor pharmacology while increasing oral bioavailability and reducing clearance to support oral dosing (Martinez-Botella et al., 2015). Despite this, few NASs have been optimized to maintain potent, multimodal GABAA receptor PAM pharmacology following oral dosing.
Here we characterize zuranolone (SAGE-217) a synthetic NAS GABAA receptor PAM currently in clinical development (Martinez-Botella et al., 2017). Zuranolone exhibited nanomolar potency at a set of recombinant human GABAA receptor configurations and enhanced inhibitory GABA currents in rodent brain slices. In addition, coapplying zuranolone and diazepam (DZP) to α1β2γ2 GABAA receptors resulted in synergistic potentiation as determined by isobolographic analysis, consistent with evidence that NASs bind to GABAA receptors at a site distinct from benzodiazepines (Hosie et al., 2007; Laverty et al., 2017; Sigel, 2002). Zuranolone exhibited pharmacokinetic properties optimized for oral dosing and produced robust pharmacodynamic effects consistent with GABAergic activity in vivo following oral dosing and CNS exposure. Together these data establish zuranolone as a potent and efficacious NAS GABAA receptor PAM with drug-like properties suitable for oral dosing.
2. Methods
2.1. In vitro
2.1.1. Heterologous expression of recombinant human GABAA receptors
The IonWorks Barracuda (Molecular Devices, Sunnyvale, CA) platform was used to obtain recordings from human embryonic kidney (HEK) cells stably expressing α1β3γ2, α2β3γ2, α3β3γ2, and α5β3γ2 GABAA receptors. Extracellular solution contained (in mM): NaCl 137, KCl 4, CaCl2 1.8, MgCl2 1, HEPES 10, Glucose 10 and maintained at pH 7.4 with NaOH. Whole cell patch clamp recordings were made using intracellular solution containing (in mM): KCl 140, MgCl 5, HEPES 10, EGTA 1 and pH 7.2 maintained with KOH. Whole cell configuration was established via patch perforation with membrane currents recorded by on-board patch clamp amplifiers. Each well on the planar patch clamp electrode was loaded with 9 μL of cell suspension and 11 μL of extracellular buffer. Following establishment of whole cell configuration, 20 μL of 2x concentrated test articles and agonist were added to the planar patch clamp wells over 2 s, to obtain a total volume of 40 μL. A submaximal concentration of GABA at each receptor (5, 2, 12.5, and 2 μM at α1β3γ2, α2β3γ2, α3β3γ2, and α5β3γ2, respectively), was applied alone and then co-applied with zuranolone (increasing half-log concentrations between 0.01 and 30 μM in 0.3% DMSO, n = 4 per concentration) in extracellular solution with Cremophor 0.02%. The concentrations of DMSO or Cremophor used for in vitro experiments were confirmed to have no effects on their own (data not shown). Cells were voltage-clamped at a holding potential of −70 mV. If current density was judged too low for measurement, the cell was discarded.
Two electrode voltage clamp was used to record currents from xenopus oocytes. Receptor subunit cDNA (Origene, Rockville, MD) for α1β1γ2 and α6β3δ was expressed in oocytes as previously described (Hogg et al., 2008). All recordings were performed at 18 °C and cells were superfused with OR2 medium containing (in mM): NaCl 88, KCl 2.5, HEPES 5, CaCl2·2H2O 1.8, MgCl2·6H2O 1, pH 7.4. Two electrode voltage clamp experiments were performed using a HiClamp platform (Multichannel Systems, Reutlingen, Germany). Membrane potentials were held at −80 mV and a submaximal concentration of GABA (10 μM for α1β1γ2 and 3 μM for α6β3δ) was applied alone for 15 s and then co-applied with individual concentrations of zuranolone (increasing half-log concentrations between 0.1 and 10 μM, in 0.3% DMSO). Data were averaged from n = 3 cells per concentration.
Effects of zuranolone on GABA-evoked currents were examined in Ltk cells stably expressing specific GABAA receptor subunit combinations (α1β2γ2 n = 7 and α2β2γ2 n = 3) or Chinese hamster ovary (CHO) cells transiently expressing α4β3δ receptors (n = 6). In order to confirm expression of the δ subunit, the δ selective PAM DS-2 was applied at the end of all experiments (data not shown). Stably expressing Ltk and transiently transfected CHO cells were obtained from B’Sys GmbH (Witterswill, Switzerland). Recordings were obtained using whole cell patch clamp as previously described (Martinez-Botella et al., 2015). Briefly, cells were voltage clamped at a holding potential of −80 mV. GABAA receptors were stimulated by a submaximal concentration of GABA (2 μM at both receptors), and zuranolone in 0.1% DMSO was sequentially applied at increasing concentrations (0.01, 0.03, 0.1, 0.3, 1, 3 and 10 μM) for 30 s prior to a 2 s stimulus with GABA. Solutions were applied using a custom gravity-fed system with an outlet placed close to the recorded cell. Complete solution exchange occurred within 300–500 ms. All concentrations of zuranolone were tested on each cell.
For all heterologous receptor experiments, the relative percentage potentiation was defined as the peak amplitude in response to GABA in the presence of zuranolone divided by the peak amplitude in response to GABA alone, multiplied by 100.
The effect of 200 nM zuranolone on the piperidine-4-sulphonic acid (P4S; Sigma, St. Louis, MO) concentration-response curve was studied in Ltk cells expressing α1β2γ2 (n = 11). Experiments were carried out as described above except instead of increasing concentrations of zuranolone, a stable concentration of 200 nM zuranolone was applied with increasing concentrations of P4S (0.1–1000 μM in log intervals).
2.1.2. Synergy with diazepam
To assess synergy between zuranolone and diazepam (DZP; Fagron, Rotterdam, The Netherlands) recordings were carried out in Ltk cells (n = 3 per experiment) expressing α1β2γ2 or CHO cells (n = 3 per experiment) expressing α4β3δ receptors as described above for zuranolone (0.01, 0.1, 1, and 10 μM; n = 3) and DZP (0.1, 1, and 10 μM; n = 4). Increasing concentrations of zuranolone were then co-applied with 100 nM DZP (n = 3). A submaximal concentration of 2 μM GABA was used for all experiments.
Isobolographic analysis was performed according to the method of Tallarida (2011). Briefly, a specific effect level (the line of additivity) was chosen and the concentrations of DZP and zuranolone required to achieve that effect in combination were compared to the concentrations required for each compound to achieve that effect individually. The percent potentiation at the EC50 of zuranolone was used to calculate the line of additivity according to the following equation:
Synergy, additivity or subadditivity was determined by the location of an experimentally determined point on the graph relative to the line of additivity. Points that fall on the line reflect additivity, below the line reflects synergy, and above the line reflects subadditivity (e.g., competition).
2.1.3. Acute hippocampal slice recording
Experiments were conducted as previously described (Modgil et al., 2017). Briefly, brain slices were prepared from postnatal day 21 C57 mice according to protocols approved by the institutional animal use committee. After recovery, slices were placed in a holding chamber where they were exposed to 0.1 or 1 μM zuranolone for 15 min and then transferred to the recording chamber. Slices in the recording chamber were submerged and perfused (2 mL/min) with oxygenated artificial cerebral spinal fluid for 30–50 min prior to visualized (infrared/differential interference contrast) whole cell patch clamp of dentate granule cells (n = 6–7). Tonic current was measured by taking the difference between the baseline current amplitude before and after application of 100 μM picrotoxin. To assess spontaneous inhibitory post-synaptic currents (sIPSCs), the recording trace was visually inspected and only events with a stable baseline, sharp rising phase, and single peak were used to negate artifacts due to event summation. Numerical data are presented as mean ± SEM. Recordings were acquired using Clampex 10.1 software and analyzed offline with Clampfit 10.1 software. Statistical significance was assessed with GraphPad Prism software using one-way ANOVA with Dunnett’s post-hoc test for multiple comparisons where p < 0.05 is considered significant.
2.2. In vivo
2.2.1. Animals
All experimental animals were treated ethically in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were group housed in a temperature- and humidity-controlled environment on a 12-h light-dark cycle with free access to food and water.
2.2.2. Pharmacokinetics
The in vivo brain and plasma pharmacokinetic parameters of zuranolone (10 mg/kg) were determined in adult male CD-1 mice following a single IP or PO dose. Zuranolone was solubilized in 15% (2-Hydroxypropyl)-β-Cyclodextrin (HP-β-CD; Adamas Reagent Co., Ltd., Shanghai, China) or 30% sulfobutyl ether β-cyclodextrin (SBECD; Ligand, San Diego, CA) for intraperitoneal (IP) or per os (PO) administration, respectively. Dose formulations were vortexed and sonicated to obtain clear solutions prior to administration. Animals were subsequently observed for clinical signs, such as emesis, to ensure the full oral dose was ingested. Mice were sampled at 0.5, 1, 2, 4, and 8 h post dose (IP, n = 2 per time point) or 0.167, 0.25, 0.05, 1, 1.5, 2, 4, 6, 8, and 12 h post dose (PO, n = 3 per time point). Blood samples were collected into tubes treated with K2 EDTA, centrifuged to obtain plasma and frozen. Brain tissues were collected and immediately snap frozen in −20 °C until ready for analysis.
For behavioral studies, plasma and brain samples were collected from a subset of test animals and from animals included solely for pharmacokinetic analysis (n = 3–4 per dose).
2.2.3. Liquid chromatography-mass spectrometry
Brain samples were first homogenized in 3 vol (v/w) of phosphate buffered saline for 2 min. Protein precipitation was utilized to extract the analyte from plasma and brain homogenates in preparation for analysis via LC-MS/MS. A 30 μL aliquot of sample was added to 100 μL of acetonitrile containing internal standard. Samples were vortex-mixed for 2 min, centrifuged for 5 min at 14,000 rpm, and supernatants ali-quoted into analytical plate. Control blanks and calibrators spiked with zuranolone were extracted concurrently with samples using the same method.
A 5 μL aliquot of supernatant was injected onto an Acquity BEH C18 2.1 × 50 mm, 1.7 μm column (Waters, Milford, MA) and elution performed using a water:methanol gradient ranging from 10% to 95% methanol at a flow rate of 0.6 μL/min. Detection was achieved on an API5500 triple quadrupole mass spectrometer (AB Sciex, Framingham, MA) in positive ion mode utilizing multiple reaction monitoring of mass transitions of the analyte and internal standard. Zuranolone concentrations were calculated using the peak area ratios and least squares linear regression of the standard calibration curve.
2.2.4. Pentylenetetrazole-induced seizures
In two separate studies, adult male CD-1 mice (25–35 g; n = 10 per group; Vital River, Beijing, China) were administered zuranolone (0.1, 0.3, or 1 mg/kg and 1, 3, or 10 mg/kg) or vehicle (15% HP-β-CD) PO 60 min prior to subcutaneous administration of 120 mg/kg pentylenetetrazole (PTZ; Sigma-Aldrich Co, St Louis, MO, USA) dissolved in 0.9% sterile saline. Data from both studies were combined, as there was no significant statistical difference between vehicle groups (p = 0.07, unpaired t-test). Immediately after PTZ injection, mice were placed into an observation chamber (25 × 15 × 15 cm) for 30 min. The latencies to clonic and tonic seizure onset were recorded. Clonic seizures were defined as seizures that persisted for ≥3 s followed by an absence of righting reflex. Tonic seizures were defined as rigid extension of all four limbs exceeding a 90° angle with the body plane. Statistical significance was assessed with GraphPad Prism software using one-way ANOVA with Dunnett’s post-hoc test for multiple comparisons.
2.2.5. PharmacoEEG
Adult male Sprague Dawley rats (369.5 ± 5.7 g; n = 7 per group; Shanghai Laboratory Animal Center, Shanghai, China) were anaesthetized (70 mg/kg pentobarbital sodium, IP; Merck, Germany) and placed in a stereotaxic frame (RWD Life Science; San Diego, CA, USA). For EEG recordings, two epidural recording screw electrodes were implanted (negative at +2.0 mm AP, −1.5 mm ML from bregma, positive at −2.5 AP, + 5 mm ML from bregma). One ground electrode was implanted over the cerebellum. Electrodes were connected to a head-stage (handmade with a double row circular hotle IC socket and electromyogram wire; DSI, St. Paul, MN, USA) which was mounted on the skull using dental acrylic. Rats were administered antibiotic (ceftriaxone sodium, 100 mg/kg IP, Shangai Xinya Pharmaceutical Guoyou Co., Ltd; Shanghai, China) and analgesia (buprenorphine, 0.3 mg/kg IP, TIPR Pharmaceutical Responsible Co., Ltd, Tianjin, China) immediately after surgery. Rats were placed on a thermal pad (35 °C) until regaining normal posture.
After two weeks of recovery, rats were randomly assigned to treatment with either zuranolone (0.3, 3, or 20 mg/kg) or vehicle (30% SBECD) PO. Rats were acclimated to the recording chamber for at least 30 min prior to one hour of continuous EEG recording to establish baseline. After compound administration, continuous EEG was recorded for 6 h. EEG was recorded using A-M Systems Differential AC Amplifier (Model 1700, A-M Systems, Carlsborg, WA, USA) and CED Micro 1401 with Spike 2 (v7.0, CED, Cambridge, UK). Rats that exhibited loss of righting reflex were provided with thermal pad heating support to prevent hypothermia.
EEG and electromyogram signals were digitized at 500 Hz without 50 Hz notch filter. The EEG signal was digitally band-pass filtered (1–70 Hz) with the digital notch filter. Changes in EEG power were analyzed in the following frequencies: Delta, 1–5.5 Hz; Theta, 5.5–8.5 Hz; Alpha, 8.5–12.5 Hz; Beta, 12.5–30 Hz; Gamma 1, 30–50 Hz; and Gamma 2, 50–70 Hz). In order to derive these powers, a spectrogram was produced using a short-time Fourier Transform with 0.5 window overlap and 1 Hz frequency bins, and then power was averaged over time and frequency to produce an average EEG power. Statistical significance was assessed with GraphPad Prism software using one-way ANOVA.
Behavioral assessment of vigilance status (sleep or wake) was automatically classified off-line by 10 s epochs with SLEEPSIGN v 3.0 (KISSEI COMTEC CO. LTD.) according to EEG and EMG properties. All of the automatically classified vigilance status data were visually inspected by a trained expert blinded to treatment conditions and adjusted for any errors in the classification made by the SLEEPSIGN algorithm. Statistical significance compared to vehicle was assessed with GraphPad Prism software using a two-way repeated measures ANOVA. The EEG analysis reported here utilizes the continuous EEG trace regardless of sleep/wake classification state.
Data are presented as mean ± S.E.M., error bars indicate S.E.M unless otherwise indicated.
3. Results
3.1. In vitro GABAA receptor pharmacology
NASs are thought to bind within a highly conserved site involving the α subunit transmembrane domain to potentiate ionotropic current at many different GABAA receptor combinations (Hosie et al., 2006, 2007, 2009; Laverty et al., 2017). NASs can also increase cell surface expression of GABAA receptors through a possible metabotropic mechanism (Abramian et al., 2014; Modgil et al., 2017). The potential ionotropic and metabotropic effects of zuranolone were examined here using whole-cell patch clamp recordings in mammalian cell lines heterologously expressing GABAA receptors and in rat brain slices.
3.1.1. Heterologous receptor modulation
Zuranolone is a PAM at human recombinant α1β2γ2 and α4β3δ GABAA receptors expressed in Ltk and CHO cells, respectively (Martinez-Botella et al., 2017). In the current study, zuranolone activity was measured in nine receptor subunit combinations and found to be a PAM at all GABAA receptors tested (Table 1). GABAA receptors containing α1β2γ2 and α4β3δ subunits are the most abundant synaptic and extrasynaptic GABAA receptors in the brain, respectively, and thus were chosen for in-depth characterization (Olsen and Sieghart, 2009). At α1β2γ2 receptors, zuranolone exhibited an EC50 of 430 nM and maximum efficacy (Emax) of 1037% (Fig. 1). At α4β3δ receptors, zuranolone exhibited an EC50 of 118 nM and Emax of 556% (Fig. 1).
Table 1.
Zuranolone exhibited PAM activity at all GABAA receptors tested.
| Receptor Type | EC50 (nM) | Emax (%) | Cell type |
|---|---|---|---|
| α4β3δ | 118 | 556 | CHO |
| α1β2γ2 | 430 | 1037 | Ltk |
| α2β2γ2 | 184 | 1343 | Ltk |
| α1β3γ2 | 390 | 684 | HEK293 |
| α2β3γ2 | 516 | 1045 | HEK293 |
| α3β3γ2 | 293 | 291 | HEK293 |
| α5β3γ2 | 298 | 534 | HEK293 |
| α1β1γ2 | 1260 | 1435 | Xenopus oocytes |
| α6β3δ | 780 | 1590 | Xenopus oocytes |
Fig. 1. Zuranolone potentiated human GABAA receptor subtypes representative of both synaptic and extrasynaptic receptors.
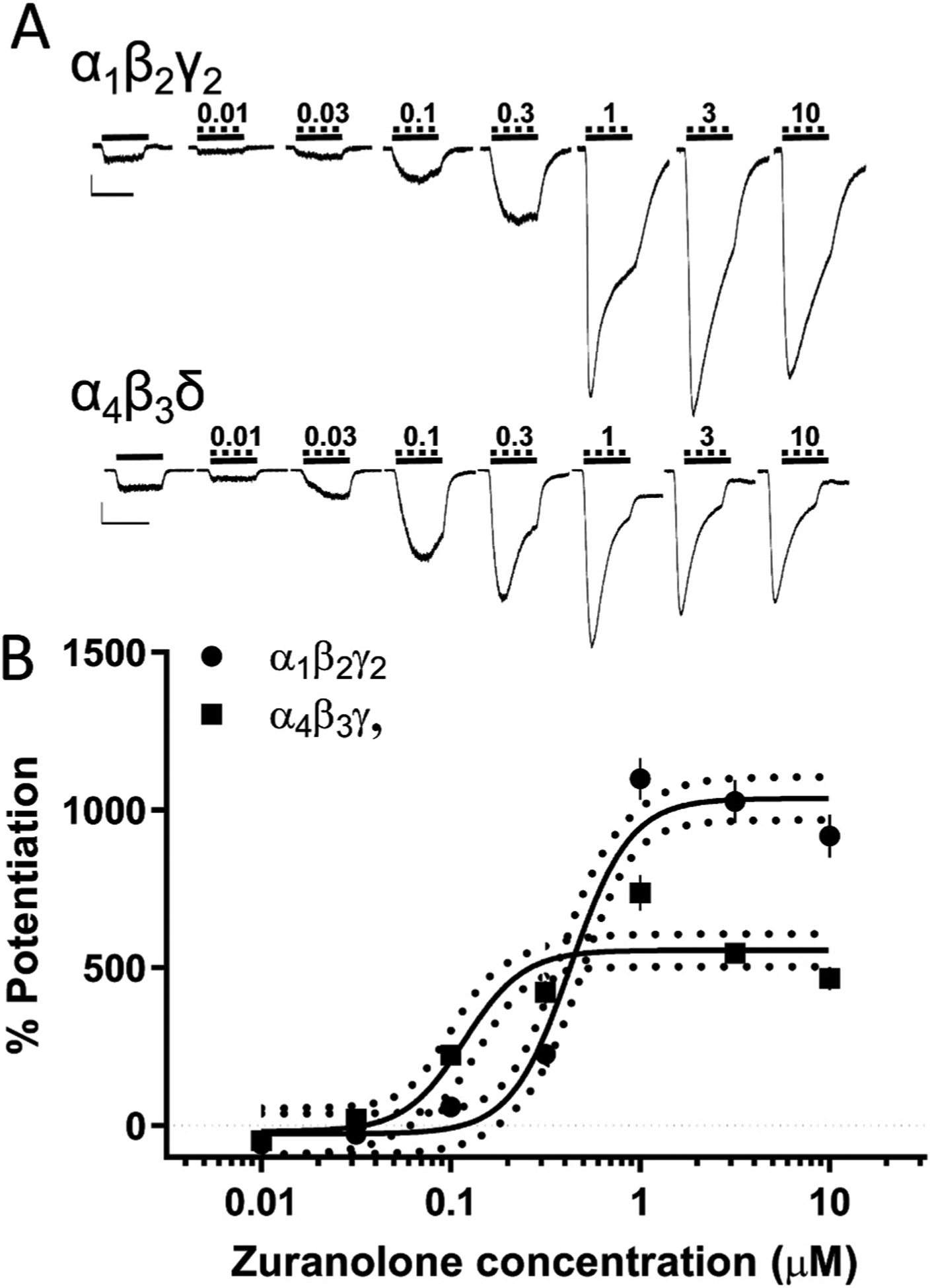
A. Representative traces from synaptic (α1β2γ2) and extrasynaptic (α4β3δ) GABAA receptors expressed in Ltk or CHO cells, respectively, showing potentiation of GABA-evoked currents. Solid lines indicate administration of a submaximal GABA concentration (2 μM), dashed lines indicate administration of zuranolone, nM. Scale bars = 500 pA, 4 sec. B. Dose-response curves for both receptor types, vertical lines indicate SEM, dotted curves indicate 95% confidence interval.
3.1.2. Effect of zuranolone on modulating orthosteric agonist activity
Allosteric modulators can alter both the binding and gating of agonists, causing a shift in potency and efficacy, respectively. Piperidine-4-sulphonic acid (P4S) was used to determine the effect of zuranolone on an orthosteric agonist. P4S is a partial agonist at GABAA receptors and is therefore less likely than GABA to induce desensitization of the receptor, allowing for better observation of allosteric effects on gating (Gielen et al., 2012; O’Shea et al., 2000). The concentration-response of P4S was measured with and without 200 nM zuranolone at α1β2γ2 GABAA receptors. Data were normalized to the maximum current evoked by a saturating concentration of GABA. P4S alone exhibited an EC50 of 45 μM and an Emax of 25.1%. Applying 200 nM zuranolone in addition to P4S, increased the agonist potency (EC50 of 25.2 μM) and efficacy (Emax 50.5%; Fig. 2), suggesting that zuranolone can also enhance potency and efficacy of GABA.
Fig. 2. Zuranolone increased potency and Emax of the orthosteric partial agonist P4S at the α1β2γ2 GABAA receptor.
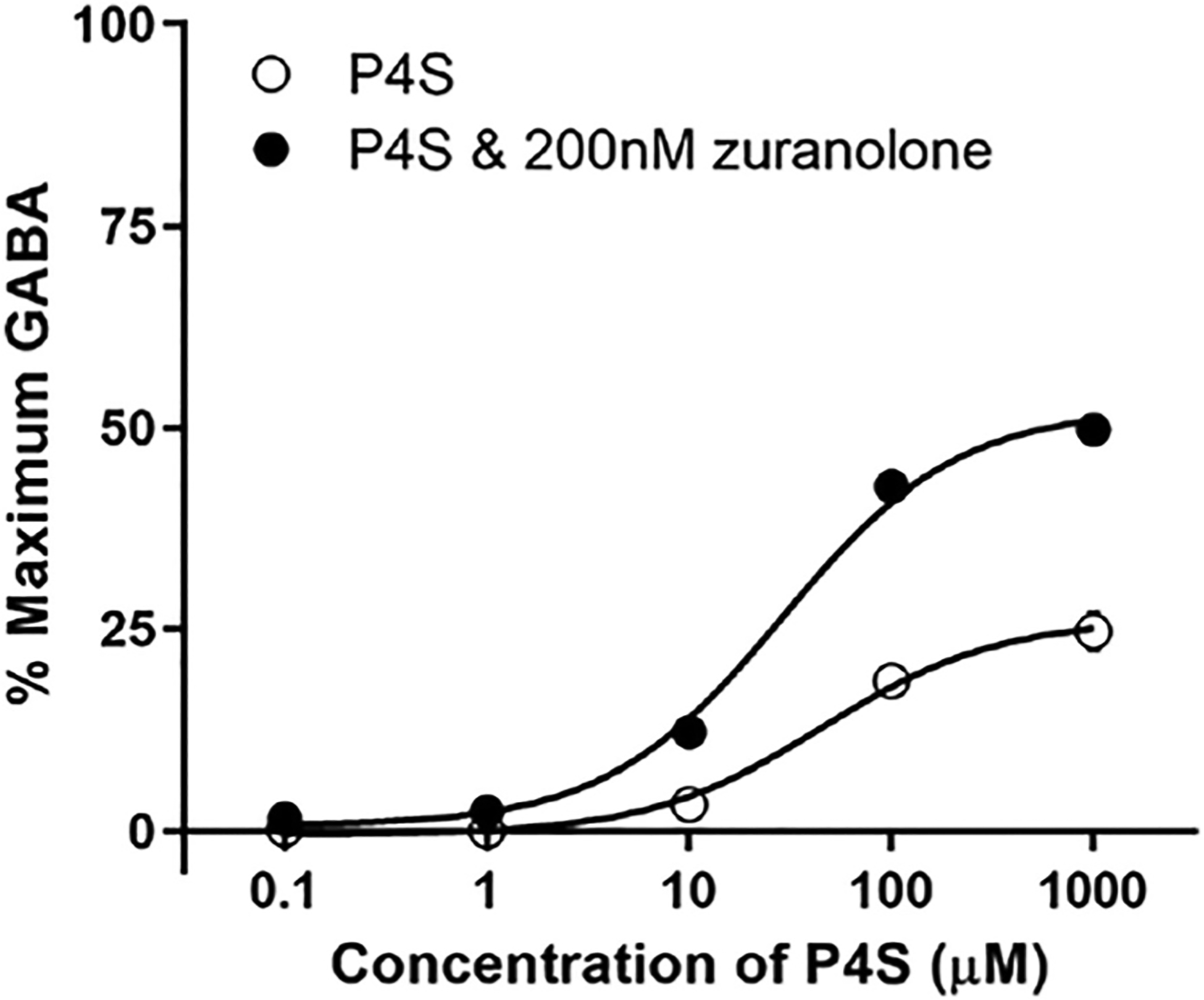
The Emax of P4S, a GABAA receptor partial agonist, was 25% of the maximal GABA (100 μM) response, which was doubled by the addition of 200 nM zuranolone to the bath solution. The EC50 for P4S was reduced by nearly half (44.5 μM–25.2 μM) by addition of 200 nM zuranolone. GABAA receptors were expressed in Ltk cells.
3.1.3. Synergism with diazepam
Allosteric modulators which bind to different sites on a given receptor may interact pharmacologically to produce either additive or synergistic effects on receptor activity. NASs are hypothesized to bind at a site on the α subunit of the GABAA receptor, while benzodiazepines bind at the interface of the α and γ subunits (Hosie et al., 2007; Sigel, 2002). The potential pharmacological interaction between the NAS zuranolone and the benzodiazepine DZP was assessed in cells expressing both α1β2γ2 and α4β3δ GABAA receptors.
In α1β2γ2 receptors, 100 nM DZP alone enhanced GABA currents by 104%. In the presence of 100 nM DZP the zuranolone concentration-response curve was shifted to the left; from an EC50 of 844 nM without DZP to an EC50 of 109 nM with DZP. The maximal potentiation of zuranolone was also increased from 511% to 1084% (Fig. 3A). Isobolographic analysis revealed that coapplying zuranolone and DZP required concentrations below the line of additivity, indicating that the two compounds acted synergistically to enhance receptor function (Fig. 3B). As expected, DZP did not potentiate current at α4β3δ receptors when applied alone, nor did it affect the concentration-response curve of zuranolone (EC50 of 135 nM without DZP and EC50 of 190 nM with DZP). These results are consistent with the inability of DZP to bind in the absence of the γ subunit (Fig. 3C) (Hosie et al., 2007; Sigel, 2002).
Fig. 3. Zuranolone exhibits synergistic activity with the benzodiazepine diazepam.
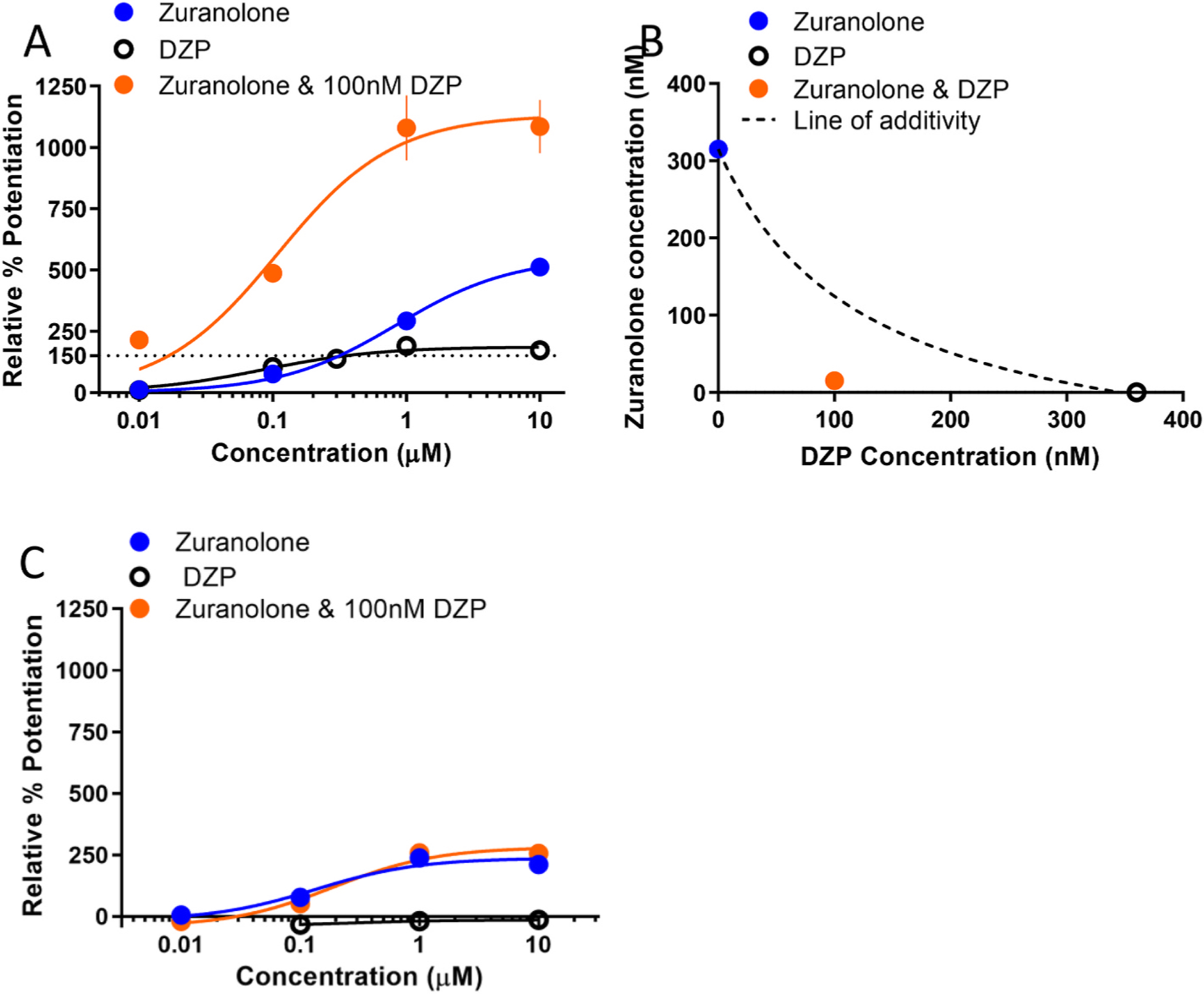
A. Concentration-response curves of zuranolone, DZP, and a combination at α1β2γ2-expressing Ltk cells. Percent potentiation is plotted relative to the amplitude of GABA (2 μM) alone. The horizontal dotted line corresponds to the effect level plotted in the isobologram. B. Isobologram of the 150% effect level. The concentration of DZP applied alone required to reach this effect was 360 nM, the concentration of zuranolone applied alone was 315 nM. When coapplied with 100 nM DZP, only 10 nM of zuranolone was required potentiate the current greater than 150%. C. Concentration-response curves of zuranolone, DZP, and a combination at α4β3δ-expressing cells. Percent potentiation is plotted relative to the maximum GABA response.
3.1.4. Tonic and phasic current recordings in rat brain slices
Some NASs, such as brexanolone and SGE-516, can produce a sustained increase in GABA current that persists even after the NAS is presumed to be washed out, which is accompanied by an increase in receptor phosphorylation and surface localization (Modgil et al., 2017). To determine whether zuranolone exhibited this effect, hippocampal slices from postnatal day 21 mice were pre-incubated in either 0.1 or 1 μM zuranolone for 15 min before a 30–50 min wash in the recording chamber. Tonic GABA currents were measured from dentate granule cells using 100 μM picrotoxin and were normalized to the cell capacitance to determine current density (Fig. 4A). Current density from control dentate granule cells was 2.17 ± 0.4 pA/pF, which was not significantly changed by 0.1 μM zuranolone (2.48 ± 0.7 pA/pF). However, after pre-incubation with 1 μM zuranolone, current density increased to 5.55 ± 1.3 pA/pF (F (2, 16) = 4.282; p = 0.0324 vs control).
Fig. 4. GABAA receptor currents were increased by acute treatment with zuranolone when measured after a 30–50 min washout period.
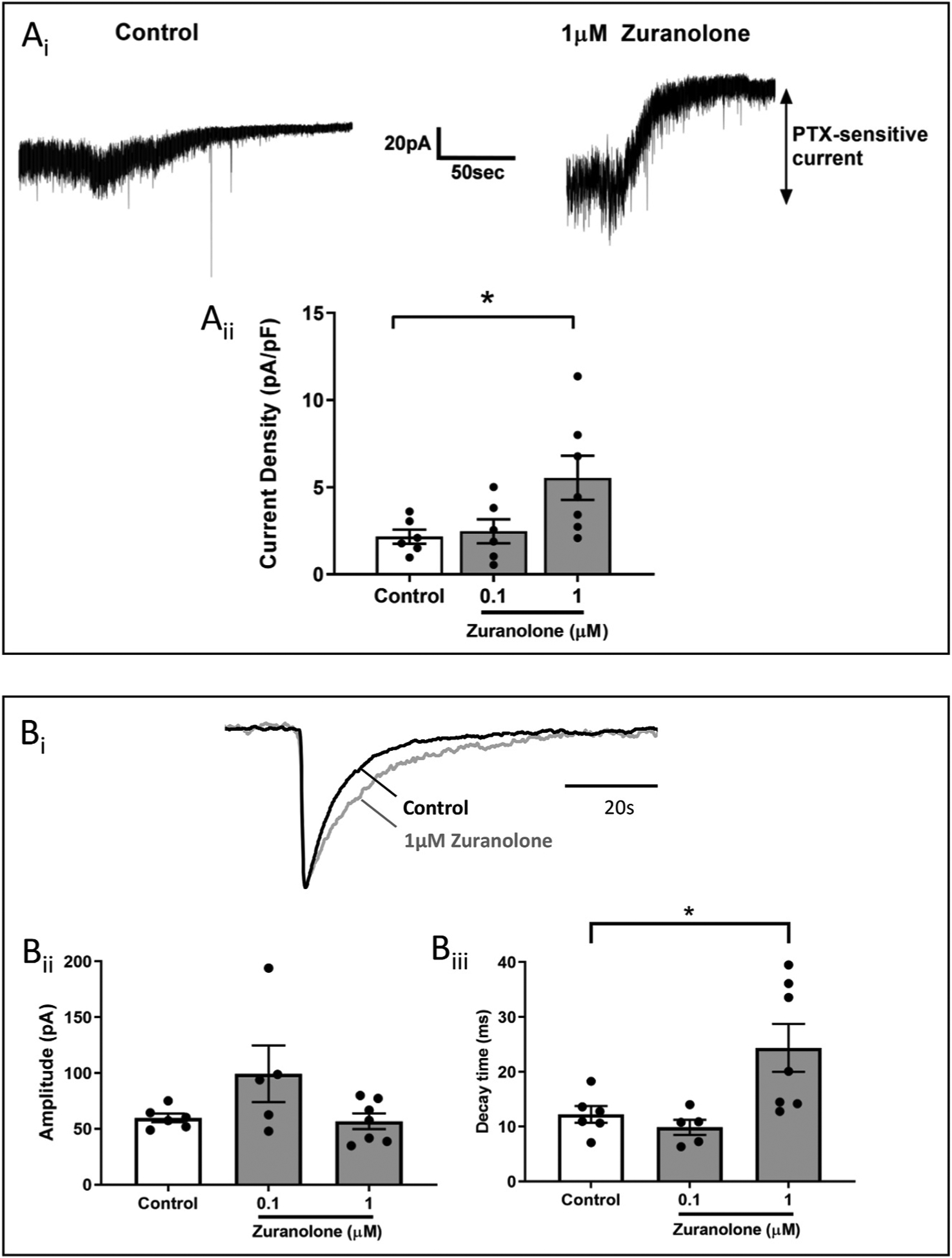
Ai. Sample recordings from dentate granule cells in mouse brain slices show larger tonic current after pre-treatment with 1 μM zuranolone compared to control treated cells. Aii. Tonic current density was approximately doubled following pre-treatment with 1 μM zuranolone. Bi. Example of averaged sIPSC recordings normalized to amplitude in vehicle control (black) or after a 15 min exposure of 1 μM zuranolone followed by 30–50 min washout (gray) demonstrating a prolongation of sIPSC decay. Bii. sIPSC amplitude was not significantly changed in cells after zuranolone pre-treatment. Biii. sIPSC decay time was significantly prolonged after pre-treatment with 1 μM zuranolone.
In parallel, the impact on sIPSC properties after the 30–50 min wash was assessed (Fig. 4B). The amplitude of sIPSCs was not significantly changed after incubation with 0.1 or 1 μM zuranolone (99 ± 26 pA and 57 ± 7 pA, respectively) compared to vehicle control (60 ± 4 pA). While there was no significant difference in sIPSC decay between 0.1 μM zuranolone and control (10 ± 1 ms, n = 5; 12 ± 2 ms, n = 6, respectively), there was a significant prolongation in sIPSC decay following incubation with 1 μM zuranolone (24 ± 4 ms, n = 7, F (2, 15) = 5.17 p = 0.02 vs control). There was no change in sIPSC frequency following zuranolone exposure (data not shown). These data, together with the previously published evidence of increased surface expression (Modgil et al., 2017), may indicate that zuranolone treatment resulted in a sustained increase in surface expression of tonic- and phasic-conducting GABAA receptors.
3.2. In vivo pharmacokinetic and pharmacodynamic activity
Drugs targeting the GABAergic system, including first generation NAS GABAA receptor PAMs have been used preclinically and clinically for decades. As a result, preclinical rodent models in which behavioral or electrophysiological endpoints are associated with activation of GABAA receptors (target engagement) are well-established and were used to evaluate the pharmacodynamic and translational activity of zuranolone in vivo.
3.2.1. Pharmacokinetics
The pharmacokinetics of zuranolone after IV and PO administration were previously reported and determined to support daily oral dosing (Martinez-Botella et al., 2017). Here, a comparison of IP and PO administration of 10 mg/kg zuranolone was performed in adult male CD1 mice (Fig. 5A). Both IP and PO administration of 10 mg/kg zuranolone achieved maximum plasma concentration (Cmax) at 30 min post dose. Oral administration resulted in a lower plasma Cmax (1335 vs 3197 ng/mL, respectively) compared to IP administration. Oral bioavailability of zuranolone in mouse was moderately high at 62%, while bioavailability was 89% after IP administration. The brain to plasma ratio following either IP or PO dosing was found to be in the range of 1.4–1.6. These data confirm previous reports of oral bioavailability and CNS exposure in mice and rats (Martinez-Botella et al., 2017).
Fig. 5. Zuranolone exhibited oral bioavailability consistent with once or twice daily dosing with exposures that cover behaviorally-active concentrations in mice.
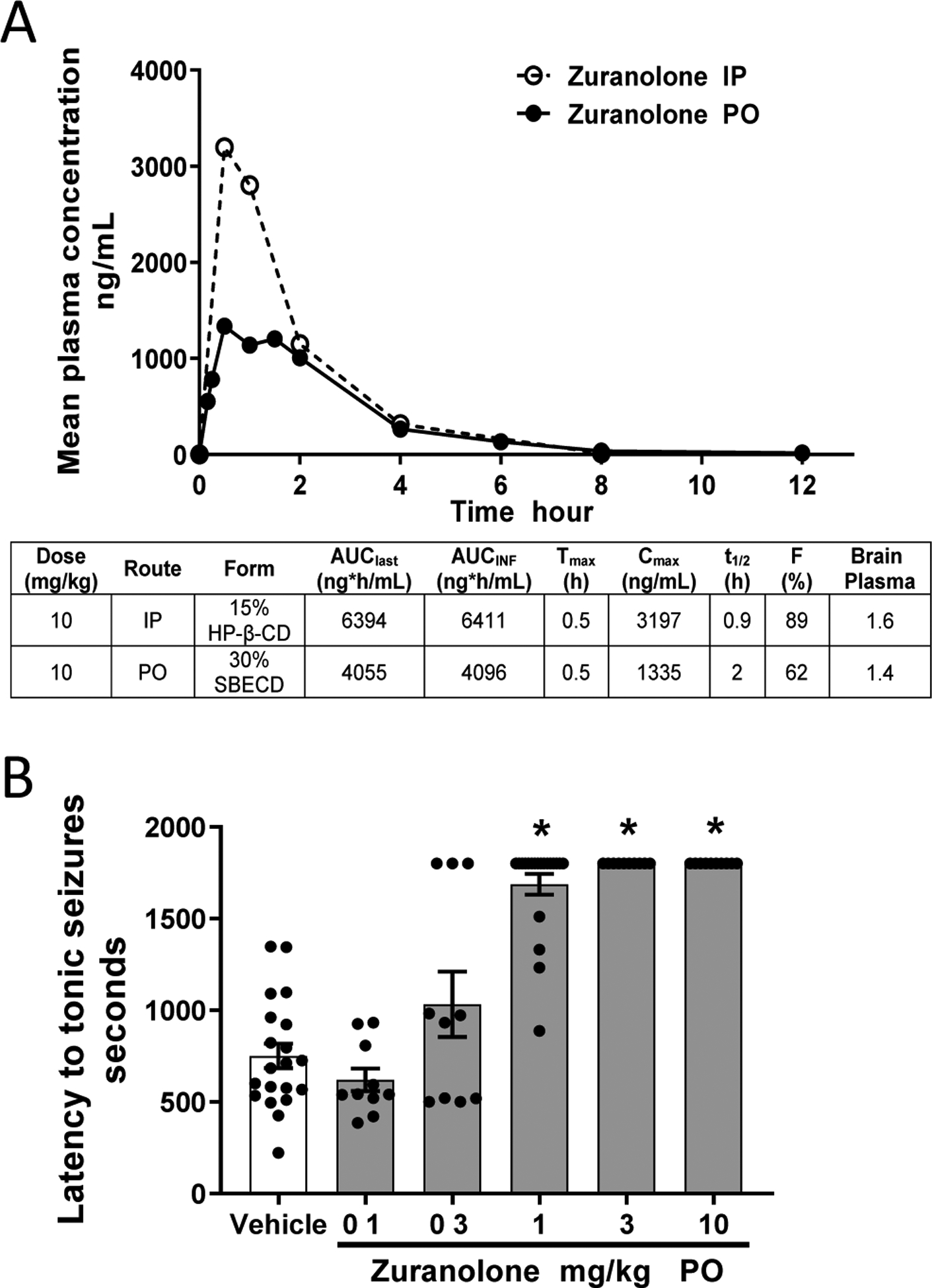
A. Mean plasma concentrations and pharmacokinetic parameters following a single IP or PO administration of 10 mg/kg zuranolone. Brain to plasma ratios were obtained by single point at 1 h post dosing. B. PO administration 60 min prior to systemic PTZ injection protected mice from exhibiting seizures with a minimum effective dose of 1 mg/kg, corresponding to a plasma concentration of 74 ng/mL and brain concentration of 143 ng/g.
3.2.2. Pharmacodynamic target engagement
To assess in vivo GABAA receptor target engagement, zuranolone was examined in the mouse PTZ acute seizure model. Prior to in vivo studies, zuranolone was confirmed to be selective for GABAA receptors vs other ion channel and hormone receptors, including AMPA and NMDA (Martinez-Botella et al., 2017). Systemic administration of PTZ evokes tonic seizures in a concentration-dependent manner consistent with its activity as a GABAA receptor inhibitor (Squires et al., 1984). Zuranolone (0.1, 0.3, 1, 3, and 10 mg/kg) was administered PO 60 min prior to PTZ to evaluate its ability to prevent seizure activity through enhancing GABAA receptor currents. At doses of 1, 3, and 10 mg/kg PO, zuranolone significantly increased the latency to tonic seizures (≥1688 ± 56 s) compared to vehicle (751 ± 67 s, F (5, 74) = 46.53; p < 0.0001; Fig. 5B). Administration of 0.3 mg/kg PO produced a non-significant trend toward increased latency to tonic seizures (1033 ± 179 s; p = 0.06). These data demonstrate that clear CNS-associated pharmacodynamic effects can be detected between 0.3 and 1 mg/kg of zuranolone in mice, which corresponds to brain concentrations of 60–143 ng/g.
3.2.3. Effects on a pharmacoEEG, a translatable biomarker
The Beta EEG band or β-band, defined as power in the 13–30 Hz frequency range, has been well-established as a robust in-vivo, translatable biomarker of GABAA PAM activity (Mandema and Danhof, 1992; Visser et al., 2003a). Multiple classes of GABAA receptor modulators produce dose-dependent changes in EEG power in the 13–30 Hz (β-band) frequency range, but there are quantitative and qualitative differences in how these drug classes affect this parameter (Christian et al., 2015). For example, NASs can maximally increase the EEG effect two to three times higher than benzodiazepines, and pharmacokinetic/pharmacodynamic studies have demonstrated a biphasic concentration-EEG effect relationship with NASs that is distinct from the monophasic relationship of benzodiazepines (Visser et al., 2002a, 2002b, 2003b).
To understand the relationship between zuranolone concentration and EEG effect, zuranolone (0.3, 3, and 20 mg/kg, PO) was administered following a brief (1 h) baseline EEG recording period. β-band EEG power increased rapidly after administration of both 3 and 20 mg/kg zuranolone. In the 3 mg/kg group, β-band EEG power peaked at approximately 0.5 h post dose (Fig. 6A, navy arrow) and slowly decayed across time and at one hour, β-band power was significantly greater than vehicle (Fig. 6B, p < 0.0005). In contrast, in the 20 mg/kg group, β-band EEG power peaked within 30 min and fell sharply to vehicle levels by one hour (Fig. 6A, turquoise arrow). At one hour, β-band EEG power with 20 mg/kg was not significantly different from vehicle (Fig. 6B, p = 0.79). This biphasic response over the tested dose and tested concentration range of zuranolone is consistent with the EEG effect-time course observed with other NASs (Visser et al., 2002a). Moreover, this response cannot be attributed to reduced zuranolone concentrations at one hour, as zuranolone levels increased dose-dependently at this time point (Fig. 6D). Administration of 0.3 mg/kg (PO) zuranolone did not elicit a significant increase in β-band EEG power relative to vehicle administration (Fig. 6A, B, p = 0.58). No changes in sleep or wake time were observed in rats after administration of 0.3 mg/kg zuranolone. Administration of 3 mg/kg zuranolone increased sleep time during the first hour (p < 0.05) while 20 mg/kg zuranolone was associated with increased time spent in sleep during all three hours (Fig. 6C, p < 0.05). Together, these data suggest that zuranolone has prominent effects on EEG β-band power that are consistent with the profile of GABA-modulating NASs and could serve as a clinical biomarker of CNS GABAA receptor target engagement.
Fig. 6. Zuranolone elicits changes in β-band EEG power in rat.
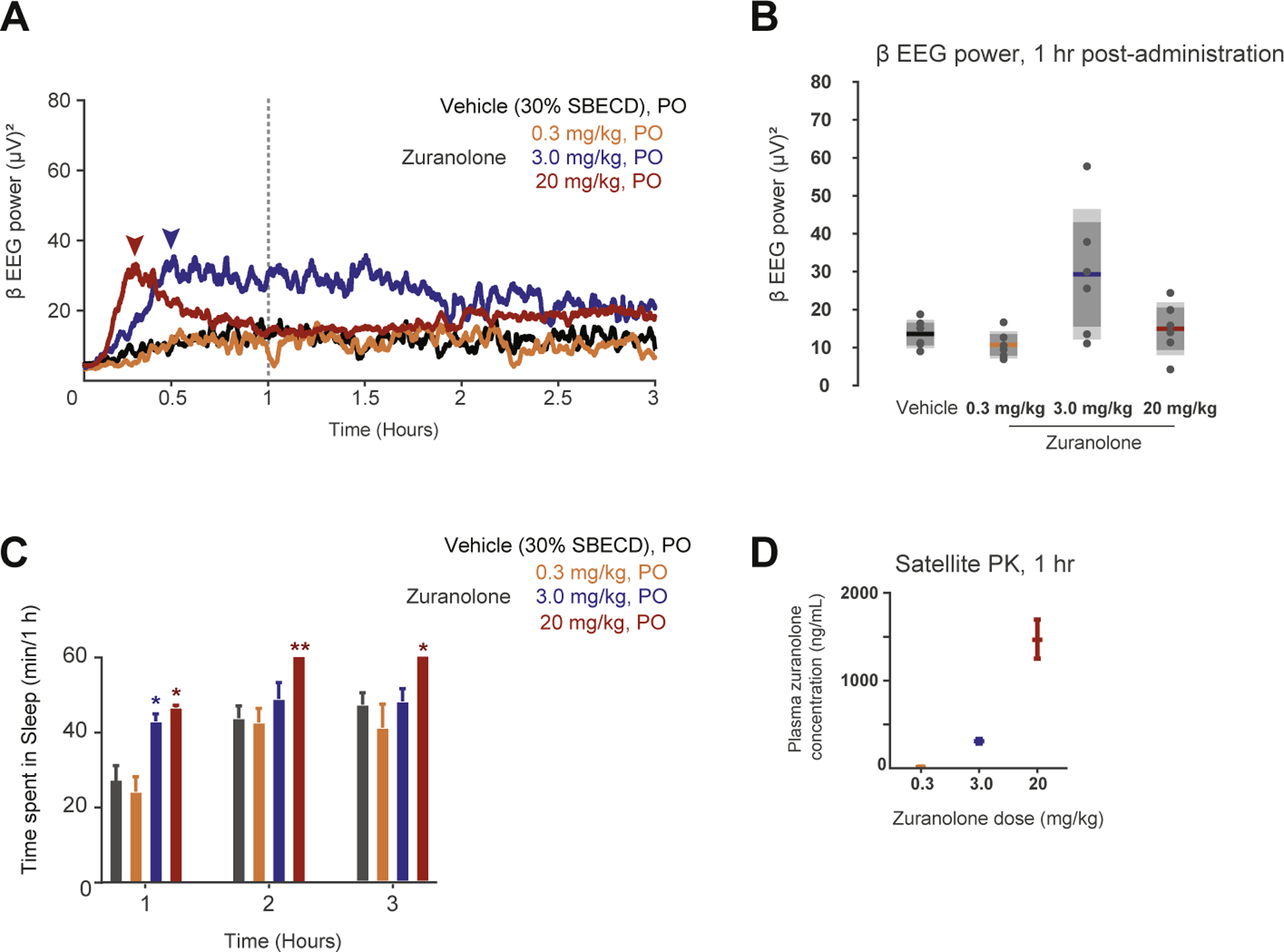
A. Averaged β-band EEG power plotted across time (post-administration) in response to vehicle, and 0.3, 3, and 20 mg/kg zuranolone in rat. Arrows indicate peak for the 3 and 20 mg/kg dose groups. Dotted line indicates 1 h time point. B. Box- and-whisker plot summarizing β-band EEG power at 1 h across distinct zuranolone concentrations. C. Cumulative time (min) spent in sleep during each hour. D. Plasma zuranolone concentrations were 22, 301, and 1466 ng/mL at 60 min post dosing for 0.3, 3.0, and 20 mg/kg zuranolone, respectively from rats designated for pharmacokinetic analysis.
4. Discussion
GABAergic neurotransmission plays a critical role in brain function, acting via multiple receptor types distributed throughout the central and peripheral nervous systems to regulate neuronal excitability in cortical and sub-cortical circuits (Engin et al., 2018). It is not surprising then that dysregulation of the excitatory/inhibitory balance in the brain contributes to the pathophysiology of many psychiatric and neurological disorders. For example, deficits in inhibitory function have been implicated in psychiatric disorders (Lüscher and Möhler, 2019), movement disorders (Blaszczyk, 2016; Gironell, 2014), neurodevelopmental disorders (Braat and Kooy, 2015) and seizure disorders (Bozzi et al., 2018; Jones-Davis and Macdonald, 2003). Pharmacological enhancement of the GABAergic system has been a productive therapeutic strategy in the treatment of multiple CNS disorders (Olsen, 2018). Interestingly, although some common properties are observed with many GABAA receptor enhancing drugs (e.g. sedative-hypnotic or anxiolytic effects), individual agents can produce very different effects on overall function (Jembrek and Vlainic, 2015; Weir et al., 2017). This diversity is attributable both to the complexity of GABAergic neurotransmission and to the specific target profiles of individual drugs.
NASs have long been hypothesized to offer clinical benefits differentiated from other GABAA receptor enhancing drugs, possibly due to their ability to modulate both synaptic and extrasynaptic GABAA receptors and to enhance tonic inhibition (Belelli et al., 2019; Zorumski, 2013). This therapeutic hypothesis has been recently confirmed in registrational clinical trials with the intravenously-delivered NAS brexanolone for the treatment of postpartum depression (Kanes et al., 2017a, 2017b; Meltzer-Brody et al., 2018). However, the ability to pair potent activity at both synaptic and extrasynaptic GABAA receptors with a readily orally available and moderately metabolized profile has, until recently, significantly limited the opportunity to deliver the pharmacological profile of NASs to broad groups of patients (Sear, 1996; Zorumski, 2013). In particular, limited absorption following oral administration coupled with rapid biotransformation and elimination have significantly limited the development of this potentially important class of drugs. Zuranolone was designed through a comprehensive structure-activity-relationship program to optimize the pharmacologic, pharmacokinetic, and pharmacodynamic properties of this class of NAS GABA modulators. Here we report the pharmacological profile of zuranolone as a potent, efficacious, and orally-bioavailable GABAA receptor PAM.
Zuranolone acted as a potent PAM at all recombinant human GABAA receptor isoforms tested (9/9), including those thought to be commonly expressed at synaptic and extrasynaptic sites on neurons (α1β2γ2 and α4β3δ receptor isoforms, respectively). In these experiments, which were conducted using different receptor expression systems, the range of potency values for zuranolone across all subunits tested (including those configurations reported by (Martinez-Botella et al., 2017)) were generally within 3- to 10-fold of each other. Differences observed for zuranolone activity both within and across different receptor expression systems are consistent with assay variability. These data are indicative of activity across numerous GABAA receptor subunit configurations, with no evidence of selectivity for or against a particular configuration.
In addition to allosteric enhancement of GABAA receptor function, zuranolone also increased tonic and phasic GABA currents in a manner consistent with the increased trafficking of extrasynaptic GABAA receptors previously reported for endogenous and synthetic neuroactive steroids (Abramian et al., 2014; Modgil et al., 2017). Previously, this trafficking effect was blocked by co-administration of the protein kinase C inhibitor, GF109203X (GFX) and was associated with increased phosphorylation of the β3 subunit and increased surface expression of β3-containing receptors, suggesting that it occurred through a metabotropic mechanism (Modgil et al., 2017). In a follow up study, the membrane progesterone receptor was identified as a potential mediator of trafficking effects (Parakala et al., 2019). More work is needed to clarify the role of membrane progesterone receptor in this process.
Efforts to characterize the NAS effects on trafficking have focused on β3-containing, presumed extrasynaptic receptors, but synaptic receptor surface expression is also dynamically regulated in a phosphorylation-dependent manner involving the β subunit (Kittler et al., 2005; Kittler and Moss, 2003). We cannot rule out a residual allosteric effect which could occur if the drug is slow to wash out. However, the observation that 1 μM zuranolone produced a sustained increase in tonic and phasic GABA currents after a ≥30 min washout period is consistent with increasing trafficking of extrasynaptic and synaptic GABAA receptors.
Tonic currents can produce a larger net inhibitory effect relative to phasic currents in neurons with both tonic (extrasynaptic) and phasic (synaptic) GABA conductance (Nusser and Mody, 2002). As a result, the ability of zuranolone to enhance receptor tonic inhibition provides further opportunities for zuranolone to enhance GABAergic current relative to compounds that only act at synaptic receptors. To date, benzodiazepines have not been shown to increase receptor trafficking; rather, benzodiazepine treatment has been associated with decreased GABAA receptor surface expression (Jacob et al., 2012).
How NASs bind at the GABAA receptor is an area of active investigation. An elegant body of work using homology modeling, site-directed mutagenesis, and a crystal structure of a chimeric GABAA receptor has unveiled NAS binding sites within the α and β-subunit transmembrane domains (Hosie et al., 2006, 2007, 2009; Laverty et al., 2017). While the precise binding location may not be the same for all compounds or under all conditions, a highly conserved site involving α-subunit transmembrane domain indicates the importance of this subunit for NAS binding (Hosie et al., 2009). The binding site for zuranolone is unknown, but the compound’s demonstrated ability to potentiate multiple receptor combinations across 6 α isoforms suggests that it could involve α transmembrane domain. Benzodiazepines, which bind to a distinct site on the γ subunit (Sigel, 2002), exhibit differentiated pharmacological properties from NASs. As such, it is reasonable to presume that co-administration of zuranolone and a benzodiazepine to receptors containing both α and γ subunits would produce physiological interactions. Indeed, there is in vitro evidence that NASs can influence the binding of the benzodiazepines [3H]flunitrazepam and [35S]TBPS (Hawkinson et al., 1994). Furthermore, positive interactions between low dose benzodiazepines and NASs have been identified in vivo in studies conducted with both rhesus monkeys (McMahon and France, 2006) and rats (Visser et al., 2003a). Nevertheless, it remains unclear from these studies whether these interactions are additive or synergistic. The formal isobolographic analysis performed here indicates that, at synaptic GABAA receptors, zuranolone synergized with the benzodiazepine diazepam, rather than producing an additive effect.
Zuranolone has been screened against multiple panels for non-GABAA receptor pharmacology as previously reported (Martinez-Botella et al., 2017). At 10 μM, it resulted in >50% inhibition of radioligand binding at two out of 73 receptors (87% at sigma, 57% at glycine), and antagonist activity (96%) at the transient receptor potential cation channel subfamily V member 1 receptor. It exhibited no significant effects at 22 nuclear hormone receptors or 8 cardiac ion channels (Martinez-Botella et al., 2017). In vivo, zuranolone exhibited 30–60% oral bioavailability in rodents and demonstrated clear pharmacodynamic effects, consistent with GABAA receptor activity, following oral administration in both a mouse PTZ seizure model and a rat EEG study. Together, these data establish the suitability of zuranolone as an orally bioavailable NAS GABAA receptor PAM with potential therapeutic utility for a variety of patient populations.
A wealth of preclinical in vivo evidence points to the potential for clinical efficacy with NAS GABAA receptor PAMs like zuranolone. Recent studies with SGE-516 have shown activity in diverse animal models of neurological and psychiatric diseases, including models of seizure and mood disorders (Althaus et al., 2017; Hammond et al., 2017; Hawkins et al., 2017; Melón et al., 2018). Interestingly, in a model of Dravet Syndrome (Hawkins et al., 2017) and a model of postpartum depression (Melón et al., 2018), SGE-516 exhibited differentiated efficacy when compared to the benzodiazepine clobazam, suggesting the distinct in vitro profile of NASs may lead to differentiated in vivo activity. Furthermore, the EEG signals recorded after oral administration of zuranolone in rat demonstrated a clear, biphasic concentration-response effect in β EEG power; the wide pharmacodynamic range of zuranolone differs from the range of benzodiazepines, which tend to have a mono-phasic concentration-effect profile (Visser et al., 2002a). Taken together, this EEG profile, the anticonvulsant activity of zuranolone in the mouse PTZ assay, and the in vitro activity of zuranolone establish the compound as a potent GABAA receptor PAM that exhibits robust pharmacological activity consistent with other NASs and differentiated from non-NAS GABAA receptor PAMs, such as benzodiazepines.
Whereas the potential therapeutic utility for NASs has been recognized for some time (Gasior et al., 1999; Gyermek and Soyka, 1975), the physicochemical and pharmacokinetic properties have, until recently, limited utility in patient populations (Sear, 1996; Zorumski, 2013). Zuranolone was designed to maintain the desirable pharmacology of neuroactive steroids but with a pharmacokinetic profile consistent with oral, daily dosing and CNS exposure (Martinez-Botella et al., 2017). Phase 1 studies in healthy volunteers have demonstrated that zuranolone was generally well tolerated and exhibited predictable pharmacokinetics following oral administration (Hoffmann et al., 2019). Zuranolone is currently under clinical investigation for the treatment of major depressive episodes in major depressive disorder, postpartum depression, and bipolar depression (Gunduz-Bruce et al., 2019; Hoffmann et al., 2019).
Funding
All experiments were paid for by Sage Therapeutics.
Abbreviations:
- AUClast
area under the curve from time 0 to last measurable
- AUCINF
area under the curve from time 0 extrapolated to infinity
- CHO
chinese hamster ovary
- Cmax
maximum concentration
- CNS
central nervous system
- DZP
diazepam
- EEG
electroencephalogram
- EC50
effective concentration (50%)
- Emax
maximum efficacy
- F
bioavailability
- HEK
human embryonic kidney
- IP
intraperitoneal
- Ltk
Mouse fibroblast cell line
- NAS
neuroactive steroid
- P4S
piperidine-4-suphonic acid
- PAM
positive allosteric modulator
- PO
per os
- PTZ
pentylenetetrazol
- SBECD
sulfobutyl ether β-cyclodextrin
- Tmax
time to reach maximum concentration
- HP-β-CD
2-Hydroxypropyl)-β-Cyclodextrin
- t1/2
half-life
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
All work described in this manuscript was paid for by Sage Therapeutics. Some authors are employees, consultants, or shareholders of Sage Therapeutics.
References
- Abramian AM, Comenencia-Ortiz E, Modgil A, Vien TN, Nakamura Y, Moore YE, Maguire JL, Terunuma M, Davies PA, Moss SJ, 2014. Neurosteroids promote phosphorylation and membrane insertion of extrasynaptic GABAA receptors. Proc. Natl. Acad. Sci. U. S. A 111, 7132–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althaus AL, McCarren HS, Alqazzaz A, Jackson C, McDonough JH, Smith CD, Hoffman E, Hammond RS, Robichaud AJ, Doherty JJ, 2017. The synthetic neuroactive steroid SGE-516 reduces status epilepticus and neuronal cell death in a rat model of soman intoxication. Epilepsy Behav. 68, 22–30. [DOI] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW, 2009. Extrasynaptic GABAA receptors: form, pharmacology, and function. J. Neurosci 29, 12757–12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Hogenkamp D, Gee KW, Lambert JJ, 2019. Realising the therapeutic potential of neuroactive steroid modulators of the GABAA receptor. Neurobiol. Stress 12, 100207. 10.1016/j.ynstr.2019.100207.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk J, 2016. Parkinson’s disease and neurodegeneration: GABA-collapse hypothesis. Front. Neurosci 10, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi Y, Provenzano G, Casarosa S, 2018. Neurobiological bases of autism-epilepsy comorbidity: a focus on excitation/inhibition imbalance. Eur. J. Neurosci 47, 534–548. [DOI] [PubMed] [Google Scholar]
- Braat S, Kooy RF, 2015. The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86, 1119–1130. [DOI] [PubMed] [Google Scholar]
- Carver CM, Reddy DS, 2013. Neurosteroid interactions with synaptic and extrasynaptic GABAA receptors: regulation of subunit plasticity, phasic and tonic inhibition, and neuronal network excitability. Psychopharmacology 230, 151–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian EP, Snyder DH, Song W, Gurley DA, Smolka J, Maier DL, Ding M, Gharahdaghi F, Liu XF, Chopra M, Ribadeneira M, Chapdelaine MJ, Dudley A, Arriza JL, Maciag C, Quirk MC, Doherty JJ, 2015. EEG-beta/gamma spectral power elevation in rat: a translatable biomarker elicited by GABA (Aalpha2/3)-positive allosteric modulators at nonsedating anxiolytic doses. J. Neurophysiol 113, 116–131. [DOI] [PubMed] [Google Scholar]
- Ellenbogen A, Raines S, Kanes S, 2016. Exploratory trial results for SAGE-547 in essential tremor. Neurology 78. P4.297–P294.297. [Google Scholar]
- Engin E, Benham RS, Rudolph U, 2018. An emerging circuit pharmacology of GABAA receptors. Trends Pharmacol. Sci 39, 710–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior M, Carter RB, Witkin JM, 1999. Neuroactive steroids: potential therapeutic use in neurological and psychiatric disorders. Trends Pharmacol. Sci 20, 107–112. [DOI] [PubMed] [Google Scholar]
- Gielen MC, Lumb MJ, Smart TG, 2012. Benzodiazepines modulate GABAA receptors by regulating the preactivation step after GABA binding. J. Neurosci. : the official journal of the Society for Neuroscience 32, 5707–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gironell A, 2014. In: The GABA Hypothesis in Essential Tremor: Lights and Shadows. Tremor and Other Hyperkinetic Movements, vol. 4, p. 254. New York, N.Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunduz-Bruce H, Silber C, Kaul I, Rothschild AJ, Riesenberg R, Sankoh AJ, Li H, Lasser R, Zorumski CF, Rubinow DR, Paul SM, Jonas J, Doherty JJ, Kanes SJ, 2019. Trial of SAGE-217 in patients with major depressive disorder. N. Engl. J. Med 381, 903–911. [DOI] [PubMed] [Google Scholar]
- Gyermek L, Soyka LF, 1975. Steroid anesthetics. Anesthesiology 42, 331–344. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Althaus AL, Ackley MA, Maciag C, Martinez Botella G, Salituro FG, Robichaud AJ, Doherty JJ, 2017. Anticonvulsant profile of the neuroactive steroid, SGE-516, in animal models. Epilepsy Res. 134, 16–25. [DOI] [PubMed] [Google Scholar]
- Hawkins NA, Lewis M, Hammond RS, Doherty JJ, Kearney JA, 2017. The synthetic neuroactive steroid SGE-516 reduces seizure burden and improves survival in a Dravet syndrome mouse model. Sci. Rep 7, 15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkinson JE, Kimbrough CL, Belelli D, Lambert JJ, Purdy RH, Lan NC, 1994. Correlation of neuroactive steroid modulation of [35S]t-butylbicyclophosphorothionate and [3H]flunitrazepam binding and gamma-aminobutyric acidA receptor function. Mol. Pharmacol 46, 977–985. [PubMed] [Google Scholar]
- Hoffmann E, Nomikos GG, Kaul I, Raines S, Wald J, Bullock A, Sankoh AJ, Doherty J, Kanes SJ, Colquhoun H, 2019. SAGE-217, A Novel GABAA Receptor Positive Allosteric Modulator: Clinical Pharmacology and Tolerability in Randomized Phase I Dose-Finding Studies. Clinical pharmacokinetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg RC, Bandelier F, Benoit A, Dosch R, Bertrand D, 2008. An automated system for intracellular and intranuclear injection. J. Neurosci. Methods 169, 65–75. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Clarke L, da Silva H, Smart TG, 2009. Conserved site for neurosteroid modulation of GABA A receptors. Neuropharmacology 56, 149–154. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva H.M.a., Smart TG, 2006. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 444, 486–489. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, Smart TG, 2007. Neurosteroid binding sites on GABAA receptors. Pharmacol. Therapeut 116, 7–19. [DOI] [PubMed] [Google Scholar]
- Jacob TC, Michels G, Silayeva L, Haydon J, Succol F, Moss SJ, 2012. Benzodiazepine treatment induces subtype-specific changes in GABA(A) receptor trafficking and decreases synaptic inhibition. Proc. Natl. Acad. Sci. U. S. A 109, 18595–18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jembrek MJ, Vlainic J, 2015. GABA receptors: pharmacological potential and pitfalls. Curr. Pharmaceut. Des 21, 4943–4959. [DOI] [PubMed] [Google Scholar]
- Jones-Davis DM, Macdonald RL, 2003. GABA(A) receptor function and pharmacology in epilepsy and status epilepticus. Curr. Opin. Pharmacol 3, 12–18. [DOI] [PubMed] [Google Scholar]
- Kanes S, Colquhoun H, Gunduz-Bruce H, Raines S, Arnold R, Schacterle A, Doherty J, Epperson CN, Deligiannidis KM, Riesenberg R, Hoffmann E, Rubinow D, Jonas J, Paul S, Meltzer-Brody S, 2017a. Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet 390, 480–489. [DOI] [PubMed] [Google Scholar]
- Kanes SJ, Colquhoun H, Doherty J, Raines S, Hoffmann E, Rubinow DR, Meltzer-Brody S, 2017b. Open-label, proof-of-concept study of brexanolone in the treatment of severe postpartum depression. Hum. Psychopharmacol 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Chen G, Honing S, Bogdanov Y, McAinsh K, Arancibia-Carcamo IL, Jovanovic JN, Pangalos MN, Haucke V, Yan Z, Moss SJ, 2005. Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proc. Natl. Acad. Sci. U. S. A 102, 14871–14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Moss SJ, 2003. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr. Opin. Neurobiol 13, 341–347. [DOI] [PubMed] [Google Scholar]
- Laverty D, Thomas P, Field M, Andersen OJ, Gold MG, Biggin PC, Gielen M, Smart TG, 2017. -receptor chimera reveal new endogenous neurosteroid-binding sites. Nat. Struct. Mol. Biol 24, 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher B, Möhler H, 2019. In: Brexanolone, a Neurosteroid Antidepressant, Vindicates the GABAergic Deficit Hypothesis of Depression and May Foster Resilience, vol. 8. F1000Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM, 1986. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232, 1004–1007. [DOI] [PubMed] [Google Scholar]
- Mandema JW, Danhof M, 1992. Electroencephalogram effect measures and relationships between pharmacokinetics and pharmacodynamics of centrally acting drugs. Clin. Pharmacokinet 23, 191–215. [DOI] [PubMed] [Google Scholar]
- Martinez-Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Blanco MJ, Belfort GM, Dai J, Loya CM, Ackley MA, Althaus AL, Grossman SJ, Hoffmann E, Doherty JJ, Robichaud AJ, 2017. Neuroactive steroids. 2. 3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1’-yl)-19-nor-5β-pregnan-20-one (SAGE-217): a clinical next generation neuroactive steroid positive allosteric modulator of the (γ-Aminobutyric Acid)A receptor. J. Med. Chem 60, 7810–7819. [DOI] [PubMed] [Google Scholar]
- Martinez-Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Shen K, Belfort GM, Loya CM, Ackley M.a., Grossman SJ, Hoffmann E, Jia S, Wang J, Doherty JJ, Robichaud AJ, 2015. Neuroactive steroids. 1. Positive allosteric modulators of the (γ-Aminobutyric acid) A receptor: structure-activity relationships of heterocyclic substitution at C-21. J. Med. Chem 58, 3500–3511. [DOI] [PubMed] [Google Scholar]
- McMahon LR, France CP, 2006. Differential behavioral effects of low efficacy positive GABAA modulators in combination with benzodiazepines and a neuroactive steroid in rhesus monkeys. Br. J. Pharmacol 147, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melón L, Hammond R, Lewis M, Maguire J, 2018. A novel, synthetic, neuroactive steroid is effective at decreasing depression-like behaviors and improving maternal Care in preclinical models of postpartum depression. Front. Endocrinol 9, 703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer-Brody S, Colquhoun H, Riesenberg R, Epperson CN, Deligiannidis KM, Rubinow DR, Li H, Sankoh AJ, Clemson C, Schacterle A, Jonas J, Kanes S, 2018. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet 392, 1058–1070. [DOI] [PubMed] [Google Scholar]
- Modgil A, Parakala ML, Ackley MA, Doherty JJ, Moss SJ, Davies PA, 2017. Endogenous and synthetic neuroactive steroids evoke sustained increases in the efficacy of GABAergic inhibition via a protein kinase C-dependent mechanism. Neuropharmacology 113, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss P, 2015. Anxiety disorders and GABA neurotransmission: a disturbance of modulation. Neuropsychiatric Dis. Treat 11, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Mody I, 2002. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J. Neurophysiol 87, 2624–2628. [DOI] [PubMed] [Google Scholar]
- O’Shea SM, Wong LC, Harrison NL, 2000. Propofol increases agonist efficacy at the GABA(A) receptor. Brain Res. 852, 344–348. [DOI] [PubMed] [Google Scholar]
- Olsen RW, 2018. GABAA receptor: positive and negative allosteric modulators. Neuropharmacology 136, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W, 2009. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 56, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parakala ML, Zhang Y, Modgil A, Chadchankar J, Vien TN, Ackley MA, Doherty JJ, Davies PA, Moss SJ, 2019. Metabotropic, but not allosteric, effects of neurosteroids on GABAergic inhibition depend on the phosphorylation of GABAA receptors. J. Biol. Chem 294, 12220–12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sear JW, 1996. Steroid anesthetics: old compounds, new drugs. J. Clin. Anesth 8, 91S–98S. [DOI] [PubMed] [Google Scholar]
- Sheean G, McGuire JR, 2009. Spastic hypertonia and movement disorders: pathophysiology, clinical presentation, and quantification. PM&R 1, 827–833. [DOI] [PubMed] [Google Scholar]
- Sigel E, 2002. Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr. Top. Med. Chem 2, 833–839. [DOI] [PubMed] [Google Scholar]
- Sigel E, Steinmann ME, 2012. Structure, function, and modulation of GABA(A) receptors. J. Biol. Chem 287, 40224–40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires RF, Saederup E, Crawley JN, Skolnick P, Paul SM, 1984. Convulsant potencies of tetrazoles are highly correlated with actions on GABA/benzodiazepine/picrotoxin receptor complexes in brain. Life Sci. 35, 1439–1444. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ, 2011. Quantitative methods for assessing drug synergism. Gene Canc. 2, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiman DM, 2001. GABAergic mechanisms in epilepsy. Epilepsia 42, 8–12. [DOI] [PubMed] [Google Scholar]
- Visser SA, Huntjens DR, van der Graaf PH, Peletier LA, Danhof M, 2003a. Mechanism-based modeling of the pharmacodynamic interaction of alphaxalone and midazolam in rats. J. Pharmacol. Exp. Therapeut 307, 765–775. [DOI] [PubMed] [Google Scholar]
- Visser SA, Smulders CJ, Reijers BP, Van der Graaf PH, Peletier LA, Danhof M, 2002a. Mechanism-based pharmacokinetic-pharmacodynamic modeling of concentration-dependent hysteresis and biphasic electroencephalogram effects of alphaxalone in rats. J. Pharmacol. Exp. Therapeut 302, 1158–1167. [DOI] [PubMed] [Google Scholar]
- Visser SAG, Gladdines WWFT, van der Graaf PH, Peletier LA, Danhof M, 2002b. Neuroactive steroids differ in potency but not in intrinsic efficacy at the GABA(A) receptor in vivo. J. Pharmacol. Exp. Therapeut 303, 616–626. [DOI] [PubMed] [Google Scholar]
- Visser SAG, Wolters FLC, Gubbens-Stibbe JM, Tukker E, Van der Graaf PH, Peletier LA, Danhof M, 2003b. Mechanism-Based pharmacokinetic/pharmacodynamic modeling of the electroencephalogram effects of GABAA receptor modulators: in vitro-in vivo correlations. J. Pharmacol. Exp. Therapeut 304, 88–101. [DOI] [PubMed] [Google Scholar]
- Weir CJ, Mitchell SJ, Lambert JJ, 2017. Role of GABAA receptor subtypes in the behavioural effects of intravenous general anaesthetics. Br. J. Anaesth 119, i167–i175. [DOI] [PubMed] [Google Scholar]
- Winsky-Sommerer R, 2009. Role of GABAA receptors in the physiology and pharmacology of sleep. Eur. J. Neurosci 29, 1779–1794. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, 2013. Neurosteroids as therapeutic leads in psychiatry. JAMA Psychiatry 70, 659. [DOI] [PubMed] [Google Scholar]
- Zorumski CF, Mennerick S, Isenberg KE, Covey DF, 2000. Potential clinical uses of neuroactive steroids. Idrugs : the investigational drugs journal 3, 1053–1063. [PubMed] [Google Scholar]