

ABSTRACT
The PorX/PorY two-component system in the periodontal pathogen Porphyromonas gingivalis controls the expression of the por genes, encoding a type IX secretion system, and the sigP gene, encoding sigma factor σP. Previous results implied that PorX/PorY and σP formed a regulatory cascade because the PorX/PorY-activated σP enhanced the por genes, including porT, via binding to their promoters. We recently showed that PorX also binds to the por promoters, thus suggesting that an alternative mechanism is required for the PorX/PorY- and σP-governed expression. Here, our in vitro assays show the PorX response regulator binds to the sigP promoter at a sequence shared with the porT promoter and enhances its transcription, mediated by a reconstituted P. gingivalis RNA polymerase holoenzyme. Merely producing σP in trans fails to reverse the porT transcription in a porX mutant, which further argues against the action of the proposed regulatory cascade. An in vitro transcription assay using a reconstituted RNA polymerase-σP holoenzyme verifies the direct role of PorX in porT transcription, since transcription is enhanced by a pure PorX protein. Accordingly, we propose that the PorX/PorY system coordinates with σP to construct a coherent regulatory mechanism, known as the feedforward loop. Specifically, PorX will not only bind to the sigP promoter to stimulate the expression of σP, but also bind to the porT promoter to facilitate the RNA polymerase-σP-dependent transcription. Importantly, mutations at the porX and sigP genes attenuate bacterial virulence in a mouse model, demonstrating that this regulatory mechanism is essential for P. gingivalis pathogenesis.
IMPORTANCE The anaerobic bacterium Porphyromonas gingivalis is not only the major etiologic agent for chronic periodontitis, but also prevalent in some common noncommunicable diseases such as cardiovascular disease, Alzheimer's disease, and rheumatoid arthritis. We present genetic, biochemical, and biological results to demonstrate that the PorX/PorY two-component system and sigma factor σP build a specific regulatory network to coordinately control transcription of the genes encoding the type IX secretion system, and perhaps also other virulence factors. Results in this study verify that the response regulator PorX stimulates the expression of the genes encoding both σP and the type IX secretion system by binding to their promoters. This study also provides evidence that σP, like the PorX/PorY system, contributes to P. gingivalis virulence in a mouse model.
KEYWORDS: PorX/PorY, Porphyromonas gingivalis, extracytoplasmic function sigma factor P, feedforward loop, in vitro transcription, transcription regulation, two-component system, type IX secretion system, virulence factors
INTRODUCTION
The Gram-negative anaerobic bacterium Porphyromonas gingivalis is the major etiologic agent for chronic periodontitis. This pathogenic bacterium produces a repertoire of virulence factors, including specific cysteine proteases, also known as gingipains (1–3). Secretion of gingipains is mediated by a type IX secretion system (T9SS) in a manner dependent on the porX and porY gene products, PorX and PorY, which were proposed to form a two-component regulatory system (TCS) by the Nakayama laboratory (4). Particularly, their results showed that PorX, the response regulator, and PorY, the histidine kinase, were able to upregulate the expression of the T9SS-encoding genes (referred to as the por genes, herein) including porT, sov, porP, porK, porL, porM, and porN (4). Furthermore, Kadowaki et al. carried out a surface plasmon resonance analysis and showed that PorY could directly interact with, and subsequently phosphorylate, PorX (5), thus experimentally demonstrating that these two proteins should be the TCS cognate pair. However, it remains elusive whether the PorX/PorY system controls transcriptional regulation of the por genes directly and, if that is the case, how the response regulator PorX interacts with these target genes.
In accordance with the observations from Kadowaki et al. (5), the Vincent and Cascales laboratory used a bacterial two-hybrid system and confirmed the in vivo interaction between the PorX and PorY proteins (6). In contrast, their results suggested that PorX should be involved in the dynamics of the T9SS system via an interaction with the cytoplasmic domain of the T9SS component PorL (6). They further argued that PorX/PorY could not regulate the por genes since they failed to observe PorX binding to the por promoters in a P. gingivalis promoter/PorX reconstitution assay performed in Escherichia coli, in which PorX was heterologously expressed for testing its role in stimulating a plasmid-borne gfp (green fluorescent protein) gene controlled by a por promoter (6). However, we realized that the α, β, and β’ subunits of P. gingivalis RNA polymerase, which are encoded by the PGN_1841, PGN_1571, and PGN_1570 genes, respectively, merely share 38%, 46%, and 50% of identity to the corresponding subunits of the E. coli RNA polymerase. Additionally, the major sigma factor σD (encoded by the PGN_0638 gene) of P. gingivalis, which is a 287-residue protein, shares just 39% identity with the C-terminal 267-aa sequence of the E. coli housekeeping sigma factor σ70 (613 aa). Therefore, it is reasonable to postulate that P. gingivalis promoters cannot be recognized by the E. coli RNA polymerase-σ70 holoenzyme. Accordingly, the gfp expression would very unlikely be enhanced in the P. gingivalis promoter/PorX reconstitution assay conducted by Vincent et al. (6), even if these P. gingivalis promoters could bind the heterologously expressed PorX protein in E. coli. Recently, our study confirmed that the PorX/PorY system exerted a regulatory effect on the transcription of its target genes (7). The PorX/PorY regulatory role was further verified by our electrophoretic mobility shift assay (EMSA) and DNase footprinting analyses, which provided evidence that a PorX protein was able to bind the promoter of a por gene, porT, by interacting with two DNA sequences, i.e., site I (5′-tattacttccataattattgttgtg-3′) and site II (5′-gattcgcgcaaaaatacaatatcttt-3′) (7).
According to the observations from Kadowaki et al. (5), the PorX/PorY system upregulates transcription of the sigP gene (i.e., PGN_0274) that encodes an extracytoplasmic function sigma factor, σP, and then σP mediates transcriptional activation of the por genes by binding to their promoters. In addition, their results suggest the function of σP is directly associated with PorX because these two proteins could be coimmunoprecipitated from P. gingivalis cell lysates (5). Based on these results, it seems reasonable to propose a regulatory cascade in which the PorX/PorY system stimulates σP and, in turn, σP enhances the por genes. However, it remains largely unknown whether the PorX/PorY system can directly or indirectly upregulate sigP transcription, and whether the PorX/PorY system and σP activate the por genes in a manner dependent on the regulatory cascade. In this study, we show that the PorX response regulator not only binds to the sigP promoter to activate transcription of this sigma factor gene, but also binds to a por promoter with a σP-RNA polymerase holoenzyme to initiate its transcription. Based on our observations from both in vitro and in vivo analyses, we propose a feedforward regulatory loop to illustrate gene regulation in a manner dependent on the PorX/PorY system and σP, which also provides an example to elucidate the coordinate interaction between two-component systems and their regulated sigma factors in gene regulation of P. gingivalis. Additionally, our results demonstrate that both the PorX/PorY system and σP are virulence factors that govern transcription of the genetic loci required for P. gingivalis virulence.
RESULTS AND DISCUSSION
PorX/PorY system and sigma factor σP coordinately regulate transcription in P. gingivalis.
The PorX/PorY two-component system and extracytoplasmic function sigma factor σP (encoded by the sigP gene) appeared to form a regulatory cascade for upregulation of the T9SS-encoding genes (i.e., the por genes) because PorX/PorY was shown to upregulate σP and, in turn, σP enhanced the transcription of the por genes (5). Particularly, it was observed that the PorX/PorY-stimulated σP bound the promoters of the por genes, including the porT gene which encodes a T9SS component (5). Besides σP, a recent result from our laboratory showed the PorX protein also bound to the porT promoter and actually interacted with two DNA regions (7). This result not only verified the DNA-binding ability of the PorX response regulator, but also provided the possibility that both PorX and σP should directly act on the por promoters. If PorX and σP must coordinately control but not build a regulatory cascade to regulate the por genes, we postulate that σP expressed in trans in the absence of PorX, or vice versa, should not stimulate por transcription. We examined this hypothesis by determining the transcription of the porT gene in a porX deletion mutant (ΔporX). As predicted, a σP protein that was expressed in trans from a plasmid (pT-COW-PsigP-sigP, referred to as p-sigP) did not exert any effect on the porT expression in the ΔporX mutant because the porT mRNA level in this mutant harboring p-sigP remained similar to that in the mutant harboring the parental plasmid pT-COW (8), both of which were ∼6.4-fold lower than that in the wild-type strain (Fig. 1A). Likewise, PorX had no effect on porT expression in the absence of σP, because a PorX protein expressed in trans from a plasmid (pT-COW-PPGN_1016-porX, referred to as p-porX) did not stimulate porT transcription in a sigP null (ΔsigP) mutant (Fig. 1A). In contrast, the alleviated porT transcription was fully reversed to a wild-type level in the ΔsigP mutant harboring p-sigP and also in the ΔporX mutant harboring p-porX (Fig. 1A), indicating the trans-expressed σP and PorX proteins were functionally active. We also determined whether this coordinate regulation was effective in controlling two other PorX/PorY- and σP-dependent genes, PGN_0341, which encodes a predicted T9SS component (4), and PGN_1639, which has been known as a σP-dependent gene (5) and recently identified as a PorX/PorY-dependent locus according to our transcriptomic and proteomic analyses (unpublished data). We confirmed that the transcription levels of PGN_0341 and PGN_1639 were upregulated by PorX and σP because their mRNA levels were significantly reduced in the ΔporX and ΔsigP mutants compared to those in the wild-type strain (Fig. 1B and 1C). Comparable to the porT regulation (Fig. 1A), the alleviated transcription of PGN_0341 and PGN_1639 was not stimulated in ΔporX mutant harboring p-sigP or in ΔsigP mutant harboring p-porX (Fig. 1B and 1C).
FIG 1.
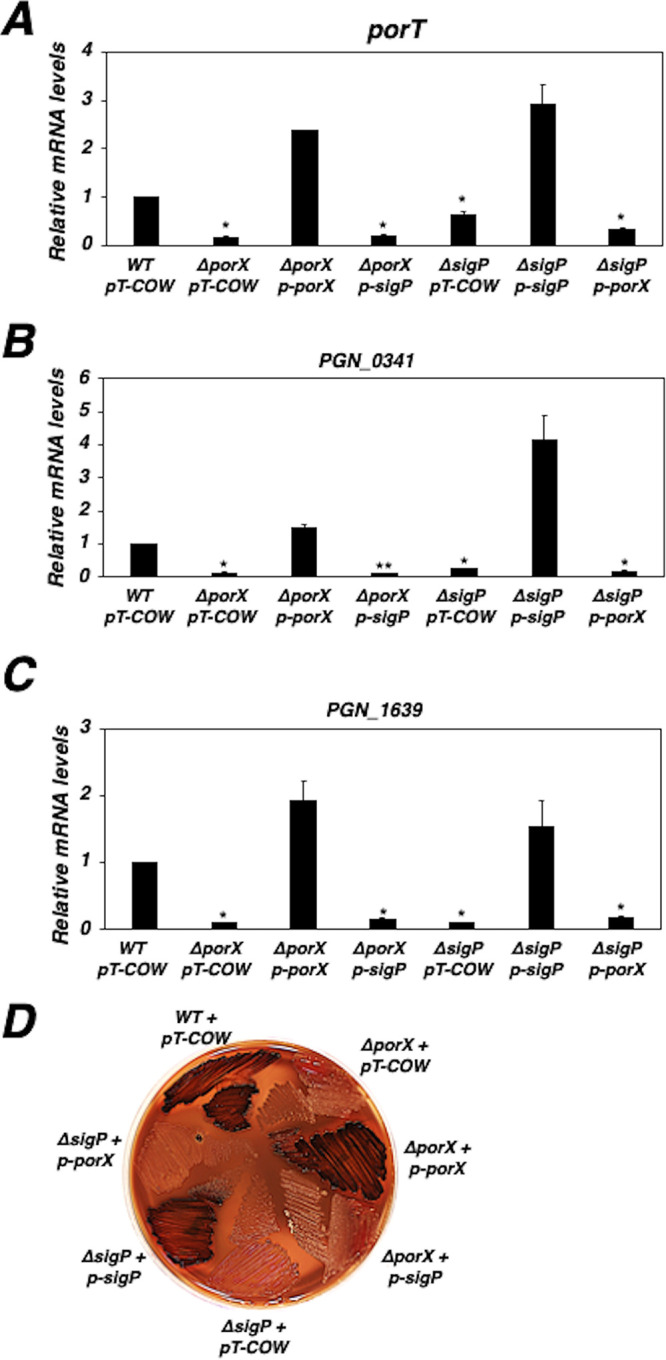
The PorX/PorY system and σP coordinate transcription in P. gingivalis. (A to C) mRNA levels of the porT gene (A), the PGN_0341 gene (B), and the PGN_1639 gene (C) in the 33277 wild-type strain, the ΔporX mutant (YS19181), and the ΔsigP mutant (YS17717) carrying pT-COW, p-porX (pYS18679, pT-COW-PPGN_1016-porX), or p-sigP (pYS19107, pT-COW-PsigP-sigP). The mRNA level in the wild-type strain was set to 1 for calculation. Results are representative of three independent experiments. *, P < 0.05; **, P < 0.01; versus wild type by t test. (D) The growth of wild-type 33277 strain with pT-COW (vector), ΔporX mutant (YS19181), and ΔsigP mutant (YS17717) carrying pT-COW, p-porX, or p-sigP, respectively, on a blood BHI plate containing tetracycline (0.5 μg/ml). Results are representative of four independent experiments.
It has been shown that T9SS mediates secretion of gingipains, which are required for pigmentation of P. gingivalis on a blood plate (for review see reference 9), and consistently both ΔporX and ΔsigP mutants display a nonpigmented phenotype (6, 10). We conducted a phenotypic analysis to evaluate the coordinate interaction between the PorX/PorY system and σP. While the ΔporX and ΔsigP mutants carrying pT-COW exhibited nonpigmented colonies on a brain heart infusion (BHI) blood plate, both the ΔporX mutant harboring p-porX and the ΔsigP mutant harboring p-sigP formed vigorous black-pigmented colonies (Fig. 1D). However, the ΔporX mutant harboring p-sigP and the ΔsigP mutant harboring p-porX exhibited a nonpigmented phenotype when they were grown on a BHI blood plate (Fig. 1D). Taken together, these genetic approaches suggest the PorX/PorY system and σP should govern transcription of the por genes via a coherent regulatory network rather than a direct regulatory cascade.
PorX response regulator directly binds to the promoter of the sigma factor gene sigP.
Evidence suggests that transcription of the sigP gene is activated by the PorX/PorY system (5). This is confirmed by our result derived from a reverse transcription-PCR, since the sigP mRNA level in the ΔporX mutant (lane 2, Fig. 2A) was 4.3-fold lower than that in the wild-type strain (lane 1, Fig. 2A). Our result also confirmed the plasmid p-sigP should be able to express the sigP gene in trans because it fully restored the sigP mRNA level in the ΔporX mutant (lane 4, Fig. 2A). To determine whether the PorX/PorY system can directly upregulate the sigP gene, we first characterized the sigP promoter region and investigated the PorX binding to this promoter by conducting an electrophoretic mobility shift assay (EMSA) using a 275-bp DNA fragment (marked as T1), including the 149-bp intergenic region of the sigP-PGN_0275 genes. We found that a PorX protein with a C-terminal His6 tag (referred to as PorX-C-His6) gel-shifted this DNA fragment in a concentration-dependent manner (Fig. 2B), thus suggesting this T1 fragment should contain the sigP promoter and also the sequence(s) that binds the PorX protein (i.e., the PorX-binding site). Therefore, we conducted a DNase footprinting assay to map the PorX-binding site in T1 and found that the PorX-C-His6 protein bound to an AT-rich DNA sequence (5′-tcgaaaaaaatgtttttctttgc-3′) in a concentration-dependent manner (Fig. 2C). This PorX-binding site, which is located −97 to −75 nucleotides (nt) upstream of the start codon (underlined nucleotides, Fig. 2D), shared a partial sequence with the PorX-binding site II (5′-gattcgcgcaaaaatacaatatcttt-3′) in the porT promoter, recently characterized by our laboratory (7). We postulate that PorX can recognize a sequence (5′-CG(A/C)AAAAA-N5-T(T/A)TCTTTGC-3′) that is conserved in these two promoters. Interestingly, the 5 nucleotides located between the conserved segments in the PorX-binding sites of the sigP and porT promoters were complementary (nucleotides labeled with arrows in Fig. 2E). Therefore, these results and our recent data (7) not only verify that PorX directly regulates transcription of the sigP gene and the por genes such as porT, but also elucidate that PorX is a DNA-binding protein and capable of recognizing specific DNA sequences in a manner similar to many other TCS response regulators.
FIG 2.

The PorX response regulator binds to the sigP promoter region. (A) The mRNA levels of the sigP gene in the 33277 wild-type strain and the ΔporX mutant (YS19181) carrying pT-COW, p-porX, or p-sigP. Results are representative of three independent experiments. (B) EMSA analysis for binding of PorX to the sigP promoter. 32P-labeled sigP DNA fragment (40 fmol) was incubated with PorX-C-His6 protein at the indicated amount. Lane 5 is the same as lane 4 but supplemented with nonlabeled (cold) sigP DNA fragment (1 pmol). The PorX/DNA mixtures were subjected to 5% PAGE. The location of DNA migration was detected by autoradiography. Arrows indicate the shifted bands after DNA fragments were associated with the PorX-C-His6 protein. The experiment was repeated twice. (C) DNase footprinting analysis of the sigP promoter fragment amplified with primers 32P-3043 and 3044 for the coding strand and increasing amounts of PorX-c-His6 protein. Products were separated in polyacrylamide DNA sequencing electrophoresis and the bands were detected by autoradiography. The bracket indicates the region protected by the PorX-C-His6 protein. Underlined DNA sequence (right of gel) indicates the PorX-protected nucleotides in the sigP promoter. The ladder M corresponds to the same 32P-labeled sigP promoter fragment and degraded by the Maxam and Gilbert reaction. Results were repeated multiple times. (D) The DNA sequence of the sigP promoter region. Underlining corresponds to the PorX-protected region characterized in (C). Capital letters represent the sigP start codon. Numbering begins from the adenine nucleotide of the start codon. Highlighted sequences are shared by the PorX-binding site in the porT promoter (also shown in panel E). (E) The homologous sequences of the PorX-binding sites in the sigP and porT promoters. Vertical lines represent the identical nucleotides in the two sequences. Arrows represent the complementary nucleotides exhibited in the two sequences. Highlighted sequences are shared in these two promoters.
PorX protein activates sigP transcription in vitro.
To further validate the direct role of the PorX/PorY system in sigP transcription, we conducted an in vitro transcription assay using a P. gingivalis RNA polymerase holoenzyme (referred to as pg-RNAP-σD) that was reconstructed from N-terminal His6-tagged subunit proteins, including α (PGN_1841), β (PGN_1571), β’ (PGN_1570), and the major sigma factor σD (PGN_0638) (for details see the Materials and Methods section). When the T1 fragment, which was tested for PorX binding (Fig. 2B and C), was used as the template for the in vitro transcription reactions supplemented with 50 nM pg-RNAP-σD, two transcripts labeled as P1 and P2, respectively, were produced (Fig. 3A). Both transcriptions were stimulated by the PorX-c-His6 protein because the amount of P1 and P2 increased in a PorX concentration-dependent manner (lanes 1 to 4, Fig. 3A). These results suggest that sigP transcription is initiated from two regions that are located at 65 to 60 nt (labeled as p1) and 99 to 94 nt (p2) upstream of the start codon, respectively (illustrated in the T1 sequence, Fig. 3B). To verify whether these transcripts were produced specifically, we compared the in vitro transcripts from the wild-type T1 template and a mutated T1 template (T1-Sub) which carried 17-nt substitutions at a 103- to 87-nt sequence located upstream of the start codon. Our results showed that levels of both P1 and P2 transcripts from a reaction using the T1-Sub template were much lower than those using the T1 template (lane 2 versus lane 1, Fig. 3C). Since this substituted sequence overlaps a partial region of the PorX-binding site for the P1 transcription and the p2 region (Fig. 3B), we reasoned that these substitutions must simultaneously interfere with transcription initiated from p1 and p2 in T1-Sub. To further verify that the transcription initiation from p1 and p2 was specific, we used another template, i.e., T2, which was a longer template (291 bp) and contained an additional 16-bp sequence extending from downstream of the T1 template (275-bp). The in vitro transcription using this T2 template could still produce two transcripts, labeled as P1’ and P2’, in a PorX concentration-dependent manner (lanes 3 and 4, Fig. 3D), and both products were exactly 16-nt longer than P1 and P2, respectively (lanes 3 and 4 versus lane 2, Fig. 3D). Therefore, the in vitro transcription of the sigP gene must be specifically initiated from two DNA regions, p1 and p2, thus allowing the T1 template to produce P1 and P2 transcripts and also the 16-bp longer T2 template to produce 16-nt longer P1’ and P2’ transcripts.
FIG 3.

PorX promotes sigP transcription in vitro, mediated by a reconstituted P. gingivalis RNA polymerase-σD. (A) In vitro transcription of a 275-bp template (T1) from the sigP promoter containing the first 29 coding nucleotides was conducted as described in the Materials and Methods. Left braces indicate the P1 and P2 transcripts synthesized by 50 nM of RNA polymerase-σD (RNAP-σD) from reactions supplemented with different amounts of the PorX-C-His6 protein. The ladder M corresponds to a PCR product generated with primers 3044 and 32P-labeled primer 3043 and degraded by the Maxam and Gilbert reaction. (B) The DNA sequence of the sigP promoter region. Underlining corresponds to the PorX-protected region. Blue dashed frames correspond to the regions labeled as p1 and p2, respectively, where transcription was initiated. The highlighted sequence corresponds to the wild-type sequence which was substituted by the sequence (Sub) in red capital letters. Numbering begins from the adenine nucleotide of the start codon (underlined capital letters). (C) In vitro transcription of the sigP templates (T1 and T1-sub) with the wild-type sequence and a substituted sequence, respectively. Blue left braces indicate the transcripts, P1 and P2, produced from the reaction with template T1. (D) In vitro transcription of the sigP templates (T1 and T2) containing the first 29 and 45 coding nucleotides, respectively. Blue right braces indicate the transcripts, P1 and P2, produced from the reaction with template T1, and red right braces indicate the transcripts P1’ and P2’, produced from the reaction with template T2. Double arrows indicate that P1 and P2 are 16 nucleotides shorter than P1’ and P2’, respectively. Results in A, C, and D were repeated two times.
PorX stimulates in vitro transcription of the porT gene carried out by a reconstructed RNA polymerase-σP holoenzyme.
Since PorX directly binds to the σP-dependent porT promoter (7), we postulated that it should be able to stimulate porT transcription in vitro. To examine this hypothesis, we conducted an in vitro transcription assay using a P. gingivalis RNA polymerase σP holoenzyme (referred to as pg-RNAP-σP) which was reconstructed from purified N-terminal His6-tagged α, β, β’ and C-terminal His6-tagged σP proteins (for details see the Materials and Methods section). When a 301-bp DNA fragment, including the porT promoter sequence, was used as the template, two transcripts labeled as S1 and S2 were produced by the reconstructed pg-RNAP-σP (at 50 nM) and both transcriptions were enhanced by PorX in a concentration-dependent manner (lanes 2 to 5, Fig. 4A). S1 transcription was initiated from the adenosine (labeled as s1, Fig. 4B) located 29 nucleotides downstream of PorX binding site II, identified in our previous study (7). S2 transcription was initiated from the guanosine (labeled as s2, Fig. 4B) located 49 nucleotides downstream of PorX binding site I in the porT promoter. Thus, we postulated that PorX should bind to site I and site II and enhance the transcription initiated at s2 and s1, respectively. Synthesis of both S1 and S2 was significantly stimulated when the pg-RNAP-σP concentration was elevated from 25 nM to 50 nM (lanes 2 and 3, left panel, Fig. 4C). In contrast, the pg-RNAP-σD holoenzyme was not as efficient as pg-RNAP-σP because only S2 could be produced to a detectable level by pg-RNAP-σD at a high concentration of 200 nM (lane 4, right panel, Fig. 4C). These observations suggest that σP should be the preferred sigma factor to mediate the porT transcription and that both PorX and σP act directly on its promoter. Interestingly, the s1 and s2 sites did not overlap the transcription initiation site (+1) detected from a primer extension assay using a total wild-type mRNA sample (7). This is probably because other factors in the bacterial cell might interact with PorX and RNA polymerase-σP holoenzyme to initiate the porT transcription from the +1 position.
FIG 4.

PorX and σP promote porT transcription in vitro. (A) In vitro transcription of a porT template containing its promoter and the first 48 coding nucleotides was conducted as described in the Materials and Methods. The left panel represents the transcripts, labeled as S1 and S2, respectively, synthesized in the reactions with different amounts of RNAP-σP with and 100 nM PorX-C-His6 protein. The right panel represents the products synthesized in the reactions with different amounts of RNAP-σD and 100 nM PorX-c-His6 protein. The ladder M corresponds to a PCR product generated with primers 4026 and 32P-labeled primer 4025 and degraded by the Maxam and Gilbert reaction. (B) The DNA sequence of the porT promoter region. Underlined sequences correspond to the PorX-protected regions and are also labeled as I and II, respectively. Bold letters, labeled as s1 and s2, correspond to the transcription initiation sites detected from the in vitro transcription. Underlined capital letters present the porT start codon. (C) In vitro transcription of porT in the reactions supplemented with 50 nM RNAP-σP and different amounts of PorX-C-His6 protein. The ladder M is the same as in A. Results in A and C were repeated two times.
PorX/PorY system is essential for the virulence of P. gingivalis in a mouse model.
According to our previous results (7), the PorX/PorY system is a virulence regulator of P. gingivalis because a virulent W83 wild-type strain, but not the ΔporX mutant, could cause infection in a mouse model described previously (11). To determine whether the PorX/PorY-activated σP contributes to bacterial virulence, we compared the pathogenesis of this wild-type strain and its isogenic ΔsigP mutant in this mouse model. Six-week-old BALB/c mice were subcutaneously injected on the dorsal surface with the strains that were grown in BHI medium for 12 h, and all five mice that were challenged by W83 wild-type cells at a dose of 4.72 × 1010 CFU died in 48 h (Fig. 5A and 5B). On the other hand, four out of the five mice challenged with the isogenic ΔsigP mutant cells at a dose of 4.58 × 1010 CFU survived the 30-day observation period (Fig. 5A and 5B), thus demonstrating that the sigma factor σP is a virulence determinant. The ΔsigP mutant was highly attenuated but not as avirulent as the ΔporX mutant, which, at a dose of 4.32 × 1010 CFU, did not kill even one mouse in the 30-day observation period (Fig. 5A and 5B). The result of the ΔporX mutant also reconfirmed that the PorX/PorY system is essential for P. gingivalis virulence (7). Accordingly, we postulated that the PorX/PorY system should also be able to activate other P. gingivalis virulence factors whose regulation is independent of σP. Based on these observations, it is reasonable to assume that the PorX/PorY system renders P. gingivalis virulent in part by activating the sigP gene in this mouse model. This assumption should be further confirmed by our ongoing RNA sequencing analysis, which compares the expression of overall PorX/PorY- and σP-regulated genes in P. gingivalis cells recovered from the animal against those grown in vitro.
FIG 5.
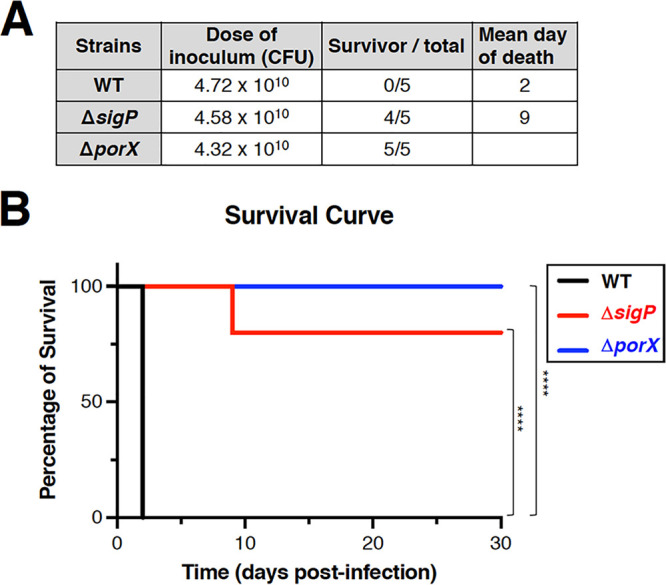
The PorX/PorY-determined virulence of P. gingivalis W83 strains. (A) Virulence test using groups of BALB/c mice (n = 5) that were subcutaneously injected with P. gingivalis W83 wild-type, ΔsigP (YS18145), and ΔporX (YS19145) strains, respectively. (B) Survival curves of the results from A (n = 5; P < 0.0001). Three sets of experiments were carried out.
In conclusion, pathogenic bacteria have developed many sophisticated mechanisms to control the expression of the genes that contribute to virulence. Growing evidence suggests that the PorX/PorY system in P. gingivalis plays an essential role in the regulation of numerous virulence determinants, exemplified by the set of por genes encoding the T9SS components. This study has revealed that the PorX/PorY system and sigma factor σP construct a regulatory pathway to coordinate the regulation of the por genes. We provide evidence that the PorX response regulator binds to the sigP promoter (Fig. 2B and 2C) and activates the sigP transcription in an in vitro transcription reaction system using a reconstructed RNA polymerase holoenzyme (Fig. 3A, C, and D), thus demonstrating that the PorX/PorY system directly regulates transcription of the sigP gene.
When two related regulators build a regulatory cascade, the first regulator regulates the second regulator, and then the second regulator regulates their target genes. Therefore, in the absence of the first regulator, the target genes will still be regulated by the second regulator when this regulator can be produced in trans. Although PorX/PorY activates σP, and then σP activates the por genes, this regulatory cascade model is inapplicable to the regulation dependent on the PorX/PorY system and σP because both the first regulator (PorX) and the second regulator (σP) are shown to bind to the por promoters (5, 7), and σP produced in trans from p-sigP is unable to activate these genes in the ΔporX mutant (Fig. 1A to C). We also show that the PorX protein can directly enhance in vitro porT transcription catalyzed by an RNA polymerase-σP holoenzyme (Fig. 4A and 4C), which further confirms the direct action of PorX on the porT promoter. Therefore, regulation of the por genes should be controlled coordinately by the PorX/PorY system and σP, in which PorX stimulates the production of σP, and both PorX and σP regulate the porT transcription. We postulate that this mechanism of action of the PorX/PorY system and σP should fall under the criteria of a regulatory motif, which is known as the feedforward loop (12) (Fig. 6). Our previous results have shown that the PorX/PorY system responds to hemin and enhances transcription of the porT gene (7). In many cases, the feedforward loop has the capability to integrate multiple signaling molecules into a gene regulation (12). It remains to be investigated whether the feedforward loop contributing to the PorX/PorY- and σP-governed signal transduction pathway is able to respond to signal molecules besides hemin.
FIG 6.

Feedforward loop model illustrating the PorX/PorY- and σP-dependent regulatory mechanism. In P. gingivalis, PorX/PorY and σP build a feedforward loop. The PorY sensor kinase phosphorylates its cognate PorX response regulator. The phosphorylated PorX protein binds to the sigP promoter at the PorX-binding site and upregulates transcription of the sigP gene. The PorX/PorY-stimulated σP protein and RNA polymerase core enzyme build a holoenzyme. Then, phosphorylated PorX protein and RNAP σP holoenzyme coordinately activate transcription of their target genes by simultaneously binding to their promoters at the PorX-binding sites and the σP recognition site, respectively. The inset illustrates the PorX/PorY σP feedforward loop that modulates por expression.
It is worth noting that the transcription of the sigP gene in the ΔporX mutant is not completely repressed (5) (Fig. 2A). According to a previous study (13), σP exerts an inhibitory effect on P. gingivalis biofilm formation, as biofilm formation is induced in a sigP null mutant in an enriched BHI medium. However, the ΔporX mutant grown in this BHI medium did not induce biofilm formation (unpublished result). We reason that the expression of the sigP gene remaining in the ΔporX mutant is sufficient to inhibit biofilm formation.
The PorX/PorY system has been shown as an essential regulator for P. gingivalis virulence since the ΔporX mutant is avirulent in mouse infection (7) (Fig. 5A and B). In this study, the murine virulence assay has demonstrated that σP contributes to P. gingivalis virulence and the ΔsigP mutant becomes attenuated. Further in vivo analysis will be needed to confirm the role of σP in the PorX/PorY-controlled mechanism required for P. gingivalis pathogenesis.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
Strains and plasmids used in this study are listed in Table 1. The P. gingivalis ATCC 33277 and W83 wild-type strains used in this study were obtained from Koji Nakayama (4). P. gingivalis cells were grown at 37°C in an anaerobic chamber (Model 2000, Coy Lab Products) that maintained 90% N2/5% CO2/5% H2 in the atmosphere. Blood agar plates (5% sheep defibrinated blood, 1.5% agar) or brain heart infusion (BHI, purchased from BD) medium supplemented with hemin (5 μg/ml) were used to culture P. gingivalis strains. When necessary, erythromycin (0.5 μg/ml) or tetracycline (0.5 μg/ml) was supplemented. P. gingivalis cells were harvested by centrifuging liquid cultures at 10,000 × g (∼8,500 rpm) in a Sorvall ST 8R centrifuge with a HIGHConic III fixed angle rotor (maximum 9,500 rpm) at 4°C for 10 min. E. coli DH5α and BL21(DE3) strains were used for cloning and protein production, respectively. E. coli cells were routinely grown in Luria broth (LB) supplemented with antibiotics when necessary (kanamycin, 50 μg/ml; ampicillin, 50 μg/ml) at 37°C. To prepare cell lysates, bacterial cells were opened with a sonicator (Misonix Sonicator 3000).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Porphyromonas gingivalis | ||
| ATCC 33277 | Wild type | |
| YS19181 | ΔporX::EmR | (7) |
| YS17717 | ΔsigP::EmR | This work |
| W83 | Wild type | |
| YS19145 | ΔporX::EmR | (7) |
| YS18145 | ΔsigP::EmR | This work |
| E. coli | ||
| DH5α | F− supE44 ΔlacU169 (ϕ80 lacZ ΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Lab collection |
| (21) | ||
| BL21(DE3) | F− ompT hsdSB (rB− mB− ) gal dcm (DE3) | (22) |
| Plasmids | ||
| pT-COW | repColE1 AmpR Tcr | (14) |
| pYS18679 | repColE1 reppB8-51 AmpR CamR TcR PPGN_1016 porXCDS | (7) |
| pYS19107 | repColE1 reppB8-51 AmpR CamR TcR | This work |
| sigPCDS | This work | |
| pET28a | repColE1 KmR lacI PT7 | Novagen |
| pET11a | repColE1 AmpR lacI PT7 | Novagen |
| pET21a | repColE1 AmpR lacI PT7 | Novagen |
| pGEM-T-Easy | reppMB1, f1 AmpR lacZ-α | Promega |
| pGEM-ermF | repColE1 AmpR ermF lacI PT7 | (16) |
| pYS17676 | repColE1 AmpR ermF lacI PT7 sigP8-305nt | This work |
| pYS18456 | repColE1 AmpR lacI PT7 porX (PGN_1019) | (7) |
| pYS18051 | repColE1 KmR lacI PT7 rpoA (PGN_1841) | This work |
| pYS18943 | repColE1 AmpR lacI PT7 rpoB (PGN_1571) | This work |
| pYS18165 | repColE1 KmR lacI PT7 rpoC (PGN_1570) | This work |
| pYS18052 | repColE1 KmR lacI PT7 rpoD (PGN_0638) | This work |
| pYS18056 | repColE1 KmR lacI PT7 sigP (PGN_0274) | This work |
Construction of plasmids and strains with chromosomal mutations.
All plasmids used in this study are listed in Table 1. Polymerase chain reactions (PCR) were performed using a Bio-Rad T100 thermal cycler with Taq DNA polymerase (New England BioLabs [NEB]). Custom oligonucleotides were synthesized by Integrated DNA Technologies (IDT) and are listed in Table 2. PCR products were isolated using a QIAquick PCR purification kit (Qiagen). Restriction enzymes were purchased from New England BioLabs and used according to the manufacturer’s instructions. Digested DNA fragments were separated in 0.8 to 1% agarose gels and then isolated using a QIAquick gel extraction kit (Qiagen). Plasmids were purified from overnight cultures of E. coli DH5α in LB at 37°C using plasmid minikit or midi kit (Qiagen). Plasmid pYS19107 for complementation assays was constructed using PCR fragments containing a 500-bp sequence of the upstream region followed by the sigP coding region, which was amplified with primers 3809 and 2827, digested with HindIII and BamHI, and ligated between the HindIII and BamHI sites of pT-COW (14). Plasmid pYS17676 for mutagenizing the sigP gene in both 33277 and W83 strains was constructed using a DNA fragment containing the 8- to 305-nt sigP coding region amplified with primers 2768 and 2769, digested with PstI, and then ligated with PstI-digested pGEM-ermF plasmid. Plasmid pYS18051 was constructed using PCR fragments containing the rpoA (PGN_1841) coding region amplified with primers 3158 and 3159, digested with NcoI and BamHI, and then ligated between the NcoI and BamHI sites of plasmid pET28a. Plasmid pYS18943 was constructed using PCR fragments containing the rpoB (PGN_1571) coding region amplified with primers 3160 and 3161, digested with NheI, and then ligated between the NheI sites of plasmid pET11a. Plasmid pYS18165 was constructed using PCR fragments containing the rpoC (PGN_1570) coding region amplified with primers 3162 and 3163, digested with NcoI and XhoI, and then ligated between the NcoI and XhoI sites of plasmid pET28a. Plasmid pYS18052 was constructed using PCR fragments containing the rpoD (PGN_0638) coding region amplified with primers 3164 and 3165, digested with NcoI and HindIII, and then ligated between the NcoI and HindIII sites of plasmid pET28a. Plasmid pYS18056 was constructed using PCR fragments containing the sigP coding region amplified with primers 3148 and 3149, digested with NcoI and XhoI, and then ligated between the NcoI and XhoI sites of plasmid pET28a. All plasmids were sequenced before use. The P. gingivalis ΔsigP mutant was constructed by introducing suicide plasmid pYS17676 into the 33277 and W83 wild-type strains, respectively, using an electroporation procedure described previously (4). Mutated sequences in these strains were confirmed by DNA sequencing.
TABLE 2.
Primers used in this studya
| Primer no. | Sequence |
|---|---|
| 2499 | gga aga gaa gac cgt agc aca agg a |
| 2500 | gag tag gcg aaa cgt cca tca ggt c |
| 2741 | ccc aag ctt gac aca gca gca gga aaa gc |
| 2742 | cgc gga tcc tta ctt ggg ttg cat cgt aat |
| 2768 | aaa act gca ggt ttc cac aag ctg act g |
| 2769 | aaa act gca gtg caa cct ggc tcc ttc c |
| 2827 | ccg ctc gag cta agc cga cat gcc cat c |
| 3043 | tca tca gtc agc ttg tgg |
| 3044 | ccg agt acg ttt acc cc |
| 3148 | cat gcc atg gca atg agc agt ttc cac aag c |
| 3149 | ccg ctc gag agc cga cat gcc cat |
| 3158 | cat gcc atg ggc cat cat cat cat cat cac gca ata tta gca ttt cag |
| 3159 | cgg gat cct tat tag tct tta tct aat tta tac |
| 3160 | cta gct agc cat cat cat cat cat cac acg ccg act aca aac aac |
| 3161 | cta gct agc tta tta gtc caa aga aaa act t |
| 3162 | cat gcc atg ggc cat cat cat cat cat cac gct ttt aga aaa gaa aat aag |
| 3163 | ccg ctc gag tta cta ttc cga tgg tgc ttc |
| 3164 | cat gcc atg ggc cat cat cat cat cat cac agg caa ctt aaa att tcc |
| 3165 | ccc aag ctt tta tta gcc gag ata acc ttt cag |
| 3264 | cgg tcg gag gca gga atg |
| 3470 | gtt cgt tcg cga ata tgc |
| 3471 | cga gga cag tag ctt tgg |
| 3764 | cca aag cta ctg tcc tcg |
| 3765 | tac gaa ggc atc gaa agg |
| 3809 | cgc gga tcc caa cta ctg cta ctg tct c |
| 3837 | atg tag gga tgc atg ccc |
| 3838 | caa agt cgg aag caa acg |
| 3912 | gtc agt tct tcc act cgg |
| 3913 | gga aga atg gtc aga tcg |
| 4025 | aga gag cga ctc tca acg |
| 4026 | cac acg ttc tat att gcg |
| 4105 | cag gcg ctg gga tcc gcg ttt ttc ttt gca ata ag |
| 4106 | cgc gga tcc cag cgc ctg aaa cag aag caa c |
| 4111 | aag gct gac caa ttc atc |
| 4193 | cgg ccg aat gcg ata tgc |
| 4194 | agc ata ttc gcc aaa agg |
All oligonucleotides were purchased from IDT (Integrated DNA Technologies)
Quantitative real-time PCR.
Bacterial cells were grown anaerobically in BHI medium at 37°C for 48 h. Total RNA was isolated from bacterial cultures using a High Pure RNA isolation kit (Roche) according to the manufacturer’s instructions. The concentration of RNA samples was determined by measuring absorbance at 260 nm using a spectrophotometer (SmartSpec Plus, BIO-RAD). The quality of RNA was evaluated in a 1.2% agarose gel. cDNAs were synthesized using random primers (IDT) and a murine leukemia virus reverse transcriptase (NEB). The amount of cDNA was quantified using PowerUp SYBR green Master Mix according to the manufacturer’s instructions with primers 3837 and 3838 for porT, 3912 and 3913 for PGN_0341, 3764 and 3765 for PGN_1639, 4193 and 4194 for sigP, and 2499 and 2500 for rpoB (Table 2) and qPCR was performed in QuantStudio 3 Real-time PCR systems (Applied Biosystems, Thermo Fisher Scientific).
Isolation of the RpoA-N-His6, RpoB-N-His6, RpoC-N-His6, σD-N-His6, σP-C-His6, and PorX-C-His6 proteins.
E. coli BL21-Gold (DE3) harboring plasmids pYS18051, pYS18943, pYS18165, pYS18052, pYS18056, and pYS18456, respectively, were grown in 500 ml of LB medium by shaking at 37°C to an optical density at 600 nm (OD600) value of 0.5, then IPTG (isopropyl-β-D-thiogalactopyranoside) was added to a final concentration of 0.4 mM, and bacterial cells were cultured for another 2 h. Bacterial cells were harvested by centrifuge at 10,000 × g for 15 min and washed with 50 ml of phosphate-buffered saline (PBS) once, suspended in 10 ml of PBS, and opened by sonication (Misonix Sonicator 3000). The cell lysate was used for purification of the RpoA-N-His6, RpoB-N-His6, RpoC-N-His6, σD-N-His6, σP-c-His6, and PorX-c-His6 proteins with Ni-NTA Affinity Gel (Qiagen) by following the manufacturer’s instructions. The purity and concentration of protein samples were determined using a Silver Staining kit (Pierce) and BCA Protein assay kit (Pierce) by following the instructions from the manufacturer.
Electrophoretic mobility shift assay.
The electrophoretic mobility shift assay (EMSA) was performed as described (15) with the following modifications. Primer 3043 was labeled using T4 polynucleotide kinase (New England BioLabs) and [γ-32P]ATP (PerkinElmer Life Sciences). Ten nanomoles of 32P-labeled DNA fragments containing the 275-bp sigP promoter region, amplified by PCR from 33277 chromosomes with primers 3044 and 32P-labeled 3043, were incubated at room temperature for 30 min with 0, 25, 50, or 100 pmol of PorX-C-His6 protein in 20 μl of an EMSA buffer consisting of 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 5 mM dithiothreitol (DTT), 10 mM NaCl, 1 mM MgCl2, and 5% glycerol. After the addition of the DNA dye solution (40% glycerol, 0.05% bromophenol blue, 0.05% xylene cyanol), the mixture was directly subjected to 4% polyacrylamide electrophoresis. Signals were detected by autoradiography.
DNase footprinting analysis.
The DNase I footprinting assay was performed as described (15) with the following modifications. 32P-labeled DNA (25 pmol, as was used for EMSA) was mixed with 0, 70, 140, or 280 pmol of the PorX-C-His6 protein in a 100 μl reaction. DNase I digestion was carried out using 0.05 units DNase I (Invitrogen) per reaction. Samples were analyzed by 6% denaturing polyacrylamide electrophoresis by comparison with a DNA sequence ladder generated by Maxam and Gilbert A+G reaction, using the same 32P-labeled PCR product. The positions of radioactive DNA fragments in the gels were detected by autoradiography.
Reconstitution of RNAP holoenzymes from isolated subunits.
A procedure for reconstitution of E. coli RNAP holoenzyme developed and described in detail (16) was successfully used for reconstitution of other bacterial RNAPs. We used a modified procedure presented in a previous study (17) to carry out reconstitution of P. gingivalis RNAP holoenzymes with the following modifications. Briefly, prior to the in vitro reconstitution, RNAP subunits isolated from the procedure above were suspended in a denaturation buffer (6 M guanidine-HCl, 50 mM Tris-HCl [pH 7.9], 10 mM MgCl2, 10 μM ZnCl2, 10% glycerol, 1 mM EDTA, and 10 mM DTT). The mixtures were left for 30 min on ice and then spun in a 4°C microcentrifuge at 10,000 × g for 30 min. The supernatants were transferred into fresh tubes and the protein concentration was determined using the BCA protein assay kit (Pierce) with bovine serum albumin (BSA) as a standard. RNAP subunits were mixed in a molar ratio of 2:8:4 (a:β:β′) and dialyzed against 250-volume reconstitution buffer (50 mM Tris-HCl [pH 7.9], 200 mM KCl, 10 mM MgCl2, 10 μM ZnCl2, 10% glycerol, 1 mM EDTA, and 10 mM 2-mecaptoethonal) at 4°C for 16 h with two changes. One molar equivalent of isolated RNAP σ subunit (σD or σP) in PBS was added to the supernatant and the mixture was incubated at 30°C for 1 h. The resulting RNAP preparations were used directly in transcription assays or stored under (NH4)2SO4 (65% saturation) until further use.
Transcription of sigP and porT in vitro.
The in vitro transcription was conducted in a 50-μl reaction mixture containing 1× in vitro transcription buffer (80 mM HEPES-KOH [pH 7.5], 24 mM MgCl2, 2 mM spermidine, 40 mM DTT with 500 μM ATP, CTP, GTP, and UTP, respectively) and 1 μg of linear double-stranded DNA (dsDNA) template with the desired amounts of PorX-c-His6 protein and an RNA polymerase holoenzyme. Reaction mixtures were incubated for 2 h at 37°C and transcripts were precipitated using three volumes of cold 100% ethanol and 1/10 volume of 3 M sodium acetate (pH 5.8) and resuspended with RNase-free water. For sigP transcription in vitro, the template T1 was amplified from 33277 chromosomal DNA using primers 3043 and 3044, while T1-Sub with substituted PorX binding sequence (from gttttgtcgaaaaaaat to caggcgctgggatccgc) was prepared with primers 3043 and 3044 and 4105 and 4106 by using an overlap extension PCR (18). The longer template T2 was amplified from 33277 chromosomal DNA using primers 3044 and 4111. For porT transcription in vitro, the template was amplified from 33277 chromosomal DNA using primers 4025 and 4026. Transcripts in vitro were monitored after being converted into cDNAs through a primer extension performed as described (19) with the following modifications. RNA pellets derived from templates T1 and T1-Sub were reverse transcribed using 2 μl of 32P-labeled primer 3043 in a 20-μl mixture containing 25 units of M-MuLV reverse transcriptase (NEB) at 42°C for 2 h. 32P-labeled primer 4111 was used for primer extension of the transcripts derived from template T2. Transcripts derived from the porT template were reverse transcribed by using 32P-labeled primer 4025. The cDNA samples were precipitated with 2.5 volumes of ethanol and 0.3 M sodium acetate (pH 5.8) and resuspended in 5 μl of Gel Loading Buffer II (Thermo Fisher), and then analyzed by a 6% denaturing polyacrylamide gel. DNA ladders were amplified from 33277 chromosomal DNA using three primer pairs (32P-labeled 3043 and 3044; 32P-labeled-4111 and 3044; and 32P-labeled-4025 and 4026) for the products from T1, T2, and the porT template, respectively, and generated by Maxam-Gilbert reaction.
Virulence assay in a mouse model.
All animal experiments conform to our animal protocols (18‐1655R) approved by the Institutional Animal Care and Use Committee (IACUC), Office of Research Integrity and Assurance, Arizona State University (ASU protocol number 18‐1655R). Groups of 6-week-old female BALB/c mice (purchased from Charles River Laboratories) were randomly allocated into different groups. Determination of virulence of the P. gingivalis W83 and mutant strains was performed using mouse subcutaneous infection experiments, as described previously (20), with slight modifications. Briefly, bacterial cells were grown in enriched BHI broth at 37°C for 12 h. The culture was diluted 20-fold in 100 ml of fresh BHI medium and grown for the time periods indicated. The cells were harvested by centrifugation at 10,000 × g for 20 min and washed once with PBS, then adjusted to a concentration of approximately 5 × 1011 CFU/ml in PBS. Resulting bacterial cultures were serially diluted and plated for bacterial CFU to determine the exact titer of all strains used for infections. Mice were challenged with subcutaneous injections of 0.1 ml at each of the two sites on the depilated dorsal surface (0.2 ml per mouse). Infected mice were examined daily for survival.
Statistics.
Each in vitro experiment was conducted at least three times independently. Mice were randomly placed into different groups before tests. A Kaplan Meier curve was used for survival analysis in this study. Comparisons between two groups were performed with Student’s t test and P ≤ 0.05 was considered significant. Statistics were calculated with GraphPad Prism version 8.0.
ACKNOWLEDGMENTS
We thank Koji Nakayama and Keiko Sato for P. gingivalis wild-type 33277 and W83 strains and plasmid pGEM-ermF, and Mary Ellen Davey for plasmid pT-COW.
This study was supported by Research Project Grant R01 DE024607 from NIDCR (Y.S. and W.K.).
W.K. and Y.S. conceived and designed the experiments. C.J., D.Y., T.H., Z.H., W.K., and Y.S. performed the experiments. C.J., D.Y., Z.H., W.K., and Y.S. analyzed data and drafted the article. All authors analyzed the results and approved the final version of the manuscript.
We declare no conflicts of interest.
Contributor Information
Wei Kong, Email: wei.kong@asu.edu.
Yixin Shi, Email: yixin.shi@asu.edu.
Craig D. Ellermeier, University of Iowa
REFERENCES
- 1.Nakayama K. 2003. Molecular genetics of Porphyromonas gingivalis: gingipains and other virulence factors. Curr Protein Pept Sci 4:389–395. doi: 10.2174/1389203033486983. [DOI] [PubMed] [Google Scholar]
- 2.Bostanci N, Belibasakis GN. 2012. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett 333:1–9. doi: 10.1111/j.1574-6968.2012.02579.x. [DOI] [PubMed] [Google Scholar]
- 3.Marcotte H, Lavoie MC. 1998. Oral microbial ecology and the role of salivary immunoglobulin A. Microbiol Mol Biol Rev 62:71–109. doi: 10.1128/MMBR.62.1.71-109.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato K, Naito M, Yukitake H, Hirakawa H, Shoji M, McBride MJ, Rhodes RG, Nakayama K. 2010. A protein secretion system linked to bacteroidete gliding motility and pathogenesis. Proc Natl Acad Sci U S A 107:276–281. doi: 10.1073/pnas.0912010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kadowaki T, Yukitake H, Naito M, Sato K, Kikuchi Y, Kondo Y, Shoji M, Nakayama K. 2016. A two-component system regulates gene expression of the type IX secretion component proteins via an ECF sigma factor. Sci Rep 6:23288. doi: 10.1038/srep23288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vincent MS, Durand E, Cascales E. 2016. The PorX response regulator of the Porphyromonas gingivalis PorXY two-component system does not directly regulate the type IX secretion genes but binds the PorL subunit. Front Cell Infect Microbiol 6:96. doi: 10.3389/fcimb.2016.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang D, Jiang C, Ning B, Kong W, Shi Y. 2021. The PorX/PorY system is a virulence factor of Porphyromonas gingivalis and mediates the activation of the Type IX secretion system. J Biol Chem 296:100574. doi: 10.1016/j.jbc.2021.100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davey ME, Duncan MJ. 2006. Enhanced biofilm formation and loss of capsule synthesis: deletion of a putative glycosyltransferase in Porphyromonas gingivalis. J Bacteriol 188:5510–5523. doi: 10.1128/JB.01685-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakayama K. 2015. Porphyromonas gingivalis and related bacteria: from colonial pigmentation to the type IX secretion system and gliding motility. J Periodontal Res 50:1–8. doi: 10.1111/jre.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato K. 2011. Por secretion system of Porphyromonas gingivalis. J Oral Biosci 53:187–196. doi: 10.1016/S1349-0079(11)80001-0. [DOI] [Google Scholar]
- 11.Neiders ME, Chen PB, Suido H, Reynolds HS, Zambon JJ, Shlossman M, Genco RJ. 1989. Heterogeneity of virulence among strains of Bacteroides gingivalis. J Periodontal Res 24:192–198. doi: 10.1111/j.1600-0765.1989.tb02005.x. [DOI] [PubMed] [Google Scholar]
- 12.Mangan S, Alon U. 2003. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A 100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Onozawa S, Kikuchi Y, Shibayama K, Kokubu E, Nakayama M, Inoue T, Nakano K, Shibata Y, Ohara N, Nakayama K, Ishihara K, Kawakami T, Hasegawa H. 2015. Role of extracytoplasmic function sigma factors in biofilm formation of Porphyromonas gingivalis. BMC Oral Health 15:4. doi: 10.1186/1472-6831-15-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner RG, Russell JB, Wilson DB, Wang GR, Shoemaker NB. 1996. Use of a modified Bacteroides-Prevotella shuttle vector to transfer a reconstructed beta-1,4-D-endoglucanase gene into Bacteroides uniformis and Prevotella ruminicola B(1)4. Appl Environ Microbiol 62:196–202. doi: 10.1128/AEM.62.1.196-202.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong W, Weatherspoon N, Shi Y. 2008. Molecular mechanism for establishment of signal-dependent regulation in the PhoP/PhoQ system. J Biol Chem 283:16612–16621. doi: 10.1074/jbc.M800547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borukhov S, Goldfarb A. 1993. Recombinant Escherichia coli RNA polymerase: purification of individually overexpressed subunits and in vitro assembly. Protein Expr Purif 4:503–511. doi: 10.1006/prep.1993.1066. [DOI] [PubMed] [Google Scholar]
- 17.Kuznedelov K, Severinov K. 2009. Recombinant bacterial RNA polymerase: preparation and applications. Methods 47:44–52. doi: 10.1016/j.ymeth.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 19.Lejona S, Aguirre A, Cabeza ML, Garcia Vescovi E, Soncini FC. 2003. Molecular characterization of the Mg2+-responsive PhoP-PhoQ regulon in Salmonella enterica. J Bacteriol 185:6287–6294. doi: 10.1128/jb.185.21.6287-6294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curtis MA, Aduse Opoku J, Rangarajan M, Gallagher A, Sterne JA, Reid CR, Evans HE, Samuelsson B. 2002. Attenuation of the virulence of Porphyromonas gingivalis by using a specific synthetic Kgp protease inhibitor. Infect Immun 70:6968–6975. doi: 10.1128/iai.70.12.6968-6975.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 22.Shuman S. 1989. Vaccinia DNA topoisomerase I promotes illegitimate recombination in Escherichia coli. Proc Natl Acad Sci U S A 86:3489–3493. doi: 10.1073/pnas.86.10.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]