

ABSTRACT
Recent work shows that the plant microbiome, particularly the initial assembly of this microbiome, influences plant health, survival, and fitness. Here, we characterize the initial assembly of the Populus microbiome across ten genotypes belonging to two poplar species in a common garden using 16S rRNA gene and ITS2 region amplicon sequencing of the leaf endosphere, leaf surface, root endosphere, and rhizosphere. We sampled these microbiomes three times throughout the first growing season and found that the composition of the microbiome changed dramatically over time across all plant-associated habitats and host genotypes. For archaea and bacteria, these changes were dominated by strong homogenizing selection (accounting for 29 to 62% of pairwise comparisons). However, fungal assembly was generally characterized by multiple ecological assembly processes (i.e., a mix of weak selective and dispersal processes). Interestingly, genotype, while a significant moderator of microbiome composition, generally explained less variation than sample date across plant-associated habitats. We defined a set of core genera that accounted for, on average, 36% of the microbiome. The relative abundance of this core community was consistent over time. Additionally, using source tracking modeling, we determined that new microbial taxa colonize from both aboveground and belowground sources, and combined with our ecological assembly null models, we found that both selective and dispersal processes explained the differences between exo- (i.e., leaf surface and rhizosphere) and endospheric microbiomes. Taken together, our results suggest that the initial assembly of the Populus microbiome is time-, genotype-, and habitat-dependent and is moderated by both selective and stochastic factors.
IMPORTANCE The initial assembly of the plant microbiome may establish the trajectory of forthcoming microbiome states, which could determine the overall future health of the plant. However, while much is known about the initial microbiome assembly of grasses and agricultural crops, less is known about the initial microbiome of long-lived trees, such as poplar (Populus spp.). Thus, a greater understanding of initial plant microbiome assembly in an ecologically and economically important plant such as Populus is highly desirable. Here, we show that the initial microbiome community composition and assembly in the first growing season of Populus is temporally dynamic and is determined by a combination of both selective and stochastic factors. Our findings could be used to prescribe ecologically informed microbial inoculations and better predict the composition of the Populus microbiome into the future and to better understand its influence on plant health.
KEYWORDS: 16S rRNA, ITS, microbial ecology, microbial community assembly, microbial source tracking, null modeling, plant endosphere, plant-microbe interactions, phyllosphere, rhizosphere
INTRODUCTION
A growing body of literature recognizes that microorganisms living inside or in close association with plant tissues, collectively known as the plant microbiome, are integral to plant health and survival (1, 2). Recent work has shown that plant microbiomes are influenced by a myriad of environmental factors, including soil type (3–6), plant metabolites (7), and climate (8). However, relatively less attention has been given to the influence of ecological assembly mechanisms on microbial community composition, especially during initial microbiome assembly. Recognizing how plant microbiomes initially assemble is essential for our understanding of plant-microbe interactions and predicting future microbiome trajectories (9) and plant survival (10).
The initial assembly of the plant microbiome involves the colonization of microorganisms from the outside (not plant-associated) environment, such as the air or surrounding soil (11, 12). It is likely that the root endosphere is colonized by microorganisms from the surrounding soil because root endosphere communities are generally a subset of the larger species pool in the bulk soil (4). Indeed, Edwards et al. (5) proposed a model of rice root endosphere colonization from the bulk soil and rhizosphere onto the rhizoplane (i.e., root surface) and into the root endosphere. The colonization source of the phyllosphere is relatively less certain, as both the air (13) and soil (14) have been proposed as the dominant source of endospheric microorganisms, although recent evidence suggests that phylloplane microorganisms are ultimately sourced from soils (15). However, it is still unclear whether dispersal or selection is the major ecological assembly process responsible for deciding which microbes can colonize endospheric microbiomes (12).
Macroecological frameworks for community assembly have recently been adapted for soil, animal, and plant microbiomes (e.g., 12–14), and such frameworks could be useful in developing models of plant microbiome assembly under field conditions. Community assembly from an ecological perspective constitutes a spectrum of deterministic selection (i.e., ecological niche theory) to stochastic selection (i.e., neutral theory). These mechanisms of community assembly have been shown to influence microbial community structure because they determine the degree of selection due to niche-based (e.g., nutrient availability, plant defense compounds) and dispersal processes (16). Prior work in grass phyllosphere bacterial microbiomes demonstrates that the phyllosphere is overall deterministically selected, but the relative influence of stochastic selection is greater during initial plant growth (13, 14). Assembly of the Phaseolus vulgaris core bacterial microbiome was characterized as relatively more selective in belowground sample types (i.e., rhizosphere and root endosphere) and more stochastic in inner aerial plant tissues (17). However, our understanding of plant microbiome assembly is still incomplete because much of what we know about the underlying plant microbiome assembly mechanisms is generally confined to annual grass species and the bacterial community. These established patterns may diverge in trees and their associated microbiomes because of structural and chemical differences among tree compartments (i.e., different plant tissues) that may impact microbial selection, drift, or dispersal. Furthermore, the assembly processes of bacterial and fungal communities may not be consistent because differences in propagule size may impact dispersal (18, 19), and bacteria and fungi respond to different selective pressures (20, 21). Indeed, across 183 sites in Scotland, soil fungal community assembly was significantly more stochastic than the assembly of bacterial communities (22). These differences are likely even more important in trees, which form both arbuscular and ectomycorrhizal symbioses (23). Hence, including novel model systems (i.e., trees), multiple plant-associated habitats, and multiple microbial domains would increase our ability to understand and predict plant microbiome trajectories holistically and better understand plant holobiont systems.
An understanding of the plant microbiome assembly processes may be particularly important for poplar species (Populus spp.) (24). Poplar trees are widely distributed throughout North America (25), and they are an important potential biofuel feedstock (26). The first poplar draft genome was sequenced in 2004 (27, 28), and since then, numerous distinct genotypes and their genomes have been described (29). These genotypes vary not only phenotypically (e.g., resistance to fungal pathogens differs among genotypes of the same poplar species [23]), but also in their microbiome composition (7, 30, 31). Genotypic influences on the plant microbiome are common across species due to differences in metabolites, elemental concentrations, and root and leaf traits (11), and these influences are generally stronger in aboveground, endospheric microbiomes (15). However, the degree to which underlying assembly mechanisms influence these microbiome compositional differences across poplar genotypes is currently unknown.
A common goal of plant microbiome research is to define and characterize the core microbial community (12, 14, 17, 32–35). Core plant microbiomes generally represent a small proportion of microbial richness but are consistently present among samples at high relative abundances (33). While it is debated whether the core microbiome confers benefits to the host (12, 33, 34), reducing the complexity of the microbiome presents a greater opportunity to investigate the impact of individual microbial members on host fitness (32, 36). Hence, developing an understanding of the core microbiome of poplar trees could lead to future targeted microbiome interventions (i.e., synthetic communities) that promote plant health and productivity. Indeed, the addition of a synthetic community consisting of Burkholderia and Pseudomonas strains isolated from Populus hosts increased lateral root formation and root hair production in Arabidopsis plate assays, and these organisms are predicted to carry out different functions related to growth and plant growth promotion in Populus (37).
To increase our understanding of the initial assembly of the Populus microbiome across genotypes, we planted ten Populus genotypes consisting of P. deltoides and P. trichocarpa in a common garden and sampled the leaf endosphere, leaf surface, root endosphere, and rhizosphere microbiomes three times throughout the first growing season—directly before field propagation (T0, 16 May 2017), 29 June 2017, and 18 September 2017. We used 16S rRNA gene and ITS2 region amplicon sequencing to characterize the archaeal and bacterial and the fungal communities, respectively. We hypothesized that microbiome community composition as well as the underlying assembly mechanisms (e.g., deterministic versus stochastic assembly) would change significantly during this time. Specifically, we hypothesized that deterministic selection would increase over time, as has been demonstrated in other successional contexts (38). Furthermore, we expected greater deterministic selection in plant endosphere microbiomes compared to the rhizosphere and leaf surface microbiomes due to the selective pressures of plant defense compounds (39). Overall, we expected the development of a dominant core community during the first growing season. We also attempted to resolve the colonization source of the root and leaf endosphere. We hypothesized that the rhizosphere would be the greatest source of root endosphere microorganisms, and the leaf surface would be the greatest source of leaf endosphere microorganisms due to spatial proximity. Our overall goals were to characterize the initial microbiome assembly across plant-associated habitats (i.e., plant tissues and immediate environment) and to determine the colonization source of endospheric microorganisms.
RESULTS
α-Diversity changes over time in multiple plant-associated habitats.
We detected a significant three-way interaction in the effect of plant-associated habitat, genotype, and sample date on archaeal and bacterial α-diversity expressed as Hill numbers (40) at both q (defined in Materials and Methods) = 0 (analogous to richness) and q = 1 (analogous to Shannon’s entropy) (analysis of variance [ANOVA] – q = 0: F42,343 = 1.921, P = 0.001; q = 1: F42,343 = 2.622, P < 0.001). When analyzed separately among plant-associated habitats, we generally found that sample date only affected α-diversity in the exospheric microbiomes (i.e., leaf surface, rhizosphere) for both q = 0 and q = 1 (Fig. 1; see Fig. S1 at https://doi.org/10.6084/m9.figshare.14251463.v1). While the leaf surface decreased in α-diversity over time, the rhizosphere increased over time. Alternatively, endospheric microbiome α-diversity was relatively stable over time. At q = 0, there was a significant effect of genotype on both the leaf endosphere and rhizosphere α-diversity, but at q = 1, the effect of genotype was only significant in the leaf endosphere (see Table S1 and Fig. S2 at https://doi.org/10.6084/m9.figshare.14251463.v1). However, for both q = 0 and q = 1, the effect of genotype interacted with sample date for leaf endosphere α-diversity such that there was only a genotype effect during the latest September sample date (see Table S1 and Fig. S2 at https://doi.org/10.6084/m9.figshare.14251463.v1).
FIG 1.

Means (and standard errors) of α-diversity based on Hill numbers (40) of archaea and bacteria and fungi across plant-associated habitats and sampling dates at q = 0 (richness). Letters represent significant differences among sample dates (P < 0.05). Note the different axis scales. T0 represents samples collected directly before field propagation.
Plant-associated habitat interacted individually with genotype and sample date for fungal alpha diversity at both q = 0 (i.e., richness; ANOVA – genotype: F27,295 = 2.989, P < 0.001; sample date: F6,316 = 26.929, P < 0.001) and q = 1 (i.e., Shannon’s entropy; ANOVA – genotype: F27,295 = 2.011, P = 0.003; sample date: F6,316 = 9.051, P < 0.001). When analyzed separately among plant-associated habitats, we generally found that fungal α-diversity in the exospheric microbiomes (i.e., leaf surface, rhizosphere) increased over time for both q = 0 and q = 1 (Fig. 1; see also Fig. S1 at https://doi.org/10.6084/m9.figshare.14251463.v1). However, we also found that α-diversity in the leaf endosphere decreased over time for both q = 0 and q = 1 (Fig. 1). Across both q = 0 and q = 1, genotype was only a significant effect of fungal alpha diversity in the rhizosphere (q = 0: F9,94 = 2.665, P = 0.038; q = 1: F9,94 = 2.806, P = 0.026; see Fig. S3 at https://doi.org/10.6084/m9.figshare.14251463.v1).
Microbial community composition consistently changes over time.
Plant-associated habitat explained 19.6% of the variation in archaeal and bacterial community composition (permutational multivariate analysis of variance [PERMANOVA]: P < 0.001, R2 = 0.196). Because there was a three-way interaction among plant-associated habitat, genotype, and sample date (P < 0.001), we analyzed each plant-associated habitat separately. When separated by plant-associated habitat, sample date was consistently a significant moderator of archaeal/bacterial community composition (Fig. 2A); however, the strength of this effect varied (Table 1). For example, the effect of sample date was, on average, a three times weaker correlate of the community composition in endospheric microbiomes (e.g., leaf and root endosphere) compared to exospheric microbiomes (e.g., leaf surface, rhizosphere).
FIG 2.

(A and B) Principal-coordinate analysis (PCoA) ordinations of archaeal and bacterial (A) and fungal (B) community composition across plant-associated habitats, sampling time, and host species. The percentages in parentheses quantify the variance explained by each axis. Note the different axis scales.
TABLE 1.
PERMANOVA results for differences in archaeal/bacterial and fungal community composition by sample date, host genotype, and their interaction among plant-associated habitatsa
| Plant part | R2date | P valuedate | R2genotype | P valuegenotype | R2interaction | P valueinteraction |
|---|---|---|---|---|---|---|
| Archaea/bacteria | ||||||
| Leaf endosphere | 0.103 | >0.001 | 0.164 | >0.001 | 0.285 | >0.001 |
| Leaf surface | 0.327 | >0.001 | 0.070 | 0.292 | 0.112 | 0.112 |
| Root endosphere | 0.090 | >0.001 | 0.098 | 0.044 | 0.140 | 0.160 |
| Rhizosphere | 0.326 | >0.001 | 0.096 | 0.016 | 0.083 | 1.000 |
| Fungi | ||||||
| Leaf endosphere | 0.240 | >0.001 | 0.240 | 0.034 | 0.189 | 0.032 |
| Leaf surface | 0.247 | >0.001 | 0.111 | >0.001 | 0.145 | >0.001 |
| Root endosphere | 0.142 | >0.001 | 0.121 | 0.134 | 0.145 | 0.131 |
| Rhizosphere | 0.251 | >0.001 | 0.145 | >0.001 | 0.099 | 1.000 |
P values have been corrected by the false-discovery rate (n = 4) (96). Bolded values represent significant differences among groups (P < 0.05).
Taxonomic succession, for the most part, varied among plant-associated habitats. For instance, in the leaf endosphere, there was a relative increase in Proteobacteria over time (particularly Gammaproteobacteria), while in the rhizosphere, the relative abundance of Proteobacteria declined (see Fig. S4A at https://doi.org/10.6084/m9.figshare.14251463.v1). Similarly, at the order level, the leaf surface and rhizosphere followed diverging patterns: the relative abundance of Sphingomonadales increased in the leaf surface and decreased in the rhizosphere over time (see Fig. S5A at https://doi.org/10.6084/m9.figshare.14251463.v1).
Genotype was also a significant moderator of archaeal and bacterial community composition in all but the leaf surface microbiome (Table 1). Genotype effects were strongest in the two endosphere microbiomes, and in these microbiomes, the effect of genotype was stronger than that of sample date (Table 1). However, in the leaf endosphere, there was a significant sample date-genotype interaction (PERMANOVA, P < 0.001), where there was only a genotype effect during the June and September sample dates (T0, P = 0.261; June, P = 0.021, R2 = 0.407; September, P = 0.003, R2 = 0.569). Interestingly, genotypic differences in the belowground microbiomes were intraspecific, as host species was not a significant moderator of community composition (root endosphere, P = 0.440; rhizosphere, P = 1.000; Fig. 2A). However, in the latter two sample dates for the leaf endosphere microbiome (when genotype was a significant moderator of community composition), there was a significant difference between communities in P. deltoides and P. trichocarpa (P = 0.026, R2 = 0.031). This difference in the microbiome composition was noted at the genus level with a greater relative abundance of Streptococcus spp. in the P. deltoides leaf endosphere (see Fig. S6A at https://doi.org/10.6084/m9.figshare.14251463.v1). Upon further multiple-comparison testing among genotypes, we failed to detect significant differences between genotypes within the same plant-associated habitat across all sample dates (P < 0.05; see Table S2 at https://doi.org/10.6084/m9.figshare.14251463.v1).
Plant-associated habitat explained 24.1% of the variation in fungal community composition (PERMANOVA, P < 0.001, R2 = 0.241). Similar to the archaeal/bacterial results described above, because there was a three-way interaction among plant-associated habitat, genotype, and sample date (P < 0.001), we analyzed each plant-associated habitat separately. When separated by plant-associated habitat, sample date was consistently a significant moderator of fungal community composition and, on average, explained 22% of the variation (Fig. 2B; full statistics in Table 1). Taxonomically, this was represented as a gradual increase in the relative abundance of Basidiomycota over time across all plant-associated habitats (see Fig. S3B at https://doi.org/10.6084/m9.figshare.14251463.v1), particularly the orders Agaricales and Termellales (see Fig. S4B at https://doi.org/10.6084/m9.figshare.14251463.v1). In the rhizosphere, there was a gradual decrease in Trichoderma spp. (see Fig. S5B at https://doi.org/10.6084/m9.figshare.14251463.v1).
Functionally, there was a shift in the root endosphere fungal community away from pathogenicity at T0 toward mycorrhization during the June and September sample dates (Fig. 3). Accordingly, at T0, fungal pathogens constituted over 60% of the fungal reads in the root endosphere, while mycorrhizal fungi were almost nonexistent (<0.02%). By the September sampling date, the relative abundance of pathogens decreased to 16% of the root endosphere fungal reads, respectively, while ectomycorrhizal (EM) fungi increased to 7%. Arbuscular mycorrhizal (AM) fungi also increased dramatically over the sampling period, although relative abundances were significantly smaller than those of both pathogens and ectomycorrhizal fungi (Fig. 3).
FIG 3.

(A and B) Relative abundance of arbuscular mycorrhizal (AM), ectomycorrhizal (EM), and pathogen fungal reads across sample dates for the root endosphere (A) and rhizosphere (B). Different letters represent significant differences in guild relative abundance among sample dates (P < 0.05). Note the different y axis scales.
Genotype was also a significant moderator of fungal community composition in all but the root endosphere microbiome (Table 1). However, in the leaf endosphere and surface microbiomes, there were significant sample date-genotype interactions (PERMANOVA – leaf endosphere, P = 0.032; leaf surface, P < 0.001), where the effect of genotype was strongest in the later sampling dates (although not significant for the leaf endosphere; see Table S3 at https://doi.org/10.6084/m9.figshare.14251463.v1). Genotypic differences could only be attributed to host species in the leaf surface microbiome (P = 0.030, R2 = 0.019), where P. trichocarpa had greater relative abundances of the plant pathogen Alternaria spp. (see Fig. S6B at https://doi.org/10.6084/m9.figshare.14251463.v1).
Microbial assembly processes differ by microbial domain.
Using a null modeling approach, we assessed ecological assembly processes of the microbial communities following Stegen et al. (41). Assembly processes were broadly categorized as (i) variable selection, whereby selective processes lead to disparate microbial communities, (ii) homogenous selection, whereby selective processes lead to similar microbial communities, (iii) dispersal limitation, whereby limitations to dispersal allow ecological drift to lead to disparate microbial communities, and (iv) homogenizing dispersal, whereby high rates of dispersal lead to similar microbial communities (see Materials and Methods for complete statistical criteria).
Across all plant-associated habitats, no single assembly process dominated archaeal and bacterial community assembly (Fig. 4A). However, when assessed for each sample date and plant-associated habitat individually (the two strongest moderators of community composition), homogenous selection was the primary driver of between‐community shifts in composition for 29 to 62% of pairwise comparisons within each plant-associated habitat-sample date combination (Fig. 4B). The relative dominance of homogenous selection as the primary assembly process varied with both plant-associated habitat and sample date (Fig. 4B). For example, homogenous selection decreased during the growing season in aboveground microbiomes but remained fairly constant in the belowground archaeal and bacterial microbiomes (Fig. 4B). Other assembly processes, such as dispersal limitation, also varied by both plant-associated habitat and sample date. While dispersal limitation increased from T0 to June and decreased from June to September in the leaf endosphere and rhizosphere, it consistently increased in the root endosphere and leaf surface archaeal and bacterial microbiome (Fig. 4B).
FIG 4.

Relative dominance of assembly processes. (A and C) Overall dominance of assembly processes for archaea and bacteria (A) and fungi (C) among all samples. (B and D) Relative dominance of assembly processes within each plant-associated habitat-sample date combination for archaea and bacteria (B) and fungi (D). Too few fungal leaf endosphere samples in September (due to insufficient read depths) prevented the characterization of assembly processes.
Interestingly, variable selection was also a relatively important moderating factor in the root endosphere throughout the first growing season, which would appear to contradict the dominance of homogenous selection. However, pairwise comparisons for the same genotype within each plant-associated habitat-sample date combination were often 100% dominated by homogenous selection (see Table S4 at https://doi.org/10.6084/m9.figshare.14251463.v1). Thus, significant variable selection among genotypes occurred in the root endosphere, particularly among P. trichocarpa genotypes (see Table S5 at https://doi.org/10.6084/m9.figshare.14251463.v1). Differences between the leaf endosphere and leaf surface and the root endosphere and rhizosphere were characterized by a combination of dispersal limitation and variable selection, while similarities were mostly characterized by homogenous selection, and these patterns were fairly consistent over time (see Fig. S7A and B at https://doi.org/10.6084/m9.figshare.14251463.v1).
Across all plant-associated habitats, sample dates, and genotypes, the fungal community was characterized by both dispersal limitation and undominated assembly processes, with selection playing a relatively minor role (Fig. 4C). When assessed for each sample date and plant-associated habitat individually, fungal community assembly was, for the most part, undominated by selective or dispersal processes (Fig. 4D). However, similar to the archaeal and bacterial communities, the dominance of assembly processes differed by sample date. In the leaf surface fungal microbiome, there was a shift from homogenizing dispersal to homogenous selection during the first growing season (Fig. 4D). Alternatively, dispersal processes, at the expense of homogenous selection, became relatively more dominant in the rhizosphere for fungi during the growing season. Unlike for archaea and bacteria, pairwise comparisons within plant-associated habitat-sample date-genotype combinations for fungi were most often characterized by assembly processes undominated by selection or dispersal (see Table S6 at https://doi.org/10.6084/m9.figshare.14251463.v1), a pattern that was consistent within host species as well (see Table S7 at https://doi.org/10.6084/m9.figshare.14251463.v1). Differences between the leaf endosphere and leaf surface were mostly controlled by weak selection and dispersal factors (i.e., “undominated”), while differences between the root endosphere and rhizosphere were characterized by a combination of undominated assembly processes and dispersal limitation (see Fig. S7C and D at https://doi.org/10.6084/m9.figshare.14251463.v1). These patterns were consistent over the growing season.
Core taxa disproportionately represent the Populus microbiome.
Using occupancy-abundance distributions of microbial genera across all time points, we detected 23 distinct bacterial (Table 2) and 12 distinct fungal (Table 3) core genera across the four plant-associated habitats based on >95% occupancy and >1% average relative abundance (see Fig. S8 at https://doi.org/10.6084/m9.figshare.14251463.v1). While we detected no core archaeal and bacterial genera in the leaf endosphere, on average, archaeal and bacterial core genera constituted 51%, 24%, and 49% of reads in the leaf surface, root endosphere, and rhizosphere microbiome, respectively. The core genera accounted for 1.4%, 0.8%, and 2.9% of archaeal and bacterial genera in the leaf surface, root endosphere, and rhizosphere microbiome, respectively. Across the growing season, the relative abundance of the core archaeal and bacterial community increased in the leaf surface (ANOVA, F2,102 = 65.74, P < 0.001), decreased in the rhizosphere (F2,103 = 13.11, P < 0.001), and did not significantly change in the root endosphere (F2,102 = 1.04, P = 0.357; Fig. 5A).
TABLE 2.
Core archaeal and bacterial genera defined by >95% occupancy and >1% average relative abundance
| Plant-associated habitat | Occupancy (%) | Relative abundance (%) | Genus |
|---|---|---|---|
| Leaf surface | 99.0 | 1.7 | Agrobacterium |
| 98.1 | 1.4 | Devosia | |
| 95.1 | 5.0 | Hymenobacter | |
| 98.1 | 3.3 | Massilia | |
| 100.0 | 13.6 | Methylobacterium | |
| 96.1 | 1.1 | Mycobacterium | |
| 98.1 | 3.5 | Pseudomonas | |
| 100.0 | 21.9 | Sphingomonas | |
| Root endosphere | 95.1 | 1.2 | Hyphomicrobium |
| 97.1 | 6.1 | Pseudomonas | |
| 99.0 | 2.7 | Rhodoplanes | |
| 100.0 | 2.2 | Sphingomonas | |
| 99.0 | 11.6 | Streptomyces | |
| Rhizosphere | 100.0 | 1.8 | Alicyclobacillus |
| 99.0 | 1.9 | Bacillus | |
| 100.0 | 3.1 | Bradyrhizobium | |
| 100.0 | 2.2 | Burkholderia | |
| 100.0 | 2.2 | Chitinophaga | |
| 100.0 | 3.5 | Devosia | |
| 100.0 | 3.2 | Dyella | |
| 100.0 | 2.6 | Hyphomicrobium | |
| 100.0 | 3.7 | Kaistobacter | |
| 100.0 | 1.8 | Mesorhizobium | |
| 100.0 | 2.7 | Mycobacterium | |
| 100.0 | 1.2 | Novosphingobium | |
| 100.0 | 3.5 | Phenylobacterium | |
| 100.0 | 1.7 | Pseudonocardia | |
| 100.0 | 9.0 | Rhodoplanes | |
| 100.0 | 2.5 | Sphingomonas | |
| 100.0 | 3.0 | Streptomyces |
TABLE 3.
Core fungal genera defined by >95% occupancy and >1% average relative abundance
| Plant-associated habitat | Occupancy (%) | Relative abundance (%) | Genus | Guilda |
|---|---|---|---|---|
| Leaf endosphere | 96.6 | 35.5 | Alternaria | Pathogen |
| 96.6 | 8.7 | Boeremia | Pathogen | |
| Leaf surface | 100.0 | 16.9 | Alternaria | Pathogen |
| 96.2 | 6.2 | Aureobasidium | Pathogen | |
| 97.1 | 11.5 | Boeremia | Pathogen | |
| 98.1 | 5.5 | Filobasidium | Saprotroph | |
| 100.0 | 2.0 | Fusarium | Pathogen | |
| 97.1 | 2.2 | Paraconiothyrium | Saprotroph | |
| Root endosphere | 98.4 | 7.4 | Fusarium | Pathogen |
| Rhizosphere | 100.0 | 1.2 | Coniochaeta | Pathogen |
| 100.0 | 5.9 | Fusarium | Pathogen | |
| 95.2 | 1.6 | Humicola | Saprotroph | |
| 99.0 | 12.2 | Kabatiella | Pathogen | |
| 100.0 | 18.7 | Phialemonium | Saprotroph | |
| 98.1 | 2.5 | Scytalidium | Saprotroph | |
| 100.0 | 24.8 | Trichoderma | Saprotroph/endophyte |
Guild is defined using the FUNGuild database (84).
FIG 5.
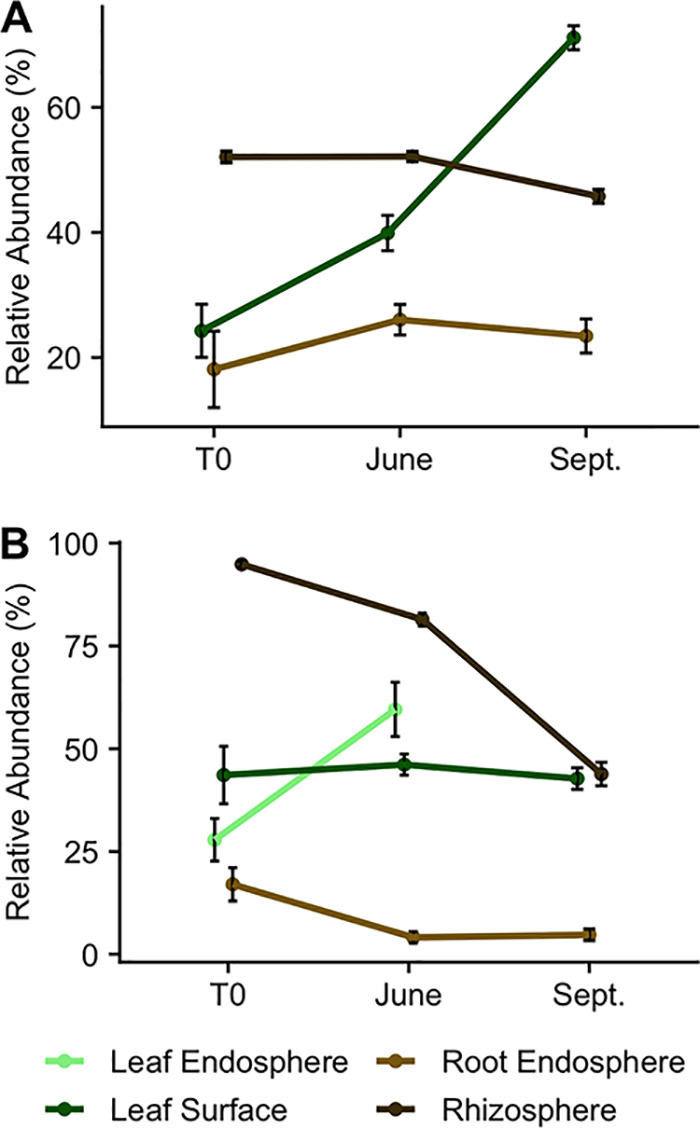
(A and B) Mean relative abundance (and standard error) of archaeal and bacterial (A) and fungal (B) core genera during the first growing season across plant-associated habitats. Note the different axis scales. Too few fungal leaf endosphere samples in September (due to insufficient read depths) prevented the characterization of a core microbiome.
For fungi, core genera constituted, on average, 44%, 44%, 7%, and 67% of reads in the leaf endosphere, leaf surface, root endosphere, and rhizosphere fungal microbiome, respectively. The core genera accounted for 0.7%, 1.4% 0.4%, and 1.8% of fungal genera in the leaf endosphere, leaf surface, root endosphere, and rhizosphere microbiome, respectively. Similar to archaea and bacteria, the relative abundance of fungal core genera across the growing season differed by plant-associated habitat. During the first growing season, the relative abundance of fungal core genera increased in the leaf endosphere (ANOVA, F1,28 = 14.06, P < 0.001), remained the same in the leaf surface (F2,103 = 0.38, P = 0.685), and decreased in the root endosphere (F2,58 = 9.46, P < 0.001) and rhizosphere (F2,103 = 105.1, P < 0.001; Fig. 5B). Half of the core fungal genera were classified as potential plant pathogens, while the other half were classified as saprotrophs. Interestingly, many core genera were shared among plant-associated habitats. For instance, the bacterium Sphingomonas and the fungal plant pathogen Fusarium were core genera in the leaf surface, root endosphere, and rhizosphere. Similarly, in addition to the aforementioned genera, Rhodoplanes and Streptomyces were core genera in both the root endosphere and rhizosphere. Additionally, the two core genera in the leaf endosphere, the fungal plant pathogens Alternaria and Boeremia, were also leaf surface core genera.
Endospheric microbiomes have both above- and belowground sources.
Across the sink microbiomes (e.g., leaf and root endosphere), we were able to determine the source for 6.9% and 3.5%, on average, of new archaeal and bacterial and fungal taxa, respectively (see Table S8 at https://doi.org/10.6084/m9.figshare.14251463.v1) using SourceTracker (42). Generally, source microbiome contributions to the sink microbiome were comparable, with each source contributing about half of the source-attributable new taxa within each sink microbiome (ANOVA, F1,124 = 8.165, P = 0.004). The singular exception was for the leaf endosphere archaeal and bacterial microbiome. Here, while model predictions estimated that the root endosphere contributed almost 12% of new taxa to the archaeal and bacterial leaf endosphere community, the leaf surface was estimated to contribute only 2.6%. Source contributions were also consistent over time for both the leaf and root endosphere across both amplicons (16S-leaf, P = 0.096; ITS-leaf, P = 0.440; 16S-root, P = 0. 213; ITS-root, P = 0.159).
DISCUSSION
The initial assembly of the plant microbiome may establish the trajectory for forthcoming microbiome states (via priority effects) and could determine the overall future health of the plant (12, 43, 44). Thus, a greater understanding of initial plant microbiome assembly in an ecologically and economically important plant such as Populus is highly desirable (25, 26). Here, we show that the initial microbiome composition and assembly in the first growing season of Populus are temporally dynamic and are determined by a combination of both selective and stochastic factors.
Sample date was consistently a strong predictor of the microbial community composition when assessed within each individual plant-associated habitat, which highlights how changing plant, soil, and climactic factors over time influence microbial community composition. First of all, increasing root and leaf biomass during the first growing season may affect the diversity of the microbial community through species-area relationships (45, 46). Additionally, seasonal internal redistribution of sugar and nitrogen among plant tissues (47, 48) would affect substrates necessary for microbial growth leading to changes in microbial community composition. Our finding that Gammaproteobacteria gradually replace Alphaproteobacteria in aboveground sample types over time contradicts previous studies in grass microbiomes (14, 49), highlighting differences between tree and grass microbiomes. Additionally, as plants grow during the first growing season, exudation of plant photosynthates into the rhizosphere from roots likely increases (50). This increase in exudation has been shown to favor Proteobacteria and Bacteroidetes at the expense of Actinobacteria in Arabidopsis (50), Avena (51), and Citrus (52). Interestingly, our results showed opposite patterns (see Fig. S4 at https://doi.org/10.6084/m9.figshare.14251463.v1), which could reflect other environmental changes that would favor Actinobacteria. Increased temperatures during the summer months could positively impact Actinobacteria (53). Furthermore, increased activity during summer months (54) may enhance nutrient availability (55, 56) and shift the microbial community composition. These results contribute to the growing literature suggesting that microbial communities are temporally dynamic (57). However, we build upon this to show that, across habitat types (e.g., leaf endosphere, rhizosphere, etc.), the initial community assembly of the plant microbiome cannot be captured through examination of a single time point and that stochastic processes as well as deterministic ones shape the plant microbiome.
The effect of sample date was stronger in exospheric microbiomes (i.e., the leaf surface and rhizosphere) than in endospheric microbiomes (i.e., the leaf and root endosphere) for archaea and bacteria. This is corroborated by multiple studies that show that the host effects dominate the endosphere microbiome, while environmental factors influence the exospheric microbiomes to a greater extent (12, 15, 17). However, host effects likely also changed during the initial growing season as leaf area increased and patterns of nutrient and sugar distribution changed (47, 48). This suggests that environmental changes over the first growing season are stronger than changes to the host. The muted community change with season in endospheric microbiomes could also be due to dispersal limitation, where microbes adapted to the changing exospheric conditions are prevented from colonizing the plant endosphere. Indeed, we found evidence for dispersal limitation to be a major factor in structuring differences between endo- and exospheric microbiomes (see Fig. S7 at https://doi.org/10.6084/m9.figshare.14251463.v1) and within the endospheric microbiomes (Fig. 5). However, this finding that exospheric archaeal and bacterial microbiomes were influenced by sample date to a greater degree than in endospheric microbiomes was not followed by fungi (Table 1). This difference could be attributed to the overall greater stochasticity or weak selective/dispersal factors found in fungal community assembly (Fig. 5). Such differences highlight the need to study the plant microbiome holistically, as different microbial domains and plant-associated habitats can follow distinct patterns.
An increased understanding of fundamental community assembly processes describes the elementary units that underpin microbial community composition. Consistent with our hypothesis, the relative dominance of assembly patterns (e.g., variable selection, homogenous selection, dispersal limitation, and homogenizing dispersal) changed over time. However, a general pattern with time across plant-associated habitats for both archaea and bacteria and fungi did not emerge. This is surprising because in other successional contexts (e.g., soils after wildfire disturbances and a salt marsh soil chronosequence), selective assembly processes tend to dominate over time (58, 59). While disturbances may lead to historical contingencies that could affect assembly mechanisms (9), the environmental pressures on the microbiome of a growing plant could also lead to large changes in assembly. Indeed, during the first 4 months of sorghum (Sorghum bicolor) growth, the relative dominance of community assembly processes of the fungal phyllosphere microbiome shifted from stochasticity (i.e., weak selective and dispersal processes) to homogenous selection and homogenizing dispersal (60). Furthermore, evidence of deterministic selection increased during successive microbial passages in the Solanum phyllosphere (61). These conflicting results among studies suggest that temporal patterns of microbial assembly may be idiosyncratic and depend on the host species. This demonstrates the need to understand plant microbiome assembly in multiple species aside from “model” grass systems.
Our finding that homogenous selection was the dominant assembly process for archaea and bacteria at each sample date across plant-associated habitats suggests that the initial Populus archaeal and bacterial microbiome assembly is repeatable and predictable even among different Populus genotypes. This is remarkable given the fact that this consistency was noted even when compared between two different Populus species. This is not to say that there were no differences among genotypes; however, the impact of genotype on microbial community composition across microbial domains was relatively similar or less than that of sample date. In the leaf and root endosphere, the archaeal and bacterial microbiome was, in fact, more structured by genotype than sample date. These genotypes vary in production of numerous metabolites, such as salicylic acid (7, 62, 63), which can correlate with changes in the microbial community composition (64). Plant phenotypic differences are even more pronounced across Populus species such as P. trichocarpa and P. deltoides, which have differential resistances to plant pathogens such as Sphaerulina musiva and Marssonina brunnea (31, 65). Differences in microbiome composition among genotypes may grow stronger, especially within the endosphere, as the plants mature and differences in pathogen infectivity begin to emerge. For example, the effects of the leaf spot and stem canker fungal pathogens Marssonina brunnea and Sphaerulina musiva are known to dramatically increase over the initial years after establishment in plantation settings (66), and similar patterns have been observed at our site in P. trichocarpa trees in the subsequent years after this study (M. Cregger and W. Muchero, unpublished data). However, we did not find differences in pathogen relative abundance among different genotypes or Populus species (Fig. 3; Fig. S6 at https://doi.org/10.6084/m9.figshare.14251463.v1), which contradicts early findings in mature Populus trees (31), further supporting out hypothesis that genotypic differences likely emerge as trees age.
With a few exceptions, our hypothesis that the dominance of a microbial core community would increase over time was unsupported by the data. Only the archaeal and bacterial leaf surface microbiome and the fungal leaf endosphere core microbiomes increased in dominance over time. However, across all time points, these 23 distinct bacterial and 12 distinct fungal core genera constituted a substantial proportion of the plant microbiome (7 to 69%). This supports earlier findings that overall beta diversity in the plant microbiome is controlled by the variation in a relatively small number of taxa (4, 14, 32, 67). This is corroborated by the fact that most of the fungal core genera in our study were considered potential plant pathogens, which can disproportionately affect the plant microbiome (36). Interestingly, although Populus plants form both EM and AM symbioses (68), none of the core fungal genera were classified as such. This is likely because at T0, mycorrhizal associations had not yet established. Indeed, the proportion of both EM and AM reads increased during the first growing season in the root endosphere and rhizosphere, which suggests that mycorrhizal colonization in the field can occur relatively rapidly (<1 month), and there is a strong preference of such a relationship with time.
The bacterial core microbiome consisted of several taxa that have also been classified as “core” in numerous other studies across vastly different plant types. For example, core bacterial genera in the leaf surface in our study included Methylobacterium, Sphingomonas, Pseudomonas, and Hymenobacter, which have been classified as core members of the Panicum virgatum, Miscanthus x giganteus, Arabidopsis thaliana, and Medicago truncatula phyllospheres (14, 69, 70). Additionally, in the rhizosphere, core Alphaproteobacteria genera in our study, such as Bradyrhizobium, Burkholderia, Mesorhizobium, and Rhodoplanes are also considered core rhizosphere bacteria across numerous plant phyla (35). This suggests that there is a consistent consortium of bacteria that frequently colonize different plant-associated habitats regardless of plant type or species. In other words, these core genera may be selected based on the plant-associated habitat (e.g., phyllosphere versus rhizosphere), and differences in plant-associated habitats among plant species may be less important. It is possible then that these bacteria perform important functions in the plant. However, in contrast to the fungal core genera, the functions of the bacterial core genera still remain unclear because metagenome prediction based on amplicon data has yet to be verified by large-scale genomic sequencing of isolates or shotgun metagenomics in plant tissues. Until then, the functional significance of the core microbiome is still debated (33, 34); however, these core members represent taxa that should be prioritized for isolation and subsequent genomic sequencing to understand their functional significance.
Consistent with previous studies, we found that the colonization of the root and leaf endosphere likely occurs from multiple sources (71–73). However, our hypothesis that the adjacent exospheric microbiome would be the dominant source of the endospheric microbiome (e.g., the rhizosphere for the root endosphere) was largely unsupported. Our results suggest that under most circumstances new taxa colonize from a combination of aboveground and belowground source environments. This finding somewhat contradicts previous studies that suggest that the soil microbiome is the dominant reservoir of plant endosphere taxa (14, 70). While it is true that highly diverse soil microbial communities, in general, share many microbial members with those in the plant endosphere, a mechanistic model of microbial colonization cannot necessarily be inferred from such observations. For example, aeolian-derived microbes from different environments may be deposited on leaf as well the soil surface (74, 75). Such microbes have been shown to enter the leaf endosphere through stomatal openings (73), so even though there is shared membership between the leaf endosphere and the soil, this does not necessarily mean that the soil is the source of said microbe. Our source tracking models attempt to remedy this by including multiple aboveground and belowground sources. Nevertheless, the lack of source attribution (<10% in many cases) limits the conclusions that can be made from these models. However, we show that it is likely that multiple sources contribute to plant endosphere colonization.
Extending these source tracking predictions to our community assembly models, we show that colonization of the plant endosphere by archaea and bacteria is influenced by both variable selection and dispersal limitation. This suggests that differences in the community composition between endo- and exo-spheric microbiomes is due to both selective pressures and physical limitations of colonization. While this has been previously hypothesized (4, 73), we are the first to demonstrate with community assembly models that this is indeed the case. Furthermore, we show that, for fungi, stochastic factors likely play a major role in influencing the colonization of the plant endosphere from its adjacent exosphere, and in the root endosphere, dispersal limitation is also a factor. The mode of microbial colonization is an important consideration of plant microbiome assembly, and these models of microbial colonization (i.e., source tracking and assembly processes) should be paired with future experiments that empirically test modeled results.
Our results comprehensively characterize the early assembly of the temporally dynamic Populus microbiome among multiple plant genotypes and species and across different plant-associated habitats. While the fundamental assembly processes and colonization sources remained largely similar throughout the first growing season, the microbiome community composition shifted dramatically. This suggests that changing seasonal factors as well as changes associated with plant growth are primarily responsible for these shifts. Future work, such as long-term, seasonal plant microbiome studies, may be able to partition the influence of season and plant growth. It is hypothesized that the initial assembly of the microbiome “sets the stage” for future microbiome states through priority effects (76). Hence, where differences exist (e.g., the modest differences among plant genotypes), our findings could be used to predict the composition of the Populus microbiome into the future and better understand its influence on plant health.
MATERIALS AND METHODS
Plant collections.
During December of 2016, eight P. trichocarpa genotypes were collected from Corvallis and Clatskanie genome-wide association study (GWAS) populations (65), and two P. deltoides genotypes were collected from a previous University of Tennessee Institute of Agriculture (UTIA) Sun Grant experiment in Blount County, TN, in the same field as the plots described below (31, 77). Cuttings were kept on ice, shipped overnight, and maintained at 4°C until propagation (March 2017). Cuttings were rinsed in a 1% Zerotol 2.0 solution for surface sterilization, rooting powder was placed on sterile cutting surfaces (0.1% indole-3-butyric acid), and plants were placed in sterile potting soil (Fafard 52 Mix, Sun Gro Horticulture, Massachusetts, USA). Once significant root growth took place and leaf buds opened, cuttings were transferred to an experimental plot in Blount County, TN, managed by the UTIA-East Tennessee Research and Education Center (ETRED) (see Cregger et al. [31] for site details).
Experimental design, harvest, and sample processing.
Soils were augured to transplant cuttings and minimize the effects of soil compaction on belowground root establishment. We planted six rows of 80 individuals spaced 1 m apart. These rows were broken up into three blocks made up of 4 to 26 replicates of each genotype. Sampling was conducted on three sampling dates in the initial growing season—day of transplant T0 (in May of 2017, representing the microbial community that was present in the cuttings and that colonized the plants in the greenhouse), June 2017, and September 2017 to assess initial microbiome community assembly across four plant-associated habitats (leaf endosphere, leaf surface, root endosphere, and rhizosphere). At each time point, three random replicate plants (one from each block) were destructively harvested per genotype, and samples were transferred to the laboratory. Roots, and the attached soil (operationally defined as rhizosphere soil), were stored at −80°C until root washing, sterilization, and genomic DNA (gDNA) extractions took place (within 2 months of sampling). Briefly, fine roots (<2 mm diameter) were sorted and surface-sterilized by sequential washing with bleach (3.125%) and then ethanol (70%) and rinsing of the roots with autoclaved water (4 times) as previously described (31); the rhizosphere was collected from an initial rinse with sterile water. Leaves were stored at 4°C after harvest and processed within 3 days. Leaves were rinsed (rinsate was collected as leaf surface samples) and surface-sterilized as previously described (31) by washing leaves with bleach and rinsing the leaves with autoclaved water (4 times). Postprocessing, leaf samples were also stored at –80°C until DNA extractions.
DNA extractions and Illumina MiSeq preparation and sequencing.
Prior to extraction, root and leaf tissues were cut into fine pieces (∼5 mm or less), and leaf and root rinsates (rhizosphere) were centrifuged at 10,000 × g, and the supernatant was removed. These pelleted soil rhizosphere samples were then extracted using the PowerSoil DNA kit (Qiagen, Venlo, The Netherlands) following the standard protocol except that a Precellys tissue homogenizer was used to bead-beat extractions (30 sec of 5,500 × g bead-beating with a 30 sec rest in triplicate). Root and leaf endosphere samples were extracted using the Qiagen PowerPlant Pro DNA kit following the standard protocol except that prior to extraction, 50 mg of tissue per extraction was bead-beaten for 1 min in liquid nitrogen blocks with one sterile steel bead two times. Extractions were quantified on a Nanodrop 1000 spectrophotometer (NanoDrop Products, Wilmington, DE, USA). Root and leaf endosphere extractions were purified and concentrated using a DNA Clean and Concentrator-5 kit (Zymo Research Corporation, Irvine, CA, USA) prior to PCR amplification.
A two-step PCR approach was used with barcode-tagged and frameshifting nucleotide primers targeting the 16S rRNA gene for archaea and bacteria and the ITS2 region for fungi (78) using pooled primer sets to increase coverage of archaeal, bacterial, and fungal taxa (31) (see Table S9 at https://doi.org/10.6084/m9.figshare.14251463.v1). The first step of PCR included 2.5 μM peptide nucleotide acid (PNA) blockers for 16S rRNA amplifications, and 2.5 μM PNA targeting plant nuclear rRNA genes for ITS2 region was used to reduce the amplification of plant material (see Table S9 at https://doi.org/10.6084/m9.figshare.14251463.v1). Thermal cycler conditions for the primary PCRs for soils were 5 cycles of 95°C for 1 min, 50°C for 2 min, and 72°C for 1 min. Primary PCR conditions for plant tissues were 5 cycles of 95°C for 1 min, 78°C for 5 s, 50°C for 2 min, and 72°C for 1 min. Primary PCR products were cleaned with 17 μl of Agencourt AMPure beads and eluted in 21 μl of nuclease-free water. Secondary PCRs had purified DNA tagged with barcoded reverse primers and forward primers (see Table S9 at https://doi.org/10.6084/m9.figshare.14251463.v1) in the 50-μl reaction mixture, except with 20 μl of purified DNA from primary PCRs. Thermal cycler conditions for secondary soil PCRs consisted of denaturation at 95°C for 45 s, followed by 32 cycles of 95°C for 15 s, annealing at 60°C for 30 s, 72°C for 30 s, and final extension at 72°C for 30 s. Secondary PCRs for plant tissue consisted of denaturation at 95°C for 45 s, followed by 32 cycles of 95°C for 15 s and 78°C for 5 s, with remaining cycle parameters the same as with soil secondary PCRs. After PCRs, all experimental units were pooled based on band intensity and purified with Agencourt AMPure XP beads (0.7:1 bead to DNA ratio; Beckman Coulter, Inc., Pasadena, CA, USA). Illumina MiSeq sequencing was carried out using a 9-pM amplicon concentration with a 15% PhiX spike (2 × 300 cycles).
Bioinformatics.
Paired-end .fastq sequence files had primers and adaptors removed individually using the Cutadapt program (79). Trimmed .fastq files were then joined using QIIME 1 and imported into the QIIME 2 environment (80). Joined sequences were demultiplexed, and median Phred quality scores were visualized. Due to poor quality on the 3′ end for 16S data, they were truncated to 190 bp. The ITS reads were not further trimmed or truncated. Both 16S and ITS2 data sets were denoised and delineated into amplicon sequence variants (ASVs) using the DADA2 algorithm in QIIME 2 (81). Representative sequences were then assigned a taxonomic classification using the naive Bayes classifier through the sklearn Python package for 16S rRNA sequences with the SILVA database (82), while ITS2 gene representative sequences were assigned a taxonomic classification in QIIME 1 using BLAST and the UNITE reference database (83). Contaminants (unassigned reads, mitochondria, chloroplasts for 16S; Protista, Chromista, Animalia, and Plantae reads for ITS2) were removed. Contaminants were a low percentage of total reads for bacteria (∼9%) and fungi (∼15%). Fungi were further classified into functional guilds using the FUNGuild database (84).
After quality and taxonomic filtering (i.e., removal of plant and plasmid DNA), we sequenced 2.35 × 107 16S reads across 386 samples (26 samples were removed due to low depths [<900 reads]), with a minimum depth of 965 reads and a maximum of 352,342 reads (see Fig. S9A for the rarefaction curve at https://doi.org/10.6084/m9.figshare.14251463.v1). For ITS2, we sequenced 1.92 × 107 reads across 323 samples (4 samples were removed due to low depths [< 900 reads] or presence of contaminant sequences) with a minimum depth of 910 reads and a maximum of 326,368 reads (see Fig. S9B for the rarefaction curve at https://doi.org/10.6084/m9.figshare.14251463.v1).
Quantifying microbial assembly processes (see below) relies on phylogenetic turnover (41). The phylogenetic tree for 16S reads was created in the QIIME2 environment using FastTree 2 (85) and was midpoint rooted. Because ITS genes do not align well across large phylogenetic distances, we used a “ghost tree” approach to create a phylogenetic tree for ITS2 reads (86). Briefly, the 18S SILVA database (82) was used to create a foundational tree, and class-level ITS branches from the UNITE database (83) were grafted onto the ends of the foundational tree. The ITS2 ASVs were then clustered into 99% operational taxonomic units (OTUs) by closed reference OTU picking using VSEARCH (87). These ITS2 OTUs were only used to quantify microbial assembly processes; other analyses were conducted using ASVs.
Statistical analysis.
All statistical analyses were conducted in R (88) using the Picante (89), phyloseq (90), and vegan (91) packages. Significance was determined at the α = 0.05 level for all statistical tests.
Differences in α-diversity over time and among genotypes were compared by means of Hill numbers (40) of samples rarified to 900 reads (for both amplicons) at orders of q = 0 and q = 1. The parameter q determines the relative weighting of rare species. At q = 0, all species are weighted equally (richness); at q = 1, species are weighted proportionally to their relative abundance (analogous to Shannon’s index). Differences in means of Hill numbers among plant-associated habitats, sample dates, and genotypes were assessed by ANOVA. Where independent variables were significant, we assessed multiple comparisons by Tukey’s test of honest significant differences. We used Q-Q plots and scale-location plots to inspect normality and homoscedasticity, respectively. Where these assumptions were unmet, independent variables were log-transformed and reanalyzed satisfying the aforementioned assumptions.
Differences in the microbial community composition among plant-associated habitats, sample dates, and genotypes were assessed by PERMANOVA (92), using Bray-Curtis distances applied to proportionally normalized data. We visualized differences in community composition using principal-coordinate analysis (PCoA).
Assembly processes were assessed using a null modeling approach following Stegen et al. (41). Assembly processes are broadly categorized as (i) variable selection, whereby selective processes lead to disparate microbial communities, (ii) homogenous selection, whereby selective processes lead to similar microbial communities, (iii) dispersal limitation, whereby limitations to dispersal allow ecological drift to lead to disparate microbial communities, and (iv) homogenizing dispersal, whereby high rates of dispersal lead to similar microbial communities. This approach uses both phylogenetic and taxonomic turnover to classify the dominant assembly process in pairwise sample comparisons. To characterize phylogenetic turnover, we use the between-community version of the (abundance-weighted) β-mean-nearest taxon distance (βMNTD) (93). Observed βMNTDs were then compared to a null model distribution of βMNTDs generated from 999 null model expectations (i.e., randomized taxa reshuffling among phylogenetic tree tips). The difference between observed βMNTD and the mean of the null distribution was measured in units of standard deviation (of the null distribution) and is referred to as the β-nearest taxon index (βNTI). Values of βNTI of ≥2 signify variable selection as the dominant assembly process, and βNTI values of ≤–2 signify homogenous selection as the dominant assembly process following Stegen et al. (41).
Taxonomic turnover was used to define the dominant assembly processes of |βNTI values| < 2. For taxonomic turnover, we use the Raup Crick index (94) modified to include species abundances (41). We calculated separate null models for archaea and bacteria and fungi using probability-based randomization. Null model microbial compositions were assembled for each sample by randomly sampling from the total ASV (or OTU for fungi, see above) pool in proportion to occupancy to define ASVs and then in proportion to abundance to define abundances into those selected ASVs, thereby maintaining similar levels of α-diversity. The community composition for each sample was probabilistically generated 999 times. For each iteration, the Bray-Curtis dissimilarity index between samples was calculated, and the proportion of iterations in which the index was smaller than or equal to the observed Bray-Curtis dissimilarity index between those pairs of samples was our resulting metric. We standardized this metric (RCBray) to range from −1 to 1 by subtracting 0.5 and multiplying by 2 (94). Values of |βNTI | < 2 and RCBray ≥ 0.95 signify dispersal limitation, and values of |βNTI | < 2 and RCBray ≤ –0.95 signify homogenizing dispersal following Stegen et al. (41). Values of |βNTI | < 2 and |RCBray| < 0.95 signify weak selection and moderate levels of dispersal such that no process dominates assembly and are therefore classified as “undominated” (95).
The core microbiome of each plant-associated habitat was characterized using occupancy-abundance distributions of microbial genera across all time points (14). Microbial genera were considered “core” when their occupancy (the proportion of presence in all samples) was at least 95% and their relative abundance was at least 1% averaged across all sampling dates within a plant-associated habitat.
We used SourceTracker (42) to determine the relative contribution of the rhizosphere and leaf surface as microbial sources of new microbial taxa (i.e., not present in the previous sample dates) in endospheric microbiomes for the June and August sampling dates. Differences in the relative contributions of sources among sample dates and genotypes were also assessed by ANOVA. These ANOVAs were analyzed using the same approach as for α-diversity.
Data availability.
The data sets presented in this study can be found in the Sequence Read Archive under BioProject number 685817. The R code for all statistics and figures as well as the final ASV, taxonomy, and sample data tables used in this analysis can be found at https://github.com/nicholascdove/Populus_microbiome_assembly.
ACKNOWLEDGMENTS
We thank the University of Tennessee Institute of Agriculture–East Tennessee AgResearch and Education Center (ETREC) for providing the site access and allowing us to establish a small plantation at their field site and assisting in its maintenance. This research was sponsored by the Genomic Science Program, U.S. Department of Energy, Office of Science, Biological, and Environmental Research, as part of the Plant Microbe Interfaces Scientific Focus Area at ORNL (Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U. S. Department of Energy under contract DEAC05-00OR22725. J.C.S. is supported by the Environmental System Science Scientific Focus Area at the Pacific Northwest National Laboratory.
This manuscript has been authored by UT-Battelle, LLC, under contract number DE-AC05-00OR22725 with the U.S. Department of Energy. The U. S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U. S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for U. S. Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Contributor Information
Melissa A. Cregger, Email: creggerma@ornl.gov.
Hideyuki Tamaki, National Institute of Advanced Industrial Science and Technology.
REFERENCES
- 1.Compant S, Duffy B, Nowak J, Clément C, Barka EA. 2005. Use of plant growth-promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl Environ Microbiol 71:4951–4959. doi: 10.1128/AEM.71.9.4951-4959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santoyo G, Moreno-Hagelsieb G, del Carmen Orozco-Mosqueda M, Glick BR. 2016. Plant growth-promoting bacterial endophytes. Microbiol Res 183:92–99. doi: 10.1016/j.micres.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Gottel NR, Castro HF, Kerley M, Yang Z, Pelletier DA, Podar M, Karpinets T, Uberbacher E, Tuskan GA, Vilgalys R, Doktycz MJ, Schadt CW. 2011. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl Environ Microbiol 77:5934–5944. doi: 10.1128/AEM.05255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shakya M, Gottel N, Castro H, Yang ZK, Gunter L, Labbé J, Muchero W, Bonito G, Vilgalys R, Tuskan G, Podar M, Schadt CW. 2013. A multifactor analysis of fungal and bacterial community structure in the root microbiome of mature Populus deltoides trees. PLoS One 8:e76382. doi: 10.1371/journal.pone.0076382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V. 2015. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci U S A 112:E911–E920. doi: 10.1073/pnas.1414592112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonito G, Benucci GMN, Hameed K, Weighill D, Jones P, Chen K-H, Jacobson D, Schadt C, Vilgalys R. 2019. Fungal-bacterial networks in the Populus rhizobiome are impacted by soil properties and host genotype. Front Microbiol 10:481. doi: 10.3389/fmicb.2019.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veach AM, Morris R, Yip DZ, Yang ZK, Engle NL, Cregger MA, Tschaplinski TJ, Schadt CW. 2019. Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome 7:76. doi: 10.1186/s40168-019-0668-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barge EG, Leopold DR, Peay KG, Newcombe G, Busby PE. 2019. Differentiating spatial from environmental effects on foliar fungal communities of Populus trichocarpa. J Biogeogr 46:2001–2011. doi: 10.1111/jbi.13641. [DOI] [Google Scholar]
- 9.Stegen JC, Bottos EM, Jansson JK. 2018. A unified conceptual framework for prediction and control of microbiomes. Curr Opin Microbiol 44:20–27. doi: 10.1016/j.mib.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Wei Z, Gu Y, Friman V-P, Kowalchuk GA, Xu Y, Shen Q, Jousset A. 2019. Initial soil microbiome composition and functioning predetermine future plant health. Sci Adv 5:eaaw0759. doi: 10.1126/sciadv.aaw0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cordovez V, Dini-Andreote F, Carrión VJ, Raaijmakers JM. 2019. Ecology and evolution of plant microbiomes. Annu Rev Microbiol 73:69–88. doi: 10.1146/annurev-micro-090817-062524. [DOI] [PubMed] [Google Scholar]
- 12.Trivedi P, Leach JE, Tringe SG, Sa T, Singh BK. 2020. Plant-microbiome interactions: from community assembly to plant health. Nat Rev Microbiol 18:607–621. doi: 10.1038/s41579-020-0412-1. [DOI] [PubMed] [Google Scholar]
- 13.Maignien L, DeForce EA, Chafee ME, Eren AM, Simmons SL. 2014. Ecological succession and stochastic variation in the assembly of arabidopsis thaliana phyllosphere communities. mBio 5:e00682-13. doi: 10.1128/mBio.00682-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grady KL, Sorensen JW, Stopnisek N, Guittar J, Shade A. 2019. Assembly and seasonality of core phyllosphere microbiota on perennial biofuel crops. Nat Commun 10:1–10. doi: 10.1038/s41467-019-11974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong C, Zhu Y-G, Wang J-T, Singh B, Han L-L, Shen J-P, Li P-P, Wang G-B, Wu C-F, Ge A-H, Zhang L-M, He J-Z. 2021. Host selection shapes crop microbiome assembly and network complexity. New Phytol 229:1091–1104. doi: 10.1111/nph.16890. [DOI] [PubMed] [Google Scholar]
- 16.Nemergut DR, Schmidt SK, Fukami T, O’Neill SP, Bilinski TM, Stanish LF, Knelman JE, Darcy JL, Lynch RC, Wickey P, Ferrenberg S. 2013. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev 77:342–356. doi: 10.1128/MMBR.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Li D, Qi J, Peng Z, Chen W, Wei G, Jiao S. 2021. Stochastic processes shape the biogeographic variations in core bacterial communities between aerial and belowground compartments of common bean. Environ Microbiol 23:949–964. doi: 10.1111/1462-2920.15227. [DOI] [PubMed] [Google Scholar]
- 18.Ingold C. 1971. Fungal spores: their liberation and dispersal. Clarendon Press, Oxford, UK. [Google Scholar]
- 19.Young KD. 2006. The selective value of bacterial shape. Microbiol Mol Biol Rev 70:660–703. doi: 10.1128/MMBR.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tedersoo L, Bahram M, Põlme S, Kõljalg U, Yorou NS, Wijesundera R, Ruiz LV, Vasco-Palacios AM, Thu PQ, Suija A, Smith ME, Sharp C, Saluveer E, Saitta A, Rosas M, Riit T, Ratkowsky D, Pritsch K, Põldmaa K, Piepenbring M, Phosri C, Peterson M, Parts K, Pärtel K, Otsing E, Nouhra E, Njouonkou AL, Nilsson RH, Morgado LN, Mayor J, May TW, Majuakim L, Lodge DJ, Lee SS, Larsson K-H, Kohout P, Hosaka K, Hiiesalu I, Henkel TW, Harend H, Guo L, Greslebin A, Grelet G, Geml J, Gates G, Dunstan W, Dunk C, Drenkhan R, Dearnaley J, Kesel AD, Dang T, et al. 2014. Global diversity and geography of soil fungi. Science 346:1256688. doi: 10.1126/science.1256688. [DOI] [PubMed] [Google Scholar]
- 21.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, Navas-Molina JA, Janssen S, Kopylova E, Vázquez-Baeza Y, González A, Morton JT, Mirarab S, Zech Xu Z, Jiang L, Haroon MF, Kanbar J, Zhu Q, Jin Song S, Kosciolek T, Bokulich NA, Lefler J, Brislawn CJ, Humphrey G, Owens SM, Hampton-Marcell J, Berg-Lyons D, McKenzie V, Fierer N, Fuhrman JA, Clauset A, Stevens RL, Shade A, Pollard KS, Goodwin KD, Jansson JK, Gilbert JA, Knight R, The Earth Microbiome Project Consortium . 2017. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature Advance Online Publication 551:457–463. doi: 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell JR, Karunaratne S, Campbell CD, Yao H, Robinson L, Singh BK. 2015. Deterministic processes vary during community assembly for ecologically dissimilar taxa. Nat Commun 6:1–10. doi: 10.1038/ncomms9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith SE, Read DJ. 2008. Mycorrhizal Symbiosis, 3rd ed. Academic Press, San Diego, CA. [Google Scholar]
- 24.Cregger M, Carper DL, Christel S, Doktycz M, Labbe J, Michener J, Dove N, Johnston ER, Moore J, Velez J, Morrell-Falvey J, Muchero W, Pelletier D, Retterer S, Tschaplinski T, Tuskan G, Weston D, Schadt C. 2021. Plant-microbe interactions: from genes to ecosystems using Populus as a model system. Phytobiomes J 5:29–38. doi: 10.1094/PBIOMES-01-20-0009-FI. [DOI] [Google Scholar]
- 25.Cooke JEK, Rood SB. 2007. Trees of the people: the growing science of poplars in Canada and worldwide. Can J Bot 85:1103–1110. doi: 10.1139/B07-125. [DOI] [Google Scholar]
- 26.Sannigrahi P, Ragauskas AJ, Tuskan GA. 2010. Poplar as a feedstock for biofuels: a review of compositional characteristics. Biofuels, Bioprod Bioref 4:209–226. doi: 10.1002/bbb.206. [DOI] [Google Scholar]
- 27.Tuskan GA, DiFazio SP, Teichmann T. 2004. Poplar genomics is getting popular: the impact of the Poplar genome project on tree research. Plant Biol (Stuttg) 6:2–4. doi: 10.1055/s-2003-44715. [DOI] [PubMed] [Google Scholar]
- 28.Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen G-L, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, Cunningham R, Davis J, Degroeve S, Déjardin A, dePamphilis C, Detter J, Dirks B, Dubchak I, Duplessis S, Ehlting J, Ellis B, Gendler K, Goodstein D, Gribskov M, Grimwood J, Groover A, Gunter L, Hamberger B, Heinze B, Helariutta Y, et al. 2006. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 29.McKown AD, Klápště J, Guy RD, Geraldes A, Porth I, Hannemann J, Friedmann M, Muchero W, Tuskan GA, Ehlting J, Cronk QCB, El‐Kassaby YA, Mansfield SD, Douglas CJ. 2014. Genome-wide association implicates numerous genes underlying ecological trait variation in natural populations of Populus trichocarpa. New Phytol 203:535–553. doi: 10.1111/nph.12815. [DOI] [PubMed] [Google Scholar]
- 30.Bálint M, Tiffin P, Hallström B, O'Hara RB, Olson MS, Fankhauser JD, Piepenbring M, Schmitt I. 2013. Host genotype shapes the foliar fungal microbiome of balsam poplar (Populus balsamifera). PLoS One 8:e53987. doi: 10.1371/journal.pone.0053987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cregger MA, Veach AM, Yang ZK, Crouch MJ, Vilgalys R, Tuskan GA, Schadt CW. 2018. The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome 6:31. doi: 10.1186/s40168-018-0413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamonts K, Trivedi P, Garg A, Janitz C, Grinyer J, Holford P, Botha FC, Anderson IC, Singh BK. 2018. Field study reveals core plant microbiota and relative importance of their drivers. Environ Microbiol 20:124–140. doi: 10.1111/1462-2920.14031. [DOI] [PubMed] [Google Scholar]
- 33.Trivedi P, Mattupalli C, Eversole K, Leach JE. 2021. Enabling sustainable agriculture through understanding and enhancement of microbiomes. New Phytol 230:2129–2147. doi: 10.1111/nph.17319. [DOI] [PubMed] [Google Scholar]
- 34.Lemanceau P, Blouin M, Muller D, Moënne-Loccoz Y. 2017. Let the core microbiota be functional. Trends Plant Sci 22:583–595. doi: 10.1016/j.tplants.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Yeoh YK, Dennis PG, Paungfoo-Lonhienne C, Weber L, Brackin R, Ragan MA, Schmidt S, Hugenholtz P. 2017. Evolutionary conservation of a core root microbiome across plant phyla along a tropical soil chronosequence. Nat Commun 8:215. doi: 10.1038/s41467-017-00262-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agler MT, Ruhe J, Kroll S, Morhenn C, Kim S-T, Weigel D, Kemen EM. 2016. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol 14:e1002352. doi: 10.1371/journal.pbio.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timm CM, Pelletier DA, Jawdy SS, Gunter LE, Henning JA, Engle N, Aufrecht J, Gee E, Nookaew I, Yang Z, Lu T-Y, Tschaplinski TJ, Doktycz MJ, Tuskan GA, Weston DJ. 2016. Two poplar-associated bacterial isolates induce additive favorable responses in a constructed plant-microbiome system. Front Plant Sci 7:497. doi: 10.3389/fpls.2016.00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ortiz-Álvarez R, Fierer N, de los Ríos A, Casamayor EO, Barberán A. 2018. Consistent changes in the taxonomic structure and functional attributes of bacterial communities during primary succession. ISME J 12:1658–1667. doi: 10.1038/s41396-018-0076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saunders M, Kohn LM. 2009. Evidence for alteration of fungal endophyte community assembly by host defense compounds. New Phytol 182:229–238. doi: 10.1111/j.1469-8137.2008.02746.x. [DOI] [PubMed] [Google Scholar]
- 40.Jost L. 2006. Entropy and diversity. Oikos 113:363–375. doi: 10.1111/j.2006.0030-1299.14714.x. [DOI] [Google Scholar]
- 41.Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, Rockhold ML, Konopka A. 2013. Quantifying community assembly processes and identifying features that impose them. ISME J 7:2069–2079. doi: 10.1038/ismej.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST. 2011. Bayesian community-wide culture-independent microbial source tracking. Nat Methods 8:761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vannette RL, Fukami T. 2017. Dispersal enhances beta diversity in nectar microbes. Ecol Lett 20:901–910. doi: 10.1111/ele.12787. [DOI] [PubMed] [Google Scholar]
- 44.Toju H, Vannette RL, Gauthier M-PL, Dhami MK, Fukami T. 2018. Priority effects can persist across floral generations in nectar microbial metacommunities. Oikos 127:345–352. doi: 10.1111/oik.04243. [DOI] [Google Scholar]
- 45.Kinkel LL, Andrews JH, Berbee FM, Nordheim EV. 1987. Leaves as islands for microbes. Oecologia 71:405–408. doi: 10.1007/BF00378714. [DOI] [PubMed] [Google Scholar]
- 46.Meyer KM, Leveau JHJ. 2012. Microbiology of the phyllosphere: a playground for testing ecological concepts. Oecologia 168:621–629. doi: 10.1007/s00442-011-2138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Landhäusser SM, Lieffers VJ. 2003. Seasonal changes in carbohydrate reserves in mature northern Populus tremuloides clones. Trees 17:471–476. doi: 10.1007/s00468-003-0263-1. [DOI] [Google Scholar]
- 48.Cooke JEK, Weih M. 2005. Nitrogen storage and seasonal nitrogen cycling in Populus: bridging molecular physiology and ecophysiology. New Phytol 167:19–30. doi: 10.1111/j.1469-8137.2005.01451.x. [DOI] [PubMed] [Google Scholar]
- 49.Ding T, Melcher U. 2016. Influences of plant species, season and location on leaf endophytic bacterial communities of non-cultivated plants. PLoS One 11:e0150895. doi: 10.1371/journal.pone.0150895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi S, Nuccio E, Herman DJ, Rijkers R, Estera K, Li J, da Rocha UN, He Z, Pett-Ridge J, Brodie EL, Zhou J, Firestone M. 2015. Successional trajectories of rhizosphere bacterial communities over consecutive seasons. mBio 6:e00746-15. doi: 10.1128/mBio.00746-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhalnina K, Louie KB, Hao Z, Mansoori N, da Rocha UN, Shi S, Cho H, Karaoz U, Loqué D, Bowen BP, Firestone MK, Northen TR, Brodie EL. 2018. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3:470–480. doi: 10.1038/s41564-018-0129-3. [DOI] [PubMed] [Google Scholar]
- 52.Xu J, Zhang Y, Zhang P, Trivedi P, Riera N, Wang Y, Liu X, Fan G, Tang J, Coletta-Filho HD, Cubero J, Deng X, Ancona V, Lu Z, Zhong B, Roper MC, Capote N, Catara V, Pietersen G, Vernière C, Al-Sadi AM, Li L, Yang F, Xu X, Wang J, Yang H, Jin T, Wang N. 2018. The structure and function of the global citrus rhizosphere microbiome. Nat Commun 9:4894. doi: 10.1038/s41467-018-07343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oliverio AM, Bradford MA, Fierer N. 2017. Identifying the microbial taxa that consistently respond to soil warming across time and space. Glob Chang Biol 23:2117–2129. doi: 10.1111/gcb.13557. [DOI] [PubMed] [Google Scholar]
- 54.Curiel Yuste J, Baldocchi DD, Gershenson A, Goldstein A, Misson L, Wong S. 2007. Microbial soil respiration and its dependency on carbon inputs, soil temperature and moisture. Global Change Biol 13:2018–2035. doi: 10.1111/j.1365-2486.2007.01415.x. [DOI] [Google Scholar]
- 55.Rustad L, Campbell J, Marion G, Norby R, Mitchell M, Hartley A, Cornelissen J, Gurevitch J, GCTE-NEWS . 2001. A meta-analysis of the response of soil respiration, net nitrogen mineralization, and aboveground plant growth to experimental ecosystem warming. Oecologia 126:543–562. doi: 10.1007/s004420000544. [DOI] [PubMed] [Google Scholar]
- 56.Melillo JM, Butler S, Johnson J, Mohan J, Steudler P, Lux H, Burrows E, Bowles F, Smith R, Scott L, Vario C, Hill T, Burton A, Zhou Y-M, Tang J. 2011. Soil warming, carbon-nitrogen interactions, and forest carbon budgets. Proc Natl Acad Sci U S A 108:9508–9512. doi: 10.1073/pnas.1018189108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N. 2013. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J 7:1493–1506. doi: 10.1038/ismej.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferrenberg S, O’Neill SP, Knelman JE, Todd B, Duggan S, Bradley D, Robinson T, Schmidt SK, Townsend AR, Williams MW, Cleveland CC, Melbourne BA, Jiang L, Nemergut DR. 2013. Changes in assembly processes in soil bacterial communities following a wildfire disturbance. ISME J 7:1102–1111. doi: 10.1038/ismej.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dini-Andreote F, Stegen JC, van Elsas JD, Salles JF. 2015. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci U S A 112:E1326–E1332. doi: 10.1073/pnas.1414261112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao C, Montoya L, Xu L, Madera M, Hollingsworth J, Purdom E, Singan V, Vogel J, Hutmacher RB, Dahlberg JA, Coleman-Derr D, Lemaux PG, Taylor JW. 2020. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat Commun 11:34. doi: 10.1038/s41467-019-13913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morella NM, Weng FC-H, Joubert PM, Metcalf CJE, Lindow S, Koskella B. 2019. Successive passaging of a plant-associated microbiome reveals robust habitat and host genotype-dependent selection. Proc Natl Acad Sci U S A 117:1148–1159. 10.1073/pnas.1908600116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen F, Liu C-J, Tschaplinski TJ, Zhao N. 2009. Genomics of secondary metabolism in Populus: interactions with biotic and abiotic environments. Critical Rev in Plant Sciences 28:375–392. doi: 10.1080/07352680903241279. [DOI] [Google Scholar]
- 63.Tschaplinski TJ, Plett JM, Engle NL, Deveau A, Cushman KC, Martin MZ, Doktycz MJ, Tuskan GA, Brun A, Kohler A, Martin F. 2014. Populus trichocarpa and Populus deltoides exhibit different metabolomic responses to colonization by the symbiotic fungus Laccaria bicolor. Mol Plant Microbe Interact 27:546–556. doi: 10.1094/MPMI-09-13-0286-R. [DOI] [PubMed] [Google Scholar]
- 64.Veach AM, Chen H, Yang ZK, Labbe AD, Engle NL, Tschaplinski TJ, Schadt CW, Cregger MA. 2020. Plant hosts modify belowground microbial community response to extreme drought. mSystems 5:e00092-20. doi: 10.1128/mSystems.00092-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muchero W, Sondreli KL, Chen J-G, Urbanowicz BR, Zhang J, Singan V, Yang Y, Brueggeman RS, Franco-Coronado J, Abraham N, Yang J-Y, Moremen KW, Weisberg AJ, Chang JH, Lindquist E, Barry K, Ranjan P, Jawdy S, Schmutz J, Tuskan GA, LeBoldus JM. 2018. Association mapping, transcriptomics, and transient expression identify candidate genes mediating plant–pathogen interactions in a tree. Proc Natl Acad Sci U S A 115:11573–11578. doi: 10.1073/pnas.1804428115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ostry ME, McNabb HS. 1985. Susceptibility of Populus species and hybrids to disease in the north central United States. Plant Dis 69:755–757. doi: 10.1094/PD-69-755. [DOI] [Google Scholar]
- 67.Ginnan NA, Dang T, Bodaghi S, Ruegger PM, McCollum G, England G, Vidalakis G, Borneman J, Rolshausen PE, Roper MC. 2020. Disease-induced microbial shifts in citrus indicate microbiome-derived responses to huanglongbing across the disease severity spectrum. Phytobiomes J 4:375–387. doi: 10.1094/PBIOMES-04-20-0027-R. [DOI] [Google Scholar]
- 68.Teste FP, Jones MD, Dickie IA. 2020. Dual-mycorrhizal plants: their ecology and relevance. New Phytol 225:1835–1851. doi: 10.1111/nph.16190. [DOI] [PubMed] [Google Scholar]
- 69.Knief C, Ramette A, Frances L, Alonso-Blanco C, Vorholt JA. 2010. Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J 4:719–728. doi: 10.1038/ismej.2010.9. [DOI] [PubMed] [Google Scholar]
- 70.Bodenhausen N, Horton MW, Bergelson J. 2013. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS One 8:e56329. doi: 10.1371/journal.pone.0056329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lindow SE, Brandl MT. 2003. Microbiology of the phyllosphere. Appl Environ Microbiol 69:1875–1883. doi: 10.1128/aem.69.4.1875-1883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barret M, Briand M, Bonneau S, Préveaux A, Valière S, Bouchez O, Hunault G, Simoneau P, Jacques M-A. 2015. Emergence shapes the structure of the seed microbiota. Appl Environ Microbiol 81:1257–1266. doi: 10.1128/AEM.03722-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frank AC, Saldierna Guzmán JP, Shay JE. 2017. Transmission of bacterial endophytes. Microorganisms 5:70. doi: 10.3390/microorganisms5040070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCartney HA, Fitt BDL, Schmechel D. 1997. Sampling bioaerosols in plant pathology. J Aerosol Science 28:349–364. doi: 10.1016/S0021-8502(96)00438-7. [DOI] [Google Scholar]
- 75.Burrows SM, Butler T, Jöckel P, Tost H, Kerkweg A, Pöschl U, Lawrence MG. 2009. Bacteria in the global atmosphere. Part 2. Modeling of emissions and transport between different ecosystems. Atmos Chem Phys 9:9281–9297. doi: 10.5194/acp-9-9281-2009. [DOI] [Google Scholar]
- 76.Dini-Andreote F, Raaijmakers JM. 2018. Embracing community ecology in plant microbiome research. Trends Plant Sci 23:467–469. doi: 10.1016/j.tplants.2018.03.013. [DOI] [PubMed] [Google Scholar]
- 77.Owens VN. 2018. Sun Grant/DOE Regional Feedstock Partnership: final technical report. DOE-SDSU-85041. South Dakota State University, Brookings, SD, USA. [Google Scholar]
- 78.Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL. 2013. Practical innovations for high-throughput amplicon sequencing. Nat Methods 10:999–1002. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- 79.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 80.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Silva RD, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, et al. 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abarenkov K, Nilsson RH, Larsson K-H, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Ursing BM, Vrålstad T, Liimatainen K, Peintner U, Kõljalg U. 2010. The UNITE database for molecular identification of fungi: recent updates and future perspectives. New Phytol 186:281–285. doi: 10.1111/j.1469-8137.2009.03160.x. [DOI] [PubMed] [Google Scholar]
- 84.Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, Schilling JS, Kennedy PG. 2016. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecology 20:241–248. doi: 10.1016/j.funeco.2015.06.006. [DOI] [Google Scholar]
- 85.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2: approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fouquier J, Rideout JR, Bolyen E, Chase J, Shiffer A, McDonald D, Knight R, Caporaso JG, Kelley ST. 2016. ghost-tree: creating hybrid-gene phylogenetic trees for diversity analyses. Microbiome 4:11. doi: 10.1186/s40168-016-0153-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.R Development Core Team. 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Australia. [Google Scholar]
- 89.Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO. 2010. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26:1463–1464. doi: 10.1093/bioinformatics/btq166. [DOI] [PubMed] [Google Scholar]
- 90.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oksanen J, Blanchet FG, Kindt R, Legendre P, Simpson GL, Minchin PR, O’Hara RB, Solymos P, Stevens MHH, Wagner H. 2013. vegan: community ecology package. http://CRAN.R-project.org/package=vegan.
- 92.Anderson MJ, Walsh DCI. 2013. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecological Monographs 83:557–574. doi: 10.1890/12-2010.1. [DOI] [Google Scholar]
- 93.Fine PVA, Kembel SW. 2011. Phylogenetic community structure and phylogenetic turnover across space and edaphic gradients in western Amazonian tree communities. Ecography 34:552–565. doi: 10.1111/j.1600-0587.2010.06548.x. [DOI] [Google Scholar]
- 94.Chase JM, Kraft NJB, Smith KG, Vellend M, Inouye BD. 2011. Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2:art24. doi: 10.1890/ES10-00117.1. [DOI] [Google Scholar]
- 95.Stegen JC, Lin X, Fredrickson JK, Konopka AE. 2015. Estimating and mapping ecological processes influencing microbial community assembly. Front Microbiol 6:370. doi: 10.3389/fmicb.2015.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets presented in this study can be found in the Sequence Read Archive under BioProject number 685817. The R code for all statistics and figures as well as the final ASV, taxonomy, and sample data tables used in this analysis can be found at https://github.com/nicholascdove/Populus_microbiome_assembly.