

Abstract
We report the synthesis and photochemical and biological characterization of the first selective and potent metal-based inhibitors of cytochrome P450 3A4 (CYP3A4), the major human drug metabolizing enzyme. Five Ru(II)-based derivatives were prepared from two analogs of the CYP3A4 inhibitor ritonavir, 4 and 6: [Ru(tpy)(L)(6)]Cl2 (tpy = 2,2′:6′,2″-terpyridine) with L = 6,6′-dimethyl-2,2′-bipyridine (Me2bpy; 8), dimethylbenzo[i]dipyrido[3,2-a:2′,3′-c]phenazine (Me2dppn; 10) and 3,6-dimethyl-10,15-diphenylbenzo[i]dipyrido[3,2-a:2′,3′-c]phenazine (Me2Ph2dppn; 11), [Ru(tpy)(Me2bpy)(4)]Cl2 (7) and [Ru(tpy)(Me2dppn)(4)]Cl2 (9). Photochemical release of 4 or 6 from 7–11 was demonstrated, and the spectrophotometric evaluation of 7 showed that it behaves similarly to free 4 (type II heme ligation) after irradiation with visible light but not in the dark. Unexpectedly, the intact Ru(II) complexes 7 and 8 were found to inhibit CYP3A4 potently and specifically through direct binding to the active site without heme ligation. Caged inhibitors 9–11 showed dual action properties by combining photoactivated dissociation of 4 or 6 with efficient 1O2 production. In prostate adenocarcinoma DU-145 cells, compound 9 had the best synergistic effect with vinblastine, the anticancer drug primarily metabolized by CYP3A4 in vivo. Thus, our study establishes a new paradigm in CYP inhibition using metalated complexes and suggests possible utilization of photoactive CYP3A4 inhibitory compounds in clinical applications, such as enhancement of therapeutic efficacy of anticancer drugs.
Graphical Abstract
INTRODUCTION
Cytochrome P450s (CYPs) are heme-containing enzymes that play a crucial role in biosynthesis and metabolism. In addition to their activity in the liver, CYPs perform biosynthetic processing and drug oxidation in many other tissues, including the gastrointestinal tract and the brain. Extrahepatic CYP activity reduces local drug bioavailability and fuels resistance and progression of diseases, such as cancer, making CYPs attractive drug targets. Better understanding of the CYP inhibitory mechanism can also help lower the risk of dangerous drug–drug interactions. Genetic diversity of human CYPs leads to pharmacokinetic differences between people of different ethnic backgrounds that make drug responses highly varied. As a result, thorough characterization of small molecule interactions with CYPs is essential; in combination with genetic sequencing, these data will one day lead to better designed and personalized therapies.1
CYP3A4 is the most abundant liver and intestinal P450 isoform that oxidizes the majority of administered drugs and other xenobiotics relevant to human health.2–9 Fast and overly extensive drug metabolism can reduce treatment efficacy by requiring higher doses to achieve the full therapeutic effect. One way to overcome fast drug metabolism is the inhibition of CYP3A4. Currently, two CYP3A4 inhibitors, ritonavir and cobicistat, are part of multidrug therapies for treating HIV and hepatitis C virus (HCV) infections, whereas ketoconazole is co-prescribed with the quickly metabolized immunosuppressants in organ transplant patients.10–14 Anticancer therapy is another field where targeted CYP3A4 inhibition holds promise. CYP3A4 clears various types of anticancer drugs via both intestinal/hepatic metabolism and enhanced expression/in situ metabolism in solid tumors.15–19 Targeted inhibition of CYP3A4 in tumors has been identified as a potential solution to improve efficacy of chemotherapy by restoring sensitivity of cancer cells.19,20 Since most anticancer drugs have a narrow therapeutic index, potent CYP3A4 inhibition (as part of drug cocktails) has great potential to improve outcomes, lower chemotherapeutic doses, and minimize adverse effects. Importantly, clinicians have already identified an urgent need for localized CYP3A4 inhibition in malignant tissues.21 Localized inhibition was postulated to be more effective than systemic inhibition in colorectal cancer because a widely prescribed class of chemotherapeutics that destabilize micro-tubules are metabolized by CYP3A4 in cancer cells but by other CYPs in the liver.21 Importantly, there are no current methods that achieve tissue-specific blockade of CYP activity. Moreover, unlike the thousands of organic small molecules characterized as CYP inhibitors, inducers, or substrates, only a small handful of metal complexes have been investigated for CYP targeting.22–24
With the potential benefits in mind, we identified photocaging as a viable strategy to achieve localized CYP inhibition. Photocaging is a powerful method for blocking the action of biologically active molecules and unleashing inhibitory compounds within desired tissues, through which highly controlled and localized CYP inhibition can be achieved.22,23 Toward this goal, Ru(II)-based photocaging can facilitate small molecule release in a noninvasive manner to provide spatial and temporal control over biological activity.25–27 Photocaging has been exploited in basic research and for drug activation during photochemotherapy (PCT),28–30 with recent in vivo validation of Ru(II)-PCT.31 In addition to PCT, Ru(II) complexes show attractive properties for photodynamic therapy (PDT) applications, including high stability and cell permeability,32,33 low inherent toxicity,34–37 and higher light-to-dark ratios for cell death compared to clinically approved PDT compounds.29,38 Due to their rich photochemistry and resistance to photobleaching,39 a common problem with current organic photosensitizers,40 ruthenium complexes are emerging as a promising new class of PDT agents,29,41–43 some of which have advanced to clinical trials.44–47 One recent example is the Ru(II) photosensitizer TLD-1433, which is currently in phase II clinical trials for the treatment of bladder cancer.48–50
Many small molecules that target CYPs contain N-donor heterocycles that coordinate to the heme iron in the active site (type II ligation) to create strong and stable enzyme–inhibitor complexes.51–53 Ru(II) photocaging is an effective strategy for blocking N-donor heterocycles from binding to their targets, including the hemes found in CYP enzymes, via strong and stable coordination between N-donors, such as imidazoyl and pyridyl groups, and the Ru(II) centers of the photocages.26 Examples include the photochemical release of the CYP17A1 inhibitor abiraterone in PC3 prostate adenocarcinoma cancer cells,23 CYP11B1 inhibitors metyrapone and etomidate caged with the Ru(bpy)2 (bpy = 2,2′-bipyridine) fragment,22 and photocaged analogs of the pan-P450 inhibitor econazole that function as photoactivated cytotoxic and emissive agents in DLD-1 colon adenocarcinoma cancer cells.24
Herein, we report the design, synthesis, and biochemical characterization of a series of photocaged CYP3A4 inhibitors. Compounds were designed as Ru(II)-caged analogs of the antiretroviral drug ritonavir,54 which is a CYP3A4 inhibitor that binds tightly to the heme iron center via its thiazole ring.51,53 Two types of Ru(II) photocaging groups were employed that show either single action PCT or dual action PCT/PDT behaviors. All compounds were highly stable in solution in the dark but released CYP3A4 inhibitors readily upon irradiation with visible light, enabling type II heme iron ligation. While the main goal of the project was to design and employ light-activated CYP inhibitory molecules, one unexpected and significant finding was that, even without light activation, some Ru(II) compounds could potently inhibit CYP3A4 by binding to the active site without heme ligation. A direct inhibitory action between a large metal complex and a CYP target was verified by X-ray crystallography. Finally, we report that photocaged CYP3A4 inhibitors can function as dual action PDT and PCT agents that can both generate 1O2 and release the inhibitor upon irradiation, respectively. It is shown that these compounds work synergistically with the microtubule-destabilizing drug vinblastine, primarily metabolized by CYP3A4 in vivo. Thus, this work establishes a new paradigm in CYP inhibition and raises the possibility that photoactive CYP3A4 inhibitory compounds can be utilized in clinical applications, such as enhancement of therapeutic efficacy of anticancer drugs.
RESULTS AND DISCUSSION
Compound Design and Synthesis.
To begin our studies, we surveyed the literature for known type II inhibitors of CYP3A4. Clinical examples include ketoconazole (1), fluconazole (2), and ritonavir (3) that contain imidazole, triazole, or thiazole N-donors, respectively (Figure 1).51,55,56 Instead, we chose to focus our efforts on CYP3A4 inhibitors containing pyridyl groups that are analogs of ritonavir (4–6).57–59 Pyridine-containing compounds show more favorable properties for Ru(II) photocaging than other heterocyclic compounds, including strong and stable binding to Ru(II) in the dark and facile release when irradiated with low-energy light.23,60–62 Compounds 4–6 inhibit CYP3A4 in the low μM to nM range in in vitro assays with a fluorogenic substrate (vide infra) and, as verified by spectroscopic and X-ray crystallography analyses, inhibit CYP3A4 by ligating directly to the heme iron via the pyridine nitrogen.57–59 Analogs 4 and 6 were chosen over 5, which showed the lowest IC50 value of the series (90 nM) but had the potential to create solubility problems in Ru(II)-caged complexes due to its hydrophobic nature. Compound 4 was obtained using a modified three-step synthetic route that used trityl protection of 3-thiopropanoic acid (Scheme S1).57 Compound 6 was synthesized from S-2-mercapto-3-phenylpropanoic acid63 following a literature protocol.58
Figure 1.
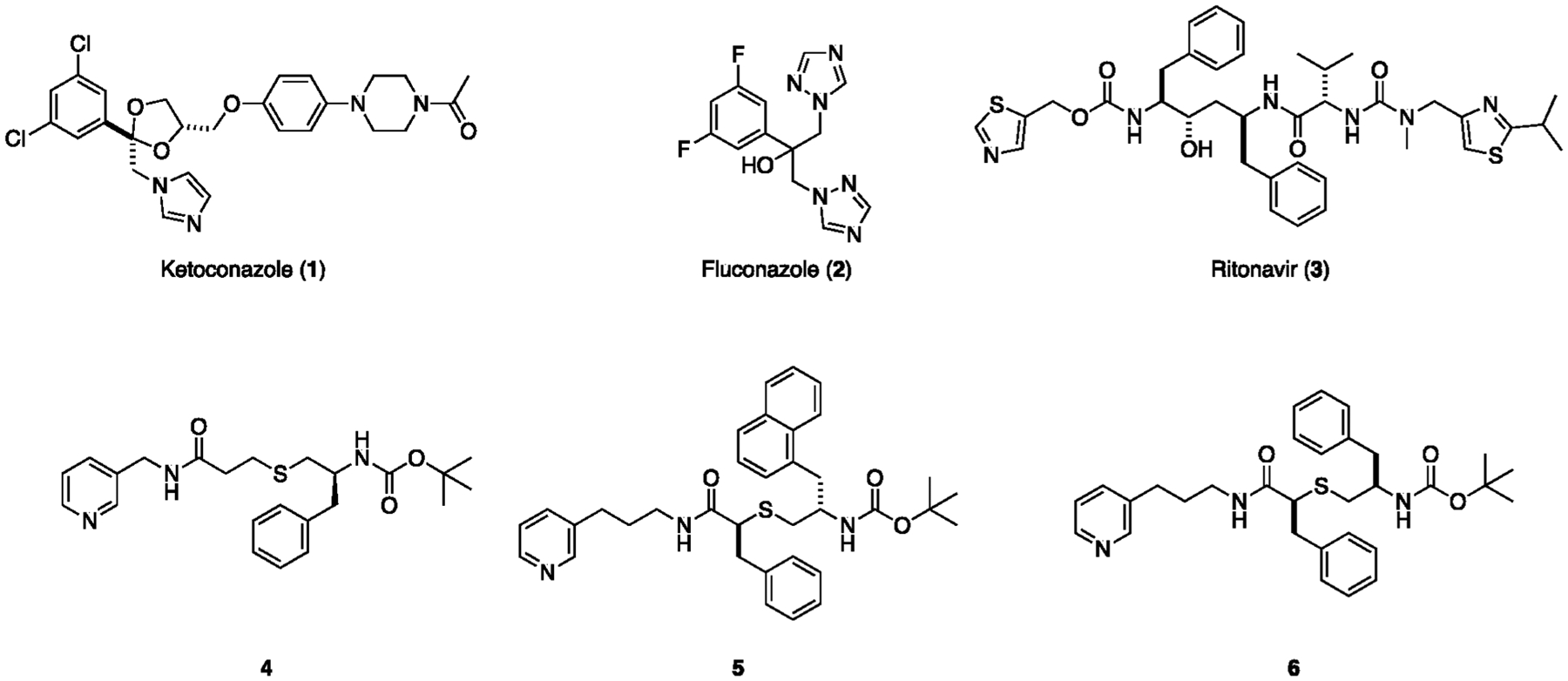
Structures of type II heme-binding CYP3A4 inhibitors including ritonavir and related analogs 4–6.
Five Ru(II) complexes containing the caged analogs of CYP3A4 inhibitors 4 and 6 were prepared as shown in Scheme 1. Complexes 7 and 8, coupled to the [Ru(tpy)(Me2bpy)] fragment as the caging group, were designed to demonstrate single action PCT behavior, similar to the pyridine model complex [Ru(tpy)(Me2bpy)(py)](PF6)2,60 as well as caged inhibitors of cysteine proteases64,65 and CYP17A123 reported by us in prior studies. Analogs 9–11, containing the [Ru(tpy)(L)] fragments as the photocaging groups, where L = dimethylbenzo[i]dipyrido[3,2-a:2′,3′-c]phenazine (Me2dppn) and 3,6-dimethyl-10,15-diphenylbenzo[i]dipyrido-[3,2-a:2′,3′-c]phenazine (Me2Ph2dppn), were synthesized to provide dual action PCT/PDT capabilities. The reaction of 4 or 6 with [Ru(tpy)(Me2bpy)(Cl)]Cl66 in a 1:1 mixture of EtOH and H2O at 80 °C gave the photocaged inhibitors 7 and 8 in 75% and 63% yield, respectively, after chromatography over alumina. Complexes 9–11 were obtained by treating 4 or 6 with [Ru(tpy)(Me2dppn)Cl]Cl67 or [Ru(tpy)(Ph2Me2dppn)Cl]Cl68 in a 1:1 mixture of EtOH and H2O at 80 °C in 48–61% yield after chromatography over alumina. The ligands Me2dppn and Me2Ph2dppn found in complexes 9–11 were included to promote ligand dissociation (PCT) from the triplet ligand field (3LF) state(s) and (1O2) generation (PDT) from the dppn-centered 3ππ* excited state(s). Importantly, we were motivated to use these ligands because our prior studies confirmed that dual action PCT/PDT behavior was necessary to achieve efficient death of triple negative breast cancer cells in 3D pathomimetic assays.65
Scheme 1.
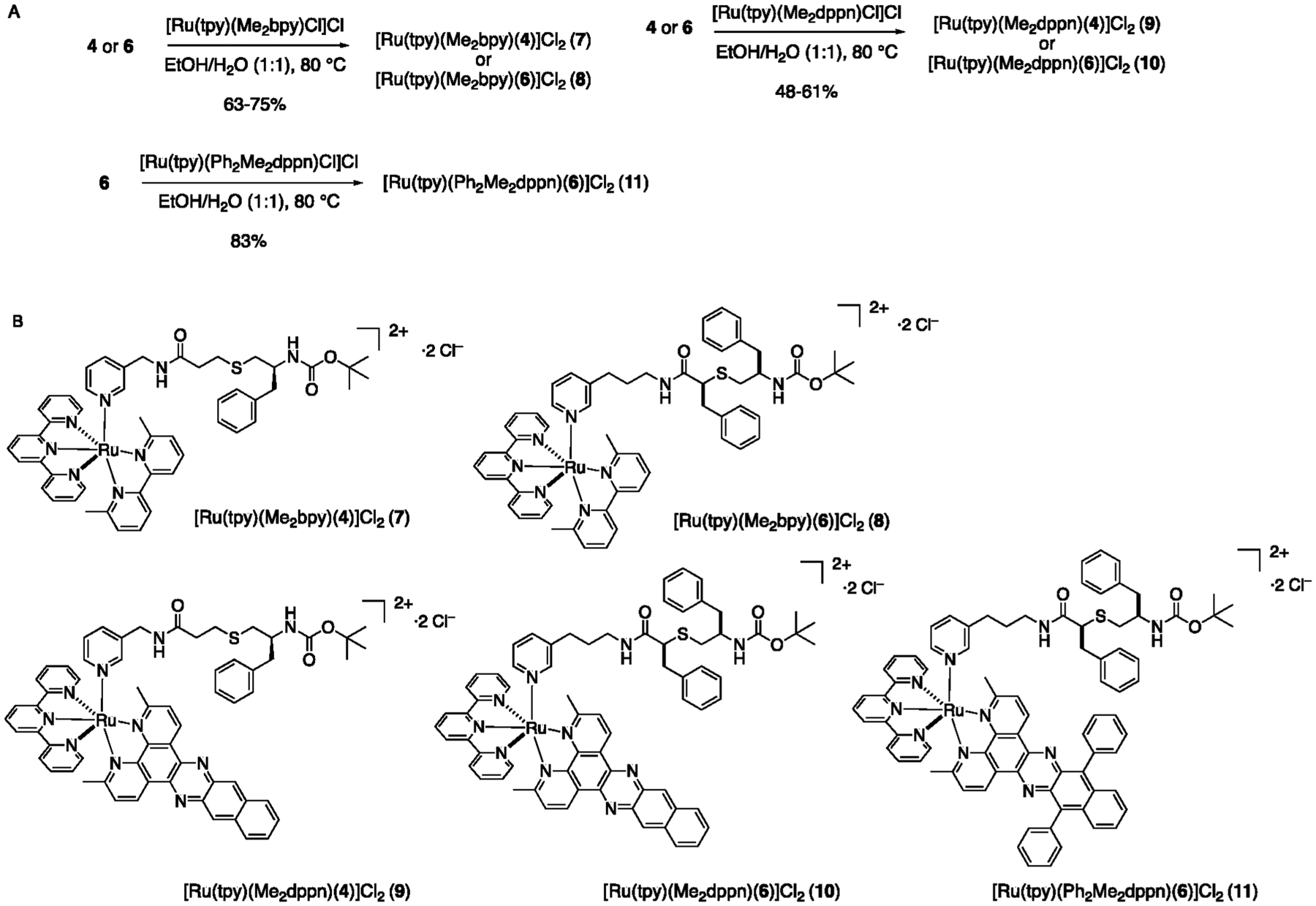
Synthesis (A) and Structures (B) of Ru(II)-Caged CYP3A4 Inhibitors 7–11
Complexes 7–11 were characterized by multiple methods, including electronic absorption, 1H NMR, COSY, and IR spectroscopies and electrospray ionization mass spectrometry (ESI-MS). The electronic absorption spectra of 7 and 8 exhibit maxima at 474 nm (ε = 7700 M−1 cm−1) and 470 nm (ε = 9700 M−1 cm−1), respectively, that are in good agreement with the corresponding pyridine model complex [Ru(tpy)(Me2bpy)(py)](PF6)2.60 Likewise, the electronic absorption spectra of 9 (λmax 485 nm, ε = 13 500 M−1 cm−1) and 10 (λmax 480 nm, ε = 12 000 M−1 cm−1) show maxima consistent with [Ru(tpy)(Me2dppn)(py)](PF6)2.69 The electronic absorption spectrum of 11 exhibits a maximum at 491 nm (ε = 13 500 M−1 cm−1) that is slightly red-shifted compared to those of 9 and 10, which agrees well with data for [Ru(tpy)(Ph2Me2dppn)(py)](PF6)2.68 NMR spectra of complexes 7–11 show resonances ranging from 10 to 1 ppm that are consistent with the presence of the Ru(II)-caging groups, as well as peaks that are attributed to inhibitors 4 and 6 present in these structures. In particular, spectra for complexes 7–11 show singlets in the region of 2.5–1.0 ppm that are consistent with the two diasterotopic methyl groups present in the ligands Me2bpy, Me2dppn, and Ph2Me2dppn. Methyl groups on the same face of the Ru(tpy) plane as the monodentate pyridyl ring are shifted upfield by ~0.7 ppm relative to resonances below that plane due to the shielding effect of the pyridyl ring; these shifts are similar to other photocaged complexes we have characterized in the past.23,64,65 Mass spectra of the photocaged complexes show major peaks with suitable isotope patterns with m/z values consistent with that expected for parent molecular dications [Ru(tpy)(Me2bpy)(4)]2+ (7, m/z = 474) and [Ru(tpy)(Me2bpy)(6)]2+(8, m/z = 526) and the monocations ([Ru(tpy)(Me2dppn)(4)]Cl)+ (9, m/z = 1159), ([Ru(tpy)(Me2dppn)(6)]Cl)+ (10, m/z = 1263), and ([Ru(tpy)(Ph2Me2dppn)(6)]Cl)+ (11, m/z = 1415). Taken together, these data are consistent with the structural assignments shown in Scheme 1.
Photochemistry.
The irradiation of 7 effectively liberates 4, resulting in ligand exchange with a solvent molecule, generating the corresponding [Ru(tpy)(Me2bpy)(L)]2+ (L = H2O or CH3CN) product in H2O or CH3CN, respectively, under N2 atmosphere. Photoactivated ligand exchange (λirr = 500 nm) of 7, with absorption maximum at 474 nm, results in a blue shift to 450 nm in CH3CN (Figure 2A) and a red shift to 495 nm in H2O (Figure 2B). The resulting absorption maxima are consistent with the formation of the corresponding product with a coordinated CH3CN or H2O molecule.23,60,70 Similarly, the irradiation of 8 with 500 nm light in CH3CN resulted in a decrease in 470 nm absorption and a concomitant increase at 455 nm. This hypsochromic shift in the metal-to-ligand charge transfer (MLCT) band is consistent with the substitution of 6 coordinated to the Ru(II) metal through a pyridine unit for a CH3CN solvent molecule (Figure S8).23,70 The presence of an isosbestic point at 463 nm indicates the formation of a single photoproduct, [Ru(tpy)(Me2bpy)(CH3CN)]2+. Comparable changes in the electronic absorption spectra of 9–11 are observed under similar experimental conditions (Figures S9–S11).
Figure 2.
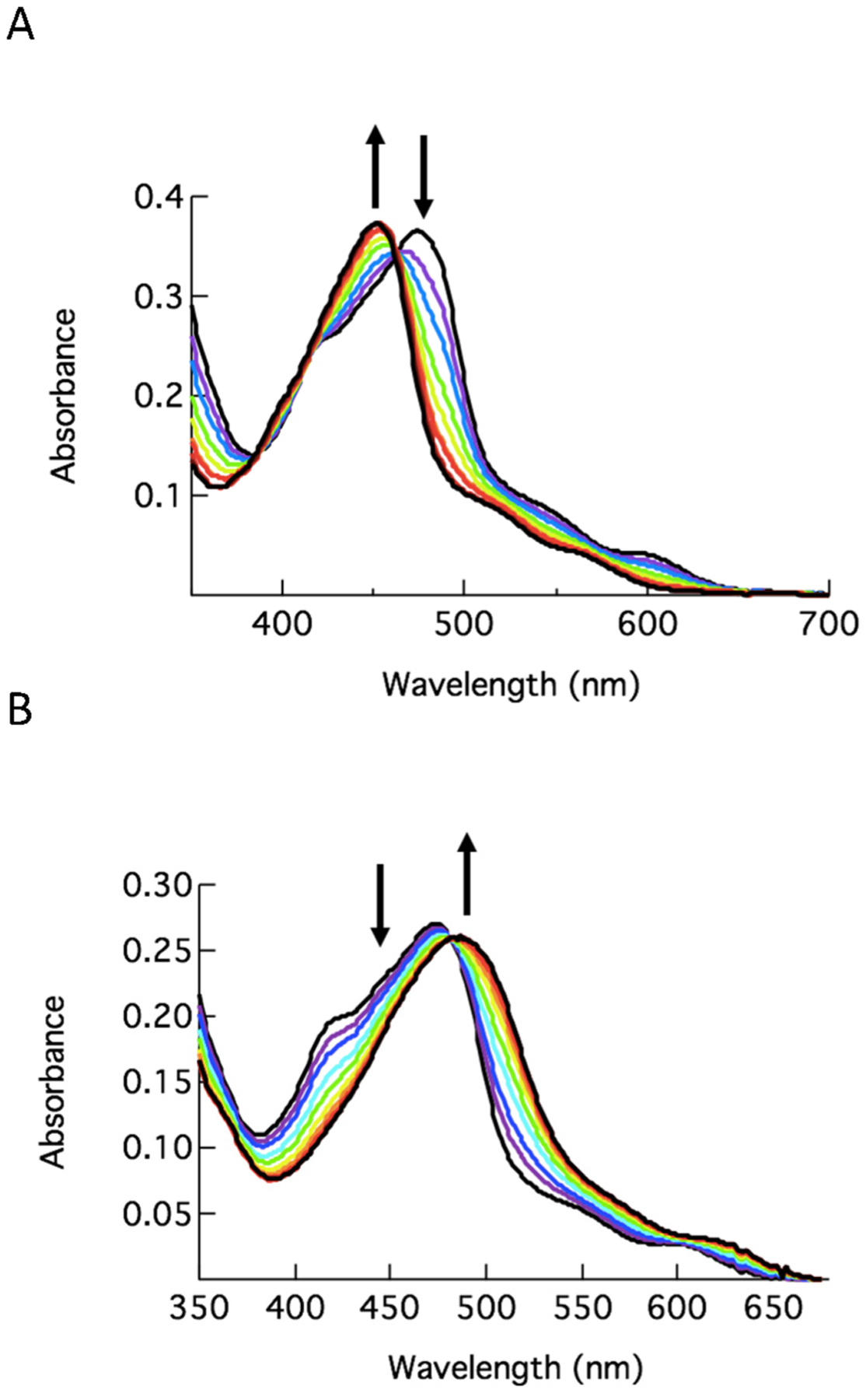
Changes to the electronic absorption spectra of 7 as a function of irradiation time (λirr = 500 nm) in CH3CN for 0–12 min (A) and in H2O for 0–20 min (B) under N2 atmosphere.
The quantum yields (ΦLE) for the ligand exchange with a solvent molecule for 7–11 are listed in Table 1. For 7, ΦLE values of 0.15(1) in H2O and 0.31(1) in CH3CN were measured upon 500 nm irradiation (Table 1). The value in H2O is lower than that observed for [Ru(tpy)(Me2bpy)(py)]2+, ΦLE = 0.41(2), but similar in CH3CN, ΦLE = 0.33(1).60 The lower quantum yield observed for 7 vs Ru(tpy)(Me2bpy)(py)]2+ in H2O can be attributed to the lower solubility of CYP3A4 inhibitor 4 in water as compared to pyridine, which reduces the ability of the former to escape the solvent cage upon release from Ru(II). Similarly, Table 1 reveals a ΦLE value lower for 8 relative to 7 in CH3CN, which likely arises from the larger size and poorer solubility of inhibitor 6 as compared to 4. Following the same trend as 7 and 8, complex 9 containing the CYP3A4 inhibitor 4 showed ~2-fold more efficient photorelease than its analog 10 containing the bulky inhibitor 6. Complex 11 showed the most efficient photorelease in the 9–11 subseries, which is consistent with our earlier observations showing that complexes containing arylated Me2dppn derivatives, such as Ph2Me2dppn, undergo more efficient photorelease than Me2dppn derivatives.68 Complex 7 exhibits the highest ligand exchange quantum yield of the five complexes. It is hypothesized that the initially populated 1MLCT excited state intersystem crosses to the triplet manifold, populating both the lowest-energy dppn 3ππ* state and the 3LF states in 9–11, and the population of the latter results in ligand dissociation. The absence of a lowest-energy long-lived dppn-centered 3ππ* excited state in 7 and 8 precludes the bifurcation of intersystem crossing, resulting in an increased population of the 3LF state and, consequently, greater photoinduced ligand exchange quantum yield as compared to 9–11.69,71
Table 1.
Quantum Yields of Ligand Exchange (ΦLE) and Singlet Oxygen (ΦΔ) Production for 7–11
| complex | ΦLEa | ΦΔb |
|---|---|---|
| 7 | 0.15(1)c | |
| 7 | 0.31(1) | |
| 8 | 0.13(2) | |
| 9 | 0.024(4) | 0.59(6) |
| 10 | 0.014(3) | 0.57(6) |
| 11 | 0.061(8) | 0.80(7) |
| [Ru(tpy)(Me2dppn)(L)]2+ d | 0.073(1) | 0.57(7) |
In CH3CN, λirr = 500 nm, N2 atmosphere.
In MeOH, λirr = 460 nm, determined with diphenylisobenzofuran (DPBF) 1O2 probe.
In H2O, λirr = 500 nm.
From ref 72; L = imatinib.
In addition to photosubstitution of the monodentate ligand, 9–11 produce cytotoxic 1O2 through the population of the lowest-energy, long-lived 3ππ* excited state upon irradiation. The quantum yields for 1O2 production, ΦΔ, by 9–11 of 0.59(6), 0.57(6), and 0.80(7), respectively, are comparable to those of other dual-activity complexes possessing dppn ligands, such as [Ru(tpy)(Ph2Me2dppn)(py)](PF6)268 and [Ru(tpy)(Me2dppn)(imatinib)]2+ (Table 1).70,72 Our prior studies established that the Ru(II) photocaging group [Ru(tpy)(Me2bpy)] found in 7 and 8 does not generate 1O2 either before or after photorelease because its excited state lifetime is too short to undergo bimolecular reactions, as is the case with other Ru(II) complexes containing the tpy ligand or those that undergo facile ligand photodissociation.69,70
The stability of 7–11 was assessed in cell growth medium at 37 °C as previously described.73,74 No spectral changes were observed for 7–9 in the dark (Figures S18–S24) over a course of 24 h, consistent with the exceptional stability of Ru(II)-caged aromatic heterocycles. Complexes 10 and 11 did show some spectral changes over the 24 h period that are consistent with compound precipitation from solution and/or thermal ligand dissociation.
CYP3A4 Inhibition Studies.
After establishing that CYP3A4 inhibitor 4 is photochemically released from its Ru(II) cage 7, the complex was evaluated against the purified CYP3A4 enzyme under dark conditions and upon irradiation. Stock solutions of 7 were left in the dark or exposed to light (λirr = 400–700 nm, tirr = 40 min) before titrating against soluble CYP3A4 (residues 3–22 deleted). Heme binding to the iron center in CYP3A4 was monitored via electronic absorption spectroscopy. Data indicated that the caged inhibitor 7 effectively released 4 from the ruthenium center upon irradiation with visible light, allowing the pyridine functional group of 4 to bind to CYP3A4 via a type II heme ligation. The difference spectra were similar to those obtained for the free inhibitor 4 and showed an increase in intensity at 427 nm and a decrease at 407 nm, consistent with type II binding (Figure 3A), where the water ligand is substituted with pyridine, converting the heme center to a low spin ferric state. Hyperbolic fitting to the titration plot resulted in Kd = 340 nM for 7 under irradiation (Figure 3B). In contrast, no spectral evidence for type II binding was observed during titration of CYP3A4 with 7 under dark conditions. Minor perturbations to the absorption spectra were attributed to the Ru(II) complex, strongly absorbing at 400–500 nm, rather than type II binding (Figure 3C). Similarly, the titration of CYP3A4 with a control compound, [Ru(tpy)(Me2bpy)(Cl)]Cl, led to minor spectral changes. Taken together, these data indicate that type II heme binding is effectively blocked by Ru(II) caging and that irradiation with visible light triggers the release of inhibitor 4, enabling its ligation to the CYP3A4 heme.
Figure 3.
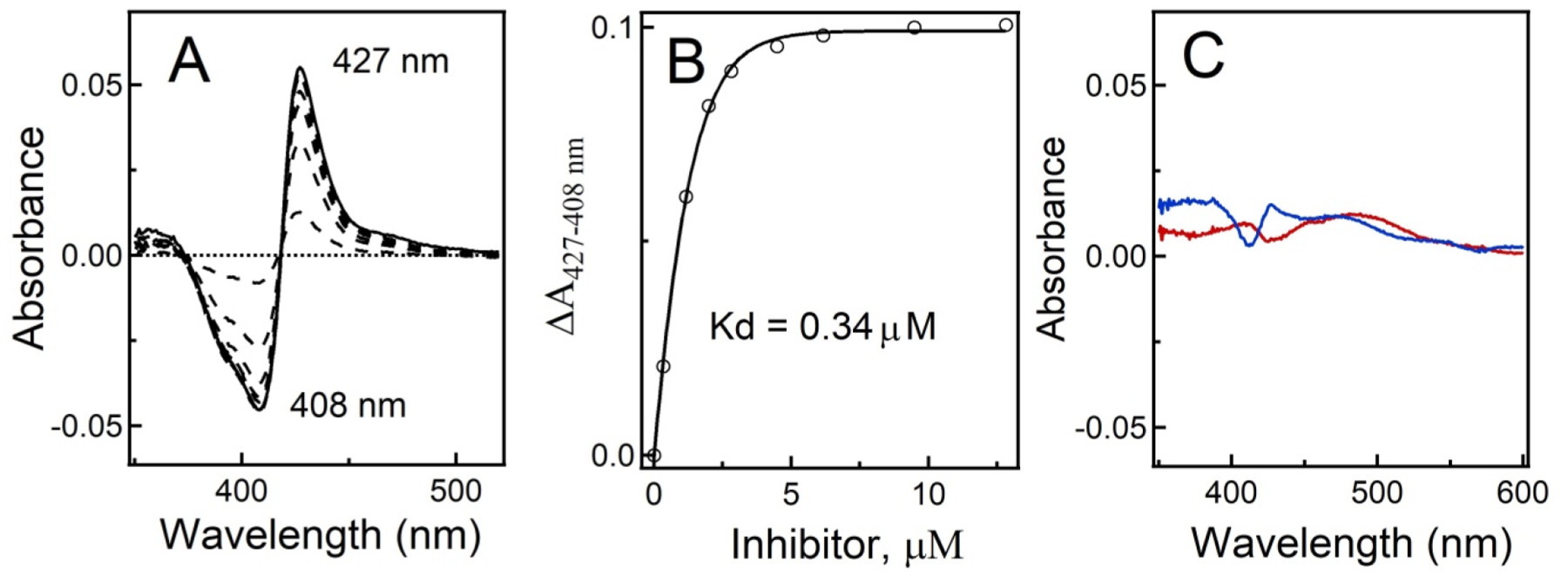
Equilibrium titration of CYP3A4 with 7 under light and dark conditions. (A) Difference spectra recorded during titration of recombinant CYP3A4 with 7 under light conditions. (B) Titration plot. The dissociation constant (Kd) was calculated by fitting the data to a hyperbolic equation: ΔA = ΔAmax[ligand]/(Kd + [ligand]), where ΔAmax is the maximal absorbance change and ΔA and [ligand] are the absorbance change and ligand concentration after each titrant addition, respectively. (C) Difference spectra recorded in control experiments, where CYP3A4 was mixed with 10 μM 7 (blue) or 10 μM [Ru(tpy)(Me2bpy)Cl]+ (red) in the dark, show the lack of spectral changes characteristic for type II N–Fe ligation.
Next, compound 7 was evaluated for its ability to inhibit CYP3A4 activity under light and dark conditions. The free inhibitor 4 and [Ru(tpy)(Me2bpy)(Cl)]Cl were included as controls. IC50 values were determined using a fluorogenic assay that monitors the O-debenzylation of 7-benzyloxy-4-trifluoromethylcoumarin (BFC), with 100% activity set at vehicle (DMSO) only. After treatment with visible light (λirr =400–700 nm, tirr = 40 min), 7 inhibited CYP3A4 nearly as well as free inhibitor 4 (IC50 of 2.2 μM and 1.5 μM, respectively), which agrees well with the spectral data (Figures 2 and 3) showing that 4 is released from 7 upon irradiation. However, to our surprise, the intact 7 was more potent in the dark (IC50 of 0.9 μM; Figure 4), suggesting that the Ru(II) complex could bind to CYP3A4 more strongly than free 4. Control experiments with [Ru(tpy)(Me2bpy)(Cl)]Cl (IC50 > 50 μM) showed that CYP3A4 inhibition was not due to just the Ru(II) fragment. Taken together, these data indicate that 7 is a stronger inhibitor when kept in the dark as compared to under irradiation.
Figure 4.
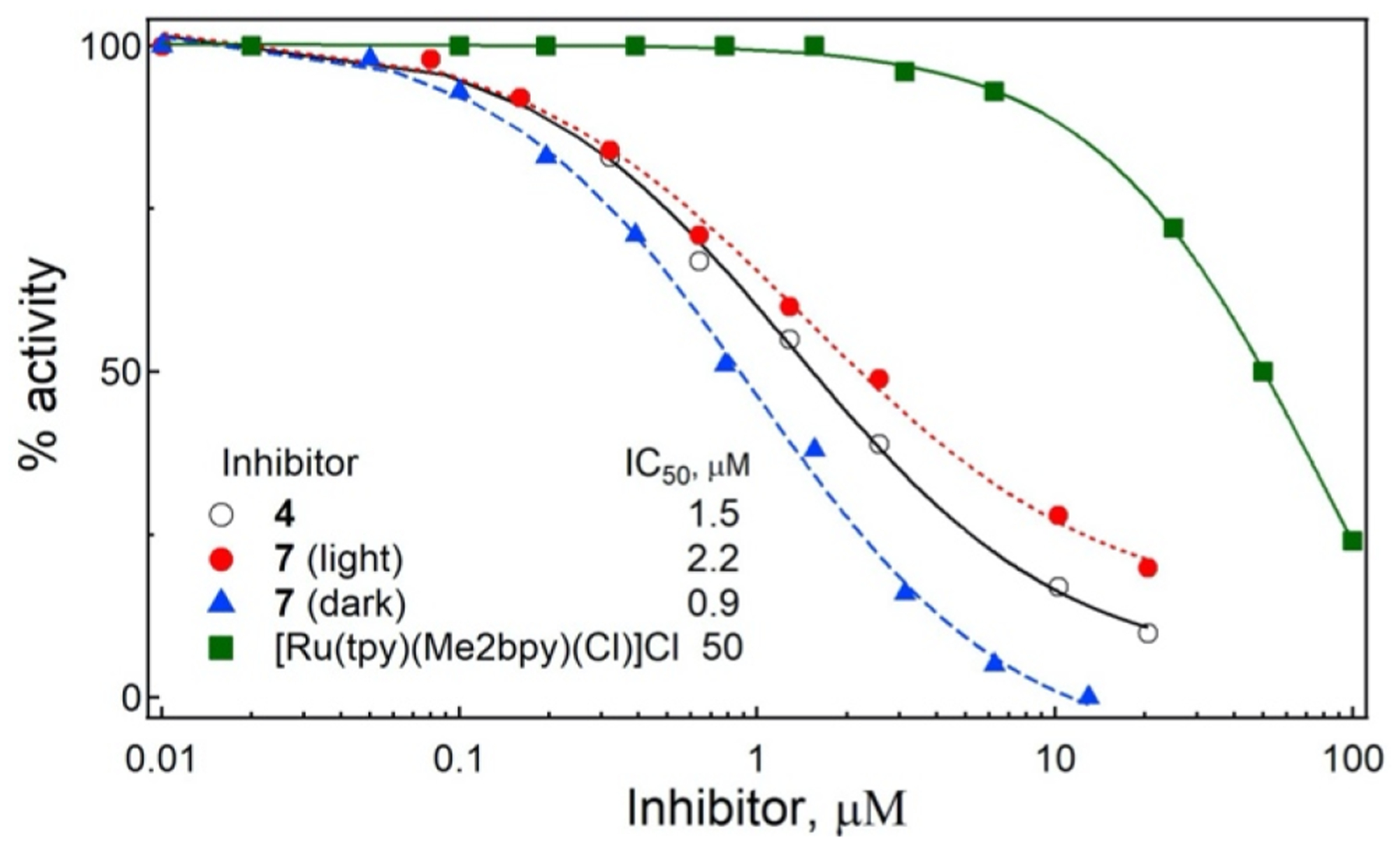
Inhibition of the BFC activity of recombinant CYP3A4 by 4, 7, and the control complex [Ru(tpy)(Me2bpy)(Cl)]Cl. Inhibitory assays were conducted at room temperature in a reconstituted system containing 0.2 μM CYP3A4 and 0.3 μM cytochrome P450 reductase by monitoring formation of a fluorescent product. The remaining activity was calculated relative to the DMSO-containing sample, used as a control (100% activity). The IC50 values were derived from fittings to the [% activity] vs [inhibitor] plots.
To confirm that the intact 7 is able to access the active site, we crystallized the CYP3A4-7 complex and solved the structure to 2.5 Å resolution. Indeed, 7 was bound in a well-defined manner within the active site (Figure 5). The inhibitor tail curls above the heme without direct binding to the iron center, while the bulky Ru(II) cage stacks inside the substrate channel. Protein–ligand interactions are predominantly hydrophobic. The inhibitor tail is surrounded by Phe241, Ile301, Phe304, and Ile369, whereas the ligands of the [Ru(tpy)(Me2bpy)] cage fragment stack with Phe108, Phe215, and Phe220 and are in close contacts with Phe57, Leu217, Met371, and Leu482. The anionic residues Asp76, Asp217, and Glu374 may also help to strengthen the inhibitory complex by creating favorable electrostatic interactions with the dicationic Ru(II) fragment. Importantly, the 7 N-pyridine does not bind to the heme iron because it is stably coordinated to Ru(II). This structure is highly valuable because it demonstrates that strong CYP3A4 inhibition by the intact, nonirradiated chimeric compound does not require Fe–N ligation.
Figure 5.
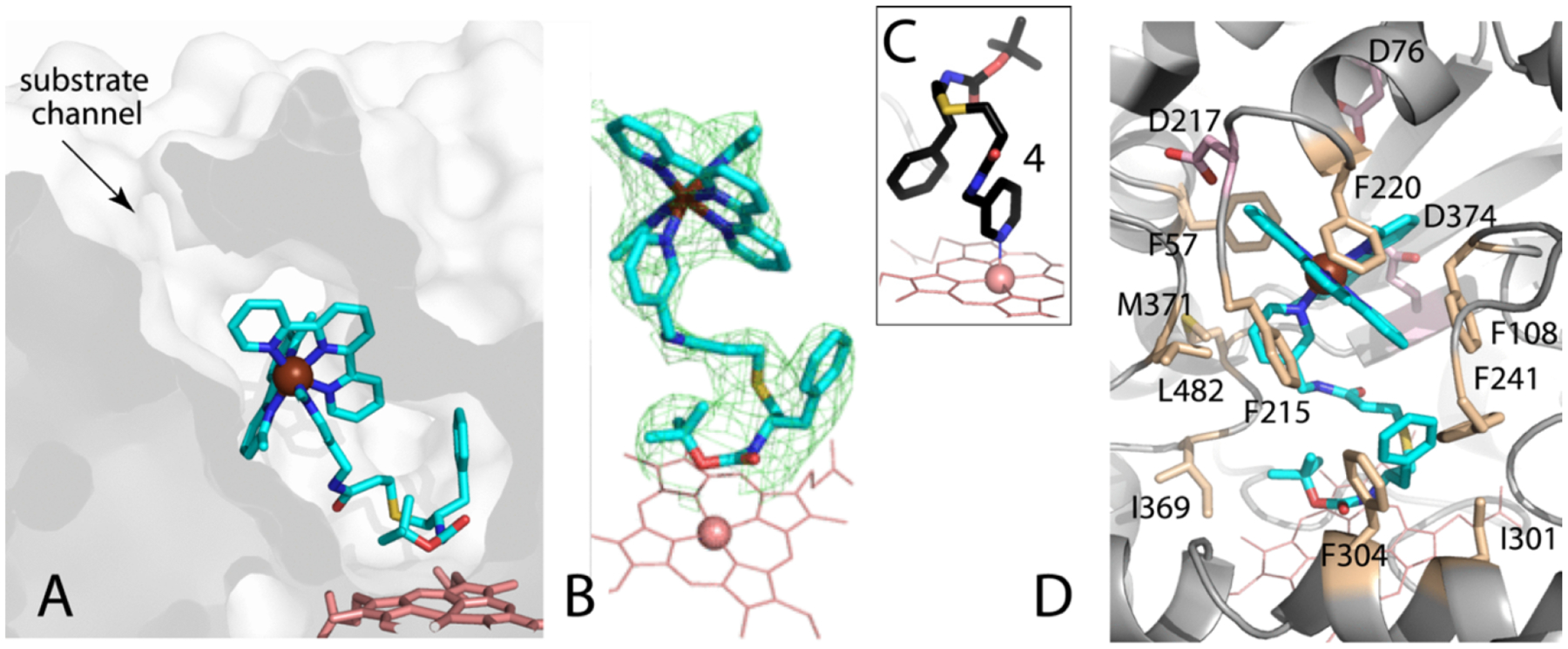
Crystal structure of CYP3A4 bound to 7 at 2.5 Å resolution: (A) slice through the CYP3A4 molecule showing orientation of 7, (B) omit electron density map for 7 at 3σ level, (C) the binding mode of free compound 4 (4D78 structure) shown for comparison, and (D) residues interacting with 7; hydrophobic in beige and acidic in pink.
On the basis of the finding that 7 potently inhibits CYP3A4 in the dark, the inhibitory assays for complexes 8–11 were conducted under both dark and light conditions (λirr = 400–700 nm, tirr = 40 min). IC50 values for the BFC activity of CYP3A4 are presented in Table 2. Complex 8, which contains inhibitor 6, inhibits CYP3A4 nearly to the same extent under dark and light conditions, giving a phototherapy index (PI) of 1.1. Interestingly, under dark conditions, 8 inhibits CYP3A4 roughly twice as potently as 7 (IC50 of ~400 nM). Since 8 willingly cocrystallized with CYP3A4, we also determined the CYP3A4-8 complex structure. Despite the fact that resolution was similar, 2.5 Å, 8 was poorly defined and the electron density around the ligand was discontinuous, which can be attributed to multiple binding modes. Nonetheless, the Ru-center and the core of the inhibitor tail could be located, allowing ligand fitting. As shown in Figure S12, the [Ru(tpy)(Me2bpy)] cage binds within the same pocket in the substrate channel. The inhibitor end-portion, in turn, similarly curls above the heme. Again, the complex is largely stabilized by aromatic stacking and hydrophobic interactions mediated by Phe57, Phe108, Phe220, Phe221, Phe241, and Phe304.
Table 2.
IC50 Values (μM) for CYP3A4 Inhibition by 4, 6, and 7–11 under Dark and Light Conditionsa
| compd | dark IC50 | light IC50b | PI |
|---|---|---|---|
| 4 | 1.54 | nd | |
| 6 | 0.40 | nd | |
| 7 | 0.9 | 2.2 | 0.41 |
| 8 | 0.40 | 0.36 | 1.1 |
| 9 | 0.25 | 0.84 | 0.30 |
| 10 | 0.28 | 0.46 | 0.61 |
| 10c | 1.02 | 0.44 | 2.3 |
| 11 | 0.40 | 0.21 | 1.90 |
Inhibitory assays for the BFC activity were conducted at room temperature in a reconstituted system containing 0.2 μM CYP3A4 and 0.3 μM cytochrome P450 reductase by monitoring formation of a fluorescent product. Stock solutions of 4, 6, 7–11 were prepared in DMSO. The activity remaining was calculated relative to the DMSO-containing sample, used as a control (100% activity). The IC50 values were derived from fittings to the [% activity] vs [inhibitor] plots. The standard error was <10%.
Light conditions (λirr = 400–700 nm, tirr = 40 min).
Assay containing 2% cyclodextrin.
Similar to 7, the Me2dppn complexes 9 and 10 inhibited CYP3A4 more potently under dark than light conditions but at lower concentrations than 7. Dark IC50 values for 9 and 10 were in the 250–280 nM range, with PI values of 0.30 and 0.61, respectively. Attempts to cocrystallize 9 and 10 with CYP3A4 were unsuccessful. Examination of inhibitors’ solutions showed that both 9 and 10 have a tendency to aggregate. Compound aggregation in solution can lead to false positives for enzyme inhibition, e.g., by trapping active enzyme within colloidal particles that block access of substrates.75 One way to distinguish between specific and nonspecific inhibition is to add detergents or other solubilizing agents to enzymatic assays. Therefore, we screened several detergents known to break up aggregates, including CHAPS, CYMAL-5, octylglucoside, and cyclodextrin. CYP3A4 was highly sensitive to detergents, with most detergents abolishing the BFC activity even in the absence of inhibitors. However, CYP3A4 preserved ~80% activity in the presence of 2% cyclodextrin. The latter agent was used for re-evaluation of 10 and, as we found, reversed the trend: dark IC50 = 1.02 μM, light IC50 = 0.44 μM, giving a PI of 2.3. Thus, aggregation was at least partially responsible for CYP3A4 inhibition by 10 in the dark. Importantly, the higher PI with 2% cyclodextrin was due to a higher IC50 for 10 in the dark; light data with and without 2% cyclodextrin were virtually the same and agreed well with those for free 6. Finally, the bulky Ph2Me2dppn-containing complex 11 showed an improved PI value, 1.90, as compared to PI = 0.61 for 10, implying that the larger caging group [Ru(tpy)(Ph2Me2dppn)] disfavors binding to the CYP3A4 active site.
In order to characterize the scope of CYP3A4 inhibition, we screened a library of 15 compounds, consisting of a diverse set of mono- and dicationic Ru(II) complexes (12–26, Figure 6; see Figure S25 for structures), against the purified enzyme. All complexes were screened against CYP3A4 under dark conditions at a concentration of 1 μM. Activities were determined using BFC as a substrate and expressed as percentage vs vehicle (DMSO) control. Thirteen complexes failed to decrease CYP3A4 activity below 75% at 1 μM concentration, confirming that potent CYP3A4 inhibition is not a general property of Ru(II) complexes. Only two compounds, [Ru(bpy)2(dppn)](PF6)2 (18) and [Ru(dppz)2(bpy)]Cl2 (19) reduced CYP3A4 activity below 75% at 1 μM concentration. Collectively, these data reveal complex structure–activity relationships for inhibition of CYP3A4 by Ru(II) complexes that warrant further investigation.
Figure 6.
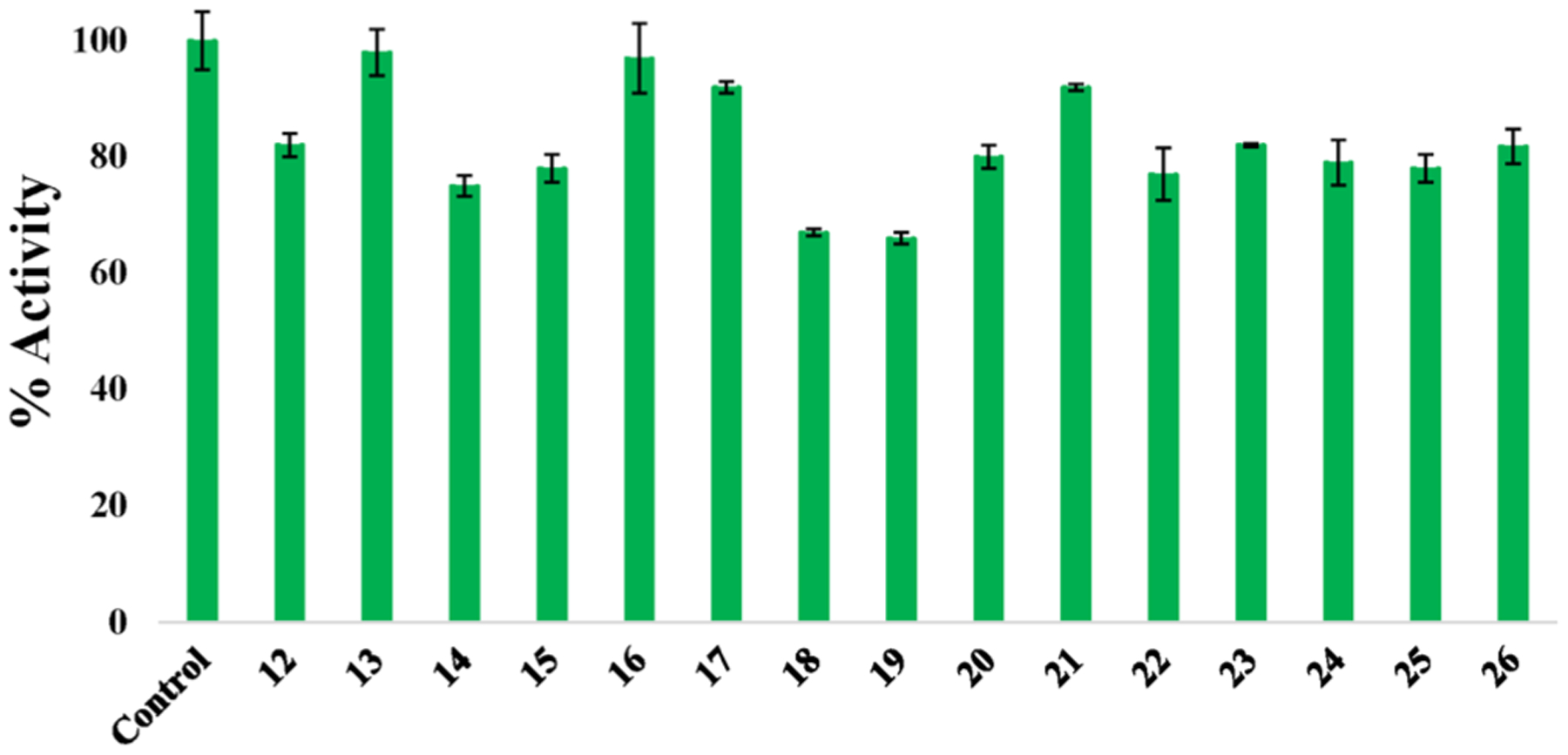
CYP 3A4 inhibition with a panel of Ru(II) complexes at 1 μM against purified enzyme. Inhibition of recombinant CYP3A4 with various complexes. Compounds are [Ru(bpy)3]Cl2 (12), [Ru(phen)3](PF6)2 (13), [Ru(bpy)2(phpy)]Cl (14), [Ru(bpy)2(acac)]PF6 (15), [Ru(bpy)2(bete)](PF6)2 (16), [Ru(bpy)2(bpte)]Cl2 (17), [Ru(bpy)2(dppn)](PF6)2 (18), [Ru(dppz)2(bpy)]Cl2 (19), [Ru(η6-p-cym)(DBM)Cl] (20), [Ru(η6-p-cym)(hfa)Cl] (21), [Ru(η6-p-cym)(bpy)Cl]Cl (22), [Ru(bpy)2(NHC-OMe)]PF6 (23), [Ru(bpy)2(NHC-COOEt)]PF6 (24), [Ru(tpy)(dppn)(py)](PF6)2 (25), [Ru(tpy)(acac)(py)]PF6 (26). See Figure S25 for structures.
To gain further insight into the potential biological applications of Ru(II)-based CYP inhibitors, we determined IC50 values for 4 and complexes 7 and 9 against microsomal CYP3A4 and two other major drug metabolizing enzymes, CYP1A2 and CYP2C9,76 using commercially available inhibitor screening kits (BioVision, Table 3). It should be pointed out that protein concentration and CYP:reductase ratios in the BioVision kits and our soluble reconstituted system were different, owing to which data in Tables 2 and 3 cannot be directly compared. For microsomal CYP3A4, inhibitor 4 was active in the nM range, with the IC50 values being nearly the same (~200 nM) under dark and light conditions. Complex 7 also inhibited CYP3A4 at nanomolar concentrations but more potently under dark vs light conditions, following the same trend as data for 7 presented in Table 2. Importantly, both 4 and 7 were much weaker inhibitors of CYP1A2 and CYP2C9. The selectivity of 4 for CYP3A4 was ~500-fold higher, whereas 7 inhibited CYP3A4 ~70-to-130-fold and 60-to-76-fold more strongly than the other CYPs under dark and light conditions (460–470 nm; 20 min), respectively. A multifold difference in IC50 measured for 4 and 7 under dark conditions suggests some influence of the released Ru(II) cage in the inhibition. The respective data were also collected for Ru(II) complex 9, which contains the same inhibitor 4 linked to the bulky and more hydrophobic photocaging group [Ru(tpy)(Me2dppn)]. Compared to 7, the inhibitory potency of 9 for microsomal CYP3A4 was ~8-and 5-fold lower under dark and light conditions, respectively, and its selectivity for other isoforms could not be accurately measured due to solubility problems. Even so, there was a common trend, as all three compounds displayed higher specificity for CYP3A4 albeit to a different extent.
Table 3.
IC50 Values (μM) for 4, 7, and 9 against Microsomal CYP3A4, CYP1A2, and CYP2C9 under Dark and Light Conditionsa
| CYP3A4 | CYP1A2 | CYP2C9 | selectivity | |||||
|---|---|---|---|---|---|---|---|---|
| compd | dark | light | dark | light | dark | light | 1A2/3A4 dark (light) | 2C9/3A4 dark (light) |
| 4 | 0.183 ± 0.01 | 0.232 ± 0.018 | >100 | ND | 87 ± 2 | ND | 566 (ND) | 475 (ND) |
| 7 | 0.217 ± 0.011 | 0.301 ± 0.001 | 28 ± 2 | 23 ± 1 | 15 ± 3 | 18 ± 1 | 129 (76) | 69 (60) |
| 9 | 1.7 ± 0.1 | 1.6 ± 0.1 | >10 | >10 | >10 | >10 | >5.9 (>6.3) | >5.9 (>6.3) |
IC50 values determined using CYP3A4, CYP1A2, or CYP2C9 inhibitor screening kits (BioVision) following the manufacturer’s protocols. Stock solutions of 4, 7, or 9 were prepared in MeCN, plated, and combined with assay buffer and irradiated with a blue LED light source (tirr = 20 min, λirr = 460–470 nm, 56 J/cm2) or left in the dark. Experiments with compound 9 did not exceed 10 μM due to solubility limitations in assay buffer. Percent activities were determined vs vehicle control. IC50 values were determined using Igor Pro graphing software. Data are the average of three experiments, and errors are standard deviations.
Biological Studies.
Studies on the interaction of 7–11 with isolated CYP3A4 showed that inhibition can be achieved via blockage of the active site by the intact caged compounds, light-activated release of the inhibitory fragment and its subsequent heme ligation, and efficient 1O2 generation. However, questions remained regarding the role of aggregation vs direct inhibition of CYP3A4 in the dark, due to sensitivity of the recombinant enzyme to common detergents. These challenges prompted us to utilize an in vitro cell-based assay to probe for CYP3A4 inhibition by our compounds. Importantly, prior studies demonstrated that CYP3A4 inhibitors work synergistically with microtubule-destabilizing drugs in cancer cells.77,78 We chose to evaluate our compounds in DU-145 prostate cancer cells because (i) they have high levels of CYP3A4 expression, (ii) prior studies showed synergism between the CYP3A4 inhibitor ketoconazole and vinblastine,77 a drug commonly used in combination therapies for various cancers, and (iii) utilization of a validated protocol for in vitro detection of synergism between a chemotherapeutic drug and CYP3A4 inhibitors would provide a reliable cell-based assay for evaluation of our compounds. Vinblastine binds to tubulin and stops production of microtubules, leading to M-phase specific cell cycle arrest. Synergism between vinblastine and ketoconazole was previously achieved by blocking CYP3A4-dependent vinblastine metabolism in several prostate cancer cell lines.77 On the basis of this knowledge, we designed experiments with DU-145 cells and our panel of compounds. First, free CYP3A4 inhibitors 4 and 6 (5 μM) were evaluated against DU-145 cells in the presence or absence of vinblastine (5 nM). Cells were treated with 4 or 6 and vinblastine or vehicle, and viability was assessed after 72 h by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. It was found that both 4 and 6 reduce viability of the DU-145 cells by up to ~40% in the presence of 5 nM vinblastine. This reduction of viability is similar to that observed with ketoconazole77 and suggests the synergy between CYP3A4 inhibition and the microtubule-destabilizing drug (Figure 7A).
Figure 7.
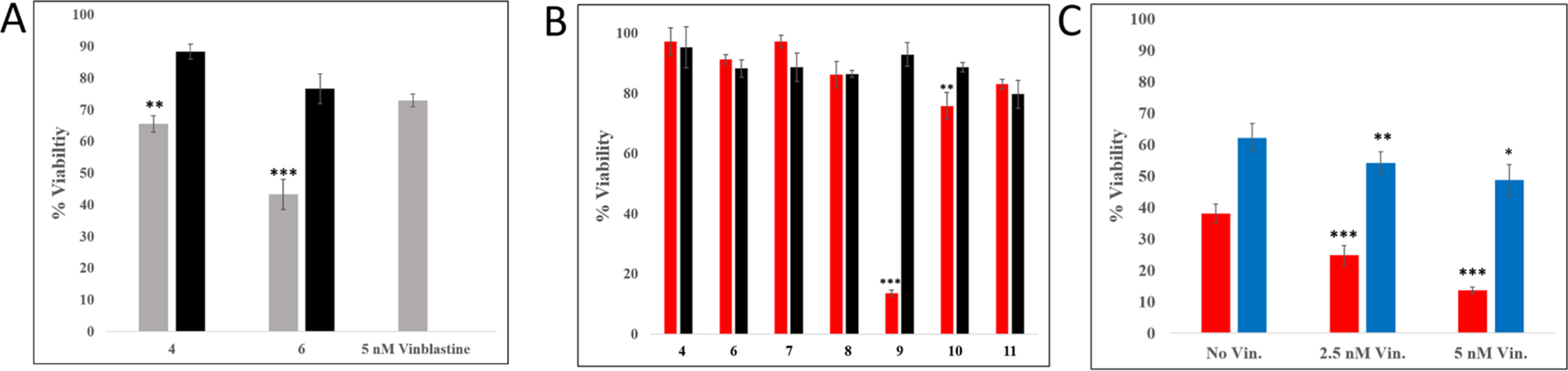
Cellular viability studies with compounds 4, 6–11, and 27 in DU-145 prostate adenocarcinoma cells. DU-145 cells were seeded in a 96-well plate at a density of 7000 cells per well and incubated overnight (~18 h). (A) The medium was aspirated from each well, and quadruplicate wells were treated with medium containing either 4 or 6 (5 μM) in 1% DMSO (black) or co-treated with vinblastine (5 nM) (gray). After 72 h of incubation at 37 °C, MTT assay was performed. Viability data were obtained by averaging blank-normalized absorbance values for control cells and expressing average absorbance for the treated samples as percent control. P-values are vs 5 nM vinblastine alone. (B) The medium was aspirated from each well, and octuplicate wells were treated with medium containing one of compounds 4 or 6–11 (5 μM) in 1% DMSO. After 1 h of incubation at 37 °C, the medium was aspirated and replaced with medium containing vinblastine (5 nM). The plates were irradiated using a blue LED light source (tirr = 20 min, λirr = 460–470 nm, 56 J/cm2) (red) or left in the dark (black) and incubated for 72 h. MTT assay was then performed. Viability data were obtained by averaging blank-normalized absorbance values for control cells and expressing average absorbance for the treated samples as percent control. P-values are vs dark viabilities for each compound. (C) The medium was aspirated from each well, and octuplicate wells were treated with medium containing either compound 9 (red) or 27 (blue) (5 μM) in 1% DMSO. After 1 h of incubation at 37 °C, the medium was aspirated and replaced with either vehicle or medium containing vinblastine (2.5 nM or 5 nM) or vehicle. The plates were irradiated using a blue LED light source (tirr = 20 min, λirr = 460–470 nm, 56 J/cm2) and then incubated for 72 h. MTT assay was then performed. Viability data were obtained by averaging blank-normalized absorbance values for control cells and expressing average absorbance for the treated samples as percent control. P-values are vs 0 nM vinblastine (No Vin.) for each compound; ***P < 0.01, **P < 0.05, *P < 0.10.
Next, inhibitors 4 and 6 were evaluated alongside the photocaged inhibitors 7–11 in the dark or with irradiation in the presence of vinblastine (5 nM). In these experiments, cells were treated with 4, 6, or 7–11 (5 μM) and kept in the dark for 1 h, then the medium was replaced with medium containing 5 nM vinblastine, and cells were irradiated with blue light (460–470 nm, 20 min) or left in the dark for 20 min. Viabilities were determined 72 h after light treatment by the MTT assay. As expected, results with the free inhibitors 4 and 6 were virtually identical under dark and light conditions, ruling out synergy between 4 or 6 and light. In these experiments, synergy with 4 and 6 was less pronounced compared to incubations where 4 or 6 was left with cells for the full 72 h without medium replacement (Figure S13), which may indicate slower uptake of these inhibitors by DU-145 cells. Among the investigated compounds, complex 9, which not only releases the CYP3A4 inhibitor 4 (PCT) but also generates 1O2 (PDT), showed the strongest response by reducing viability to ~10% in the light compared to ~90% in the dark (Figure 7B). Again, results were less pronounced when 7–11 were left with DU-145 cells for the full 72 h without replacement of medium (Figure S13), further indicating that cell uptake is slower for some of the Ru(II) complexes. Nonetheless, complex 9 showed a strong response with replacement of medium after only 1 h, which supports the ability of 9 to penetrate DU-145 cells within that time frame.
Next, we probed the impact of CYP3A4 inhibition in cell-induced toxicity with vinblastine. Complex 9 was compared side-by-side with the [Ru(tpy)(Me2dppn)(py)](PF6)2 complex (27), which generates 1O2 just as efficiently69 but serves as a control by releasing pyridine rather than the CYP3A4 inhibitor 4. Experiments with 27 were important to carry out because prior studies demonstrated that PDT can work synergistically with microtubule-targeting drugs.79 The results in Figure 7C show that 9 (5 μM) produces a strong, dose-dependent synergy with vinblastine (0–5 nM), whereas less toxic 27 (5 μM) does not. These data suggest that CYP3A4 inhibition is synergistic with PDT and vinblastine.
In order to quantify drug synergy, the Chou–Talalay method was applied, which is the field standard for assessing the synergy of a drug combination.80–83 DU-145 cells were treated with either 4, 7, 9, 27, or vinblastine alone over a range of concentrations to determine EC50 (Table 4). For only 9, 27, and vinblastine, the EC50 values were <25 μM under light conditions (tirr = 20 min, λirr = 460–470 nm, 72 h MTT); in the dark, EC50 for all compounds was >25 μM. Next, DU-145 cells were treated with a combination of single concentrations of 9 or 27 and vinblastine over a range of concentrations spanning the EC50 values under light conditions (tirr = 20 min, λirr = 460–470 nm, 56 J/cm2, 72 h MTT), resulting in a panel of 16 distinct combinations (Figure 8 for 9, Figure S14 for 27). Viabilities for the single drug and combination treatments were compared against the vehicle control to measure the % effectiveness as the proportion between live and dead cells in a given treatment. By use of the dose and effect for each monotreatment and each combination, the combination index (CI) values for each treatment pair were calculated using Compusyn software (Figure 8).80 CI values less than 1 indicate synergy, equal to 1 indicate an additive effect, and greater than 1 indicate antagonism. For compound 9, 12 out of 16 combinations surveyed showed CI values <1, with the other four combinations showing CI values near 1, indicating high synergism between 9 and vinblastine under light conditions. In contrast, 27 showed weaker synergism under all concentrations surveyed under light conditions (Figure S14). Taken together, these data suggest that (i) 9 blocks intracellular metabolism of vinblastine via CYP3A4 inhibition and (ii) the CYP3A4 inhibition, PDT, and vinblastine act together and produce a stronger cytotoxic response in the DU-145 cells than the combination of PDT and vinblastine. Because microtubule-destabilizing drugs have deleterious side effects and narrow therapeutic indexes, the combination of localized CYP3A4 inhibition and PDT may prove to be a promising approach to achieve synergy and lower the doses of chemotherapeutic drugs like vinblastine.
Table 4.
EC50 Values for 4, 7, 9, and 27 in DU-145 Cellsa
| EC50 (μM) | |||
|---|---|---|---|
| entry | compd | light | dark |
| 1 | 4 | >25 | >25 |
| 2 | 6 | ND | 17 ± 3 |
| 3 | 7 | >25 | >25 |
| 4 | 9 | 2.8 ± 1.0 | >25 |
| 5 | 27 | 5.5 ± 0.8 | >25 |
| 6 | vinblastine | (8.3 ± 1.1) × 10−3 | ND |
EC50 determination for compounds 4, 6, 7, 9, 27, and vinblastine was performed on DU-145 cells. Data are the average of three independent experiments using quadruplicate wells; errors are standard deviations. After treatment, cells were incubated at 37 °C and 5% CO2 for 1 h. Medium was aspirated and replaced with vehicle. Cells were irradiated using a blue LED light source (tirr = 20 min, λirr = 460–470 nm, 56 J/cm2) and incubated for 72 h. After that, viability was assessed by MTT assay. EC50 values were obtained using Igor Pro graphing software for 4, 6, 7, 9, and 27 and with Compusyn software for vinblastine.
Figure 8.
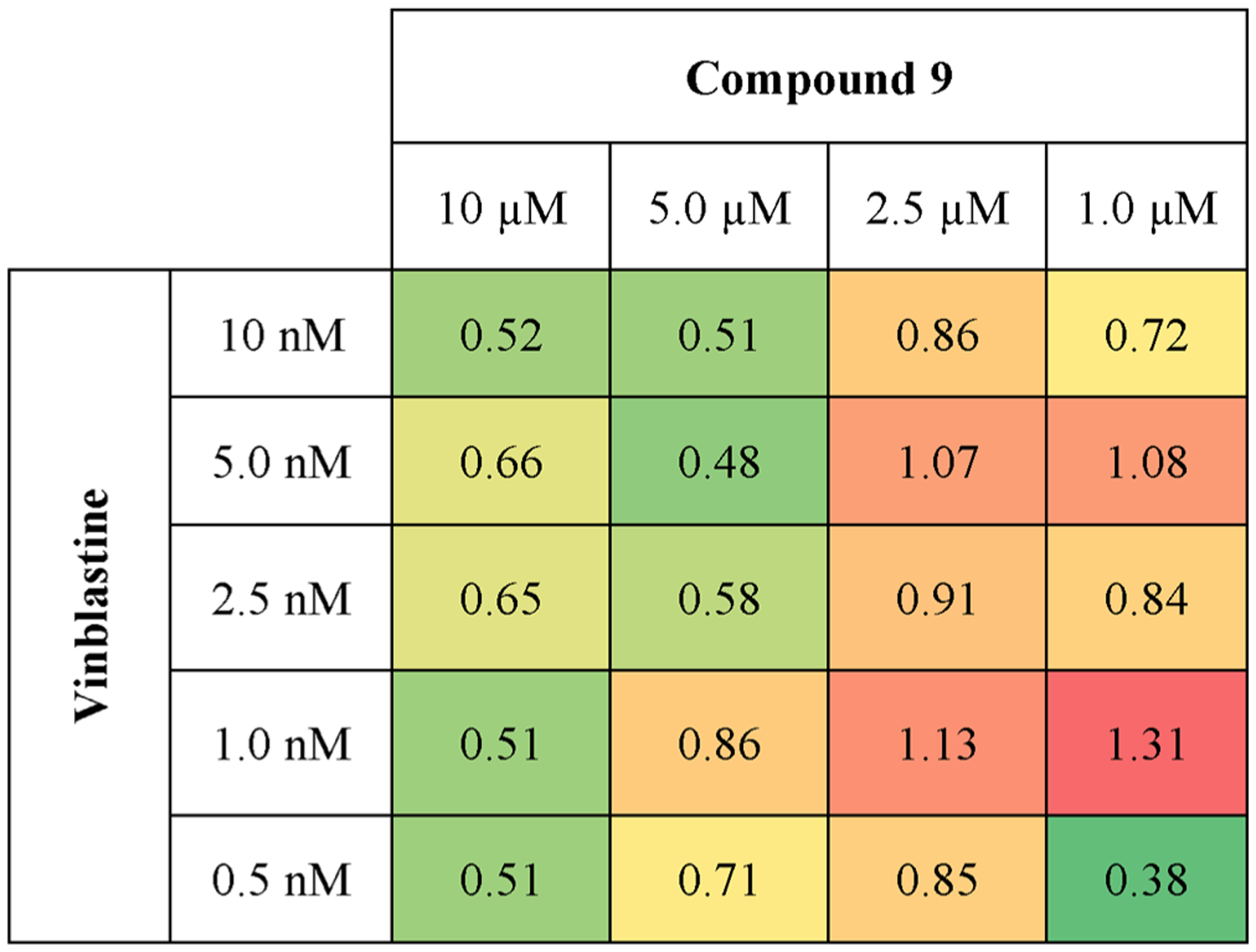
Chou–Talalay combination index heat map. Chou–Talalay determination of drug synergy between 9 and vinblastine under light conditions (tirr = 20 min, λirr = 460–470 nm, 56 J/cm2). Effects on cell killing were determined by MTT 48 h after light treatment. Values shown in colored boxes denote combination indices (CI). CI > 1: antagonism. CI = 1: additive effect. CI < 1: synergy. CI values were obtained using Compusyn software.
CONCLUSION
This is the first report on the synthesis and biological evaluation of metal-based inhibitors of the major human drug metabolizing enzyme CYP3A4. Using two analogs of ritonavir, we synthesized and characterized five Ru(II)-caged CYP3A4 inhibitors (7–11) that showed either single action PCT or dual action PCT/PDT behavior. Serendipitously, we demonstrated that CYP inhibition can be enhanced through inhibitor metalation, as the caged complexes can tightly and selectively bind to the CYP3A4 active site without heme ligation. Moreover, compound 9 was identified as a dual-action PCT/PDT lead compound, which effectively generates 1O2 and releases the CYP3A4 inhibitor to act synergistically with the common chemotherapeutic drug vinblastine in DU-145 adenocarcinoma cells. These findings warrant further studies on photoactive CYP inhibitory compounds to determine their potential use for clinical applications, such as enhancement of therapeutic efficacy of chemotherapeutic drugs.
EXPERIMENTAL SECTION
Materials. General Procedure for Synthesis of Ru(II) Complexes.
Some reactions were performed under ambient atmosphere unless otherwise noted. Anaerobic reactions were performed by purging the reaction solutions with Ar or nitrogen. Complexes 12 and 13 were purchased (Strem Chemicals). Complexes 14,84 15,85 16,86 17,86 18,87 19,88 20–22,89 23 and 24,90 25,70 and 2691 were prepared following literature protocols. For synthesis of 7–11, a solution of [Ru(tpy)(L1)Cl]Cl in EtOH was treated with pyridine. Water was added, and the mixture was deoxygenated by bubbling Ar through a submerged needle for 20 min. The pressure tube was sealed and heated to 80 °C for 16 h. The reaction mixture was cooled to room temperature, concentrated, and the residue was purified by column chromatography on neutral alumina to give [Ru(tpy)(L1)(L2)](Cl)2 complexes.
Synthesis of [Ru(tpy)(Me2bpy)(4)]Cl2 (7).
[Ru(tpy)(Me2bpy)Cl]Cl67 (19.0 mg, 0.0300 mmol) was added to a solution of 4 (28 mg, 0.070 mmol) in a 1:1 mixture of EtOH and H2O (3.0 mL each) under inert atmosphere in a pressure flask. The pressure flask was wrapped with aluminum foil. The solution was purged with argon for 10 min at room temperature. The pressure flask was sealed, and the reaction mixture was refluxed at 80 °C for 16 h under an inert atmosphere. The color of the reaction mixture turned from purple to brown. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The crude product was purified over neutral alumina (5% MeOH/DCM) in the dark to give 7 as a brown solid (25 mg, 75%): 1H NMR (400 MHz CD3OD) δ 8.75–8.72 (m, 1H), 8.69 (d, 2H, J = 8.4 Hz), 8.63 (d, 2H, J = 8.0 Hz), 8.48–8.45 (m, 1H), 8.29 (t, 1H, J = 8.0 Hz), 8.24–8.18 (m, 3H), 8.14 (t, 2H, J = 8.0 Hz), 7.81–7.72 (m, 2H), 7.66–7.60 (m, 4H), 7.55 (d, 1H, J = 5.6 Hz), 7.28–7.17 (m, 5H), 7.08 (t, 1H, J = 7.6 Hz), 7.01 (d, 1H, J = 7.6 Hz), 4.04–3.93 (m, 2H), 3.86–3.79 (m, 1H), 2.94–2.88 (m, 1H), 2.72 (t, 2H, J = 7.2 Hz), 2.68–2.65 (m, 1H), 2.60–2.55 (m, 2H), 2.34 (t, 2H, J = 7.2 Hz), 2.03 (s, 3H), 1.50 (s, 3H), 1.36–1.29 (m, 9H); IR νmax (cm−1) 3372, 2926, 2830, 1740, 1711, 1536, 1447, 1371, 1223, 1022, 519; ESMS calcd for C50H54N8O3RuS (M+2) 474, found 474; UV–vis λmax = 474 nm (ε = 7700 M−1 cm−1).
Synthesis of [Ru(tpy)(Me2bpy)(6)]Cl2 (8).
Compound 8 was prepared by following the general procedure by treating [Ru(tpy)(Me2bpy)Cl]Cl67 (13 mg, 0.022 mmol) with 6 (23 mg, 0.044 mmol) in EtOH (3 mL) and water (3 mL). The residue was purified by column chromatography on neutral alumina (4–6% MeOH in DCM) to give a red solid (15 mg, 63%). 1H NMR (400 MHz, methanol-d4) δ 8.76 (d, J = 8.0 Hz, 1H), 8.73–8.62 (m, 3H), 8.57 (t, J = 7.3 Hz, 1H), 8.50 (d, J = 8.1 Hz, 1H), 8.27 (dd, J = 9.2, 6.9 Hz, 3H), 8.18 (ddd, J = 12.3, 6.1, 3.6 Hz, 2H), 8.07 (ddt, J = 7.9, 3.9, 2.0 Hz, 1H), 7.77 (dt, J = 13.1, 7.7 Hz, 2H), 7.68–7.60 (m, 1H), 7.60–7.54 (m, 1H), 7.48 (dq, J = 8.7, 6.0, 4.8 Hz, 3H), 7.28–7.15 (m, 8H), 7.15–6.99 (m, 4H), 3.82 (ttd, J = 8.9, 6.7, 3.0 Hz, 1H), 3.55 (dd, J = 9.7, 5.7 Hz, 1H), 3.08 (ddd, J = 13.7, 9.7, 6.6 Hz, 1H), 2.97–2.81 (m, 2H), 2.78–2.60 (m, 5H), 2.10 (t, J = 8.1 Hz, 5H), 1.51 (d, J = 3.1 Hz, 3H), 1.39–1.25 (m, 9H), 1.25–1.18 (m, 2H); IR (KBr) 3395, 3242, 3058, 3027, 2974, 2927, 2859, 1698, 1660, 1602, 1542, 1523, 1496, 1447, 1388, 1364, 1282, 1248, 1168, 1119, 1078, 1016, 916, 778, 748, 701, 672, 646; UV–vis λmax = 470 nm (ε = 9700 M−1 cm−1); ESMS calculated for C58H62N8O3RuS [M2+] 526, found 526.
Synthesis of [Ru(tpy)(Me2dppn)(4)]Cl2 (9).
Compound 9 was prepared by following the general procedure by treating [Ru(tpy)(Me2dppn)Cl]Cl65 (22 mg, 0.029 mmol) with 4 (25 mg, 0.058 mmol) in EtOH (4 mL) and water (4 mL). The residue was purified by column chromatography on neutral alumina (3–4% MeOH in DCM) to give a red solid (20 mg, 57%). 1H NMR (400 MHz, methanol-d4) δ 9.93 (dd, J = 8.4, 1.6 Hz, 1H), 9.33 (d, J = 8.2 Hz, 1H), 8.98 (d, J = 12.1 Hz, 2H), 8.78 (d, J = 8.1 Hz, 1H), 8.78–8.68 (m, 2H), 8.65 (d, J = 8.2 Hz, 1H), 8.33–8.23 (m, 3H), 8.26–8.17 (m, 1H), 8.16 (t, J = 7.8 Hz, 2H), 8.11 (h, J = 3.8 Hz, 2H), 7.75–7.68 (m, 2H), 7.68–7.58 (m, 3H), 7.61–7.50 (m, 1H), 7.50 (t, J = 6.7 Hz, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.26–7.09 (m, 7H), 4.03 (d, J = 4.6 Hz, 2H), 3.85–3.74 (m, 1H), 3.34 (d, J = 3.6 Hz, 1H), 2.86 (ddd, J = 15.9, 10.9, 5.5 Hz, 1H), 2.74 (t, J = 7.1 Hz, 2H), 2.66–2.58 (m, 1H), 2.58 (q, J = 2.7, 2.3 Hz, 2H), 2.37 (t, J = 7.0 Hz, 2H), 2.34 (s, 3H), 1.79 (s, 3H), 1.31 (d, J = 7.6 Hz, 9H); IR (KBr) 3394, 3056, 3027, 2924, 2853, 1966, 1697, 1662, 1542, 1520, 1446, 1363, 1247, 1167, 1056, 1017, 880, 776, 703; UV–vis λmax = 485 nm (ε = 13 500 M−1 cm−1); ESMS calculated for C62H58ClN10O3RuS [M+] 1159, found 1159.
Synthesis of [Ru(tpy)(Me2dppn)(6)]Cl2 (10).
Compound 10 was prepared by following the general procedure by treating [Ru(tpy)(Me2dppn)Cl]Cl65 (17 mg, 0.022 mmol) with 6 (23 mg, 0.044 mmol) in EtOH (5 mL) and water (5 mL). The residue was purified by column chromatography on neutral alumina (3–4% MeOH in DCM) to give a red solid (17 mg, 61%). 1H NMR (400 MHz, methanol-d4) δ 9.96 (dd, J = 8.3, 6.4 Hz, 1H), 9.45 (dd, J = 8.2, 3.8 Hz, 1H), 9.14–9.02 (m, 2H), 8.83–8.72 (m, 2H), 8.68 (d, J = 8.0 Hz, 1H), 8.59–8.50 (m, 1H), 8.35 (t, J = 6.3 Hz, 1H), 8.26 (dtd, J = 15.0, 9.2, 5.4 Hz, 4H), 8.15 (dd, J = 18.4, 6.0 Hz, 2H), 8.05–7.98 (m, 1H), 7.77–7.66 (m, 3H), 7.61–7.53 (m, 3H), 7.57–7.30 (m, 2H), 7.18 (s, 4H), 7.27–7.12 (m, 1H), 7.16–7.04 (m, 5H), 3.76–3.46 (m, 1H), 3.47 (dt, J = 9.1, 6.0 Hz, 1H), 3.13–2.98 (m, 1H), 2.80 (dtd, J = 43.5, 15.3, 6.5 Hz, 1H), 2.67 (s, 2H), 2.67–2.58 (m, 1H), 2.56 (s, 1H), 2.36 (dd, J = 11.6, 3.3 Hz, 3H), 2.16 (dp, J = 21.1, 7.0 Hz, 1H), 1.80 (s, 3H), 1.29 (d, J = 8.1 Hz, 11H); IR (KBr) 3365, 3256, 3056, 3025, 2974, 2922, 2852, 2360, 2342, 1868, 1792, 1760, 1733, 1698, 1653, 1558, 1542, 1522, 1447, 1388, 1362, 1243, 1161, 1056, 881, 841, 775, 752, 700, 669; UV–vis λmax = 480 nm (ε = 12 000 M−1 cm−1); ESMS calculated for C70H66ClN10O3RuS [M+] 1263, found 1263.
Synthesis of [Ru(tpy)(Ph2Me2dppn)(4)]Cl2 (11).
Compound 11 was prepared by following the general procedure by treating [Ru(tpy)(Ph2Me2dppn)Cl]Cl68 (19 mg, 0.021 mmol) with 6 (22 mg, 0.042 mmol) in EtOH (3 mL) and water (3 mL). The residue was purified by column chromatography on neutral alumina (3–4% MeOH in DCM) to give a red solid (14 mg, 48%). 1H NMR (400 MHz, methanol-d4) δ 9.42–9.30 (m, 1H), 8.88–8.58 (m, 4H), 8.50 (dt, J = 6.6, 3.3 Hz, 1H), 8.32–7.89 (m, 9H), 7.83–7.55 (m, 14H), 7.46–7.29 (m, 2H), 7.29–6.96 (m, 12H), 3.83–3.68 (m, 1H), 3.46 (td, J = 9.2, 6.7 Hz, 1H), 2.99 (dd, J = 13.7, 9.6 Hz, 1H), 2.93–2.47 (m, 7H), 2.28 (d, J = 10.6 Hz, 3H), 2.14 (tdd, J = 16.6, 8.0, 3.7 Hz, 2H), 1.73 (s, 3H), 1.33 (d, J = 4.0 Hz, 1H), 1.27 (d, J = 2.2 Hz, 10H); IR (KBr) 3255, 3057, 3025, 2973, 2924, 2853, 2360, 2330, 1698, 1684, 1653, 1558, 1542, 1522, 1496, 1490, 1448, 1420, 1387, 1362, 1246, 1166, 1073, 1014, 839, 773, 701, 670; UV–vis λmax = 491 nm (ε = 13 500 M−1 cm−1); ESMS calculated for C82H74ClN10O3RuS [M+] 1415, found 1415.
Instrumentation and Methods.
NMR spectra were recorded on a Varian FT-NMR Mercury 400 MHz spectrometer. UV–vis spectra were recorded on a Varian Cary 60 spectrophotometer. Steady state electronic absorption spectra were collected using an Agilent Cary 8453 diode array spectrometer, and emission data were collected using a Horiba FluoroMax-4 fluorimeter. All experiments involving DU-145 cells were carried out in Dulbecco’s modified Eagle’s medium containing 10% FBS and 1000 units/mL penicillin/streptomycin. The irradiation source for quantum yield measurements was a 150 W Xe arc lamp (USHIO) in a MilliArc lamp housing unit, powered by an LPS-220 power supply and an LPS-221 igniter (PTI). The emission wavelengths were selected using a CVI Melles Griot long-pass filter, and the appropriate irradiation wavelengths for photolysis experiments were selected with a bandpass filter (Thorlabs) and long-pass filter (CVI Melles Griot). The quantum yields (Φ) for ligand dissociation were determined in CH3CN with an irradiation wavelength of 500 nm. The rate of moles reacted at early irradiation times was determined by monitoring the decrease in the MLCT absorption maximum as a function of time. The photon flux of the lamp with a 435 nm long-pass filter and a 500 nm bandpass filter was determined using ferrioxalate actinometry as previously described in detail.92 Singlet oxygen quantum yields were performed using [Ru(bpy)3]2+ as a standard (ΦΔ = 0.81 in MeOH) and 1,3-diphenylisobenzofuran (DPBF) as a 1O2 trapping agent and following a previously established procedure.93
Studies on Recombinant CYP3A4.
Full-length and truncated (Δ3–22) human CYP3A4 was expressed and purified as described previously.94
Spectral Binding Titrations.
Equilibrium ligand binding to Δ3–22 CYP3A4 was monitored in a Cary 300 spectrophotometer at ambient temperature in 0.1 M phosphate buffer, pH 7.4, supplemented with 20% glycerol and 1 mM dithiothreitol. Inhibitors and caged compounds, with or without visible light irradiation (λirr = 400–700 nm, tirr = 40 min), were dissolved in DMSO and added to a 2 μM protein solution in small aliquots, with the final solvent concentration of <2%. Spectral dissociation constants (Kd) were determined from hyperbolic fits to titration plots.
Inhibitory Potency Assays.
Inhibitory potency for the 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) O-debenzylase activity of CYP3A4 was evaluated fluorometrically in a soluble reconstituted system. Full-length CYP3A4 and rat cytochrome P450 reductase (40 μM and 60 μM, respectively) were preincubated at room temperature for 1 h before 10-fold dilution with the reaction buffer consisting of 0.1 M potassium phosphate, pH 7.4, catalase, and superoxide dismutase (2 units/mL each). Prior to measurements, 85 μL of the reaction buffer was mixed with 10 μL of the NADPH-regenerating system (10 mM glucose, 0.2 mM NADP+, and 2 units/mL glucose 6-phosphate dehydrogenase), 5 μL of the protein mixture (0.2 μM final CYP3A4 concentration), and 2 μL of the cage/inhibitor solution or DMSO. The mixture was incubated for 2 min, after which 1 μL of 2 mM BFC and 1 μL of 7 mM NADPH were added to initiate the reaction. Accumulation of the fluorescent product, 7-hydroxy-4-(trifluoromethyl)coumarin, was monitored for 2 min at room temperature in a Hitachi F400 fluorimeter (λex = 404 nm; λem = 500 nm). Within this time interval, fluorescence changes were linear. The average of three measurements was used to calculate the remaining activity, with the DMSO-containing sample used as a control (100% activity). The IC50 values were derived from four-parameter logistic fittings to the [% activity] vs [inhibitor] plots.
Crystallization of 7- and 8-Bound CYP3A4.
Both complexes were crystallized using a microbatch method under oil. Prior to crystallization setup, Δ3–22 CYP3A4 (50–60 mg/mL in 75–100 mM phosphate, pH 7.4) was incubated with a 2-fold ligand excess for 15 min and centrifuged to remove the precipitate. The supernatant (0.4 μL) was mixed with 0.4–0.5 μL of the crystallization solution containing 10% PEG 3350 and 80 mM tribasic ammonium citrate, pH 7.0, for 7 and 8% PEG 3350 and 70 mM dl-malate, pH 7.0, for 8. Crystals were grown at room temperature for 2–3 days and cryoprotected with Paratone-N before freezing in liquid nitrogen.
Determination of the X-ray Structures.
X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource beam-lines 9-2 and 12-2. Crystal structures were solved by molecular replacement with PHASER95 and 5VCC as a search model. Ligands were built with eLBOW96 and manually fit into the density with COOT.97 The initial models were rebuilt and refined with COOT and PHENIX.96 Polder omit electron density maps were calculated with PHENIX. Data collection and refinement statistics are summarized in Table S1. The atomic coordinates and structure factors for the 7- and 8-bound CYP3A4 were deposited to the Protein Data Bank with the identifier codes 7KS8 and 7KSA, respectively.
IC50 Determination Studies.
Cytochrome P450 inhibitor screening kits for CYP3A4, CYP1A2, and CYP2C9 were obtained from BioVision. Stock solutions of compounds 4, 7, and 9 were prepared at 5× concentrations in the provided assay buffer. Stock solutions were dispensed into triplicate wells of a 96-well plate and irradiated (tirr = 20 min, λirr = 460–470 nm) or left in the dark. Compounds 4 and 7 (100 μM to 100 nM) and compound 9 (100 μM to 100 nM) were evaluated following the manufacturer’s protocols. Percentage of enzyme activities was calculated from the initial linear slopes of the fluorescence vs time plots (first 5 min), using solvent control (no inhibitor, 1% MeCN in assay buffer) as 100% activity. The slope of the blank plot (no enzyme, 1% MeCN in assay buffer) was subtracted from each experimental slope value. Percent inhibition was expressed as the quotient of the blank subtracted experimental slopes over the blank subtracted solvent control slope. Igor Pro graphing software was used to produce % activity vs log(molarity) dose–response plots (Figures S15–S17), from which IC50 values were determined.
Biological Studies.
General Viability Assays.
DU-145 cells were seeded in a 96-well plate at a density of 7000 cells per well in 100 μL of Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS and 1000 units/mL penicillin/streptomycin. Each plate was incubated in a 37 °C humidified incubator ventilated with 5% CO2 overnight (16 h). The medium was aspirated from each well, and octuplicate wells were treated with medium containing 4 or 6–12 (5 μM) in DMEM medium with 1% DMSO. Plates also contained blank wells with no cells and control wells with DMEM medium containing 1% DMSO (vehicle). After 1 h of incubation at 37 °C, plates were removed from the incubator and the medium was aspirated and replaced with either vehicle or medium containing vinblastine (2.5–5 nM). The plates were then irradiated using a blue LED light source (tirr = 20 min, λirr = 460–470 nm) or left in the dark and incubated for 72 h in a 37 °C humidified incubator ventilated with 5% CO2. After incubation, MTT reagent (10 μL, 5 mg/mL in PBS) was added to each well, and plates were kept at 37 °C and 5% CO2 for 2 h. The medium was aspirated from each well, and DMSO (100 μL) was added. The wells were shaken for 30 min to allow solvation of the formazan crystals. Absorbance at 570 nm was measured in each well. Average absorbance values for the blank wells were subtracted from absorbance values for each sample to eliminate the background. Viability data were obtained by averaging blank-normalized absorbance values for control cells and expressing average absorbance for the treated samples as percent control.
EC50 Determination.
DU-145 human prostate cancer cells were seeded in a 96-well plate at a density of 7000 cells per well in 100 μL of Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS and 1000 units/mL penicillin/streptomycin. Each plate was incubated in a 37 °C humidified incubator ventilated with 5% CO2 overnight (16 h). The medium was aspirated from each well, and quadruplicate wells were treated with medium containing 4, 7, 9, 12 (25 μM–0.5 μM) or vinblastine (10 nM–0.5 nM) in 1% DMSO. Plates also contained blank wells with no cells and control wells with medium containing 1% DMSO. After 1 h of incubation at 37 °C, wells containing 4,7, 9, or 12 were aspirated and replaced with fresh medium. Wells with vinblastine were left alone. Plates were then irradiated with blue light (460–470 nm; 20 min) or left in the dark and incubated for 72 h in a 37 °C humidified incubator ventilated with 5% CO2. After incubation, MTT reagent (10 μL, 5 mg/mL in PBS) was added to each well, and plates were kept at 37 °C and 5% CO2 for 2 h. The medium was aspirated from each well, and DMSO (100 μL) was added. The wells were shaken for 30 min to allow for the solvation of the formazan crystals. Absorbance at 570 nm was measured in each well. Average absorbance values for the blank wells were subtracted from absorbance values for each sample to eliminate the background. Viability data were obtained by averaging normalized absorbance values for untreated cells and expressing absorbance for the treated samples as percent control. EC50 values were determined using Igor Pro graphing software or Compusyn software.
Chou–Talalay Synergy Determination.
DU-145 human prostate cancer cells were seeded in a 96-well plate at a density of 7000 cells per well in 100 μL of Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS and 1000 units/mL penicillin/streptomycin. Each plate was incubated in a 37 °C humidified incubator ventilated with 5% CO2 overnight (16 h). The medium was aspirated from each well and replaced with treatment medium containing compound 9 (10−1 μM) or vehicle (medium with 1% DMSO). Plates were than incubated for 1 h. After incubation the medium from each well was aspirated and replaced with medium containing vinblastine (10−0.5 nM) or vehicle, resulting in vinblastine alone and 9 alone monotreatments as well as combination treatments at each compound concentration, all in quadruplicate. Plates were then irradiated with blue light (460–470 nm; 20 min). After irradiation the plates were incubated in a 37 °C humidified incubator ventilated with 5% CO2 for 72 h. After incubation, MTT reagent (10 μL, 5 mg/mL in PBS) was added to each well, and plates were kept at 37 °C and 5% CO2 for 2 h. The medium was aspirated from each well, and DMSO (100 μL) was added. The wells were shaken for 30 min to allow for the solvation of the formazan crystals. Absorbance at 570 nm was measured in each well. Average absorbance values for the blank wells were subtracted from absorbance values for each sample to eliminate the background. Viability data were obtained by averaging normalized absorbance values for untreated cells and expressing absorbance for the treated samples as percent effect. Dose and effect data points were then inserted into the Compusyn software, which solved for the EC50 for both the monotreatments and the combination as well as the CI values for each treatment combination (Figure 8).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health Grants EB016072 (J.J.K. and C.T.) and ES025767 (I.F.S). C.T. and J.J.K. also thank the National Science Foundation (Grant CHE 1800395) and The Ohio State University and Wayne State University for partial support of this work. For instrumentation support we thank the NSF (Grant 0840413) and Penrose TherapeuTx. This work involves research carried out at the Stanford Synchrotron Radiation Lightsource. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (Grant P30GM133894).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c04155.
Spectral data for 4 and 6–11, inhibitory assays for other compounds, structural and cell viability data, and 7–11 stability studies (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c04155
The authors declare no competing financial interest.
Contributor Information
Nicholas Toupin, Department of Chemistry, Wayne State University, Detroit, Michigan 48202, United States.
Sean J. Steinke, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, United States
Thomas N. Rohrabaugh, Jr., Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, United States
Eric R. Samuels, Pharmaceutical Sciences, University of California, Irvine, California 92697, United States;
Claudia Turro, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, United States;.
Irina F. Sevrioukova, Molecular Biology and Biochemistry, University of California, Irvine, California 92697, United States;.
Jeremy J. Kodanko, Department of Chemistry, Wayne State University, Detroit, Michigan 48202, United States; Barbara Ann Karmanos Cancer Institute, Detroit, Michigan 48201, United States;.
REFERENCES
- (1).Chen Q; Wei D Human cytochrome P450 and personalized medicine. Adv. Exp. Med. Biol 2015, 827, 341–51. [DOI] [PubMed] [Google Scholar]
- (2).Guengerich FP; Shimada T Oxidation of toxic and carcinogenic chemicals by human cytochrome P-450 enzymes. Chem. Res. Toxicol 1991, 4 (4), 391–407. [DOI] [PubMed] [Google Scholar]
- (3).Li AP; Kaminski DL; Rasmussen A Substrates of human hepatic cytochrome P450 3A4. Toxicology 1995, 104 (1–3), 1–8. [DOI] [PubMed] [Google Scholar]
- (4).Guengerich FP Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol 1999, 39, 1–17. [DOI] [PubMed] [Google Scholar]
- (5).Mehmood Z; Kelly DE; Kelly SL Cytochrome P450 3A4 mediated metabolism of 2,4-dichlorophenol. Chemosphere 1997, 34 (11), 2281–2291. [DOI] [PubMed] [Google Scholar]
- (6).Chae YH; Yun CH; Guengerich FP; Kadlubar FF; el-Bayoumy K Roles of human hepatic and pulmonary cytochrome P450 enzymes in the metabolism of the environmental carcinogen 6-nitrochrysene. Cancer. Res 1993, 53 (9), 2028–2034. [PubMed] [Google Scholar]
- (7).Hodgson E In vitro human phase I metabolism of xenobiotics I: pesticides and related compounds used in agriculture and public health. J. Biochem. Mol. Toxicol 2003, 17 (4), 201–206. [DOI] [PubMed] [Google Scholar]
- (8).Mehmood Z; Williamson MP; Kelly DE; Kelly SL Human cytochrome P450 3A4 is involved in the biotransformation of the herbicide 2,4-dichlorophenoxyacetic acid. Environ. Toxicol. Pharmacol 1996, 2 (4), 397–401. [DOI] [PubMed] [Google Scholar]
- (9).Mehmood Z; Williamson MP; Kelly DE; Kelly SL Metabolism of organochlorine pesticides: the role of human cytochrome P450 3A4. Chemosphere 1996, 33 (4), 759–769. [DOI] [PubMed] [Google Scholar]
- (10).Xu L; Desai MC Pharmacokinetic enhancers for HIV drugs. Curr. Opin. Invest. Drugs 2009, 10 (8), 775–786. [PubMed] [Google Scholar]
- (11).Chapman SA; Lake KD; Solbrack DF; Milfred SK; Marshall PS; Kamps MA Considerations for using ketoconazole in solid organ transplant recipients receiving cyclosporine immunosuppression. J. Transpl. Coord 1996, 6 (3), 148–154. [DOI] [PubMed] [Google Scholar]
- (12).Kempf DJ; Marsh KC; Denissen JF; McDonald E; Vasavanonda S; Flentge CA; Green BE; Fino L; Park CH; Kong XP; et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. U. S. A 1995, 92 (7), 2484–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Brayer SW; Reddy KR Ritonavir-boosted protease inhibitor based therapy: a new strategy in chronic hepatitis C therapy. Expert Rev. Gastroenterol. Hepatol 2015, 9 (5), 547–558. [DOI] [PubMed] [Google Scholar]
- (14).Kempf DJ; Marsh KC; Kumar G; Rodrigues AD; Denissen JF; McDonald E; Kukulka MJ; Hsu A; Granneman GR; Baroldi PA; Sun E; Pizzuti D; Plattner JJ; Norbeck DW; Leonard JM Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother 1997, 41 (3), 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).van Eijk M; Boosman RJ; Schinkel AH; Huitema ADR; Beijnen JH Cytochrome P450 3A4, 3A5, and 2C8 expression in breast, prostate, lung, endometrial, and ovarian tumors: relevance for resistance to taxanes. Cancer Chemother. Pharmacol 2019, 84 (3), 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lolodi O; Wang YM; Wright WC; Chen T Differential Regulation of CYP3A4 and CYP3A5 and its Implication in Drug Discovery. Curr. Drug Metab 2018, 18 (12), 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Thummel KE; Wilkinson GR In vitro and in vivo drug interactions involving human CYP3A. Annu. Rev. Pharmacol. Toxicol 1998, 38, 389–430. [DOI] [PubMed] [Google Scholar]
- (18).Teo YL; Ho HK; Chan A Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: current understanding, challenges and recommendations. Br. J. Clin. Pharmacol 2015, 79 (2), 241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Breslin S; Lowry MC; O’Driscoll L Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br. J. Cancer 2017, 116 (5), 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ikezoe T; Hisatake Y; Takeuchi T; Ohtsuki Y; Yang Y; Said JW; Taguchi H; Koeffler HP HIV-1 protease inhibitor, ritonavir: a potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004, 64 (20), 7426–31. [DOI] [PubMed] [Google Scholar]
- (21).Martínez C; García-Martín E; Pizarro RM; García-Gamito FJ; Agúndez JAG Expression of paclitaxel-inactivating CYP3A activity in human colorectal cancer: implications for drug therapy. Br. J. Cancer 2002, 87 (6), 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zamora A; Denning CA; Heidary DK; Wachter E; Nease LA; Ruiz J; Glazer EC Ruthenium-containing P450 inhibitors for dual enzyme inhibition and DNA damage. Dalton Trans. 2017, 46 (7), 2165–2173. [DOI] [PubMed] [Google Scholar]
- (23).Li A; Yadav R; White JK; Herroon MK; Callahan BP; Podgorski I; Turro C; Scott EE; Kodanko JJ Illuminating cytochrome P450 binding: Ru(II)-caged inhibitors of CYP17A1. Chem. Commun 2017, 53 (26), 3673–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Karaoun N; Renfrew AK A luminescent ruthenium(ii) complex for light-triggered drug release and live cell imaging. Chem. Commun 2015, 51 (74), 14038–14041. [DOI] [PubMed] [Google Scholar]
- (25).White JK; Schmehl RH; Turro C An overview of photosubstitution reactions of Ru(II) imine complexes and their application in photobiology and photodynamic therapy. Inorg. Chim. Acta 2017, 454, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li A; Turro C; Kodanko JJ Ru(II) polypyridyl complexes as photocages for bioactive compounds containing nitriles and aromatic heterocycles. Chem. Commun 2018, 54, 1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hopkins SL; Bonnet S Ligand Photosubstitution Reactions with Ruthenium Compounds. In Applications in Chemical Biology and Medicinal Chemistry; Browne WR, Holder AA, Lawrence MA, Bullock JL, Lothar L Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2017; pp 91–116. [Google Scholar]
- (28).Farrer NJ; Salassa L; Sadler PJ Photoactivated chemotherapy (PACT): the potential of excited-state d-block metals in medicine. Dalton Trans. 2009, 48, 10690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Howerton BS; Heidary DK; Glazer EC Strained ruthenium complexes are potent light-activated anticancer agents. J. Am. Chem. Soc 2012, 134 (20), 8324–8327. [DOI] [PubMed] [Google Scholar]
- (30).Mari C; Pierroz V; Ferrari S; Gasser G Combination of Ru(II) complexes and light: new frontiers in cancer therapy. Chem. Sci 2015, 6 (5), 2660–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).van Rixel VHS; Ramu V; Auyeung AB; Beztsinna N; Leger DY; Lameijer LN; Hilt ST; Le Devedec SE; Yildiz T; Betancourt T; Gildner MB; Hudnall TW; Sol V; Liagre B; Kornienko A; Bonnet S Photo-Uncaging of a Microtubule-Targeted Rigidin Analogue in Hypoxic Cancer Cells and in a Xenograft Mouse Model. J. Am. Chem. Soc 2019, 141 (46), 18444–18454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Puckett CA; Ernst RJ; Barton JK Exploring the cellular accumulation of metal complexes. Dalton Trans. 2010, 39 (5), 1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Puckett CA; Barton JK Mechanism of Cellular Uptake of a Ruthenium Polypyridyl Complex. Biochemistry 2008, 47 (45), 11711–11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mulcahy SP; Li S; Korn R; Xie X; Meggers E Solid-phase synthesis of tris-heteroleptic ruthenium(II) complexes and application to acetylcholinesterase inhibition. Inorg. Chem 2008, 47 (12), 5030–5032. [DOI] [PubMed] [Google Scholar]
- (35).Meggers E Targeting proteins with metal complexes. Chem. Commun 2009, 9, 1001–1010. [DOI] [PubMed] [Google Scholar]
- (36).Respondek T; Sharma R; Herroon MK; Garner RN; Knoll JD; Cueny E; Turro C; Podgorski I; Kodanko JJ Inhibition of Cathepsin Activity in a Cell-Based Assay by a Light-Activated Ruthenium Compound. ChemMedChem 2014, 9, 1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ramalho SD; Sharma R; White JK; Aggarwal N; Chalasani A; Sameni M; Moin K; Vieira PC; Turro C; Kodanko JJ; Sloane BF Imaging Sites of Inhibition of Proteolysis in Pathomimetic Human Breast Cancer Cultures by Light-Activated Ruthenium Compound. PLoS One 2015, 10 (11), e0142527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hidayatullah AN; Wachter E; Heidary DK; Parkin S; Glazer EC Photoactive Ru(II) Complexes With Dioxinophenanthroline Ligands Are Potent Cytotoxic Agents. Inorg. Chem 2014, 53 (19), 10030–10032. [DOI] [PubMed] [Google Scholar]
- (39).Lee J; Udugamasooriya DG; Lim H-S; Kodadek T Potent and selective photo-inactivation of proteins with peptoid-ruthenium conjugates. Nat. Chem. Biol 2010, 6 (4), 258–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Allison RR; Downie GH; Cuenca R; Hu X-H; Childs CJH; Sibata CH Photosensitizers in clinical PDT. Photodiagn. Photodyn. Ther 2004, 1 (1), 27–42. [DOI] [PubMed] [Google Scholar]
- (41).Smith NA; Sadler PJ Photoactivatable metal complexes: from theory to applications in biotechnology and medicine. Philos. Trans. R. Soc., A 2013, 371 (1995), 20120519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Schatzschneider U Photoactivated Biological Activity of Transition-Metal Complexes. Eur. J. Inorg. Chem 2010, 2010 (10), 1451–1467. [Google Scholar]
- (43).Frei A; Rubbiani R; Tubafard S; Blacque O; Anstaett P; Felgentrager A; Maisch T; Spiccia L; Gasser G Synthesis, Characterization, and Biological Evaluation of New Ru(II) Polypyridyl Photosensitizers for Photodynamic Therapy. J. Med. Chem 2014, 57 (17), 7280–7292. [DOI] [PubMed] [Google Scholar]
- (44).Clarke MJ Ruthenium Metallopharmaceuticals. Coord. Chem. Rev 2003, 236 (1–2), 209–233. [Google Scholar]
- (45).Alessio E; Mestroni G; Bergamo A; Sava G Ruthenium anticancer drugs. Met. Ions Biol. Syst 2004, 42, 323–351. [PubMed] [Google Scholar]
- (46).Hartinger CG; Zorbas-Seifried S; Jakupec MA; Kynast B; Zorbas H; Keppler BK From bench to bedside - preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem 2006, 100 (5–6), 891–904. [DOI] [PubMed] [Google Scholar]
- (47).Alessio E; Mestroni G; Bergamo A; Sava G Ruthenium antimetastatic agents. Curr. Top. Med. Chem 2004, 4 (15), 1525–1535. [DOI] [PubMed] [Google Scholar]
- (48).Arenas Y; Monro S; Shi G; Mandel A; McFarland S; Lilge L Photodynamic inactivation of Staphylococcus aureus and methicillin-resistant Staphylococcus aureus with Ru(II)-based type I/type II photosensitizers. Photodiagn. Photodyn. Ther 2013, 10 (4), 615–625. [DOI] [PubMed] [Google Scholar]
- (49).Fong J; Kasimova K; Arenas Y; Kaspler P; Lazic S; Mandel A; Lilge L A novel class of ruthenium-based photosensitizers effectively kills in vitro cancer cells and in vivo tumors. Photochem. Photobiol. Sci 2015, 14 (11), 2014–2023. [DOI] [PubMed] [Google Scholar]
- (50).Kaspler P; Lazic S; Forward S; Arenas Y; Mandel A; Lilge L A ruthenium(II) based photosensitizer and transferrin complexes enhance photo-physical properties, cell uptake, and photodynamic therapy safety and efficacy. Photochem. Photobiol. Sci 2016, 15 (4), 481–495. [DOI] [PubMed] [Google Scholar]
- (51).Sevrioukova IF; Poulos TL Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (43), 18422–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Sevrioukova IF; Poulos TL Pyridine-Substituted Desoxyritonavir Is a More Potent Inhibitor of Cytochrome P450 3A4 than Ritonavir. J. Med. Chem 2013, 56 (9), 3733–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Samuels ER; Sevrioukova I Inhibition of Human CYP3A4 by Rationally Designed Ritonavir-Like Compounds: Impact and Interplay of the Side Group Functionalities. Mol. Pharmaceutics 2018, 15 (1), 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rock BM; Hengel SM; Rock DA; Wienkers LC; Kunze KL Characterization of Ritonavir-Mediated Inactivation of Cytochrome P450 3A4. Mol. Pharmacol 2014, 86 (6), 665. [DOI] [PubMed] [Google Scholar]
- (55).Ekroos M; Sjögren T Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (37), 13682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sevrioukova I Interaction of Human Drug-Metabolizing CYP3A4 with Small Inhibitory Molecules. Biochemistry 2019, 58 (7), 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kaur P; Chamberlin AR; Poulos TL; Sevrioukova IF Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J. Med. Chem 2016, 59 (9), 4210–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Samuels ER; Sevrioukova IF An increase in side-group hydrophobicity largely improves the potency of ritonavir-like inhibitors of CYP3A4. Biorg. Bioorg. Med. Chem 2020, 28 (6), 115349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Samuels ER; Sevrioukova IF Rational Design of CYP3A4 Inhibitors: A One-Atom Linker Elongation in Ritonavir-Like Compounds Leads to a Marked Improvement in the Binding Strength. Int. J. Mol. Sci 2021, 22 (2), 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Knoll JD; Albani BA; Durr CB; Turro C Unusually Efficient Pyridine Photodissociation from Ru(II) Complexes with Sterically Bulky Bidentate Ancillary Ligands. J. Phys. Chem. A 2014, 118 (45), 10603–10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Loftus LM; White JK; Albani BA; Kohler L; Kodanko JJ; Thummel RP; Dunbar KR; Turro C New RuII Complex for Dual Activity: Photoinduced Ligand Release and 1O2 Production. Chem. - Eur. J 2016, 22 (11), 3704–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Li A; White JK; Arora K; Herroon MK; Martin PD; Schlegel HB; Podgorski I; Turro C; Kodanko JJ Selective Release of Aromatic Heterocycles from Ruthenium Tris(2-pyridylmethyl)amine with Visible Light. Inorg. Chem 2016, 55 (1), 10–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Samuels ER; Sevrioukova IF Direct synthesis of α-thio aromatic acids from aromatic amino acids. Tetrahedron Lett. 2018, 59 (12), 1140–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Huisman M; White JK; Lewalski VG; Podgorski I; Turro C; Kodanko JJ Caging the uncageable: using metal complex release for photochemical control over irreversible inhibition. Chem. Commun 2016, 52 (85), 12590–12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Arora K; Herroon M; Al-Afyouni MH; Toupin NP; Rohrabaugh TN; Loftus LM; Podgorski I; Turro C; Kodanko JJ Catch and Release Photosensitizers: Combining Dual-Action Ruthenium Complexes with Protease Inactivation for Targeting Invasive Cancers. J. Am. Chem. Soc 2018, 140 (43), 14367–14380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Bahreman A; Limburg B; Siegler MA; Koning R; Koster AJ; Bonnet S Ruthenium Polypyridyl Complexes Hopping at Anionic Lipid Bilayers through a Supramolecular Bond Sensitive to Visible Light. Chem. - Eur. J 2012, 18 (33), 10271–10280. [DOI] [PubMed] [Google Scholar]
- (67).Bahreman A; Limburg B; Siegler MA; Bouwman E; Bonnet S Spontaneous Formation in the Dark, and Visible Light-Induced Cleavage, of a Ru–S Bond in Water: A Thermodynamic and Kinetic Study. Inorg. Chem 2013, 52 (16), 9456–9469. [DOI] [PubMed] [Google Scholar]
- (68).Toupin NP; Nadella S; Steinke SJ; Turro C; Kodanko JJ Dual-Action Ru(II) Complexes with Bulky π-Expansive Ligands: Phototoxicity without DNA Intercalation. Inorg. Chem 2020, 59 (6), 3919–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Knoll JD; Albani BA; Turro C New Ru(II) Complexes for Dual Photoreactivity: Ligand Exchange and 1O2 Generation. Acc. Chem. Res 2015, 48 (8), 2280–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Knoll JD; Albani BA; Turro C Excited state investigation of a new Ru(II) complex for dual reactivity with low energy light. Chem. Commun 2015, 51 (42), 8777–8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Albani BA; Pena B; Leed NA; de Paula NABG; Pavani C; Baptista MS; Dunbar KR; Turro C Marked Improvement in Photoinduced Cell Death by a New Tris-heteroleptic Complex with Dual Action: Singlet Oxygen Sensitization and Ligand Dissociation. J. Am. Chem. Soc 2014, 136 (49), 17095–17101. [DOI] [PubMed] [Google Scholar]
- (72).Rohrabaugh TN Jr.; Rohrabaugh AM; Kodanko JJ; White JK; Turro C Photoactivation of imatinib-antibody conjugate using low-energy visible light from Ru(II)-polypyridyl cages. Chem. Commun 2018, 54 (41), 5193–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Li A; Yadav R; White JK; Herroon MK; Callahan BP; Podgorski I; Turro C; Scott EE; Kodanko JJ Illuminating cytochrome P450 binding: Ru (II)-caged inhibitors of CYP17A1. Chem. Commun 2017, 53 (26), 3673–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Li A; White JK; Arora K; Herroon MK; Martin PD; Schlegel HB; Podgorski I; Turro C; Kodanko JJ Selective Release of Aromatic Heterocycles from Ruthenium Tris (2-pyridylmethyl) amine with Visible Light. Inorg. Chem 2016, 55 (1), 10–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Feng BY; Shoichet BK A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc 2006, 1 (2), 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Paine MF; Hart HL; Ludington SS; Haining RL; Rettie AE; Zeldin DC The human intestinal cytochrome P450 “pie”. Drug Metab. Dispos 2006, 34 (5), 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Blagosklonny MV; Dixon SC; Figg WD Efficacy of microtubule-active drugs followed by ketoconazole in human metastatic prostate cancer cell lines. J. Urol 2000, 163 (3), 1022–1026. [PubMed] [Google Scholar]
- (78).Ikezoe T; Hisatake Y; Takeuchi T; Ohtsuki Y; Yang Y; Said JW; Taguchi H; Koeffler HP HIV-1 Protease Inhibitor, RitonavirA Potent Inhibitor of CYP3A4, Enhanced the Anticancer Effects of Docetaxel in Androgen-Independent Prostate Cancer Cells In vitro and In vivo. Cancer Res. 2004, 64 (20), 7426. [DOI] [PubMed] [Google Scholar]
- (79).Zhong D; Wu H; Wu Y; Li Y; Yang J; Gong Q; Luo K; Gu Z Redox dual-responsive dendrimeric nanoparticles for mutually synergistic chemo-photodynamic therapy to overcome drug resistance. J. Controlled Release 2021, 329 (10), 1210–1221. [DOI] [PubMed] [Google Scholar]
- (80).Chou T-C Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70 (2), 440–446. [DOI] [PubMed] [Google Scholar]
- (81).Duarte D; Vale N New Trends for Antimalarial Drugs: Synergism between Antineoplastics and Antimalarials on Breast Cancer Cells. Biomolecules 2020, 10, 1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Gallant J-N; Allen JE; Smith CD; Dicker DT; Wang W; Dolloff NG; Navaraj A; El-Deiry WS Quinacrine synergizes with 5-fluorouracil and other therapies in colorectal cancer. Cancer Biol. Ther 2011, 12 (3), 239–251. [DOI] [PubMed] [Google Scholar]
- (83).Chi R.-p. A.; van der Watt P; Wei W; Birrer MJ; Leaner VD Inhibition of Kpnβ1 mediated nuclear import enhances cisplatin chemosensitivity in cervical cancer. BMC Cancer 2021, 21, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Bomben PG; Robson KCD; Sedach PA; Berlinguette CP On the Viability of Cyclometalated Ru(II) Complexes for Light-Harvesting Applications. Inorg. Chem 2009, 48 (20), 9631–9643. [DOI] [PubMed] [Google Scholar]
- (85).Thomas RA; Tsai CN; Mazumder S; Lu IC; Lord RL; Schlegel HB; Chen YJ; Endicott JF Energy Dependence of the Ruthenium(II)-Bipyridine Metal-to-Ligand-Charge-Transfer Excited State Radiative Lifetimes: Effects of ππ*(bipyridine) Mixing. J. Phys. Chem. B 2015, 119 (24), 7393–7406. [DOI] [PubMed] [Google Scholar]
- (86).Garner RN; Joyce LE; Turro C Effect of electronic structure on the photoinduced ligand exchange of Ru(II) polypyridine complexes. Inorg. Chem 2011, 50 (10), 4384–4391. [DOI] [PubMed] [Google Scholar]
- (87).McConnell AJ; Lim MH; Olmon ED; Song H; Dervan EE; Barton JK Luminescent Properties of Ruthenium(II) Complexes with Sterically Expansive Ligands Bound to DNA Defects. Inorg. Chem 2012, 51 (22), 12511–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Pena B; Leed NA; Dunbar KR; Turro C Excited state dynamics of two new Ru(II) cyclometallated dyes: relation to cells for solar energy conversion and comparison to conventional systems. J. Phys. Chem. C 2012, 116 (42), 22186–22195. [Google Scholar]
- (89).Habtemariam A; Melchart M; Fernández R; Parsons S; Oswald IDH; Parkin A; Fabbiani FPA; Davidson JE; Dawson A; Aird RE; Jodrell DI; Sadler PJ Structure–Activity Relationships for Cytotoxic Ruthenium(II) Arene Complexes Containing N,N-, N,O-, and O,O-Chelating Ligands. J. Med. Chem 2006, 49 (23), 6858–6868. [DOI] [PubMed] [Google Scholar]
- (90).Kender WT; Turro C Unusually Slow Internal Conversion in N-Heterocyclic Carbene/Carbanion Cyclometallated Ru(II) Complexes: A Hammett Relationship. J. Phys. Chem. A 2019, 123, 2650–2660. [DOI] [PubMed] [Google Scholar]
- (91).Dovletoglou A; Adeyemi SA; Meyer TJ Coordination and Redox Chemistry of Substituted-Polypyridyl Complexes of Ruthenium. Inorg. Chem 1996, 35 (14), 4120–4127. [DOI] [PubMed] [Google Scholar]
- (92).Montalti M; Credi A; Prodi L; Gandolfi MT Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, FL, 2006. [Google Scholar]
- (93).Rohrabaugh TN Jr.; Collins KA; Xue C; White JK; Kodanko JJ; Turro C New Ru(II) complex for dual photochemotherapy: release of cathepsin K inhibitor and 1O2 production. Dalton Trans. 2018, 47, 11851–11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Sevrioukova IF High-Level Production and Properties of the Cysteine-Depleted Cytochrome P450 3A4. Biochemistry 2017, 56 (24), 3058–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40 (4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macro-molecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.