

Abstract
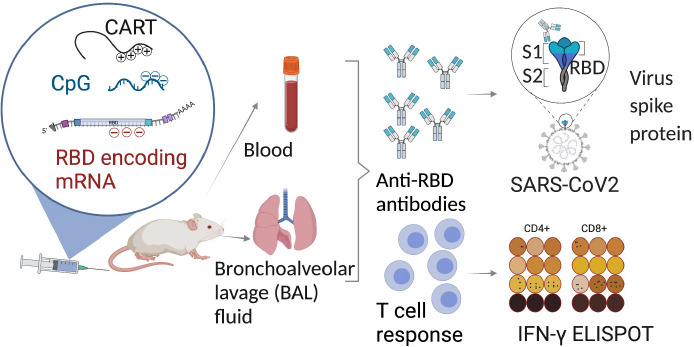
The SARS-CoV-2 pandemic has necessitated the rapid development of prophylactic vaccines. Two mRNA vaccines have been approved for emergency use by the FDA and have demonstrated extraordinary effectiveness. The success of these mRNA vaccines establishes the speed of development and therapeutic potential of mRNA. These authorized vaccines encode full-length versions of the SARS-CoV-2 spike protein. They are formulated with lipid nanoparticle (LNP) delivery vehicles that have inherent immunostimulatory properties. Different vaccination strategies and alternative mRNA delivery vehicles would be desirable to ensure flexibility of future generations of SARS-CoV-2 vaccines and the development of mRNA vaccines in general. Here, we report on the development of an alternative mRNA vaccine approach using a delivery vehicle called charge-altering releasable transporters (CARTs). Using these inherently nonimmunogenic vehicles, we can tailor the vaccine immunogenicity by inclusion of coformulated adjuvants such as oligodeoxynucleotides with CpG motifs (CpG-ODN). Mice vaccinated with the mRNA-CART vaccine developed therapeutically relevant levels of receptor binding domain (RBD)-specific neutralizing antibodies in both the circulation and in the lung bronchial fluids. In addition, vaccination elicited strong and long-lasting RBD-specific TH1 T cell responses including CD4+ and CD8+ T cell memory.
Short abstract
Charge-altering releasable transporters effectively deliver coformulated SARS-CoV-2 RBD mRNA and adjuvant. Prime-boost immunization leads to robust antigen-specific B and T cell responses in mice.
Introduction
Coronavirus pandemics have been a growing concern for more than a decade, and several attempts have been made to develop vaccines against SARS-CoV-1 and the Middle Eastern Respiratory Syndrome (MERS).1 The global spread of the SARS-CoV-2 virus stimulated worldwide efforts to leverage previous insights from coronavirus vaccines to develop safe, effective, and scalable vaccines to alleviate the COVID-19 pandemic. While a variety of vaccine candidates and approaches are being investigated worldwide,2 the extraordinary pace of development and implementation of mRNA vaccines3−6 illustrates the potential of this emerging technology. The mRNA vaccines granted emergency use authorization by the FDA against SARS-Cov-2 represent a triumph of basic and applied science as these advances enabled the most rapid clinical translation from concept to clinical trial ever for a vaccine.5,6 mRNA is transiently expressed, does not integrate into the genome, and is eliminated through natural degradation mechanisms in the body. mRNA vaccines offer a flexible and fast design that will allow for subsequent generations of products to address the emergence of new virus variants. The currently approved mRNA vaccines5,6 generated by in vitro transcription use chemically modified nucleotides incorporated in mRNAs encoding the full viral spike protein, usually containing 2 structural epitope mutations, formulated in lipid nanoparticles (LNPs) and are administered intramuscularly. Despite their extraordinary success, the underlying science that contributes to the most effective, safe, and scalable vaccine against COVID-19 continues to evolve. Modifications and sequence optimization of the mRNA and the particular components of the delivery vehicle can influence the immunological response.7 Previous studies of SARS-CoV and MERS have shown that the proper choice of encoded antigen is critical8 to avoid potential complications from the antibody-dependent enhancement (ADE) of disease.9 The chemistry of the delivery vehicle is also important as the ionizable lipids that are a component of LNPs act as adjuvants but can induce adverse events,10 and the use of polyethylene glycol (PEG) in the LNP formulations can contribute to allergic reactions.11,12
These continuing challenges and the degree to which the global scale of the COVID-19 pandemic has strained supply chains for the existing LNP technologies highlight the need to develop alternative approaches, (i) to enhance the resilience of the global response in the face of this and future pandemics, (ii) to test alternative approaches that might lead to the most effective and durable immunological responses, as well as (iii) to be able to respond rapidly to emerging new virus variants.13
In this study, we present an alternative 3-component mRNA vaccine utilizing mRNA encoding the receptor binding domain (RBD) of the SARS-CoV-2 spike protein formulated with a highly efficient, nontoxic, PEG-free mRNA delivery platform called charge-altering releasable transporters (CARTs)14−16 and a TLR9 agonist (CpG) as a coformulated adjuvant16,17 (Figure 1A). Prior studies had indicated that the RBD is a promising antigen target18−20 as antibodies elicited against RBD are often strongly neutralizing. CARTs offer a promising new gene delivery platform and have proven to be effective deliverers of mRNA vaccines in preclinical mouse studies.16,21 CARTs are single-component amphiphilic diblock oligomers containing a sequence of lipid monomers and a sequence of cationic monomers (Figure 1B, Figure S1A). They are readily produced on scale in a two-step organocatalytic oligomerization. CARTs electrostatically encapsulate mRNA (or other coformulated nucleotides like CpG) and deliver the genetic cargo into cells (Figure S1B). A unique feature of CARTs is their ability to undergo a charge-altering rearrangement to produce neutral diketopiperazine small molecules (DKPs). This transformation facilitates the release of mRNA (Figure 1C) and eliminates any toxicity associated with persistent cations.22 Previous observations had shown that, upon intravenous (IV) injection, CARTs containing hydroxyethyl glycine repeating cation units selectively deliver mRNA to the spleen,14−16 whereas intramuscular (IM) injections of these same products result in mRNA translation locally in the injected muscle (Figure 1D). Bioluminescence studies with Firefly luciferase (fLuc) mRNA indicate that in vivo protein expression is greater in the spleen after IV injection than in the muscle after IM injection. In either site, expression peaks after 4–6 h (Figure 1E) and decreases over a period of 3–4 days. Moreover, CARTs containing unsaturated lipid blocks exhibit the enhanced transfection of antigen presenting cells15 motivating our choice of such a CART for the COVID-19 vaccine. The CART delivery vehicle does not induce nonspecific immune stimulation by itself.16,21 This property allows the coformulation of oligodeoxynucleotide adjuvants such as the TLR9 agonist CpG-ODN to tune the induced immune response.
Figure 1.

CART delivery platform methodology effectively complexes, delivers, and releases mRNA via both systemic and local administration. (A) CART electrostatic formulation, cellular uptake, endosomal escape, and translation of SARS-CoV-2 RBD mRNA. (B) CART synthesis via ring-opening polymerization (DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TU = 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea). (C) CART chemical structure, degradation products, and charge-altering mechanism. (D) In vivo luciferase reporter gene expression via systemic IV administration (left, 5 μg of fLuc mRNA), and local IM administration (2.5 μg of fLuc mRNA each flank). (E) Quantification of in vivo mRNA expression at 4 h postadministration.
Here, we show that the coformulation of a CART with mRNA encoding SARS-CoV-2 RBD with the TLR9 agonist CpG (RBD mRNA + CpG-CART) induces robust neutralizing antibodies and RBD-specific T cell responses in mice. Moreover, we detect significant levels of these antibodies and memory T cells in the spleen and lung of vaccinated animals.
Results
RBD mRNA + CpG-CART Vaccination Elicits Anti-RBD-Specific Antibodies at Day 4 after Immunization
An mRNA encoding the receptor binding domain (RBD)23 of SARS-CoV-2 was made by in vitro transcription and based on the published sequence of the virus. A His tag was included to allow for protein detection of the translated mRNA product as a quality control step. The resulting mRNA contained the optimal CAP-1 structure; uridines were replaced with modified N1-methyl-pseudouridine, and cytidines were replaced with 5-methylcytidine to maximize mRNA stability and translation. Protein expression was verified by Western Blot in transfected 293F and HeLa cells (Figure S2A). To demonstrate that the administration of mRNA-CART complexes does not lead to nonspecific immune stimulatory effects when formulated with optimally modified mRNA,24 mice were injected intravenously (IV) with mRNA-CART and monitored for the activation of innate immune cells, a test that we have found to be most sensitive. mRNA-CART complexes were free of such nonspecific stimulatory effects when formulated with modified mRNA. However, when formulated with unmodified mRNA, nonspecific activation of innate immune cell subsets could be observed, demonstrating that the property of nonspecific immune stimulation is dependent on the cargo and not the CARTs (Figure S2B). We were then able to direct the immune response of our candidate vaccine by coformulating the mRNA-CART complexes with CpG oligonucleotides that trigger endosomal TLR9 receptors in antigen presenting cells. These CpG entities are known to be safe and effective vaccine adjuvants.17
We assessed the effects of the RBD mRNA-CART vaccination with or without the addition of CpG as the adjuvant. IV injection of CARTs effectively delivers the cargo to antigen presenting cells (APCs) such as B cells, macrophages, and dendritic cells in the spleen15,16 (Figure 1D). This route of administration of the mRNA-CART vaccination had proven effective in previous studies of therapeutic cancer vaccination.16 Based on this knowledge, we evaluated our vaccine first by IV administration. Mice were primed on day 0 and received two boosts on day 4 and on day 8 with 3 μg of RBD mRNA-CART formulated with or without 3 μg of CpG (Figure 2A). As a control, a group that received 3 μg of CpG-CART alone with no mRNA and a control group treated with phosphate buffered saline (PBS) were included. Mice vaccinated with RBD mRNA-CART plus CpG developed detectable levels of anti-RBD IgG and IgM as early as 4 days after vaccination (Figure S3A). Over time, we observed an increase in the levels of anti-RBD antibodies in the serum of both mRNA-treated groups compared to controls (Figure 2B). Importantly, the antibodies induced by our vaccine were specific for RBD and did not cross react to an irrelevant His tagged protein (GFP-His) (Figure S3B). Notably, RBD-specific antibody responses in mice vaccinated with the formulation including CpG consistently exceeded those observed in groups treated without CpG (Figure 2B,D).
Figure 2.

Addition of CpG to RBD mRNA-CART elicits a stronger anti-RBD immunoglobulin response and leads to earlier isotype switching. (A) BALB/c mice were immunized intravenously with either 3 μg of RBD mRNA-CART (n = 5), 3 μg of RBD mRNA-CART plus 3 μg of CpG (n = 5), 3 μg of CpG CART (n = 5), or Naïve untreated (n = 5) and boosted on day 4 and day 8 after priming. (B) Serum levels of RBD-specific IgGs from RBD mRNA-CART (black), RBD mRNA + CpG-CART (blue), CpG CART (orange), and Naïve (red) mice were monitored over the course of 60 days postpriming by ELISA. (C) On day 14 and day 60 after priming, the distribution of IgG isotypes specific to RBD was analyzed using antimouse IgG1 (blue), IgG2a (black), IgG2b (red), and IgG3 (gray) monoclonal antibodies by ELISA. (D) On day 14 and day 60 after priming, the absolute concentration of the anti-RBD IgG was evaluated. * = P < 0.05 unpaired Student’s t test (two-tailed). Data representative of 2 individual experiments.
Addition of CpG Results in Early Isotype Switching of RBD-Specific Antibody Responses
By day 14, we observed that the CpG coformulated vaccine induced an RBD-specific Ig response that had undergone isotype switching from IgG1 to IgG2a, IgG2b, and IgG3. By contrast, mice receiving the vaccine formulation without coformulated CpG produced predominantly unswitched IgG1 (Figure 2C and Figure S4A,B). By day 60, these differences were less apparent; however, we detected higher levels of all classes of RBD-specific antibodies in the serum of mice vaccinated with the CpG-containing vaccine (Figure 2C and Figure S4B).
Antibodies Induced by RBD mRNA + CpG-CART Vaccination Inhibit RBD-ACE2 Binding and Neutralize Pseudotyped Viral Entry
The induced antisera were next tested for their ability to inhibit the RBD-ACE2 interaction (Figure 3A) and to block pseudotyped viral entry into ACE2-expressing target cells (Figure 3B). Functional RBD-ACE2 receptor blocking was assayed both against the binding of RBD protein to the ACE2-expressing target cell by flow cytometry (data not shown) and by binding to the solid phase coated with ACE2 protein. On day 28, we observed a striking difference in the level of neutralizing antibodies between the group that had received the vaccine containing the CpG as compared to the group vaccinated without the adjuvant. While antisera from RBD mRNA + CpG-CART vaccinated mice displayed a high degree of receptor blocking and pseudotyped viral neutralization with IC50 of 1:500 and 1:18 000, respectively, the antisera from mice vaccinated with RBD mRNA-CART were only barely positive above the background (Figure 3A,B). However, by day 60 antisera from mice vaccinated with RBD mRNA-CART were able to block RBD-ACE2 receptor binding and neutralize pseudotyped viral entry, although to a lesser extent than antisera from mice vaccinated with RBD mRNA + CpG-CART (Figure 3A,B). The results from the receptor interaction blocking assay and the pseudovirus neutralization assay were well correlated.
Figure 3.

RBD mRNA + CpG-CART generates early high levels of RBD neutralizing antibodies. BALB/c mice (n = 5) were immunized as described in Figure 2A. (A) Sera from mice immunized with RBD mRNA-CART (black), RBD mRNA + CpG-CART (blue), and CpG CART (orange) were collected on D28 and D60 and tested in a commercially available RBD-ACE-2 inhibition assay. The same set of serum samples was tested in a pseudotyped virus neutralization assay. RBD-expressing pseudovirus particles containing a zsGreen and firefly luciferase vectors were coincubated with titrated concentrations of heat-inactivated mouse serum. (B) The pseudovirus particle–serum mix was then added to wells containing ACE-2-overexpressing 293F cells. Firefly luciferase expression was measured at 48 and 72 h after the start of the experiment. On D60, BAL was harvested from RBD mRNA-CART (black), RBD mRNA + CpG-CART (blue), and CpG-CART (orange) immunized mice. (C) RBD-specific total IgG was assayed by ELISA. (D) BAL containing immunoglobulins was tested for their ability to inhibit binding of RBD to hACE-2 using a commercial ACE-2 inhibition kit. (E) Lung single-cell suspensions from naive mice (gray, n = 3) or mice vaccinated on D0 and D21 IV with 3 μg of RBD-mRNA + 3 μg of CpG (blue, n = 3) were collected on D28 and incubated with media alone, RBD protein, or an irrelevant protein (CD81-His) [5 μg/mL] for 48 h and stained for T cell activation markers CD134, CD137, and intracellular TNFα on CD4+ T cells. Data are shown as mean ± SD. Data representative of 3 independent experiments (B–D) and 1 experiment (E). * = P < 0.05, ** = P < 0.01 *** = P < 0.001, **** = P < 0.0001 one-way ANOVA (Tukey’s multiple comparison test) (D) or two-way ANOVA (Tukey’s multiple comparison test) (E).
RBD-Specific Antibodies in Bronchoalveolar Lavage of Vaccinated Mice
To evaluate the presence of RBD-specific immunoglobulin in the lungs of vaccinated mice, we collected bronchoalveolar lavage (BAL) on D60 after treatment. RBD-specific Ig was detected in BAL from both RBD mRNA + CpG-CART and RBD mRNA-CART vaccinated mice by ELISA (Figure 3C). Notably, as BAL is collected by flushing lungs with 2 mL of sterile PBS, the Ig titers represent highly diluted samples. Importantly, although diluted, these immunoglobulins from BAL of both vaccinated groups blocked RBD-ACE2 binding on D60 (Figure 3D).
RBD-Specific CD4+ T Cells in the Lung of Vaccinated Mice
In mice, Ig class switching is linked to TH1 T cell responses.25,26 To evaluate the vaccine induced T cell responses, we prepared single-cell suspensions from the lungs of IV vaccinated mice on D28 and cultured the cells for 48 h in media alone or in the presence of soluble RBD-His protein or a control protein (hCD81-His). Cells were then collected and assayed by flow cytometry for T cell activation using fluorochrome conjugated monoclonal antibodies for memory and activation-specific surface proteins and intracellular cytokines. Remarkably, upon RBD protein restimulation, a defined RBD-specific CD4+/CD44high/CD134+ and CD4+/CD44high/TNFα+ activated T cell subset could be identified in lung cell suspensions from mice vaccinated with RBD mRNA + CpG-CART that could not be detected when cells were cultured with media alone or in the presence of the control protein (Figure 3E). TNFα secretion by CD4+ T cells is associated with a TH1 polarization.
RBD mRNA + CpG-CART Vaccine Elicits Robust Anti-RBD Ig Responses by an Intravenous, Intramuscular, and Subcutaneous Route of Administration
To test the efficacy of our vaccine in relation to the route of administration (ROA), we compared RBD-specific Ig responses induced by immunizations given IV, IM, or SC. Mice were primed with 3 μg of RBD mRNA and 3 μg of CpG formulated in CARTs on day 0 and received a boost on day 8 (Figure S5A). All routes of administration led to detectable neutralizing antibody titers at the analyzed time points D14 and D28, with no significant differences between IM and SC administration (Figure S5B). There was a tendency toward higher titers in IV vaccinated mice. Isotype switching occurred independent of the route of administration (Figure S5C).
RBD mRNA + CpG-CART Vaccine Induces Robust RBD-Specific Ig Responses in a Clinically Relevant Prime-Boost Regimen and Is Independent of CpG Source
Guided by the immunization regimen chosen by the currently approved SARS-CoV-2 vaccines,27 mice were primed with the vaccine on D0 and boosted on D21. Mice were immunized either IV or IM with 3 μg of RBD mRNA + CpG-CART (Figure 4A). Confirming previous observations, robust responses were observed for both groups. IV immunized mice showed higher—statistically not significant—titers of anti-RBD antibodies in serum and in the BAL on both D21 and D28 (Figure 4B–D). Antisera from both groups effectively inhibited RBD-ACE2 binding on D21 (Figure S6B), although substantially more effective on D28 reflecting the difference observed in total RBD-specific IgGs between the two groups (Figure 4C). Moreover, robust antibody responses against the complete spike protein were observed in both groups, although higher in the intravenous group (Figure S6C). Thus, the vaccine induced anti-RBD response is primarily directed toward exposed RBD epitopes in the complete spike protein. Importantly, although CpG is required for robust isotype switched anti-RBD immunoglobulin, the response is independent of the source of CpG. We tested 4 different sources of CpG, three of the C-subclass of CpGs (CpG-C) and one of the B-subclass (CpG-B). No significant difference was observed between the different CpG-Cs (Figures S6 and S7), while the B-subclass CpG underperformed compared to the CpG-Cs (data not shown).
Figure 4.

RBD mRNA + CpG-CART elicits neutralizing anti-RBD immunoglobulin responses after IV and IM vaccination. (A) BALB/c mice (n = 5 per group) were immunized intravenously (IV) or intramuscularly (IM) with 3 μg of RBD mRNA plus 3 μg of CpG and boosted on day 21 after priming. (B, C) RBD-specific immunoglobulin titers in serum were measured and quantified on day 21 and day 28. (D) On day 28, BAL was harvested from both IV and IM treated mice, and anti-RBD immunoglobulins were assayed by ELISA. Data are shown as mean ± SD. Data representative of 2 independent experiments. Statistical significance was assessed by a Student’s t test (two-tailed, unpaired) ns = P > 0.05.
RBD mRNA + CpG-CART Vaccine Induced Long-Lasting TH1 CD4+ and CD8+ T Cell Memory
Splenocytes from mice that had received 3 μg of RBD-mRNA + 3 μg of CpG-CART or 3 μg of Ctrl mRNA + 3 μg of CpG-CART either IV or IM on D1 and D21 were harvested on day 105 after vaccination and characterized for T cell responses by an IFNγ enzyme-linked immunosorbent spot assay (ELISpot). In this assay, pooled splenocytes were enriched for either CD4+ or CD8+ T cells and cultured overnight with a SARS-CoV-2 RBD peptide pool or media alone. Significant IFNγ responses in CD4+ and CD8+ T cells were detected by both IV and IM vaccination. Since the route of administration of IV vaccinated mice targets the spleen, it is expected that spleen T cells would give a stronger response. On the other hand, it is remarkable to detect responding T cells 105 days after vaccination (Figure 5A,B).
Figure 5.

RBD mRNA plus CpG vaccination induces long-lasting memory TH1 CD4+ and CD8+ T cell responses. (A, B) BALB/c mice (n = 5 per group) were immunized intravenously (IV) or intramuscularly (IM) with 3 μg of ctrl mRNA + 3 μg of CpG or 3 μg of RBD mRNA + 3 μg of CpG on D1 and boosted on D21 after priming. On day 105, pooled splenocytes were harvested, enriched for CD4+ and CD8+ T cells, and stimulated separately for 16 h with an RBD peptide mix for a direct ex vivo IFNγ ELISpot assay. For ELISpot analysis, splenocytes from the respective groups were measured in triplicates. Additionally, whole splenocytes of individual mice (n = 5 per group) were incubated with media or an RBD peptide pool for 18 h. (C, D) After incubation, cells were collected and stained for T cell memory marker CD44 as well as intracellular cytokines IFNγ, TNFα, and IL-4. Each dot represents the measurement of an individual mouse. ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001 two-way ANOVA (B) or one-way ANOVA (D) (Tukey’s multiple comparisons test).
To further assess the functionality and polarization of the vaccine induced T cells, we incubated splenocytes of individual mice from the same experiment in the presence of an RBD peptide pool or media alone. After 18 h of incubation, CD4+ and CD8+ T cells were assayed separately by flow cytometry for their expression of memory markers CD44 and for the intracellular cytokines IFNγ, TNFα, and IL-4. Even at this late time point after vaccination, a significant population of RBD-specific IFNγ producing CD4+ and CD8+ T cells and TNFα producing CD8+ T cells could be identified in the RBD-mRNA + CpG-CART IV vaccinated group. There was no increase in IL-4 producing CD4+ T cells, indicating that T cell memory was predominately TH1 (Figure 5C,D, Figure S8). In contrast to the IFNγ ELISpot results, we were not able to identify these low-frequency populations (mean 28 and 55 IFNγ spots per 5 × 105 cells for CD4+ and CD8+ enriched conditions, respectively) in IM vaccinated mice by flow cytometry.
Neutralizing Antibody Levels of Immunized Mice Are Comparable to Those Achieved in Vaccinated Humans
Results from clinical trials indicate that the Pfizer/BioNTech mRNA vaccine and the Moderna vaccine can both confer protection from symptomatic infection prior to administration of their second booster vaccine doses.28,29 This implies that the antibody levels in humans at that early preboost time point are sufficient to confer disease protection. Accordingly, we compared the levels of neutralizing antibodies achieved in our vaccinated mice to those in immunized humans both prior to and after their boosters. BALB/c mice were vaccinated with 3 μg of RBD mRNA + 3 μg of CpG-CART either IM or IV on D1 and D21, and serum was collected on D28. These mice sera were then compared to sera from 13 individual Pfizer/BioNTech mRNA-LNP vaccinated humans collected 15–21 days after their priming vaccination and then again 15 ± 4 days after their booster vaccinations. The level of RBD-ACE2 inhibition achieved with postboost sera from our IM and IV vaccinated mice was similar to or higher than that of the human preboost sera. The inhibitory antibody levels in the mice receiving the IV vaccination equaled those in humans after boosting (Figure 6A,B).
Figure 6.

Neutralizing antibody levels of immunized mice are comparable to those achieved in vaccinated humans. Serum from immunized mice (IM in red, IV in blue, n = 5) was harvested on day 28. Serum from blood donors (n = 13) who were vaccinated with the Pfizer/BioNTech mRNA vaccine was collected either within 7 days before (preboost, black) or 15 ± 4 days after the boost (postboost, green) and was tested for the ability to inhibit RBD/ACE-2 binding using a commercially available surrogate Virus Neutralization Test. * = P < 0.05, **** = P < 0.0001 one-way ANOVA (Tukey’s multiple comparison test).
RBD mRNA + CpG-CART Vaccination Shows a Favorable Safety Profile
To evaluate the safety profile of the vaccine, mice were treated on D0 and D21 with either PBS, 3 μg of GFP mRNA-CART, 3 μg of GFP mRNA + 3 μg of CpG-CART or 3 μg of RBD mRNA + 3 μg of CpG-CART (Figure S9A). No differences in body weight were observed after treatment (Figure S9B,C). IV administration of CpG-containing formulations induced a transient decrease in white blood cell (WBC) count 24 h after treatment that recovered by D2. This was driven by CpG; injection of mRNA-CART without CpG did not alter the WBC count (Figure S9D–F). Serum levels of TNFα and IL-6 measured 1 day and 1 week after treatment were not affected by vaccination. IM and IV vaccination led to a transient increase of serum IP10 and IFNα levels 1 day after prime. Serum cytokine levels were within a normal range at the second analyzed time point 7 days after prime. Again, transient effects were mediated by CpG; mice treated with mRNA-CART without CpG showed an unaltered cytokine profile after treatment compared to untreated mice (Figure S9G–J). Similar dynamics for both WBC count and cytokine profile were observed after the boost treatment (data not shown). Treatment did not induce liver toxicity as assessed by serum liver enzyme levels of alanine transferase (ALT), aspartate transferase (AST) (Figure S9K–N), and alkaline phosphatase (AP) (data not shown). In addition, gross histopathology performed on day 1 and 5 after booster treatment revealed no pathologic findings in the gross assessment of the main organs.
Discussion
For the first time, mRNA-based therapeutics have been approved by the FDA, and the success of both mRNA-based SARS-CoV-2 vaccines is both remarkable and mutually validating. However, challenges in production, deployment, and availability of SARS-CoV-2 vaccines remain. One important aspect for the continuous success and refinement of mRNA-based therapeutics in general will be to create access to diverse choices of safe delivery vehicles with varying chemical and biological properties. Here, we demonstrate an effective mRNA vaccination strategy against the clinically relevant RBD antigen of SARS-CoV-2 using an alternative mRNA delivery platform to the clinically used lipid nanoparticles. We observed an effective SARS-CoV-2 specific immune response that was enhanced by the inclusion of the TLR9 agonist CpG as an adjuvant. We further showed that the resulting anti-RBD sera were able to neutralize pseudoviral entry into ACE2-expressing cells. Focusing on the ACE2 receptor binding domain of the SARS-CoV-2 virus, we were able to generate high amounts of isotype switched RBD-specific neutralizing antibodies in both the serum and lungs of immunized animals. In addition, we compared serum levels of neutralizing antibodies from sera of humans vaccinated with approved Pfizer/BioNTech mRNA-LNP vaccines with those of mRNA-CART vaccinated mice. The levels of neutralizing antibodies in fully immunized mice were similar (IM) or higher (IV) than the levels of neutralizing antibodies measured in preboost blood samples from humans vaccinated with the Pfizer/BioNTech mRNA vaccine. Since it has been shown that this approved mRNA vaccine confers protection as early as day 12 post prime, this is an indication that our vaccine induced neutralizing antibody levels can be sufficient to confer protection.28 Of course, for further validation of these results, vaccination studies in larger animals such as nonhuman primates are needed. Finally, the RBD mRNA + CpG-CART vaccine induced RBD-specific CD4+ and CD8+ T cell responses with the induction of long-lasting T cell memory of TH1 polarization. In addition, the IgG isotype profile dominated by IgG2a and IgG2b confirms a TH1 polarized T cell response.
When compared to LNPs, CARTs have a unique biodistribution, selectively delivering mRNA to the spleen or other organs without the need for targeting ligands, simply through changes in the CART structure. CARTs can be readily prepared and formulated with multiple mRNAs in any desired nucleotide combination,21 only require a single structural component that is mixed with mRNA, and do not require specialized microfluidics instruments for their manufacture. This allows for alternative drug application strategies. Preliminary experiments show that CARTs formulated with mRNA are stable for 11 days at −20 °C (Figure S11). However, since formulation does not require specific equipment, a mix-and-shoot administration of the vaccine could be used. Our initial experiments indicate that unformulated CARTs in DMSO are stable for more than 12 months at −20 °C. CARTs and mRNA could therefore be divided into two separate chambers of a two-chamber syringe, allowing for mixing and mRNA-CART formation at the point of administration and avoiding thermostability issues of preformulated complexes. In contrast to LNPs, CARTs have no unspecific immunostimulatory effects, which allows more flexibility for vaccine design and the option to vary oligodeoxynucleotide adjuvant quantity when codelivered with mRNA, rather than relying on the inherent immunogenicity of LNPs. When CARTs are injected IM, gene expression localizes exclusively at the site of injection and does not spread to other organs. In a SARS-CoV-2 vaccine study using IM injection of LNPs, the liver showed the highest level of reporter gene and antigen expression.19 In addition, IV injection of CARTs confers mRNA expression exclusively to the spleen which could explain the higher potency of IV vaccination compared to IM. While IV administration of the vaccine induced a stronger antibody and T cell response, significant responses could be induced via both routes of administration. Interestingly, it has been shown recently that IV vaccination with a BCG vaccine against tuberculosis profoundly altered the protective outcome in nonhuman primates with an increase of antigen responsive CD4+ and CD8+ T cells in blood, spleen, BAL, and lung lymph nodes when compared to the established intradermal or aerosol administration.30 Additionally, IV administered mRNA lipid nanoparticles have demonstrated potency in preclinical mouse models and a clinical phase I study of therapeutic cancer vaccination.31,32
We chose to direct our vaccine specifically against the RBD rather than the whole spike protein sequence. Potent humoral and cellular immune responses have been observed in clinical trials with both the SARS-CoV-2 full-length spike protein and RBD.6 Moreover, an RBD mRNA directed vaccine was proven safe in a phase 1/2 clinical trial tested in the US and in Germany.27,33 In addition, RBD provides essential targetability for humoral and cellular immune responses. Piccoli et al. showed that 90% of the neutralizing activity of serum from an exposed patient targets the RBD.34 RBD is also an epitope for T cell responses against SARS-CoV-2 S protein.35 A fast-spreading mutant with a mutation in the RBD (N501Y) has been identified in the UK (B.1.1.7) raising concerns about coverage of the current mRNA-based vaccines.6 This N501Y mutation does not seem to affect the efficacy of an RBD vaccination since mice vaccinated against the original RBD sequence were able to clear a SARS-CoV-2 variant containing the specific mutation.36 However, recent data indicates that other clinically relevant RBD and non-RBD mutations can mediate escape from vaccine induced humoral immunity,13,37 highlighting the urgent need of flexible and rapidly adaptable vaccine platforms.
The safety data of mRNA + CpG-CART vaccination seems to be favorable. The detected changes in the white blood cell count and cytokine profile were mediated by CpG. However, CpG has a well-known safety record in clinical studies of other vaccines. We believe that the ability to formulate TLR activating molecules like CpG into our vaccine will aid in inducing a protective immune response in populations with less competent immune systems and that are more at risk for severe COVID19 symptoms. CpG directly activates pDCs and B cells, contributing to the induction of both innate and adaptive immune responses. The cascade of events initiated by CpG indirectly supports maturation, differentiation, and proliferation of natural killer cells, T cells, and monocytes/macrophages.38−41 B cells activated by CpG upregulate expression of their Fc receptor (FcR) and costimulatory molecules including MHC class II, CD40, CD80, and CD86.42−44 Subsequently, the CpG-stimulated B cells proliferate and differentiate into plasma cells and memory B cells.45 The adjuvant effects of CpG are supported by our study where the addition of CpG resulted in a more rapid immune response, higher anti-RBD titers in the serum and bronchoalveolar lavage, more effective ACE2-RBD inhibition and pseudotyped virus neutralization, an increased T cell response, and more pronounced isotype switching.
In conclusion, our study demonstrates the potency and flexibility of this mRNA-CART vaccine platform against the clinically relevant SARS-CoV-2 RBD antigen. The robust induction of both B and T cell responses via different routes of administration warrants further exploration and its use as an alternative to the clinically approved lipid nanoparticles in the general development of mRNA-based therapeutics against infectious diseases.
Material and Methods
Recombinant Proteins
The pCAGGS plasmids coding for soluble RBD-His, residues 319–541, and spike-His, residues 1–1213 from the Wuhan-Hu-1 genome sequence (GenBank MN9089473), were a gift from Prof. Florian Krammer (Icahn School of Medicine at Mount Sinai).23 The pcDNA3 plasmid coding for a soluble ACE2-hIgA FC fusion protein was purchased from addgene (ID 145154). Plasmids were expanded using One Shot TOP10 Chemically Competent Escherichia coli (ThermoFisher Scientific) and the ZymoPURE II Plasmid Maxiprep kit (Zymo Research). Recombinant proteins were produced using the Expi293F cells (Thermo Fisher Scientific) by transfecting 200 × 106 of these cells with purified DNA using the ExpiFectamine 293 transfection kit (Thermo Fisher Scientific). Supernatants from transfected cells were harvested 3 days post-transfection by centrifugation at 300g for 10 min and filtration through a 22 μm filter. RBD-His and Spike-His containing supernatants were batch purified using the HisPur Ni-NTA resin (ThermoFisher Scientific). Supernatants were incubated with 6 mL of resin for 1 h at room temperature (RT). Then, the resin was recovered by centrifugation (2 min at 700g), washed, and finally eluted per manufacturer recommendation. Elution fractions were analyzed by SDS-PAGE and Western blot to confirm RBD-His or Spike-His purification, and the positive fractions were pooled.
In Vitro Transcription
The RBD-6his was cloned from the pCAGGS expression plasmid into the LF-pLMCT plasmid that contains a T7 promoter and a polyA sequence required for mRNA synthesis. The LF-pLMCT plasmid was a gift from Dr. Kris Thielemans (Free University of Belgium). The RBD-6his coding sequence was amplified by PCR using PHUSION polymerase (NEB) and TAAACTTAAGACAACCATGGTCGTGTTTCTGGTGC as a forward primer and GGGGATCCcGTCTTCCTCGAGTTATCAATGGTGATGGTGA as a reverse primer. The PCR product was then inserted into pLMCT by NcoI and XhoI. mRNA coding for RBD was synthesized per manufacturer recommendation using Hiscribe T7 (NEB) with cotranscriptional CleanCap AG (Trilink), N1-methyl-pseudouridine (Trilink), and 5-methylcytidine (Trilink). The template for in vitro transcription was a PCR amplicon from the pLMCT-RBD-6His produced using the PHUSION high-fidelity DNA polymerase (NEB) and TGTGGAATTGTGAGCGGATA as a forward primer and CTTCACTATTGTCGACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA as a reverse primer.
CART Preparation and Characterization
CART O6-stat-N6: A9, consisting of a first block of a 1:1 statistical mixture of oleyl and nonenyl-substituted carbonate monomers, followed by a block of α-amino ester monomer was prepared as previously reported.14,15 Briefly, to a mixture of nonenyl (29 mg, 1 mmol) and oleyl carbonate (40.5 mg, 1 mmol) in toluene (150 μL) were added TU, DBU, and BnOH (5 mol % TU/DBU, 0.2 mmol BnOH) in 50 μL of toluene. The reaction was stirred for 1.5 h; then, the morpholinone monomer (33.4 mg, 0.16 mmol) was added as a solid and then stirred for an additional 2.5 h. The reaction was quenched with AcOH then dialyzed overnight in DCM/MeOH (3.5 kDa Mw, cutoff). Concentration after dialysis afforded 85 mg of clear residue which was deprotected with TFA (0.85 mL) in dry DCM (8.5 mL) overnight. End group analysis of the deprotected polymer showed block lengths of 6 nonenyl and 6 oleyl carbonate units and 9 cationic aminoester units.
CART Oligonucleotide Formulation
To prepare the CART-vaccine, CARTs were formulated with a mixture of CpG and RBD mRNA at a 10:1 cation:anion ratio assuming full protonation of the CART and full deprotonation of the oligonucleotides (1:1 mass ratio of CpG and mRNA nucleotides). Formulations were prepared by mixing the reagents for 20 s in acidic PBS (pH adjusted to 5.5 by addition of 0.1 M HCl) in a total volume of 50–100 μL, followed by a brief spin in a tabletop centrifuge. The formulation was used within 5 min for in vitro or in vivo experiments.
Mouse Vaccination
Female BALB/c mice (8- to 12-week-old) were purchased from The Jackson Laboratory and housed in the Laboratory Animal Facility of the Stanford University Medical Center. All experiments were approved by the Stanford Administrative Panel on Laboratory Animal Care and were conducted in accordance with Stanford University Animal Facility and NIH guidelines. RBD-mRNA and CpG were formulated with CART polymer in PBS at pH 5.5 as described above. Mice were injected with 3 μg of RBD-mRNA formulated with 2.6 μL of CART (5 mM) or 3 μg of CpG formulated with 2.6 μL of CART (5 mM) or 3 μg of RBD-mRNA plus 3 μg of CpG formulated with 5.2 μL of CART (5 mM). Mice were vaccinated by IV, IM, or SC injection and were boosted as described in the experiment. CARTs are formulated at indicated concentrations of mRNA in 50–100 μL total volume. For IV administration, 100 μL of formulated CART was administered per tail vein injection. For IM injections, 50 μL of formulated CART was injected in the thigh muscle. SC injections were administered on the back of the mouse near the tail. At indicated time points, mice were bled, and serum was collected.
HeLa and 293F Transfection
HeLa cells and 293F cells were plated at 106 cells per well in a 12-well plate in Opti-MEM media (ThermoFisher Scientific). 2 μg of the RBD-his mRNA or GFP mRNA (Trilink) was formulated in 6.6 μL of PBS pH 5.5 with 1.37 μL of 5 mM CART and added to the cells. After 4 h of transfection, Opti-MEM media was replaced by RPMI media containing 10% FCS and penicillin–streptomycin 1000 U/mL. 12 hours post-transfection, RBD and GFP expression were monitored by Western blot and fluorescence microscopy, respectively.
Western Blot
15 μL of media from HeLa or 293F transfected cells was mixed with 4× sample loading buffer (Invitrogen) and was loaded on a 4–12% NuPAGE gel (Invitrogen). Electrophoresis was performed in an MES buffer at 200 V for 35 min. Proteins were transferred to a cellulose membrane using the iBLOT system (Invitrogen). The membrane was stained with Ponceau red to verify protein transfer, and then, the membrane was blocked for 1 h in TBS buffer containing 0.1% Tween 20 (TBST) containing 5% nonfat dry milk. The membrane was washed 3 times in TBST and incubated in TBST containing 5% nonfat dry milk and 1:1000 mouse anti-His (Biolegend) overnight. After 3 washes in TBST, the membrane was incubated with 1:10 000 antimouse Ig (Southern Biotech) in TBST containing 5% nonfat dry milk for 1 h. After 3 washes in TBST, the blot was revealed using the EC Prime Western blotting system (Sigma). The membrane was imaged using a Chemidoc MP imaging system from BioRad.
Serum Preparation
For human samples, informed consent was obtained from the subjects prior to blood draw. Blood was collected in Eppendorf tubes and allowed to coagulate for 60 min at room temperature. After 10 min of centrifugation at 1000g, the supernatant was collected. Serum was heat-inactivated at 56 °C for 30 min.
ELISA
Nunc-Immuno MicroWell 96-well ELISA plates (MilliporeSigm) were coated overnight with 50 μL per well of 2 μg/mL RBD-His or Spike-His protein in carbonate buffer pH 9. After 3 washes in ELISA wash buffer (PBS with 0.1% Tween 20), plates were blocked using 100 μL of 5% nonfat dry milk diluted in TBS buffer containing 0.1% Tween 20 (TBST) for 1 h at room temperature. Serum, BAL, and antibody dilutions were prepared in TBST containing 1% nonfat dry milk. The blocking solution was removed, and 50 μL of each serial dilution was added to the plate for 1 h at room temperature. Plates were washed three times and incubated with HRP conjugated antihuman Ig (1:5000, BioSource), antimouse Ig (1:5000, Cell Signaling), antimouse IgG2a (1:5000, Southern Biotech), antimouse IgG2b (1:5000, Southern Biotech), antimouse IgG1 (1:5000, Southern Biotech), antimouse IgG3 (1:5000, Southern Biotech), antimouse IgA (1:5000, Invitrogene), or antimouse IgM (1:5000, Southern Biotech). Plates were washed three times, and 100 μL of TMB ELISA substrate (Abcam) was added to each well. ELISA was developed for 10 min, and then, the reaction was stopped by adding 50 μL of Stop Solution for TMB Substrates (ThermoFisher Scientific) to each well. In some assays, a human anti-RBD (Invivogene) of known antibody concentration was used as a standard. Optical density at 450 nm (OD450) was measured using a SpectraMax Paradigm microplate reader (Molecular devices).
RBD-ACE2 Interaction Blocking Assay ELISA
The RBD-ACE2 interaction blocking assay was evaluated using three methods: a commercial kit from Genescript, an in house developed ELISA, and flow cytometry.
For the commercial kit, we used the SARS-CoV-2 surrogate virus neutralization test (sVNT) kit (Genescript) following the manufacturer’s instructions. In short, samples and controls were diluted at indicated ratios with dilution buffer and preincubated with HRP-RBD in a 1:1 ratio for 30 min at 37 °C. Samples were then added to the capture plate in wells precoated with hACE2. After 15 h of incubation at 37 °C, wells were washed four times with wash buffer. TMB solution was added and incubated for 15 h at room temperature in the dark. After 15 h, stop solution was added to the wells and promptly analyzed. Optical density at 450 nm (OD450) was measured using a SpectraMax Paradigm microplate reader (Molecular devices).
For the in-house developed ELISA, Nunc-Immuno MicroWell 96-well ELISA plates (Millipore) were coated overnight with 50 μL per well of 2 μg/mL RBD-His or Spike-His protein in carbonate buffer pH 9. After 3 washes in ELISA wash buffer (PBS with 0.1% Tween 20), plates were blocked using 100 μL of 5% nonfat dry milk diluted in TBS buffer containing 0.1% Tween 20 (TBST) for 1 h at room temperature. Serum, BAL, and antibody dilutions were prepared in TBST containing 1% nonfat dry milk. The blocking solution was removed, and 50 μL of each serial dilution was added to the plate for 1 h at room temperature. Plates were washed three times, and 50 μL of 2 times diluted ACE2-hIgA supernatant was added to each well for 1 h. After 3 washes, the plate was incubated with HRP conjugated antihuman IgA (1:1000, Thermo Scientific) for 1 h in TBST with 1% nonfat dry milk. Plates were washed three times, and 100 μL of TMB ELISA Substrate (Abcam) was added to each well. ELISA was allowed to develop for 10 min, and then, the reaction was stopped by adding 50 μL of Stop Solution for TMB Substrates (ThermoFisher Scientific) to each well. In some assays, human anti-RBD (Invivogen) of known antibody concentration was used as a standard. Optical density at 450 nm (OD450) was measured using a SpectraMax Paradigm microplate reader (Molecular devices).
For the flow cytometry assay, RBD-His 2μg/mL was incubated with sera for 1 h. Then, the 4 × 105 ACE2-expressing HEK293T cells were added to the RBD-His/sera mix and incubated at RT for 30 min. Cells were then washed 2 times in PBS containing 1% BSA. RBD was then detected using an Alexa Fluor 488 conjugated anti-His antibody (clone J099B1, Biolegend). Cells were analyzed by flow cytometry (BD).
Pseudovirus Assay
Pseudotyped lentivirus expressing the Sars-Cov-2 spike protein and the luciferase was produced in HEK293T cells as previously described.46,47 One day before transfection, 6 × 106 HEK293T cells were seeded in a 10 cm culture plate in RPMI containing 10% FCS, 2 mM l-glutamine, streptomycin, and penicillin. Using TransIT (Mirus), cells were then transfected with 10 μg of the lentiviral packaging vector (pHAGE_Luc2_IRES_ZsGreen), the 3.4 μg of SARS-CoV-2 spike, and lentiviral helper plasmids (2.2 μg of HDM-Hgpm2, 2.2 μg of HDM-Tat1b, and 2.2 μg of pRC-CMV_Rev1b). The spike vector contained the full-length wild-type spike sequence from the Wuhan-Hu-1 strain of SARS-CoV-2 (GenBank NC_045512). These 5 plasmids were kindly provided by Dr. Jesse Bloom (Fred Hutch Seattle, University of Washington). 72 h after transfection, virus-containing supernatant was harvested, centrifuged at 300g for 5 min, filtered on a 0.45 μm filter, aliquoted, and frozen at −80 °C.
For viral neutralization assays, ACE2-expressing HEK293T47 cells were plated in poly-l-lysine-coated, white-walled, clear-bottom 96-well plates at 12 500 cells/well 1 day prior to infection. Mouse serum was centrifuged at 2000g for 15 min, heat-inactivated for 30 min at 56 °C, and diluted in D10 media (DMEM medium supplemented with 10% FCS). Virus was diluted in D10 medium, supplemented with polybrene (5 μg/mL), and then added to serum dilutions. The virus/serum mix was preincubated for 1 h at 37 °C before it was added to the cells and incubated at 37 °C for ∼48 h. Cells were lysed by adding BriteLite assay readout solution (PerkinElmer), and luminescence values were measured with a SpectraMax Paradigm microplate reader (Molecular devices). As a positive control, a neutralizing human anti-SARS-Cov-2 IgG1 antibody was used (Acro).
Bronchoalveolar Lavage
Mice were sacrificed, and lungs were inflated 2 times with 1 mL of PBS following a previously described procedure.48 Lavage fractions were pooled and centrifuged at 1200 rpm for 5 min. Supernatant was collected and assayed for anti-RBD antibodies by ELISA.
T Cell Response Assay on Lungs
Mouse lungs were harvested at indicated days after vaccination. To prepare lung single-cell suspensions, lungs were cut into small pieces and incubated at 37 °C in RPMI containing Collagenase D (2 mg/mL, Sigma) and DNase (50 μg/mL, Sigma) for 30 min. Then, digestion mix was diluted 5 times, and the lung preparations were processed through a 70 μm cell strainer. Red blood cells present in the spleen or lung single-cell suspensions were lysed using ACK buffer (ThermoFisher Scientific). Single-cell suspensions were kept on ice until further processing for the T cell response assay. Cells were cultured in 96-well plates (Corning, V-bottom) at 1 × 106 cells/well and stimulated for 48 h with 5 μg/mL RBD-His or hCD81-His or media alone (RPMI + 5% FCS) in the presence of antimouse CD28 antibody [0.5 μg/mL] (Southern Biotech). As a positive control, cells were stimulated with antimouse CD3 [0.05 μg/mL] (Southern Biotech). For intracellular staining, cells were treated with GolgiStop (BD Biosciences) for 5 h prior to staining. Following stimulation, cells were washed, stained with Aqua live/dead viability dye (Thermo Fisher) in PBS, washed two additional times, and stained with a cocktail of monoclonal antibodies and Fc block: CD16/32, CD4 BV605 RM4-5, CD8 FITC 53-6.7, CD44 APC IM7, CD69 PE-Cy7 H1.2F3, CD134 BV786 OX-86, CD137 PE 1AH2, and CD45R/B220 Per-CP 5.5 RA3-6B2 (all BD Bioscience). Cells were fixed and permeabilized according to the manufacturer’s protocol (BD Biosciences) and stained with aTNFα BV650 MP6-XT22 (BD Bioscience). Cells were washed, fixed with 2% formaldehyde, acquired on a BD LSR II, and analyzed using Cytobank V7.3.0.
T Cell Response Assay on the Spleen
Mouse spleens were harvested on D105 after vaccination, and single-cell suspensions were prepared by processing them through a 70 μm cell strainer (BD Biosciences). Cells were then incubated in FACS tubes at 6 × 105 cells per tube and stimulated for 18 h with 2 μg/mL RBD peptide mix [PepMix SARS-CoV-2 (S-RBD) Protein ID: P0DTC2 PM-WCPV-S-RBD-1, JPT] or media alone. As a positive control, cells were stimulated with antimouse CD3 [0.05 μg/mL] (Southern Biotech) and antimouse CD28 antibody [0.5 μg/mL] (Southern Biotech). GolgiStop (BD Biosciences) was added for the last 10 h of the assay. Following stimulation, cells were washed, stained with Aqua live/dead viability dye (Thermo Fisher) in PBS, washed two additional times, and stained with a cocktail of monoclonal antibodies and Fc block: CD16/32, CD4 Ax700 RM4-5, CD8 APC-H7 53-6.7, and CD44 APC IM7 (all BD Bioscience). Cells were fixed and permeabilized according to the manufacturer’s protocol (BD Biosciences) and stained for intracellular cytokines with IFNγ PE-Cy7 XMG1.2, TNFα BV650 MP6-XT22, and IL-4 BV786 11B11 (BD Bioscience). Cells were washed, fixed with 2% formaldehyde, acquired on a Cytek Aurora (Northern Lights), and analyzed using Cytobank V7.3.0.
IFNγ ELISpot
The assay was performed following the manufacturer’s instructions (R&D systems, mouse IFNγ kit cat. EL485). In short, IFNγ ELISpot analysis was performed ex vivo (without further in vitro culturing for expansion) using PBMCs depleted of CD4+ and enriched for CD8+ T cells or depleted of CD8+ and enriched for CD4+ T cells by MACS sort (Miltenyi CD4+ or CD8+ microbeads following the manufacturer instructions). Tests were performed in triplicates and with a positive control [anti-CD3 monoclonal antibody (0.05 μg/mL; Southern Biotech)]. PVDF backed microplates precoated with IFNγ-specific antibodies (R&D systems, mouse IFNγ kit cat. EL485) were washed with PBS and blocked with RPMI medium (Corning) containing 5% FCS for 20 min at room temperature. Per well, 5 × 105 effector cells were stimulated for 16 h with 2 μg/mL RBD peptide mix [PepMix SARS-CoV-2 (S-RBD) Protein ID: P0DTC2 PM-WCPV-S-RBD-1, JPT]. After stimulation, wells were washed and incubated with a biotinylated anti-IFNγ antibody (R&D systems, mouse IFNγ kit cat. EL485) overnight at 4 °C. The next day, wells were washed and incubated with streptavidin-AP (R&D systems, mouse IFNγ kit cat. EL485) for 2 h at RT. After washing, wells were incubated with a 5-bromo-4-chloro-3′-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) substrate (R&D systems, mouse IFNγ kit cat. EL485). Plates were scanned and analyzed using an ImmunoSpot microanalyzer.
In Vivo Bioluminescence Imaging
For the bioluminescence assessment, mice were anesthetized with isoflurane gas (2% isoflurane in oxygen, 1 L/min) during injection and imaging procedures. Intraperitoneal injections of D-luciferin (Biosynth AG) were done at a dose of 150 mg/kg, providing a saturating substrate concentration for Fluc enzyme (luciferin crosses the blood–brain barrier). Mice were imaged in a light-tight chamber using an in vivo optical imaging system (AMI HT; Spectral Instruments imaging) equipped with a cooled charge-coupled device camera. During image recording, mice inhaled isoflurane delivered via a nose cone, and their body temperature was maintained at 37 °C in the dark box of the camera system. Bioluminescence images were acquired between 10 and 20 min after luciferin administration. Mice usually recovered from anesthesia within 2 min of imaging.
White Blood Cell Count
5 μL of blood was harvested and mixed with 45 μL of 3% acetic acid with methylene blue (Stemcell), and nuclei were counted using a hematocytometer.
Cytokine Analysis
IP10, IFNa, TNFa, and IL6 were measured in serum samples using the LEGENDplex bead-based immunoassays from Biolegend per manufacturer protocol. The assay was analyzed on a BD FACSCalibur instrument.
Complete Blood Count
A complete blood count (CBC) analysis was performed by the animal diagnostic lab at Stanford. Automated hematology was performed on an Sysmex XN-1000 V analyzer system. Blood smears were made for all CBC samples and reviewed by a clinical laboratory scientist. Manual differentials were performed as indicated by species and automated analysis.
Liver Enzyme Analysis
Liver enzymes were analyzed by the animal diagnostic lab at Stanford. The chemistry analysis was performed on the Siemens Dimension EXL200/LOCI analyzer. A clinical laboratory scientist performed all testing, including dilutions and repeat tests as indicated, and reviewed all data.
Safety Statement
For all mouse experiments, no unexpected or unusually high safety hazards were encountered.
Acknowledgments
We thank the Dynavax Corporation for the gift of the CpG formulation SD101. This work was supported by Fast Grants (to R.L), Stanford University Innovative Medicines Accelerator (IMA) (R.L., R.M.W., and P.A.W.); The National Science Foundation Grant NSF CHE-2002933 (to R.M.W.); NIH R01 CA031845 (P.A.W.); The Child Health Research Institute at Stanford University, and the SPARK Translational Research Program in the Stanford University School of Medicine (R.M.W., P.A.W., R.L.). The support of German cancer aid (Deutsche Krebshilfe-Mildred Scheel postdoctoral fellowship to J.J.K.L.) and the Stanford Cancer Translational Nanotechnology Training T32 Training Grant T32 CA196585 funded by the National Cancer Institute (T.R.B.) is gratefully acknowledged. B.M. is grateful for financial support from a Cancer-TNT Fellowship. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award F32CA254128. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00361.
CART characterization, NMR spectrum, ζ potential, IVT mRNA quality control and immunogenicity, mRNA expression by Western Blot, upregulation of activation markers upon in vivo administration by flow cytometry, early RBD-specific immunoglobulin responses on D4 and 14, ELISA results, route of administration—intravenous, subcutaneous, and intramuscular effects, induction of neutralizing antibodies independent of CpG source, ELISA D21 and D28 results, intracellular cytokine staining (IFNγ, TNFα, IL-4) in CD4+ and CD8+ T cells on D105, mouse in vivo safety studies, white blood cell count, liver enzymes, serum cytokines after prime and boost, and mRNA-CART stability D11 at room temperature, 4 °C, and −20 °C (PDF)
Author Contributions
⊥ O.A.W.H., J.J.K.L., and A.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Padron-Regalado E. Vaccines for SARS-CoV-2: Lessons from Other Coronavirus Strains. Infect Dis Ther 2020, 9, 255–274. 10.1007/s40121-020-00300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Zhao S.; Ou J.; Zhang J.; Lan W.; Guan W.; et al. COVID-19: Coronavirus Vaccine Development Updates. Front. Immunol. 2020, 11, 602256. 10.3389/fimmu.2020.602256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberer M.; Gnad-Vogt U.; Hong H. S.; Mehr K. T.; Backert L.; Finak G.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. 10.1016/S0140-6736(17)31665-3. [DOI] [PubMed] [Google Scholar]
- Feldman R. A.; Fuhr R.; Smolenov I.; Mick Ribeiro A.; Panther L.; Watson M.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. 10.1016/j.vaccine.2019.04.074. [DOI] [PubMed] [Google Scholar]
- Anderson E. J.; Rouphael N. G.; Widge A. T.; Jackson L. A.; Roberts P. C.; Makhene M.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020, 383, 2427–2438. 10.1056/NEJMoa2028436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh E. E.; Frenck R. W.; Falsey A. R.; Kitchin N.; Absalon J.; Gurtman A.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. 10.1056/NEJMoa2027906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ura T.; Yamashita A.; Mizuki N.; Okuda K.; Shimada M. New vaccine production platforms used in developing SARS-CoV-2 vaccine candidates. Vaccine 2021, 39, 197–201. 10.1016/j.vaccine.2020.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L.; Gao G. F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. S.; Wheatley A. K.; Kent S. J.; DeKosky B. J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol 2020, 5, 1185–1191. 10.1038/s41564-020-00789-5. [DOI] [PubMed] [Google Scholar]
- Liang F.; Lindgren G.; Lin A.; Thompson E. A.; Ols S.; Röhss J.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. 10.1016/j.ymthe.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; Gao P.; Hu Y.; Wang J.; Wang H.; Yang J.; et al. PEGylated versus non-PEGylated drugs: A cross-sectional analysis of adverse events in the FDA Adverse Event Reporting System (FAERS) Database. Int. J. Clin. Pharmacol. Ther. 2020, 58, 332–342. 10.5414/CP203735. [DOI] [PubMed] [Google Scholar]
- Castells M. C.; Phillips E. J. Maintaining Safety with SARS-CoV-2 Vaccines. N. Engl. J. Med. 2021, 384, 643–649. 10.1056/NEJMra2035343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. E.; Zhang X.; Case J. B.; Winkler E. S.; Liu Y.; VanBlargan L. A.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinlay C. J.; Vargas J. R.; Blake T. R.; Hardy J. W.; Kanada M.; Contag C. H.; et al. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E448–E456. 10.1073/pnas.1614193114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinlay C. J.; Benner N. L.; Haabeth O. A.; Waymouth R. M.; Wender P. A. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E5859–E5866. 10.1073/pnas.1805358115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haabeth O. A. W.; Blake T. R.; McKinlay C. J.; Waymouth R. M.; Wender P. A.; Levy R. mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E9153–E9161. 10.1073/pnas.1810002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C.; Zhao G.; Steinhagen F.; Kinjo T.; Klinman D. M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat F.; Krammer F. SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N.-N.; Li X.-F.; Deng Y.-Q.; Zhao H.; Huang Y.-J.; Yang G.; et al. A Thermostable mRNA Vaccine against COVID-19. Cell 2020, 182, 1271–1283. 10.1016/j.cell.2020.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q.; Ji K.; Tian S.; Wang F.; Huang B.; Tong Z.; et al. A single-dose mRNA vaccine provides a long-term protection for hACE2 transgenic mice from SARS-CoV-2. Nat. Commun. 2021, 12, 12. 10.1038/s41467-021-21037-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haabeth O. A. W.; Blake T. R.; McKinlay C. J.; Tveita A. A.; Sallets A.; Waymouth R. M.; et al. Local Delivery of Ox40l, Cd80, and Cd86 mRNA Kindles Global Anticancer Immunity. Cancer Res. 2019, 79, 1624–1634. 10.1158/0008-5472.CAN-18-2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H. Y.; Liu S.; Wong H. L. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine (London, U. K.) 2014, 9, 295–312. 10.2217/nnm.13.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanat F.; Stadlbauer D.; Strohmeier S.; Nguyen T. H. O.; Chromikova V.; McMahon M.; et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020, 26, 1033–1036. 10.1038/s41591-020-0913-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan S.; Azizian K. T.; Haque A. K. M. A.; Henderson J. M.; Hendel A.; Shore S.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther.--Nucleic Acids 2018, 12, 530–542. 10.1016/j.omtn.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toellner K.-M.; Luther S. A.; Sze DM-Y; Choy RK-W; Taylor D. R.; MacLennan I. C. M.; et al. T Helper 1 (Th1) and Th2 Characteristics Start to Develop During T Cell Priming and Are Associated with an Immediate Ability to Induce Immunoglobulin Class Switching. J. Exp. Med. 1998, 187, 1193–1204. 10.1084/jem.187.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt R. L.; Liang H.-E.; Locksley R. M. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 2009, 10, 385–393. 10.1038/ni.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan M. J.; Lyke K. E.; Kitchin N.; Absalon J.; Gurtman A.; Lockhart S.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. 10.1038/s41586-020-2639-4. [DOI] [PubMed] [Google Scholar]
- Polack F. P.; Thomas S. J.; Kitchin N.; Absalon J.; Gurtman A.; Lockhart S.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baden L. R.; El Sahly H. M.; Essink B.; Kotloff K.; Frey S.; Novak R.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah P. A.; Zeppa J. J.; Maiello P.; Hackney J. A.; Wadsworth M. H.; Hughes T. K.; et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 2020, 577, 95–102. 10.1038/s41586-019-1817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwitz C.; Salomon N.; Vascotto F.; Selmi A.; Bukur T.; Diken M.; et al. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology 2019, 8, e1629259. 10.1080/2162402X.2019.1629259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U.; Oehm P.; Derhovanessian E.; Jabulowsky R. A.; Vormehr M.; Gold M.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. 10.1038/s41586-020-2537-9. [DOI] [PubMed] [Google Scholar]
- Sahin U.; Muik A.; Derhovanessian E.; Vogler I.; Kranz L. M.; Vormehr M.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. 10.1038/s41586-020-2814-7. [DOI] [PubMed] [Google Scholar]
- Piccoli L.; Park Y.-J.; Tortorici M. A.; Czudnochowski N.; Walls A. C.; Beltramello M.; et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell 2020, 183, 1024–1042. 10.1016/j.cell.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. R. F.; Patel A.; Ramos S.; Elwood D.; Zhu X.; Yan J.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. 10.1038/s41467-020-16505-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H.; Chen Q.; Yang G.; He L.; Fan H.; Deng Y.-Q.; et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. 10.1126/science.abc4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Beltran W. F.; Lam E. C.; St. Denis K.; Nitido A. D.; Garcia Z. H.; Hauser B. M.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372. 10.1016/j.cell.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas Z. K.; Rasmussen W. L.; Krieg A. M. Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J. Immunol 1996, 157 (5), 1840–1845. [PubMed] [Google Scholar]
- Klinman D. M.; Yi A. K.; Beaucage S. L.; Conover J.; Krieg A. M. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 2879–2883. 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Zhang X.; Tough D. F.; Sprent J. Type I interferon-mediated stimulation of T cells by CpG DNA. J. Exp. Med. 1998, 188, 2335–2342. 10.1084/jem.188.12.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey K. J.; Sweet M. J.; Hume D. A. Macrophages ingest and are activated by bacterial DNA. J. Immunol 1996, 157 (5), 2116–2122. [PubMed] [Google Scholar]
- Krieg A. M.; Yi A. K.; Matson S.; Waldschmidt T. J.; Bishop G. A.; Teasdale R.; et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- Davis H. L.; Weeratna R.; Waldschmidt T. J.; Tygrett L.; Schorr J.; Krieg A. M.; et al. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J. Immunol 1998, 160 (2), 870–876. [PubMed] [Google Scholar]
- Kobayashi H.; Horner A. A.; Takabayashi K.; Nguyen M. D.; Huang E.; Cinman N.; et al. Immunostimulatory DNA pre-priming: a novel approach for prolonged Th1-biased immunity. Cell. Immunol. 1999, 198, 69–75. 10.1006/cimm.1999.1572. [DOI] [PubMed] [Google Scholar]
- Jung J.; Yi A.-K.; Zhang X.; Choe J.; Li L.; Choi Y. S. Distinct response of human B cell subpopulations in recognition of an innate immune signal, CpG DNA. J. Immunol. 2002, 169, 2368–2373. 10.4049/jimmunol.169.5.2368. [DOI] [PubMed] [Google Scholar]
- Crawford K. H. D.; Eguia R.; Dingens A. S.; Loes A. N.; Malone K. D.; Wolf C. R.; et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers T. F.; Zhao F.; Huang D.; Beutler N.; Burns A.; He W.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, 369, 956–963. 10.1126/science.abc7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Xiao G.; Qu Z. Murine Bronchoalveolar Lavage. Bio Protoc 2017, 7, 7. 10.21769/BioProtoc.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.