

Introduction
Approximately 80% of newborns worldwide have some degree of hyperbilirubinemia and jaundice.1 Severe cases of hyperbilirubinemia can progress to kernicterus, leading to permanent developmental disorders. Several risk factors contribute to hyperbilirubinemia and kernicterus, including G6PD deficiency, which is one of the most common human enzymopathies. In the developing world, lack of access to common treatments for hyperbilirubinemia, along with a high rate of G6PD deficiency, leads to high incidence of kernicterus. Below, we highlight the need for a novel therapy to prevent kernicterus, and discuss the pharmacological activation of G6PD as a promising therapeutic strategy.
Hyberbilirubinemia and kernicterus
Hyperbilirubinemia results from increased bilirubin production coupled with inefficient bilirubin excretion. Hemolysis and subsequent heme breakdown produces bilirubin, which can be conjugated and excreted in the liver. However, lower liver function in newborns leads to reduced bilirubin removal and increased levels of serum bilirubin.
Low to moderate levels of serum bilirubin (<20 mg/dL) are non-toxic and can be reduced by non-invasive treatment such as phototherapy, which uses light in a narrow wavelength band with a peak around 460-490 nm to break down bilirubin into excretable byproducts. In cases of severe hyperbilirubinemia, exchange transfusion is used to reduce bilirubin concentration in the blood. Other therapies, less commonly used, include intravenous immune globulin (mechanism of action unknown) and pharmacologic therapies to reduce bilirubin production or increase bilirubin conjugation.2
If severe hyperbilirubinemia is left untreated, bilirubin that crosses the blood-brain barrier can rise to toxic levels in the brain, leading to acute bilirubin encephalopathy (kernicterus). Kernicterus is characterized by lethargy, decreased feeding, high-pitched cry, fever, seizures, and even death. Up to 84% of infants with kernicterus will develop chronic bilirubin encephalopathy, characterized by permanent movement disorders, mental retardation, and hearing loss.3
The incidence of kernicterus is difficult to estimate due to delayed diagnosis, an error in the diagnosis code, under-reporting, or lack of reporting in third-world countries.4 The prevalence of severe hyperbilirubinemia is estimated to be 2-45 per 100,000 births, and of kernicterus 0.4-2.7 per 100,000 births in developed countries.5 The incidence of kernicterus in developing countries is higher due to a variety of factors, such as genetic differences, prematurity, low birth weight, underfeeding, and sepsis.5,6
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
G6PD deficiency is one of the most common human enzymopathies, estimated to affect 400 million people worldwide.7 The deficiency is caused by single nucleotide polymorphisms (SNPs) leading to single amino acid changes in the protein glucose-6-phosphate dehydrogenase (G6PD). Over 400 SNPs, responsible for 160 different amino acid changes, have been observed in G6PD deficiency.8,9 These mutations can cause deficiency of varying severity, and are classified into four clinical categories (Table 1). The majority of G6PD deficiency can be accounted for by a few common and relatively mild (Class II or III) mutations, while severe (Class I) mutations are more rare.10 As G6PD is X-linked, heterozygous females can carry severe mutations and remain symptomless, whereas hemizygous males with Class I mutations suffer from chronic nonspherocytic hemolytic anemia (CNSHA).11,12 Class II and III mutations lead to episodes of hemolytic anemia after stressors such as ingestion of fava beans (favism), ingestion of certain prescription drugs, or infection.
Table 1:
Clinical classification of G6PD mutations. (adapted from [10])
| Classification of G6PD mutations | Clinical outcome | |
|---|---|---|
| Class I | < 10% activity | Severe; CNSHA |
| Class II | < 10% activity | Severe episodes of hemolytic anemia |
| Class III | 10 - 60% activity | Mild episodes of hemolytic anemia |
| Class IV | 60 - 150% activity | Asymptomatic |
Recent completion of the 1000 Genomes Project13 has allowed us to update the estimation of the prevalence of G6PD deficiency (Fig 1). The 1000 Genomes Project sequenced the entire genomes of ~3,500 individuals from five main human populations, revealing the presence of 22 single mutations and 5 double mutations in the G6PD protein. The majority of mutations were Class III, with few Class II and no known Class I mutations observed. Extrapolation of these numbers using data from the 2015 Population Reference Bureau14 estimates that 7.6% of the world population, or 560 million people, carries a mutated G6PD allele. On average, weighted by the world population distribution, 5.3% of males have G6PD deficiency.
Figure 1:
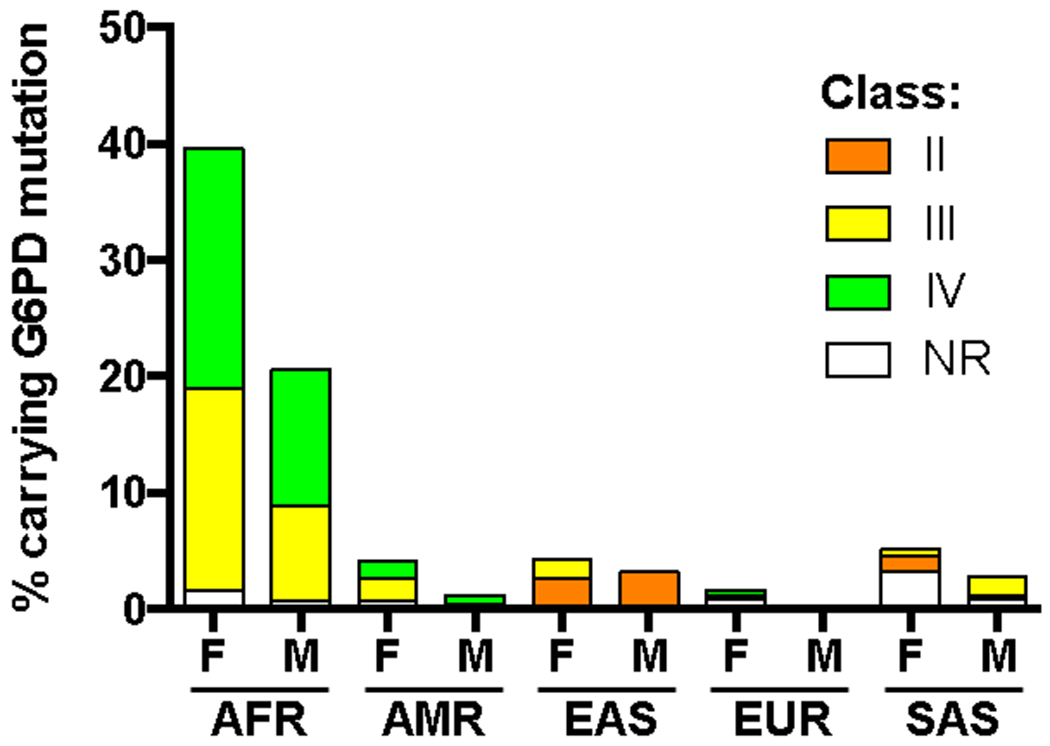
Percent of females (F) and males (M) from the 1000 Genomes Project carrying a G6PD mutation. No Class I mutations were observed. AFR: African; AMR: Mixed American; EAS: East Asian; EUR: European; SAS: South Asian; NR: not reported.
G6PD deficiency increases the risk of neonatal hyperbilirubinemia and kernicterus
Infants with G6PD deficiency are significantly more likely to develop hyperbilirubinemia;15 a recent meta-analysis of five studies including over 20,000 subjects found that G6PD-deficient infants are almost four times more likely to develop hyperbilirubinemia and three times more likely to receive phototherapy compared to G6PD-normal infants.16
Moreover, G6PD-deficient infants are more likely to develop kernicterus. In the United States, 20% of infants who develop kernicterus have G6PD deficiency, compared to an estimated 4-7% prevalence of G6PD deficiency in the average population.17,18 Of infants with kernicterus, 15% of those with G6PD-deficiency died, compared to 1% mortality rate in the G6PD-normal infants.18 A summary of kernicterus and G6PD deficiency in other countries is summarized in Table 2. Enrichment of G6PD deficiency in kernicterus is seen in almost all cases.
Table 2:
Kernicterus is over seven fold more likely to occur in G6PD deficient newborns worldwide.
| Summary of known kernicterus and G6PD deficiency rates in various countries | |||||
|---|---|---|---|---|---|
| Country | Incidence of kernicterus (per 100,000) | % G6PD deficiency in infants with kernicterus | Expected % G6PD deficiency, based on general population | Fold over-representation of G6PD deficiency in infants with kernicterus | Ref |
| Canada | 2.3 - 7.9 | 39 - 58 | 0 - 3 | 30 X | 7,75–77 |
| Cuba | 4.6 | 5 | 2 - 4 | 1.6 X | 78,79 |
| Denmark | 1.8 - 9 | 2 | 0 - 3 | 1 X | 7,80,81 |
| Hong Kong | 269 | 55 | 3 - 6 | 12 X | 82 |
| Nigeria | 800 - 1600 | 61 - 80 | 15 | 4.7 X | 83–86 |
| Oman | 346 | 70 | 18 | 3.9 X | 87 |
| Singapore | N/A | 43 | 2 | 22 X | 88 |
| Turkey | N/A | 18 | 13 | 1.4 X | 89 |
| USA | 1.5 | 20 | 1 - 7 | 5.6 X | 7,18,90 |
| UK/Ireland | 0.9 | 21 | 0 - 3 | 13 X | 7,91 |
| Average | 7.5 X | ||||
Interestingly, African-American neonates exhibit lower peak levels of serum bilirubin compared to Caucasian neonates, yet are more likely to develop kernicterus after discharge from the hospital, accounting for 25% of all the kernicterus cases in the USA.19 As G6PD deficiency is highly common in African-Americans (up to 21% of males in certain regions), this suggests that G6PD-deficient neonates develop signs of kernicterus at lower levels of serum bilirubin than G6PD-normal neonates.
In many Asian, African, Mediterranean, and Middle Eastern countries, where G6PD deficiency is common, all newborns are screened for G6PD deficiency. This has been associated with reduction of the instance of severe hyperbilirubinemia and kernicterus in several countries. In regions where G6PD deficiency is historically less common, an increase in global population movement has raised the question of whether G6PD deficiency screening should be implemented everywhere.17
How does G6PD deficiency contribute to kernicterus?
G6PD is the rate-limiting enzyme in the pentose phosphate pathway, catalyzing the oxidation of glucose-6-phosphate to 6-phospho-gluconate while reducing NADP+ to NADPH. NADPH then fuels the regeneration of reduced glutathione (GSH), which in turn neutralizes reactive oxygen species (ROS) (Fig. 2). G6PD is a major source of NADPH, along with mitochondrial enzymes isocitrate dehydrogenase and malic enzyme.20 However, in erythrocytes, which lack mitochondria, G6PD is the sole source of NADPH and therefore plays a critical role in protection against ROS. Indeed, Class I G6PD mutations cause CNSHA due to lack of protection against ROS in red blood cells. Similarly, G6PD deficiency in infants leads to an increase in hemolysis and subsequent rise in serum bilirubin levels from heme breakdown, and partially explains the higher prevalence of hyperbilirubinema in G6PD-deficient babies.
Figure 2:
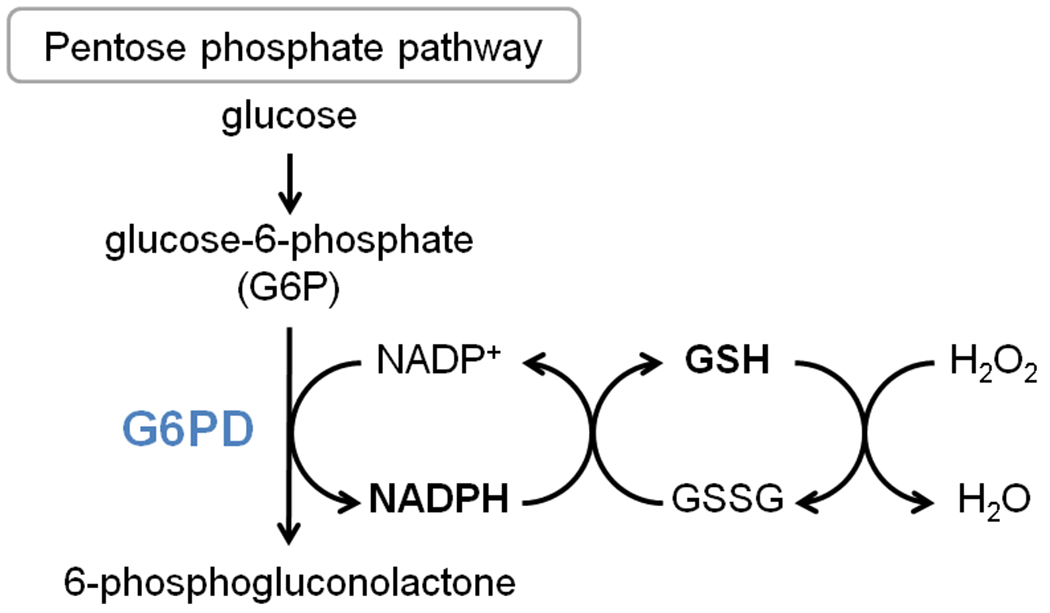
Scheme of G6PD activity.
The mechanism by which G6PD deficiency contributes to kernicterus is less well understood.21 Unconjugated bilirubin accumulates in the brain, specifically in neurons, neuronal processes, and microglia.22 A high rate of hemolysis may cause serum bilirubin to be produced more quickly than it can be conjugated or diffuse into skin and other body tissues; during this spike in which serum bilirubin concentrations are higher than the diffusion threshold, bilirubin penetrates the blood-brain barrier and enters brain cells.23 Furthermore, because bilirubin is lipophilic, it preferentially accumulates in fatty tissue and therefore does not diffuse out of the brain even at high concentrations.
Bilirubin toxicity begins at the cell membranes, as bilirubin is lipophilic and is highly concentrated in membrane compartments.24 This bridges membrane permeability, leads to lipid peroxidation, and inhibits the functions of membrane-bound proteins, such as ATPases.25 Similarly, bilirubin also targets mitochondrial membranes, leading to disruption of the electron transport chain and membrane-bound proteins, which in turn results in mitochondrial swelling, membrane permeability, depolarization, cytochrome c release and cell death by apoptosis and necrosis.26–30
Bilirubin itself has antioxidant properties, yet generation of ROS is a hallmark of bilirubin toxicity in the brain.30–32 At high bilirubin concentrations, increased production of superoxide radical anion,33 depletion of reduced glutathione (GSH), and increase in the oxidized disulfide form of glutathione (GSSG) are observed.34 High concentrations of ROS lead to protein oxidation, lipid peroxidation, and DNA damage, which activates pathways signaling for neuroinflammation, cell cycle arrest and apoptosis.21,35 In G6PD-deficient infants, kernicterus and increased bilirubin-induced neurotoxicity can be partially attributed to the lack of GSH regeneration due to decreased G6PD activity.
In general, the brain is sensitive to damage from increased ROS because of its high utilization of oxygen and high concentration of oxidizable polyunsaturated fatty acids.36 Additionally, infants (especially pre-term) have lower levels of antioxidant enzymes and scavengers such as vitamin E.37–39 Furthermore, the developing brain exhibits very low mitochondrial antioxidant activity and relies primarily on cytoplasmic enzymes (such as G6PD) to maintain redox homeostasis.36
Although the mechanism of bilirubin-induced neurotoxicity is not completely understood, it is clear that G6PD deficiency contributes to kernicterus via at least two mechanisms: initially, through increased hemolysis-induced spike in serum bilirubin levels and subsequent accumulation of bilirubin in the brain, and secondly, by reduced buffering capacity against bilirubin-induced ROS (Fig. 3). This second mechanism may explain why G6PD-deficient infants develop kernicterus even at lower levels of serum bilirubin.
Figure 3:
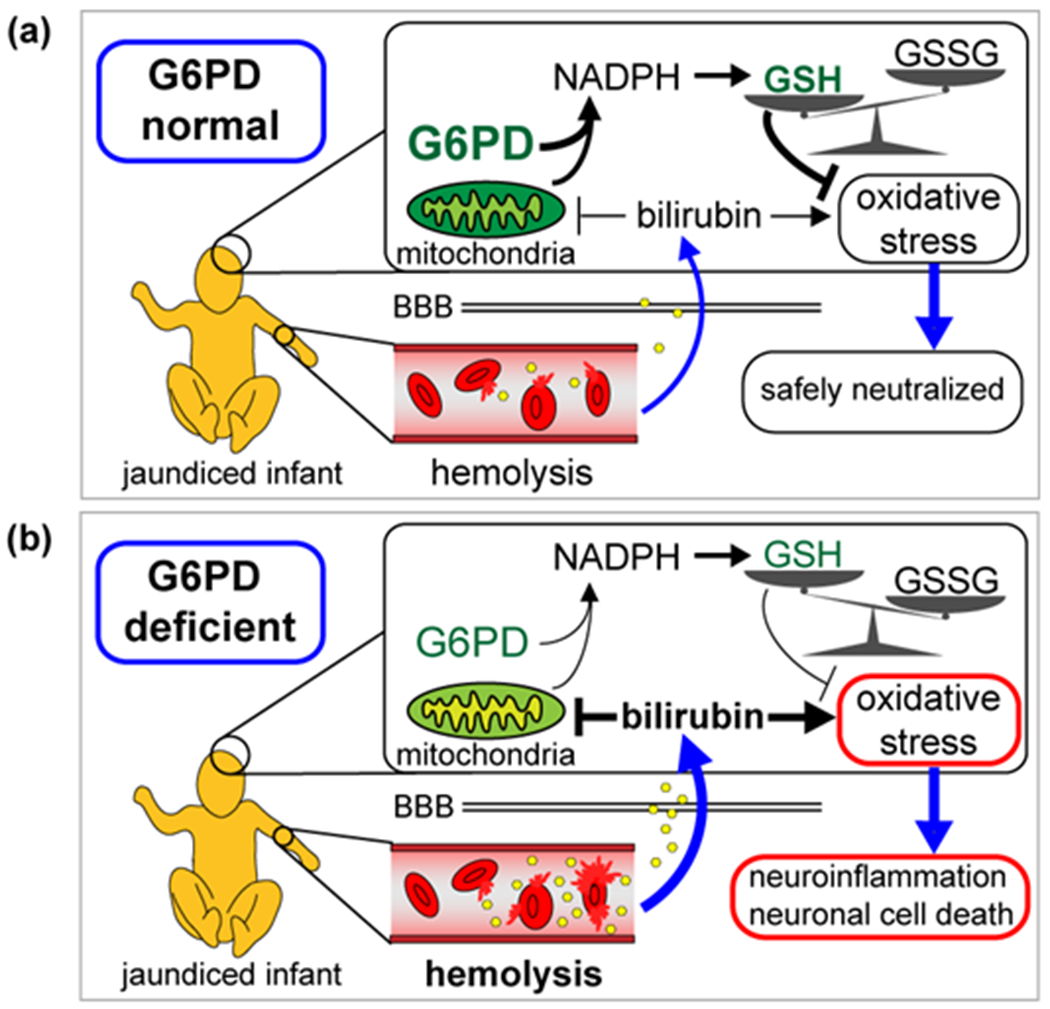
Contribution of G6PD deficiency to bilirubin-induced neurotoxicity. (a) Neonatal jaundice results from production of bilirubin (shown as yellow hexagons) following hemolysis. In G6PD-normal infants, GSH levels are properly maintained and low levels of bilirubin-induced oxidative stress are safely neutralized. (b) In G6PD-deficient infants, higher levels of hemolysis lead to higher concentration of bilirubin in the brain, which inhibits mitochondrial activity (indicated by yellowed mitochondria). Reduced G6PD activity leads to low NADPH levels, and GSH is depleted in favor of GSSG. Buildup of ROS leads to neuroinflammation, cell death, and kernicterus. BBB; blood-brain-barrier
How do we prevent kernicterus?
The incidence of kernicterus has risen in recent years due to a variety of factors.5,40,41 Infants are often discharged from the hospital within 24-48 hours of birth, whereas bilirubin levels often peak four to five days after birth; lack of proper monitoring at home allows the development of kernicterus, which might have otherwise been prevented if the infant were to remain at the hospital.42 Increasing popularity of breastfeeding has also contributed to hyperbilirubinemia and kernicterus.40,43
In developing regions, access to phototherapy and exchange transfusions is often limited.44 Even in developed nations, despite these simple and accessible treatments, up to 6.6% of G6PD-deficient babies will develop kernicterus45 and 12-50% of G6PD-deficient infants with kernicterus will die.46 Especially in cases where an acute hemolytic event in G6PD-deficient infants triggers rapid rise of bilirubin concentration in the brain, kernicterus may be impossible to prevent by conventional treatments.47 Furthermore, exchange transfusion leads to adverse events in 5% of infants, and death in 0.4% of infants.48
There is a clear need for a novel therapy to prevent kernicterus and its complications, especially in G6PD-deficient newborns. Below, we propose possible approaches to treat or prevent kernicterus.
Strategies to reduce serum bilirubin levels
One approach is to reduce the amount of circulating bilirubin by increasing bilirubin conjugation, increasing the binding capacity of albumin for serum bilirubin, or by binding bilirubin with an exogenous therapeutic agent before it can cross the blood-brain barrier. Phenobarbital increases expression of UGT1A1, which enhances bilirubin conjugation in the liver, and has been shown to reduce peak serum bilirubin levels. However, the use of phenobarbital is no longer recommended due to its slow onset of effect, long duration of effect, and ineffectiveness when given in the first 12 hours of life.49
Previously, therapeutic attempts to increase binding of bilirubin by albumin via a small molecule have been unsuccessful.50,51 The normal range of bilirubin concentration in the blood is 6 mg/dL (102 uM), and at least 25 mg/dL (427 uM) in cases of severe hyperbilirubinemia.42 With this extremely high concentration of circulating bilirubin, a therapeutic bilirubin-binding agent would need to reach an unreasonably high concentration in the blood in order to have a physiologically significant effect.
Treatment with antioxidants
Major bilirubin toxicity stems from GSH depletion and imbalanced redox equilibrium. Although bilirubin induces upregulation of genes involved in GSH homeostasis, NADPH homeostasis, and antioxidant defense,52,53 the temporal delay in upregulation still allows ample time for spikes in bilirubin-related ROS to exert neuronal damage. Studies have shown that levels of antioxidant vitamins A, C, and E are reduced in hyperbilirubinemic infants;54 however, a clinical study showed that treating infants with hyperbilirubinemia with the antioxidant vitamin E was unsuccessful.55
A more recent study showed that total antioxidant capacity remains unchanged or may increase in hyperbilirubinemia, possibly due to the antioxidant activity of bilirubin.56 This interplay between antioxidant activity and ROS is characteristic of other ROS-related diseases, and although bilirubin-induced ROS is a main driver of kernicterus, the use of antioxidants to prevent kernicterus is likely to be ineffective. Antioxidant treatment has proven ineffective in many other ROS-related diseases,57,58 possibly because these antioxidants may be scavenging both ‘good’ and ‘bad’ ROS.59 Further, the transient, spike-like nature of bilirubin-induced ROS is difficult to pinpoint, and administration of antioxidants too early or too late will not be effective.
Therefore, restoring redox equilibrium by improving endogenous antioxidant defense represents a more promising treatment for kernicterus and ROS-related diseases in general.
Restoring endogenous GSH: activating G6PD
Specifically, GSH depletion and increase in GSSG are hallmarks of bilirubin toxicity; a therapy directed at restoring the GSH/GSSG balance may be more helpful than general treatment with antioxidants. For example, pre-treatment with N-acetylcysteine (NAC), a GSH precursor, reduced bilirubin toxicity in rat neuronal cells in culture.34,52 NAC has been administered to preterm infants with no ill effects, but its effect on development of hyperbilirubinemia and/or kernicterus has not been reported.60 However, supplementing with GSH or GSH precursors does not bypass the issue of timing, and sudden increase in GSH may have unwanted impacts on glucose and/or iron metabolism.58,61 Moreover, GSH does not efficiently cross the blood-brain barrier,62 and it us unknown whether NAC can cross the blood-brain barrier.63 Rather, a therapy directed at maintaining endogenous control of the GSH/GSSG balance is desirable.
G6PD is the rate-limiting producer of cytoplasmic NADPH, which in turn converts GSSG to GSH (Fig. 2). As discussed above, in the developing brain mitochondrial production of NADPH is low, and is made even lower by the toxic effect of bilirubin on the mitochondrial membrane. Therefore, G6PD is the main driver of GSH regeneration. The loss of this major source of GSH in G6PD deficiency explains why higher rates and worse outcomes of kernicterus are observed in G6PD-deficient babies. Therefore, we propose a small-molecule activator or chaperone of G6PD as a novel treatment for kernicterus. Discovery of molecular chaperones and activators is uncommon compared to the discovery of small-molecule enzyme inhibitors, but a few small-molecule chaperones have been used successfully.
One example is a pharmacological chaperone therapy for Gaucher disease, a lysosomal storage disease. The disease is caused by mutations in acid β-glucosidase leading to significant protein misfolding and subsequent degradation of the protein before it can be transported to the lysosome. Interestingly, binding of small-molecule inhibitors to the mutated protein rescues protein folding and allows the protein to be transported to the lysosome; these inhibitors have been termed “pharmacological chaperones”. The pharmacological chaperone is competitively replaced by the highly concentrated substrate in the lysosome, leading to a net gain in acid β-glucosidase activity. Pharmacological chaperones have entered Phase II clinical trials to treat Gaucher disease in humans.64
Several common G6PD variants, such as G6PD A-, which is present in up to 45% of the population in some regions of Africa,7 and G6PD Mediterranean, which is the most common G6PD mutation in Caucasians,65 have been found to exhibit misfolding66,67 and reduced half-life in erythrocytes.68 Moreover, many Class I G6PD mutations exhibit highly decreased thermostability69 and inefficient protein folding.70 A pharmacological chaperone similar to the one found for Gaucher disease may be an effective strategy for correcting these types of G6PD mutations (Fig. 4a).
Figure 4.
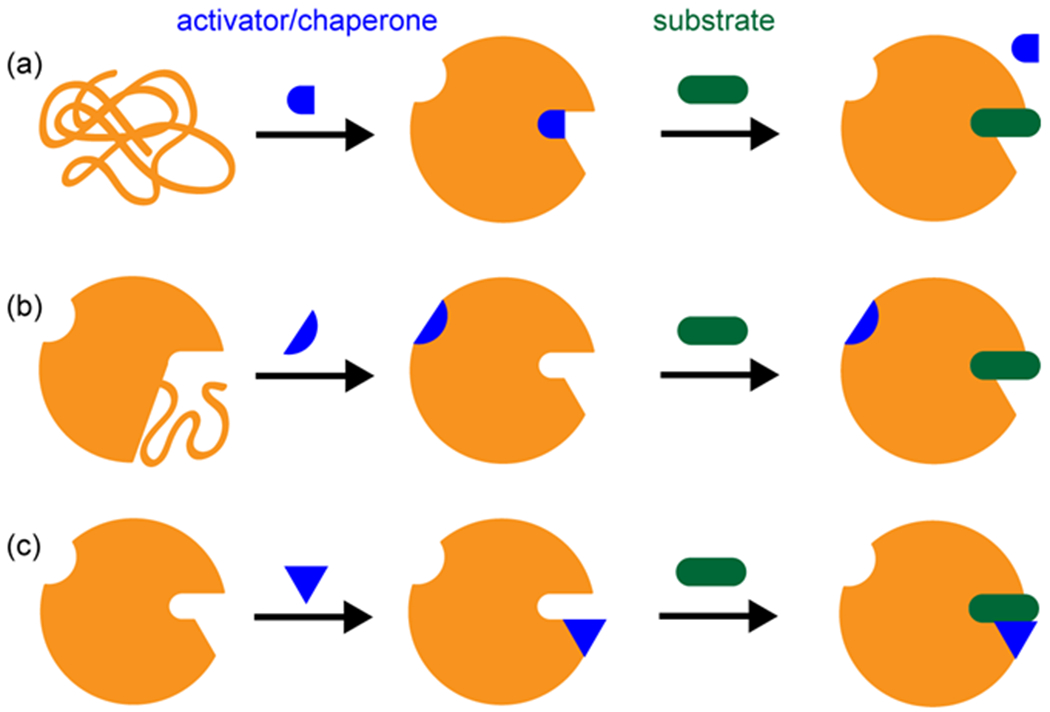
Mechanisms of pharmacological chaperone or small-molecule activator. (a) A pharmacological chaperone rescues misfolding. In the case of pharmacological chaperones for acid β-glucosidase, the chaperone is competitively replaced by substrate. (b) A small-molecule activator correcting a disordered region allosterically. (c) A small-molecule activator binding in the active site, increasing productive interaction between substrate and catalytic residues.
Another example of a small-molecule activator is Alda-1, an activator of aldehyde dehydrogenase 2 (ALDH2). A common ALDH2 variant, referred to as ALDH2*2, shows disordered folding in a small region of the protein caused by a single amino acid mutation. The ALDH2*2 variant remains viable, but exhibits <5% activity of ALDH2. Alda-1 increases the activity of ALDH2*2 by 11-fold and allosterically restores folding to the disordered region as demonstrated by a co-crystal structure.71,72 Moreover, Alda-1 also increases the activity of wild-type ALDH2 by two-fold. Crystallography shows that Alda-1 binds near the active site, increasing productive encounters between the substrate and catalytic residues.71
Most G6PD mutants have not been crystallized, but if mutations induce disordered regions similar to ALDH2*2, they could potentially be corrected allosterically by a small molecule in a mechanism similar to Alda-1 (Fig. 4b). Canton G6PD, the most common mutation in East Asia, has been crystallized but shows no gross structural differences compared to wild-type G6PD.73,74 In this case, a small molecule activator could be identified that binds in the catalytic site of G6PD and facilitates substrate and/or cofactor binding (Fig. 4c). Such a small molecule could also potentially activate wild-type G6PD in the same fashion; this activator could serve as a treatment to prevent kernicterus even in G6PD-normal infants with hyperbilirubinemia.
Best Practices Box.
What is the current practice?
Jaundiced infants with serum bilirubin level above 20 mg/dL are subjected to phototherapy, and generally are given exchange transfusion at serum bilirubin level above 25 mg/dL.
Phototherapy and exchange transfusion may not be available in developing nations where G6PD deficiency is most common.
What changes in current practice are likely to improve outcomes?
We propose development of a novel treatment to prevent kernicterus in addition to current practice.
A G6PD activator or pharmacological chaperone may reduce hemolysis and increase protection against bilirubin-induced ROS in the developing brain.
Screening for G6PD deficiency even in regions where G6PD deficiency is uncommon may help identify infants at higher risk for developing kernicterus.
Summary Statement
Preventable kernicterus and the subsequent sequelae still occur with a regular frequency in both developed and developing nations. We propose activation of G6PD as a novel treatment for kernicterus in both G6PD-deficient and G6PD-normal infants.
KEY POINTS.
G6PD deficiency increases the risk of kernicterus in jaundiced newborns.
G6PD is a major source of protection against bilirubin-induced oxidative stress in the developing brain.
There is need both in developed and developing nations for a novel treatment for kernicterus, especially in regions with a high rate of G6PD deficiency.
We propose a small-molecule activator or pharmacological chaperone for G6PD as a therapy for kernicterus in both G6PD-deficient and G6PD-normal infants with hyperbilirubinemia.
SYNOPSIS.
Hyperbilirubinemia occurs frequently in newborns, and in severe cases can progress to kernicterus and permanent developmental disorders. G6PD deficiency, one of the most common human enzymopathies, is a major risk factor for hyperbilirubinemia and greatly increases the risk of kernicterus even in the developed world. Lack of access to common treatments for hyperbilirubinemia, coupled with a high rate of G6PD deficiency, leads to high incidence of kernicterus in the developing world. Therefore, a novel treatment for kernicterus is needed, especially for G6PD-deficient newborns. Oxidative stress is a hallmark of bilirubin toxicity in the brain. We propose activation of G6PD via a small-molecule chaperone as a strategy to increase endogenous defense against bilirubin-induced oxidative stress and prevent kernicterus.
Footnotes
DISCLOSURE STATEMENT
The Authors have nothing to disclose
References
- 1.Maisels MJ. Managing the jaundiced newborn: a persistent challenge. CMAJ Can Med Assoc J J Assoc Medicale Can. 2015;187(5):335–343. doi: 10.1503/cmaj.122117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watchko JF, Tiribelli C. Bilirubin-Induced Neurologic Damage — Mechanisms and Management Approaches. N Engl J Med. 2013;369(21):2021–2030. doi: 10.1056/NEJMra1308124. [DOI] [PubMed] [Google Scholar]
- 3.Kaplan M, Hammerman C. Understanding severe hyperbilirubinemia and preventing kernicterus: adjuncts in the interpretation of neonatal serum bilirubin. Clin Chim Acta Int J Clin Chem. 2005;356(1-2):9–21. doi: 10.1016/j.cccn.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Bhutani VK, Johnson LH, Jeffrey Maisels M, et al. Kernicterus: Epidemiological Strategies for Its Prevention through Systems-Based Approaches. J Perinatol. 2004;24(10):650–662. doi: 10.1038/sj.jp.7211152. [DOI] [PubMed] [Google Scholar]
- 5.Olusanya BO, Ogunlesi TA, Slusher TM. Why is kernicterus still a major cause of death and disability in low-income and middle-income countries? Arch Dis Child. 2014;99(12):1117–1121. doi: 10.1136/archdischild-2013-305506. [DOI] [PubMed] [Google Scholar]
- 6.Bhutani VK, Wong RJ. Bilirubin neurotoxicity in preterm infants: risk and prevention. J Clin Neonatol. 2013;2(2):61–69. doi: 10.4103/2249-4847.116402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42(3):267–278. doi: 10.1016/j.bcmd.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Beutler E G6PD: Population genetics and clinical manifestations. Blood Rev. 1996;10(1):45–52. doi: 10.1016/S0268-960X(96)90019-3. [DOI] [PubMed] [Google Scholar]
- 9.Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B, Capoluongo E. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the “old” and update of the new mutations. Blood Cells Mol Dis. 2012;48(3):154–165. doi: 10.1016/j.bcmd.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371(9606):64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 11.Luzzatto L Glucose 6-phosphate dehydrogenase deficiency: from genotype to phenotype. Haematologica. 2006;91(10):1303–1306. [PubMed] [Google Scholar]
- 12.Corrons JV, Feliu E, Pujades MA, et al. Severe-glucose-6-phosphate dehydrogenase (G6PD) deficiency associated with chronic hemolytic anemia, granulocyte dysfunction, and increased susceptibility to infections: description of a new molecular variant (G6PD Barcelona). Blood. 1982;59(2):428–434. [PubMed] [Google Scholar]
- 13.Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kristin B, Kaneda T. 2014 World Population Data Sheet. Population Reference Bureau; 2015. http://www.prb.org/wpds/2015/. Accessed October 6, 2015.
- 15.Kaplan M, Hammerman C. Glucose-6-phosphate dehydrogenase deficiency and severe neonatal hyperbilirubinemia: a complexity of interactions between genes and environment. Semin Fetal Neonatal Med. 2010;15(3):148–156. doi: 10.1016/j.siny.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Liu W, Tang X, Wang T. Association between G6PD deficiency and hyperbilirubinemia in neonates: a meta-analysis. Pediatr Hematol Oncol. 2015;32(2):92–98. doi: 10.3109/08880018.2014.887803. [DOI] [PubMed] [Google Scholar]
- 17.Watchko JF, Kaplan M, Stark AR, Stevenson DK, Bhutani VK. Should we screen newborns for glucose-6-phosphate dehydrogenase deficiency in the United States? J Perinatol. 2013;33(7):499–504. doi: 10.1038/jp.2013.14. [DOI] [PubMed] [Google Scholar]
- 18.Johnson L, Bhutani VK, Karp K, Sivieri EM, Shapiro SM. Clinical report from the pilot USA Kernicterus Registry (1992 to 2004). J Perinatol Off J Calif Perinat Assoc. 2009;29 Suppl 1:S25–S45. doi: 10.1038/jp.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Watchko JF. Hyperbilirubinemia in African American neonates: clinical issues and current challenges. Semin Fetal Neonatal Med. 2010;15(3):176–182. doi: 10.1016/j.siny.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Tomanek L Proteomic responses to environmentally induced oxidative stress. J Exp Biol. 2015;218(12):1867–1879. doi: 10.1242/jeb.116475. [DOI] [PubMed] [Google Scholar]
- 21.Stevenson DK, Maisels MJ, Watchko JF, eds. Bilirubin Toxicity. In: Care of the Jaundiced Neonate. The McGraw-Hill Companies, Inc.; 2012:115–143. [Google Scholar]
- 22.Martich-Kriss V, Kollias SS, Ball WS. MR findings in kernicterus. Am J Neuroradiol. 1995;16(4):819–821. [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan M, Bromiker R, Hammerman C. Hyperbilirubinemia, hemolysis, and increased bilirubin neurotoxicity. Semin Perinatol. 2014;38(7):429–437. doi: 10.1053/j.semperi.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Hansen T, Tommarello S, Allen J. Subcellular localization of bilirubin in rat brain after in vivo i.v. administration of [3H]bilirubin. Pediatr Res. 2001;49(2):203–207. doi: 10.1203/00006450-200102000-00012. [DOI] [PubMed] [Google Scholar]
- 25.Brito MA, Brites D, Butterfield DA. A link between hyperbilirubinemia, oxidative stress and injury to neocortical synaptosomes. Brain Res. 2004;1026(1):33–43. doi: 10.1016/j.brainres.2004.07.063. [DOI] [PubMed] [Google Scholar]
- 26.Rodrigues CMP, Solá S, Castro RE, Laires PA, Brites D, Moura JJG. Perturbation of membrane dynamics in nerve cells as an early event during bilirubin-induced apoptosis. J Lipid Res. 2002;43(6):885–894. [PubMed] [Google Scholar]
- 27.Keshavan P, Schwemberger SJ, Smith DLH, Babcock GF, Zucker SD. Unconjugated bilirubin induces apoptosis in colon cancer cells by triggering mitochondrial depolarization. Int J Cancer J Int Cancer. 2004;112(3):433–445. doi: 10.1002/ijc.20418. [DOI] [PubMed] [Google Scholar]
- 28.Rodrigues CM, Solá S, Silva R, Brites D. Bilirubin and amyloid-beta peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol Med Camb Mass. 2000;6(11):936–946. [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigues CMP, Solá S, Brites D. Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatol Baltim Md. 2002;35(5):1186–1195. doi: 10.1053/jhep.2002.32967. [DOI] [PubMed] [Google Scholar]
- 30.Oakes GH, Bend JR. Early steps in bilirubin-mediated apoptosis in murine hepatoma (Hepa 1c1c7) cells are characterized by aryl hydrocarbon receptor-independent oxidative stress and activation of the mitochondrial pathway. J Biochem Mol Toxicol. 2005;19(4):244–255. doi: 10.1002/jbt.20086. [DOI] [PubMed] [Google Scholar]
- 31.Seubert JM, Darmon AJ, El-Kadi AOS, D’Souza SJA, Bend JR. Apoptosis in Murine Hepatoma Hepa 1c1c7 Wild-Type, C12, and C4 Cells Mediated by Bilirubin. Mol Pharmacol. 2002;62(2):257–264. doi: 10.1124/mol.62.2.257. [DOI] [PubMed] [Google Scholar]
- 32.Cesaratto L, Calligaris SD, Vascotto C, et al. Bilirubin-induced cell toxicity involves PTEN activation through an APE1/Ref-1-dependent pathway. J Mol Med. 2007;85(10):1099–1112. doi: 10.1007/s00109-007-0204-3. [DOI] [PubMed] [Google Scholar]
- 33.Loftspring MC, Johnson HL, Feng R, Johnson AJ, Clark JF. Unconjugated bilirubin contributes to early inflammation and edema after intracerebral hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2011;31(4):1133–1142. doi: 10.1038/jcbfm.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaz AR, Silva SL, Barateiro A, et al. Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol Cell Neurosci. 2011;48(1):82–93. doi: 10.1016/j.mcn.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh H-L, Yang C-M, Hsieh H-L, Yang C-M. Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases, Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases. BioMed Res Int BioMed Res Int. 2013;2013, 2013:e484613. doi: 10.1155/2013/484613, 10.1155/2013/484613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikonomidou C, Kaindl AM. Neuronal Death and Oxidative Stress in the Developing Brain. Antioxid Redox Signal. 2010;14(8):1535–1550. doi: 10.1089/ars.2010.3581. [DOI] [PubMed] [Google Scholar]
- 37.Lindeman JHN, Van Zoeren-Grobben D, Schrijver J, Speek AJ, Poorthuis BJHM, Berger HM. The Total Free Radical Trapping Ability of Cord Blood Plasma in Preterm and Term Babies. Pediatr Res. 1989;26(1):20–24. [DOI] [PubMed] [Google Scholar]
- 38.Sullivan JL, Newton RB. Serum antioxidant activity in neonates. Arch Dis Child. 1988;63(7 Spec No):748–750. doi: 10.1136/adc.63.7_Spec_No.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McElroy MC, Postle AD, Kelly F J. Catalase, superoxide dismutase and glutathione peroxidase activities of lung and liver during human development. Biochim Biophys Acta BBA - Gen Subj. 1992;1117(2):153–158. doi: 10.1016/0304-4165(92)90073-4. [DOI] [PubMed] [Google Scholar]
- 40.Watchko JF. Identification of Neonates at Risk for Hazardous Hyperbilirubinemia: Emerging Clinical Insights. Pediatr Clin North Am. 2009;56(3):671–687. doi: 10.1016/j.pcl.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 41.Ives K Preventing kernicterus: a wake-up call. Arch Dis Child Fetal Neonatal Ed. 2007;92(5):F330–F331. doi: 10.1136/adc.2006.112342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhutani VK, Johnson L. Kernicterus in the 21st century: frequently asked questions. J Perinatol. 2009;29(S1):S20–S24. doi: 10.1038/jp.2008.212. [DOI] [PubMed] [Google Scholar]
- 43.Gourley GR. Breast-feeding, neonatal jaundice and kernicterus. Semin Neonatol SN. 2002;7(2):135–141. [DOI] [PubMed] [Google Scholar]
- 44.Olusanya BO, Slusher TM. Reducing the burden of severe neonatal jaundice in G6PD-deficient populations in low-income countries: are we doing enough? Int Health. 2010;2(1):22–24. doi: 10.1016/j.inhe.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Weng Y-H, Chiu Y-W. Clinical Characteristics of G6PD Deficiency in Infants With Marked Hyperbilirubinemia: J Pediatr Hematol Oncol. 2010;32(1):11–14. doi: 10.1097/MPH.0b013e3181c09aec. [DOI] [PubMed] [Google Scholar]
- 46.Kaplan M, Bromiker R, Hammerman C. Severe Neonatal Hyperbilirubinemia and Kernicterus: Are These Still Problems in the Third Millennium? Neonatology. 2011;100(4):354–362. doi: 10.1159/000330055. [DOI] [PubMed] [Google Scholar]
- 47.Kaplan M, Hammerman C. Glucose-6-phosphate dehydrogenase deficiency: a hidden risk for kernicterus. Semin Perinatol. 2004;28(5):356–364. [DOI] [PubMed] [Google Scholar]
- 48.Muchowski KE. Evaluation and treatment of neonatal hyperbilirubinemia. Am Fam Physician. 2014;89(11):873–878. [PubMed] [Google Scholar]
- 49.Hyperbilirubinemia S on. Management of Hyperbilirubinemia in the Newborn Infant 35 or More Weeks of Gestation. Pediatrics. 2004;114(1):297–316. doi: 10.1542/peds.114.1.297. [DOI] [PubMed] [Google Scholar]
- 50.Woods JT, Bryan LE, Chan G, Schiff D. Gentamicin and albumin-bilirubin binding. An in vivo study. J Pediatr. 1976;89(3):483–486. [DOI] [PubMed] [Google Scholar]
- 51.Brossard Y, Larsen M, Mesnard G, et al. [Bilirubin-albumin-binding function of 2 human albumin preparations (placental and plasma). Comparison of their efficacy in the icteric premature infant]. Arch Fr Pédiatrie. 1988;45(2):91–97. [PubMed] [Google Scholar]
- 52.Qaisiya M, Coda Zabetta CD, Bellarosa C, Tiribelli C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell Signal. 2014;26(3):512–520. doi: 10.1016/j.cellsig.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 53.Deganuto M, Cesaratto L, Bellarosa C, et al. A proteomic approach to the bilirubin-induced toxicity in neuronal cells reveals a protective function of DJ-1 protein. PROTEOMICS. 2010;10(8):1645–1657. doi: 10.1002/pmic.200900579. [DOI] [PubMed] [Google Scholar]
- 54.Turgut M, Başaran O, Çekmen M, Karataş F, Kurt A, Aygün A. Oxidant and antioxidant levels in preterm newborns with idiopathic hyperbilirubinaemia. J Paediatr Child Health. 2004;40(11):633–637. doi: 10.1111/j.1440-1754.2004.00489.x. [DOI] [PubMed] [Google Scholar]
- 55.Fischer AF, Inguillo D, Martin DM, Ochikubo CG, Vreman HJ, Stevenson DK. Carboxyhemoglobin concentration as an index of bilirubin production in neonates with birth weights less than 1,500 grams: a randomized double-blind comparison of supplemental oral vitamin E and placebo. J Pediatr Gastroenterol Nutr. 1987;6(5):748–751. [DOI] [PubMed] [Google Scholar]
- 56.Bélanger S, Lavoie JC, Chessex P. Influence of bilirubin on the antioxidant capacity of plasma in newborn infants. Biol Neonate. 1997;71(4):233–238. [DOI] [PubMed] [Google Scholar]
- 57.Guallar E, Stranges S, Mulrow C, Appel LJ, Miller I, Edgar R. Enough Is Enough: Stop Wasting Money on Vitamin and Mineral Supplements. Ann Intern Med. 2013;159(12):850–851. doi: 10.7326/0003-4819-159-12-201312170-00011. [DOI] [PubMed] [Google Scholar]
- 58.Kawagishi H, Finkel T. Unraveling the truth about antioxidants: ROS and disease: finding the right balance. Nat Med. 2014;20(7):711–713. doi: 10.1038/nm.3625. [DOI] [PubMed] [Google Scholar]
- 59.Carocho M, Ferreira ICFR. A review on antioxidants, prooxidants and related controversy: Natural and synthetic compounds, screening and analysis methodologies and future perspectives. Food Chem Toxicol. 2013;51:15–25. doi: 10.1016/j.fct.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 60.Soghier LM, Brion LP. Cysteine, cystine or N-acetylcysteine supplementation in parenterally fed neonates. In: The Cochrane Collaboration, ed. Cochrane Database of Systematic Reviews. Chichester, UK: John Wiley & Sons, Ltd; 2006. 10.1002/14651858.CD004869.pub2. Accessed October 6, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar C, Igbaria A, D’Autreaux B, et al. Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control: GSH crucial for iron metabolism, minor for redox. EMBO J. 2011;30(10):2044–2056. doi: 10.1038/emboj.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smeyne M, Smeyne RJ. Glutathione metabolism and Parkinson’s disease. Free Radic Biol Med. 2013;62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta BBA - Gen Subj. 2013;1830(8):4117–4129. doi: 10.1016/j.bbagen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 64.Benito JM, Fernández JMG, Mellet CO. Pharmacological chaperone therapy for Gaucher disease: a patent review. Expert Opin Ther Pat. 2011;21(6):885–903. doi: 10.1517/13543776.2011.569162. [DOI] [PubMed] [Google Scholar]
- 65.Oppenheim A, Jury CL, Rund D, Vulliamy TJ, Luzzatto L. G6PD Mediterranean accounts for the high prevalence of G6PD deficiency in Kurdish Jews. Hum Genet. 1993;91(3):293–294. [DOI] [PubMed] [Google Scholar]
- 66.Gómez-Gallego F G-P A. Unproductive folding of the human G6PD-deficient variant A-. FASEB J Off Publ Fed Am Soc Exp Biol. 1996;10(1):153–158. [DOI] [PubMed] [Google Scholar]
- 67.Gómez-Gallego F, Garrido-Pertierra A, Bautista JM. Structural Defects Underlying Protein Dysfunction in Human Glucose-6-phosphate Dehydrogenase A– Deficiency. J Biol Chem. 2000;275(13):9256–9262. doi: 10.1074/jbc.275.13.9256. [DOI] [PubMed] [Google Scholar]
- 68.Piomelli S, Corash LM, Davenport DD, Miraglia J, Amorosi EL. In vivo lability of glucose-6-phosphate dehydrogenase in GdA- and GdMediterranean deficiency. J Clin Invest. 1968;47(4):940–948. doi: 10.1172/JCI105786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gómez-Manzo S, Terrón-Hernández J, De la Mora-De la Mora I, et al. The stability of G6PD is affected by mutations with different clinical phenotypes. Int J Mol Sci. 2014;15(11):21179–21201. doi: 10.3390/ijms151121179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X-T, Engel PC. Clinical mutants of human glucose 6-phosphate dehydrogenase: impairment of NADP(+) binding affects both folding and stability. Biochim Biophys Acta. 2009;1792(8):804–809. doi: 10.1016/j.bbadis.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Perez-Miller S, Younus H, Vanam R, Chen C-H, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17(2):159–164. doi: 10.1038/nsmb.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen C-H, Budas GR, Churchill EN, Disatnik M-H, Hurley TD, Mochly-Rosen D. An Activator of Mutant and Wildtype Aldehyde Dehydrogenase Reduces Ischemic Damage to the Heart. Science. 2008;321(5895):1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kotaka M, Gover S, Vandeputte-Rutten L, Au SWN, Lam VMS, Adams MJ. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 5):495–504. doi: 10.1107/S0907444905002350. [DOI] [PubMed] [Google Scholar]
- 74.Au SW, Gover S, Lam VM, Adams MJ. Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure. 2000;8(3):293–303. doi: 10.1016/S0969-2126(00)00104-0. [DOI] [PubMed] [Google Scholar]
- 75.Sgro M. Incidence and causes of severe neonatal hyperbilirubinemia in Canada. Can Med Assoc J. 2006;175(6):587–590. doi: 10.1503/cmaj.060328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sgro M, Campbell DM, Kandasamy S, Shah V. Incidence of Chronic Bilirubin Encephalopathy in Canada, 2007–2008. Pediatrics. 2012;130(4):e886–e890. doi: 10.1542/peds.2012-0253. [DOI] [PubMed] [Google Scholar]
- 77.AlOtaibi SF, Blaser S, MacGregor DL. Neurological complications of kernicterus. Can J Neurol Sci J Can Sci Neurol. 2005;32(3):311–315. [DOI] [PubMed] [Google Scholar]
- 78.Henny-Harry C, Trotman H. Epidemiology of neonatal jaundice at the University Hospital of the West Indies. West Indian Med J. 2012;61(1):37–42. [PubMed] [Google Scholar]
- 79.Monteiro WM, Val FF, Siqueira AM, et al. G6PD deficiency in Latin America: systematic review on prevalence and variants. Mem Inst Oswaldo Cruz. 2014;109(5):553–568. doi: 10.1590/0074-0276140123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bjerre JV, Petersen JR, Ebbesen F. Surveillance of extreme hyperbilirubinaemia in Denmark. A method to identify the newborn infants. Acta Pædiatrica. 2008;97(8):1030–1034. doi: 10.1111/j.1651-2227.2008.00879.x. [DOI] [PubMed] [Google Scholar]
- 81.Ebbesen F, Andersson C, Verder H, et al. Extreme hyperbilirubinaemia in term and near-term infants in Denmark. Acta Paediatr. 2005;94(1):59–64. doi: 10.1080/08035250410022170. [DOI] [PubMed] [Google Scholar]
- 82.Lai HC, Lai MP, Leung KS. Glucose-6-phosphate dehydrogenase deficiency in Chinese. J Clin Pathol. 1968;21(1):44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Battistuzzi G, Esan GJ, Fasuan FA, Modiano G, Luzzatto L. Comparison of GdA and GdB activities in Nigerians. A study of the variation of the G6PD activity. Am J Hum Genet. 1977;29(1):31–36. [PMC free article] [PubMed] [Google Scholar]
- 84.Ogunlesi TA, Ogunfowora OB. Predictors of Acute Bilirubin Encephalopathy Among Nigerian Term Babies with Moderate-to-severe Hyperbilirubinaemia. J Trop Pediatr. 2011;57(2):80–86. doi: 10.1093/tropej/fmq045. [DOI] [PubMed] [Google Scholar]
- 85.Williams O, Gbadero D, Edowhorhu G, Brearley A, Slusher T, Lund TC. Glucose-6-phosphate dehydrogenase deficiency in Nigerian children. PloS One. 2013;8(7):e68800. doi: 10.1371/journal.pone.0068800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Owa JA, Ogunlesi TA, Ogunlesi TA. Why we are still doing so many exchange blood transfusion for neonatal jaundice in Nigeria. World J Pediatr WJP. 2009;5(1):51–55. doi: 10.1007/s12519-009-0009-2. [DOI] [PubMed] [Google Scholar]
- 87.Nair AK, Khusaiby SMA. Kernicterus and G6PD Deficiency—a Case Series from Oman. J Trop Pediatr. 2003;49(2):74–77. doi: 10.1093/tropej/49.2.74. [DOI] [PubMed] [Google Scholar]
- 88.Wong Hock Boon null. Singapore kernicterus--the position in 1965. J Singapore Paediatr Soc. 1965;7(2):35–43. [PubMed] [Google Scholar]
- 89.Katar S Glucose-6-Phosphate Dehydrogenase Deficiency and Kernicterus of South-East Anatolia: J Pediatr Hematol Oncol. 2007;29(5):284–286. doi: 10.1097/MPH.0b013e31805180dc. [DOI] [PubMed] [Google Scholar]
- 90.Burke BL, Robbins JM, Bird TM, Hobbs CA, Nesmith C, Tilford JM. Trends in Hospitalizations for Neonatal Jaundice and Kernicterus in the United States, 1988–2005. Pediatrics. 2009;123(2):524–532. doi: 10.1542/peds.2007-2915. [DOI] [PubMed] [Google Scholar]
- 91.Manning DJ, Maxwell MJ, Todd PJ, Platt MJ. Prospective surveillance study of severe hyperbilirubinaemia in the newborn in the United Kingdom and Ireland. Arch Dis Child - Fetal Neonatal Ed. October 2006. doi: 10.1136/adc.2006.105361. [DOI] [PMC free article] [PubMed] [Google Scholar]