

Abstract
Background
Traumatic injury and the associated acute bleeding are leading causes of death in people aged 1 to 44 years. Acute bleeding in pathological and surgical settings also represents a significant burden to the society. Yet there are no approved hemostatic drugs currently available. While clinically proven as an effective pro‐coagulant, activated factor VII (FVIIa) use in acute bleeding has been hampered by unwanted thromboembolic events. Enhancing the ability of FVIIa to quickly stop a bleed and clear rapidly from circulation may yield an ideal molecule suitable for use in patients with acute bleeding.
Objectives
To address this need and the current liability of FVIIa, we produced a novel FVIIa molecule (CT‐001) with enhanced potency and shortened plasma residence time by cell line engineering and FVIIa protein engineering for superior efficacy for acute bleeding and safety.
Methods
To address safety, CT‐001, a FVIIa protein with 4 desialylated N‐glycans was generated to promote active recognition and clearance via the asialoglycoprotein receptor. To enhance potency, the gamma‐carboxylated domain was modified with P10Q and K32E, which enhanced membrane binding.
Results
Together, these changes significantly enhanced potency and clearance while retaining the ability to interact with the key hemostatic checkpoint proteins antithrombin and tissue factor pathway inhibitor.
Conclusions
These results demonstrate that a FVIIa molecule engineered to combine supra‐physiological activity and shorter duration of action has the potential to overcome the current limitations of recombinant FVIIa to be a safe and effective approach to the treatment of acute bleeding.
Keywords: blood coagulation, factor VIIa, hemorrhage, hemostasis, protein engineering
Essentials.
Currently, there are no approved pharmaceutical interventions for acute bleeding.
Short‐acting activated factor VII (FVIIa) may effectively treat acute bleeds with reduced thrombogenicity.
CT‐001 is a FVIIa molecule engineered to have supra‐physiological and short‐acting activity.
In vitro studies of CT‐001 support further in vivo evaluation as a treatment for acute bleeds.
1. INTRODUCTION
Traumatic injury and associated bleeding are the major causes of death in people aged 1 to 44 years. 1 Acute bleeding in pathological and surgical settings also leads to significant morbidity and mortality. 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 There are no approved pharmaceuticals in current intervention protocols, though many have been tested clinically, including tranexamic acid, 19 , 20 fibrinogen concentrate (RiaSTAP), 21 , 22 and prothrombin complex concentrates (activated or nonactivated). 23 Recombinant FVIIa is approved for treating individuals with hemophilia A and B with autoantibodies against factor VIII and factor IX, 24 individuals with acquired hemophilia, 25 individuals with congenital FVII deficiency, 26 , 27 and individuals with Glanzmann thrombasthenia with refractoriness to platelet transfusion. 28 This has led to clinical trials in a diverse set of indications such as intracerebral hemorrhage, 2 , 3 traumatic bleeding, 4 , 5 cardiovascular bypass surgery, 6 traumatic brain injury, 7 liver surgery, 8 , 9 , 10 liver cirrhosis, 11 idiopathic thrombocytopenic purpura, 12 spinal surgery, 13 postpartum hemorrhage, 14 burns, 15 hematopoietic stem cell transplantation, 16 reversal of bleeding related to anticoagulants, 23 and diffuse alveolar hemorrhage. 17 , 18 While activity was seen in some of these indications, adverse thromboembolic events have limited its use. 29 In early studies, wild‐type (WT) FVIIa did not appear to increase thromboembolic events. 30 However, the thrombotic liability of WT FVIIa became evident with more studies. Thromboembolic events have been reported for people with trauma 31 and intracerebral hemorrhage 2 . A study based on the US Food and Drug Administration’s (FDA) Adverse Event Reporting System indicated that of the 144 reports with event timing information, 73 events (52%) occurred within 24 hours after the last dose of WT FVIIa, with 30 events within 2 hours. 32 In a meta‐analysis of randomized controlled trials, arterial event rate was significantly higher with WT FVIIa treatment, with the higher risk associated with higher doses and older patients. 33 The FDA has required a boxed warning on thromboembolic complications to the prescribing information. 34 Given the proven clinical hemostatic activity of FVIIa in the bleeding setting, efforts to address the safety limitations of the current molecule are warranted.
Factor VIIa is a molecule composed of a gamma‐carboxylated (Gla) domain, an aromatic amino acid stack, 2 epidermal growth factor (EGF) domains, and a protease domain (PD). 35 , 36 It also has two N‐glycosylation sites (N145, N322) and two O‐glycosylation sites (S52, S60). To address bleeding in the acute setting and the current liabilities of the native FVIIa molecule, we have engineered two additional N‐linked glycan sites (T106 N, V253 N) and used desialylation to actively shorten the protein half‐life for safety while compensating for shortened half‐life by increasing the potency through Gla domain engineering (P10Q, K32E). The resultant molecule, CT‐001, demonstrates enhanced potency and a faster clearance in vitro and suggest that further in vivo studies are warranted to understand if CT‐001 can serve as a stand‐alone or complementing therapy to current treatments of acute bleeding.
2. MATERIALS AND METHODS
2.1. Materials
Plasma human factor X, activated factor X (FXa), thrombin, antithrombin (AT) and recombinant full‐length tissue factor (flTF) were purchased from Haematologic Technologies (Essex, VT, USA); neuraminidase from New England BioLabs (Ipswich, MA, USA), anti–activated factor VII (FVIIa) monoclonal antibody (AD‐1), biotinylated anti–recombinant FVIIa antibody (AA‐3), HRP‐streptavidin 1‐Step Ultra TMB ELISA, William’s E medium, primary hepatocyte thawing and plating supplements, and primary hepatocyte maintenance supplements from Thermo Fisher Scientific (Waltham, MA, USA); S‐2765 and S‐2288 from DiaPharma (West Chester, OH, USA); ferric chloride, and chromozym tPA from Sigma‐Aldrich (St. Louis, MO, USA); soluble tissue factor (sTF) and tissue factor pathway inhibitor (TFPI) from BioLegend (San Diego, CA, USA); soluble endothelial protein C receptor (sEPCR) from R&D Systems (Minneapolis, MN, USA); convulxin from Cayman Chemicals (Ann Arbor, MI, USA); Ornithodoros moubata tick anticoagulant peptide (TAP) from Biomatik Corp. (Wilmington, DE, USA); C57Bl/6 mouse plasma, cynomolgus monkey plasma, and cryopreserved human hepatocytes (BioIVT, Westbury, NY, USA); STA‐PTT Automate 5, STA Neoplastin CI Plus 5 from Stago (North York, Toronto, ON, Canada); nonactivated thromboelastometry and intrinsic pathway thromboelastometry (INTEM) from Instrumentation Laboratory (Parsippany, NJ, USA), BIOPHEN FVIIa Assay from HYPHEN BioMed (Neuville Sur Oise, Ile‐de‐France, France); Dade Innovin from Siemens (Malvern, PA, USA); mouse thrombin‐antithrombin ELISA from AbCam (Cambridge, England); World Health Organization (WHO) FVIIa Second International Standard (07/228) from the National Institute for Biological Standards and Control; WT FVIIa (NovoSeven, eptacog alfa activated) from Novo Nordisk (Bagsvaerd, Denmark); and phosphatidylserine (PS):phosphatidylcholine (PC) liposomes from Encapsula Nanosciences (Brentwood, TN, USA).
2.2. Expression and purification of FVIIa variants
FVIIa variants were produced in serum‐free CHO‐K1 cells and purified from conditioned media through sequential application of Q‐Sepharose FF, CaFVII‐22 immunoaffinity, and POROS HQ chromatography or through sequential application of Q‐Sepharose FF, hydroxyapatite, and anionic exchange chromatography. The purified and activated CT‐001(Sial) was equilibrated in 10 mM glycyl‐glycine (pH 5.5) containing 50 mM NaCl, 10 mM CaCl2, and 0.01% polysorbate 80 (PS80). CT‐001 was created through enzymatic desialylation of purified CT‐001(Sial) with neuraminidase followed by Q HP‐Sepharose purification for neuraminidase removal. CT‐001 was formulated in 20 mM histidine, 10 mM calcium chloride, 230 mM trehalose, 10 mM methionine, and 0.01% polysorbate 20 (PS20), pH 6.4. Desialylated wild‐type (dWT) FVIIa was produced by the CHO‐K1 cell line expressing both WT FVII and the catalytic domain of Artherobacter urefaciens sialidase, purified as described above, and formulated in 10 mM histidine, 10 mM glycylglycine, 10 mM calcium chloride, 40 mM sodium chloride, 3.4 mM (0.5 mg/mL) methionine, 10 mg/ml sucrose, 25 mg/ml D[‐]mannitol, 0.07 mg/ml PS80, pH 6.0. For comparison, another batch of dWT FVIIa was created through enzymatic desialylation of purified WT FVIIa with bacterial sialidase followed by Q HP‐Sepharose purification for sialidase removal. All desialylated recombinant FVIIa preparations resulted in <0.2 mole sialic acid per mole of protein.
2.3. Flow cytometry analysis of activated platelet surface binding
Human washed platelets were prepared as previously described. 37 Platelets in Tyrode’s buffer with 10 mg/mL bovine serum albumin (BSA), and 5 mM CaCl2 were activated with 2 nM thrombin and 100 ng/mL convulxin for 15 minutes before being exposed to FVIIa variants (10 and 30 nM) for another 20 minutes at room temperature. The binding of FVIIa to cell surfaces was fixed for 10 minutes (0.5% formaldehyde) followed by three washes of Tyrode’s buffer with 10 mg/mL BSA and 5 mM CaCl2. The activation of platelets was monitored by PE‐anti‐CD62P (clone AC1.2; BD Biosciences, San Jose, CA, USA) and the binding of FVIIa to activated platelet surfaces was detected by Alexa488‐anti‐FVII (clone AA3). Donor informed consent and collection protocols are approved by the Institutional Review Board of AllCells.
2.4. Inhibition by antithrombin and tissue factor pathway inhibitor
Inhibition by AT (125 nM) was evaluated with 10 nM FVIIa variants, 15 U/mL unfractionated heparin, and 100 nM sTF at 37°C in 20 mM HEPES, 150 mM NaCl, 5 mM CaCl2, and 0.1% BSA, pH 7.4. Residual FVIIa activity was measured by adding 1 mM Chromozym tissue‐type plasminogen activator (tPA). Inhibition by TFPI was evaluated with 1 nM FVIIa variants, 4 nM FXa, and TFPI (0.316, 1, 3.16, 10, and 31.6 nM) incubated in Innovin (lipidated TF) at 37°C for 30 minutes. Residual FVIIa activity (OD405/min) was measured in the presence of 1 μM TAP by 0.9 mM S‐2288 in 20 mM HEPES, 150 mM NaCl, 5 mM CaCl2, and 0.1% BSA, pH 7.4.
2.5. Surface plasmon resonance analysis of TF and EPCR binding
CM5 sensor chip (GE Healthcare, Chicago, IL, USA) was activated with 50 mM of N‐hydroxysuccinimide and 200 mM of 1‐ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide hydrochloride for 420 seconds (10 µL/min). sEPCR, flTF, and sTF diluted in 10 mM NaAC (pH 4.5) were injected into the flow cells (10 µL/min) to achieve conjugation of 581, 72, and 60 response units, respectively. After the amine coupling, the remaining active sites were blocked with 1 M of ethanolamine hydrochloride. A surface without conjugation served as a blank. Binding was performed at 25°C with HBS‐EP (150 mM NaCl, 5 mM CaCl2 and 1 mM MgCl2, 0.005% surfactant P20, pH7.4). Diluted FVIIa variants were injected over the cells at the association phase (180 seconds), followed by running buffer at the dissociation phase (420 seconds for sEPCR; and 660 s for flTF and sTF). In flTF and sTF studies, CT‐001 and CT‐001(Sial) were injected at 2.19, 4.37, 8.75, 17.5, 35.0, 70.0, and 140 nM, while dWT FVIIa and WT FVIIa were injected at 1.56, 3.12, 6.25, 12.5, 25.0, 50.0, 100 nM. In sEPCR studies, all four FVIIa variants were injected at 62.5, 125, 250, 500, and 1000 nM. Binding data were fitted to a 1:1 Langmuir binding model (T2000 Evaluation 3.1; Biacore, Uppsala, Sweden).
2.6. Specific activity
The specific activities were evaluated by the BIOPHEN FVIIa chromogenic assay. WHO FVIIa Second International Standard (07/228) was used for calibration.
2.7. FXa generation enzyme kinetic assays
Ten micromolar PS:PC liposomes (30%:70%, w/w, 100 nm), 10 nM FVIIa variants, and factor X (0.0156 to 0.5 nM) in 25 mM HEPES, 100 mM NaCl, 5 mM CaCl2, and 0.1% BSA, pH 7.4, were incubated at 37°C for 2 to 5 minutes. Reactions were quenched by EDTA and FXa generation was determined using S‐2765. In the TF‐dependent FXa generation assay, 0.33 μl/L Innovin (lipidated TF), 2 nM FVIIa variants and factor X (0.0625–2 nM) in 25 mM HEPES, 100 mM NaCl, 5 mM CaCl2, and 0.1% BSA, pH 7.4, were incubated at 37°C for 2 to 5 minutes. Reactions were quenched, and FXa generation was determined.
2.8. Thrombin generation assay
Thrombin generation was measured in normal human platelet‐poor plasma in the presence of FVII variants using MP‐Reagents (containing 4 μM phospholipid) with a Fluoroskan Ascent reader (Stago).
2.9. Activated partial thromboplastin time and prothrombin time clotting assays
The effect of FVIIa variants on activated partial thromboplastin time (aPTT) was evaluated with citrated human and cynomolgus monkey plasma pools with Stago STA‐PTTA following the manufacturer’s protocol. C57Bl/6 plasma was diluted (1 plasma:2 Owren‐Koller buffer) before incubation with aPTT reagents. Prothrombin time (PT) was performed with STA‐Neoplastine CI Plus 5 (rabbit brain thromboplastin) with normal pooled human plasma.
2.10. Whole blood rotational thromboelastogram
FVIIa variants (1–1000 nM) were diluted in 20 mM HEPES and 154 mM NaCl, pH 7.4, and incubated in citrated whole blood from normal volunteers. ROTEM INTEM (ellagic acid) assays were performed following the manufacturer’s protocol.
2.11. Clearance of FVIIa variants by hepatocyte in vitro
Cryopreserved female human hepatocytes were prepared as previously described with modifications. 38 Then, 2.5 ng/mL of CT‐001, CT‐001(Sial), dWT FVIIa, or WT FVIIa in William’s E medium with CM4000 supplement containing 0.25% BSA was incubated for 60 minutes with one million viable hepatocytes/mL at 37°C. The suspensions were gently mixed using a rotator. At 10, 20, 40, or 60 minutes, a 0.2 mL suspension was removed and centrifuged at 94g for 3 minutes for supernatant. FVIIa variants in supernatant were determined by ELISA. Microtiter plates were coated with 100 μL/well of anti‐FVIIa (clone AD‐1) at 1 μg/mL in PBS for 1 hour. Plates were blocked with 200 μL of 1% casein (in 50 mM Tris, 100 mM NaCl, 0.05% PS20, pH 7.2) for 2 hours. Plates were washed four times with PBS, 0.05% PS20. Samples diluted into assay buffer (50 mM Tris, 100 mM NaCl, 0.05% Tween20, 0.1% casein, pH 7.2) were loaded into the wells for incubation (100 μL/well; 1 hour at room temperature). Plates were washed four times and biotin‐conjugated anti‐FVIIa (clone AA‐3) was added (100 μL/well). After 1 hour, plates were washed four times, and 100 μL of streptavidin‐horseradish peroxidase was added into each well for 1 hour. After four washes, FVIIa was detected by TMB. The reaction was quenched after 20 minutes with H2SO4. Three independent studies were performed.
3. RESULTS
3.1. Generation of FVIIa variants
dWT FVIIa has a WT FVII amino acid sequence with N‐glycans desialylated either enzymatically or during protein expression. Both methods removed sialic acid to nearly undetectable levels when measured by liquid chromatography–mass spectrometry analysis (99.95% desialylated, data not shown). CT‐001(Sial) is engineered for superior activity through amino acid substitutions at P10Q and K32E, positions known to increase the affinity of FVIIa to negatively charged surfaces. 39 CT‐001(Sial) was also engineered to prolong circulating half‐life through new N‐glycosylation sites. Two factors were considered in selecting new N‐glycosylation sites: (A) sequence motif for N‐linked glycosylation, and (B) exposure of sites on the protein surface. Sites with the sequence pattern N‐X‐S/T‐Z (N, asparagine; X, any amino acid residues but proline; S, serine; T, threonine; Z, any residues) will potentially be glycosylated on the asparagine residue. 40 , 41 , 42 New N‐glycosylation sites can be introduced by substitution of one residue to generate a new motif at a position with the N residue side chain exposed. Among the potential sites (data not shown), T106 and V253 were selected for the introduction of new N‐glycans, (Figure 1). CT‐001 is a desialylated variant of CT‐001(Sial).
FIGURE 1.
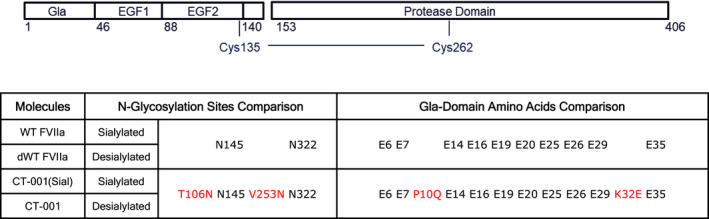
Structures of FVIIa variants. Wild‐type FVIIa (WT FVIIa) is composed of a Gla domain, 2 EGF domains, and a protease domain (PD). It is glycosylated with 2 N‐glycosylation sites (N145, N322). The N145 and N322 sites are composed of di‐ and triantennary carbohydrate chains with terminal GalNac capped with sialic acid. Highlighted in red are the amino acid substitutions relative to WT FVIIa. CT‐001(Sial) is engineered with P10Q and K32E in the Gla domain for enhanced affinity to negatively charged phospholipids and two additional N‐glycan structures T106 N and V253 N capped with sialic acid for providing a prolonged circulating half‐life to the molecule. CT‐001 is a desialylated variant of CT‐001(Sial) with additional terminal Gal and GalNAc moieties in comparison to dWT FVIIa to further accelerate plasma clearance and enhanced activity versus wild‐type sequences. dWT, desialylated wild‐type; EGF, epidermal growth factor; FVIIa, activated factor VII
3.2. FVIIa variants with Gla domain engineering show increased binding to activated human platelets
The binding of FVIIa variants was tested with platelets from three independent healthy donors by flow cytometry. As expected, due to their engineered Gla domain, CT‐001 and CT‐001(Sial) showed approximately threefold higher binding to activated human platelets. Ten nanomoles of CT‐001 and CT‐001(Sial) provided a similar amount of FVIIa binding to activated platelets by MFI in comparison to 30 nM of dWT FVIIa and WT FVIIa. No binding of FVIIa variants was observed with resting platelets (Figure 2).
FIGURE 2.
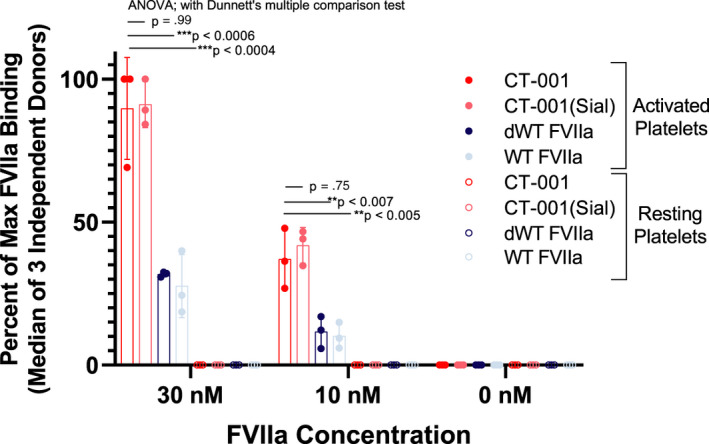
CT‐001 and CT‐001(Sial) demonstrate enhanced binding to human platelets activated with human thrombin and convulxin. Activated platelets from three healthy independent donors were washed and exposed to FVIIa variants, and the binding to cell surfaces was detected by Alexa488‐anti‐FVII (clone AA3) using flow cytometry. The mean FL1 of platelet population was measured and expressed as a percent of maximum FL1 achieved by each study (n=3 independent healthy donors). CT‐001 showed significantly increased binding to activated platelets in comparison to dWT FVIIa and WT FVIIa. ANOVA, analysis of variance; dWT, desialylated wild‐type; FVIIa, activated factor VII
3.3. CT‐001 interaction with physiological inhibitors AT and TFPI
CT‐001, CT‐001(Sial), and dWT FVIIa showed similar sensitivity to inhibition by AT/heparin39,40 (Figure 3A). The inhibition of the FVIIa variants by TFPI in the presence of lipidated TF and FXa was monitored using S‐2288 (Figure 3B). The Gla domain P10Q/K32E amino acid substitutions and the desialylation of FVIIa variants did not affect their inhibition by AT as well as TFPI in the presence of TF and FXa.
FIGURE 3.
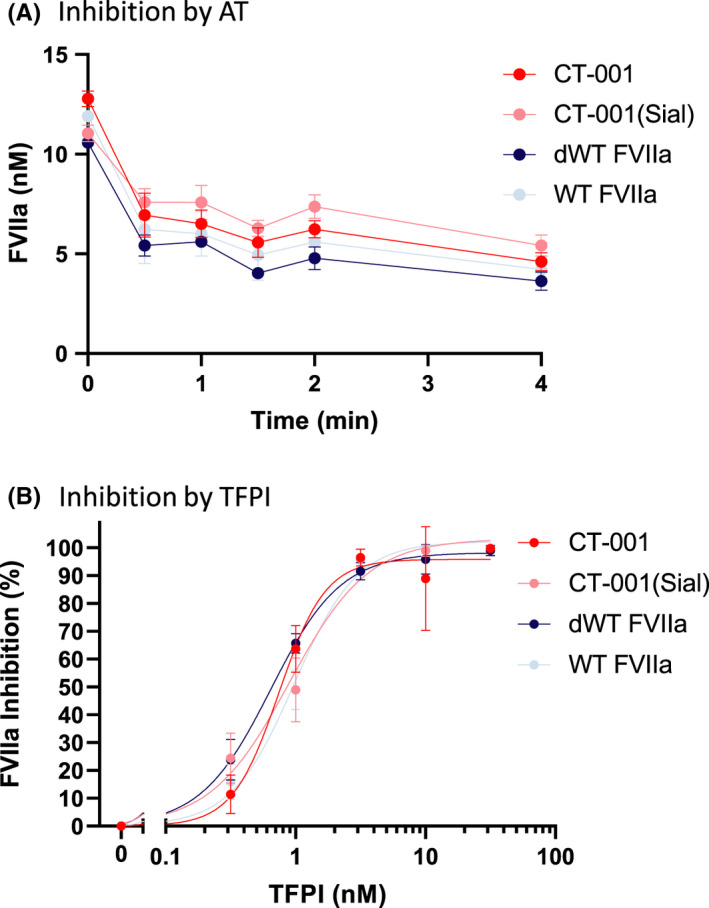
Inactivation of FVIIa variants by AT and TFPI. (A) There is no significant difference among CT‐001, CT‐001(Sial), dWT FVIIa, and WT FVIIa in response to inhibition by the presence of 125 nM of AT and heparin. Data are displayed as an average of three independent experiments. (B) CT‐001, CT‐001(Sial), dWT FVIIa, and WT FVIIa exhibited similar sensitivity to TFPI inhibition. Data are displayed as an average of three independent experiments. AT, antithrombin; dWT, desialylated wild‐type; FVIIa, activated factor VII; TFPI, tissue factor pathway inhibitor
3.4. Effect of protein engineering (P10Q, K32E, T106 N, V253 N) and desialylation of FVIIa variants on binding affinity for sEPCR and flTF but weaker binding to sTF
The binding of FVIIa variants to EPCR and TF was evaluated by surface plasmon resonance. The different combinations of amino acid substitutions in Gla domain (P10Q, K32E); introduction of new N‐glycans (T106 N, V253 N); and desialylation of CT‐001, CT‐001(Sial), and dWT FVIIa had led to some differences in their equilibrium dissociation constants (KDs) with sEPCR relative to WT FVIIa (Figure 4, Figure S1). CT‐001(Sial) was 1.9‐fold weaker than the WT FVIIa (1.05 × 10−7 M vs 5.52 × 10−8 M). This weaker KD of CT‐001(Sial) versus WT FVIIa was also observed with binding to flTF (2‐fold) and sTF (3.5‐fold). Interestingly, desialylation of CT‐001(Sial) to become CT‐001 restored some of the lowered affinity of the molecule toward sEPCR, flTF, and sTF. This increase in affinity by desialyation was also observed in dWT FVIIa in comparison to WT FVIIa, albeit to a much lesser extent.
FIGURE 4.
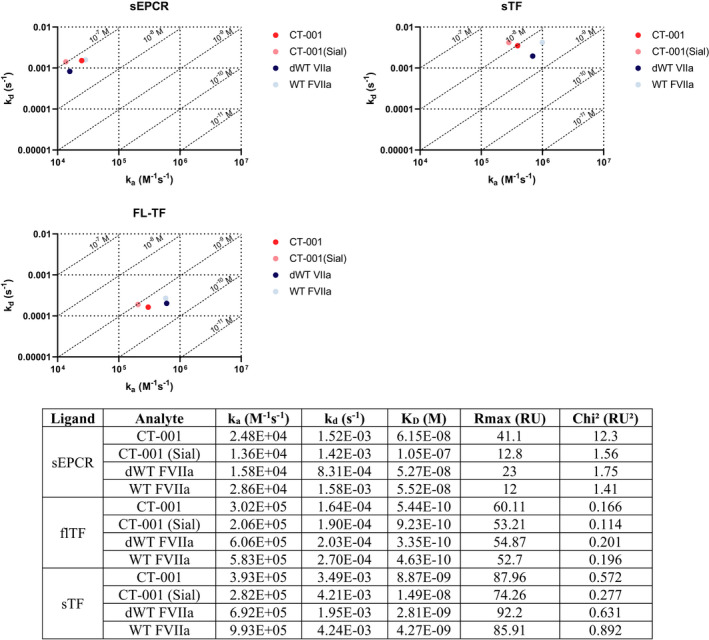
CT‐001 binding affinities to sEPCR and flTF are not altered but are lowered toward sTF versus WT FVIIa. sEPCR, flTF, and sTF sEPCR were immobilized on sensor chips, and FVIIa variants of varying concentrations were injected over the surface of flow cells. The substitution of amino acids (P10Q, K32E, T106 N, V253 N) in CT‐001(Sial) led to lower KDs toward sEPCR, flTF, and sTF relative to WT FVIIa. The desialylation of CT‐001(Sial) to become CT‐001appears to restore some of the lowered affinity of the molecule towards sEPCR, flTF, and sTF. This increase in affinity by desialyation was also observed in dWT FVIIa in comparison to WT FVIIa. dWT, desialylated wild‐type; flTF, full‐length tissue factor; FVIIa, activated factor VII; sEPCR, soluble endothelial protein C receptor; sTF, soluble tissue factor; WT, wild‐type
3.5. In vitro characterization of CT‐001 activity
The specific activities of FVIIa variants were measured by BIOPHEN FVIIa assay (Table 1). The combination of negatively charged phospholipids with recombinant truncated soluble TF in this assay overcomes the limitation of maximum TF association in assays that utilize membrane‐bound TF. Therefore, the assay is able to differentiate FVIIa with enhanced photoluminescence binding in the presence of TF. As expected, the specific activity of CT‐001 is higher than WT FVIIa (n = 3‐6; analysis of variance [ANOVA]; *P < 0.0001).
TABLE 1.
CT‐001 has enhanced specific activity relative to WT FVIIa
| Specific activities (IU/mg) | ||||
|---|---|---|---|---|
| CT−001 | CT−001(Sial) | dWT FVIIa | WT FVIIa | |
| Average | 246 000* | 219 000* | 50 800 | 55 100 |
| SD | 6280 | 5230 | 1290 | 1590 |
The specific activities of FVIIa variants were evaluated by the BIOPHEN FVIIa chromogenic assay which measured activities with recombinant truncated human tissue factor in the presence of synthetic phospholipids. CT‐001 and CT‐001(Sial) were revealed to have significantly higher specific activities than dWT FVIIa and WT FVIIa (n = 3–6; ANOVA; *P < 0.0001). WHO FVIIa Second International Standard (07/228) was used as a calibrator.
Abbreviations: ANOVA, analysis of variance; dWT, desialylated wild‐type; FVIIa, activated factor VII; WHO, World Health Organization; WT, wild‐type.
In the presence of PS:PC liposomes, CT‐001 and CT‐001(Sial) had more than two times higher catalytic rate constant (kcat), maximum reaction rate (Vmax), and specificity constant (kcat/Km) than WT FVIIa (ANOVA; *P < 0.01, **P < 0.0005). dWT FVIIa and WT FVIIa have similar kinetic constants, demonstrating that desialyation of N‐glycans did not affect FXa generation (Figure 5A). In the presence of lipidated TF (Innovin), as expected, all FVIIa variants bind flTF directly and with similarly high affinity (Figure 5B). Therefore, the phospholipid‐binding affinity of FVIIa was not the dominant contributor for TF/FVIIa complex‐mediated Fxa generation in this assay. The interaction of FVIIa with TF being the primary determinant of FXa generation in an assay with lipidated TF has also been described previously. 39
FIGURE 5.
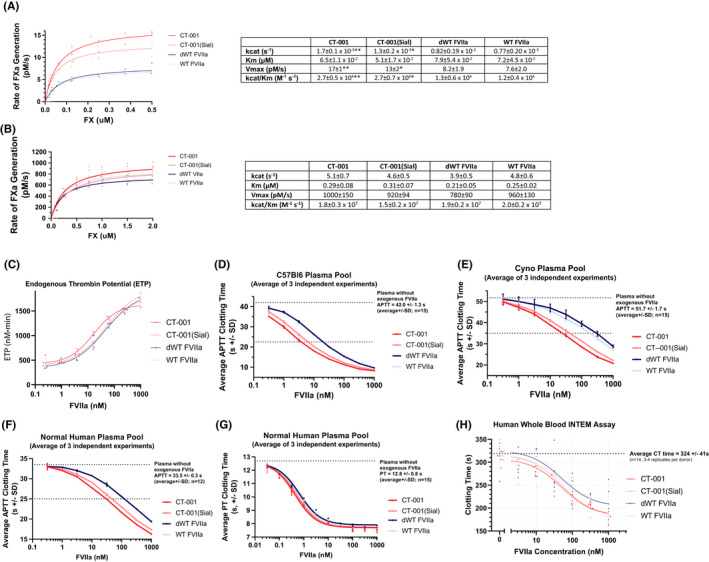
In vitro and ex vivo activity characterization of CT‐001. FXa generation was measured in both (A) TF independent, and (B) TF dependent reactions to determine the ability of the FVIIa variants to generate FXa. FXa generation was determined using chromogenic substrate S‐2765 and a purified FXa standard curve. Independent of TF, CT‐001 had two times higher catalytic rate constant (kcat), maximum reaction rate (Vmax), and specificity constant (kcat/KM) in comparison to WT FVIIa. dWT FVIIa and WT FVIIa have similar kcat, Michaelis constant (KM), and Vmax under this experimental condition. The data are displayed as the average of three experiments (ANOVA; *P < 0.01; **P < 0.0005). Under TF‐dependent conditions, similar kcat, KM, and Vmax were observed among CT‐001, CT‐001(Sial), dWT FVIIa, and WT FVIIa. The data are displayed as the average of three to six experiments. (C) CT‐001 has enhanced thrombin generation activity relative to other FVIIa variants. Thrombin generation was measured in normal human plasma with 0.024 – 1000 nM of FVIIa in the presence 4 μM phospholipid. Thrombin generation was initiated with CaCl2, and in the presence of thrombin fluorogenic substrate. The endogenous thrombin potential (ETP) was measured. CT‐001 had two times higher potency than CT‐001(Sial), dWT FVIIa, and WT FVIIa. The data are represented by the average of three independent experiments using a normal human plasma pool. (D‐G) CT‐001 has superior clotting activity to WT FVIIa across species. aPTT (STA‐PTT A) assays were performed in normal plasma pool from C57Bl/6 mice (D), cynomolgus monkeys (E), and humans (F). For all species, CT‐001 and CT‐001(Sial) were similar and demonstrated superior clotting time relative to dWT FVIIa or WT FVIIa. (G) In PT assays (STA‐Neoplastine CI Plus 5, rabbit brain thromboplastin), human plasma has an average PT clotting time of 12.8 seconds. CT‐001, CT‐001(Sial), dWT FVIIa, and WT FVIIa had similar potency, requiring 0.5, 0.6, 0.8, and 0.7 nM respectively to shorten PT clotting time to 10 seconds. (H) CT‐001 and CT‐001(Sial) showed enhanced clotting activity in rotational thromboelastometry assay in comparison to dWT FVIIa and WT FVIIa. INTEM assays were performed with ellagic acid for activating the intrinsic pathway. While all FVIIa variants shortened the average clotting time, CT‐001 and CT‐001(Sial) were superior to either dWT FVIIa or WT FVIIa (n=4 independent normal donors). ANOVA, analysis of variance; aPTT, activated partial thromboplastin time; dWT, desialylated wild‐type; FVIIa, activated factor VII; FXa, activated factor X; INTEM, intrinsic pathway thromboelastometry; PT, prothrombin time; TF, tissue factor; WT, wild‐type
Thrombin generation was measured in human plasma in the presence of 4 μM phospholipid (Figure 5C). At 15 nM, CT‐001 provided an endogenous thrombin potential (ETP) of 1000 nm/min. This is significantly lower than the 32 nM WT FVIIa needed to provide the same level of ETP. In comparison, 26 nM of CT‐001(Sial) and 31 nM dWT FVIIIa were needed to provide an ETP of 1000 nm·min. Overall, CT‐001 has a faster time to peak (ANOVA, Dunnett’s multiple comparisons; P < 0.001) and higher ETP (ANOVA, Dunnett’s multiple comparisons; P < 0.02) at 15.6 and 62.5 nM than WT FVIIa variants (Figure S2).
In C57Bl/6 plasma, 3.6 ± 0.2 nM of CT‐001 and 6.0 ± 0.6 nM of CT‐001(Sial) shortened clotting time 23 seconds. In comparison, a significantly higher amount of dWT FVIIa (18.6 ± 0.8 nM) and WT FVIIa (19.9 ± 0.7 nM) was needed for the same shortened clotting time (ANOVA; *P < 0.0001; Figure 5D). Similar enhanced activity of CT‐001 and CT‐001(Sial) over dWT FVIIa and WT FVIIa was seen in cynomolgus (ANOVA; *P < 0.007; Figure 5E), and human plasma (ANOVA; *P < 0.0001; Figure 5F). Overall, P10Q/K32E substitutions enhanced activity for CT‐001 and CT‐001(Sial). Desialylation did not enhance dWT FVIIa activity in comparison to WT FVIIa. However, desialylation provided enhanced activity for CT‐001 in comparison to CT‐001(Sial). This increased activity associated with desialylation in CT‐001 could be due to a subtle conformational change to the molecule that could be detected by the aPTT assay. In PT clotting assays, all four variants shortened prothrombin clotting time and showed essentially the same potency (Figure 5G).
INTEM assays with ellagic acid and calcium chloride for activating the intrinsic pathway were used to evaluate the FVIIa variants (Figure 5H). CT‐001 demonstrated an almost twofold higher potency than dWT FVIIa and WT FVIIa in whole blood.
3.6. Desialylation enhanced clearance of CT‐001 by hepatocytes
CT‐001 is designed to have enhanced clearance through desialylation and the subsequent exposure of galactose residues on N‐glycans, which are recognized by the asialoglycoprotein receptor (ASGPR) highly expressed on human hepatocytes. 43 A human hepatocyte clearance assay was used to test this design and evaluate the effect of desialylated N‐glycans on the clearance of FVIIa variants in vitro. In this assay, the majority of CT‐001(Sial) and WT FVIIa was recovered in the supernatant after 60 minutes of incubation with hepatocytes (Figure 6). In comparison, over 80% of CT‐001, which has four desialylated N‐glycans, was taken up by hepatocytes after 20 minutes of incubation. dWT FVII, which has two desialylated N‐glycans, was removed at a slower rate, with 40% of the dWT FVIIa cleared after 20 minutes of incubation. This study demonstrates the dramatic effect of desialylation on the clearance of the molecule by hepatocytes and signals that the rate of clearance is dependent on the number of desialylated glycans.
FIGURE 6.
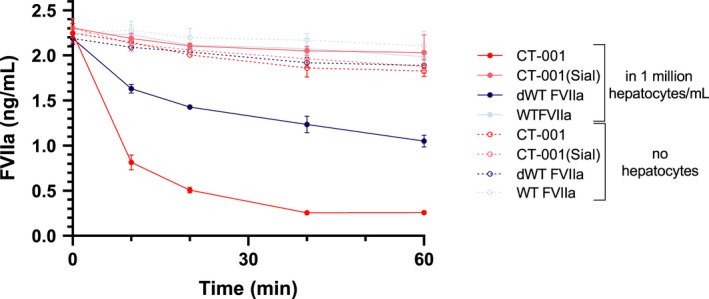
Desialylated FVIIa variants have increased clearance by hepatocytes in vitro. CT‐001, which has four desialylated N‐glycans, was cleared by hepatocytes from supernatant significantly faster than CT‐001(Sial) and WT FVIIa. dWT FVII, which has two desialylated N‐glycans, was removed at a slower rate than CT‐001. The majority of CT‐001(Sial) and WT FVIIa was recovered in the supernatant after 60 minutes of incubation with hepatocytes. Three independent studies were performed. dWT, desialylated wild‐type; FVII, factor VII; FVIIa, activated factor VII; WT, wild‐type
4. DISCUSSION
CT‐001 is developed to address thromboembolic events associated with FVIIa as a treatment for acute bleeds. We postulated that a shortened circulating half‐life would address the thrombogenicity risk. To shorten the half‐life of the protein in circulation, we took advantage of naturally occurring and engineered N‐glycosylation and the significant role it plays in pharmacokinetics. 44 Terminally sialylated N‐glycans enable longer serum circulation by masking galactose residues responsible for protein uptake and degradation via the ASGPR. 43 To generate an active clearance mechanism while not compromising the physical protective benefits afforded by N‐glycans, we engineered CT‐001, a desialylated protein, where exposed galactose residues enable the recognition and rapid clearance via ASGPR. In this article, we have demonstrated that these desiaylated glycans led to rapid clearance of CT‐001 by hepatocytes in vitro (Figure 6). This rapid clearing property by hepatocytes may allow administration of CT‐001 to achieve the peak plasma concentration needed for therapeutic effect yet reduce the total systemic exposure to FVIIa activity, thus reducing thrombogenicity. In a study on WT FVIIa, the terminal half‐life was determined to be around 2.6 hours with the formation of WT FVIIa‐AT complex identified as being the major pathway for FVIIa clearance, which is responsible for the clearance of 65% of WT FVIIa activity. 45 Since CT‐001 and WT FVIIa have similar sensitivity toward inhibition by AT (Figure 5a), the desialylated glycans engineered to CT‐001 are expected to provide a clearance rate significantly faster than that of WT FVIIa based on the heavy influence of sialic acid on the clearance of glycoproteins. The lack of terminal sialic acid has been shown to reduce the half‐life of many desialylated glycoproteins down to minutes through the removal by the liver. 46 Therefore, the desialylated glycans on CT‐001 will likely be the dominant clearance mechanism. It is important to note that other FVIIa‐binding partners such as alpha2‐macroglobulin, 47 EPCR, 48 , 49 , 50 low‐density lipoprotein receptor–related protein 1, 51 as well as megalin and cubilin on renal cells 52 could also be potential modulators of CT‐001 clearance. Thus, the hypothesis of desialylated glycans on CT‐001 being the dominant mechanism for activity clearance will need to be confirmed by in vivo pharmacokinetic and pharmacodynamic studies.
To compensate for the rapid clearance, supra‐physiological activity was introduced by two amino acid substitutions (P10Q and K32E) in the Gla domain. These changes improve membrane‐binding affinity of these variants as seen on activated platelets (Figure 2). This results in enhanced activity as demonstrated in specific activity, FXa generation, thrombin generation, plasma clotting, and whole blood clotting assays (Table 1, Figure 5). Importantly, this did not compromise the protein’s function or ability to interact with physiologically relevant molecules, AT and TFPI, necessary for hemostatic control (Figure 3). Some binding affinity differences were noted between FVIIa variants. The modulation of FVIIa activity by different glycosylation patterns has been reported previously. 53 , 54 We postulate the addition of N‐linked glycosylation at T106 N and V253 N on the protein surface and the reduction of negative charges on these glycans of the FVIIa variants to be potential contributors of subtle conformation changes and binding affinity shifts. Overall, these data demonstrate that desialylation of protein glycans to achieve rapid clearance could be an effective pharmacological approach in lowering thrombogenicity of FVIIa treatment by limiting systemic exposure to procoagulant activity.
CT‐001 has four newly engineered amino acids (P10Q K32E T106 N V253 N). Genetically modified proteins can lead to unwanted immune responses in patients. 55 , 56 Immunogenicity has been reported for two different recombinant FVIIa molecules, vatreptacog alfa and BAY 86–6150, tested for individuals with hemophilia A or B with inhibitors. 57 , 58 , 59 Vatreptacog alfa and BAY 86–6150 differ from the native FVIIa by three amino acids (V158D, E296 V, M298Q) 60 and six amino acids (P10Q, K32E, A34E, R36E, T106 N, V253 N), respectively. 59 Of the 72 patients enrolled in the phase III vatreptacog alfa trial (treated at 80 µg/kg), eight patients (11%) developed antibodies, including four with low‐titer cross‐reactivity against recombinant human FVIIa (rFVIIa). 57 , 58 Similarly, of the 10 patients treated with BAY 86–6150, 1 patient developed neutralizing antibodies to BAY 86–6150 that was confirmed to cross react with FVIIa. 59 Importantly, this readministration played a critical role in antibody development. The binding antibodies to vatreptacog alfa were detected following 1 to 8 exposure days. The four individuals who developed low titers to FVIIa occurred only at later time points after treatment. The FVIIa cross reactivity ended after exposure to vatreptacog alfa ceased and while they continued to receive rFVIIa. 58 Similarly, antibodies to BAY 86–6150 were detected only after three different exposures, and subsequent bleeding episodes were controlled using FEIBA. The four amino acid substitutions in CT‐001 (P10Q, K32E, T106 N, V253 N) are within the six amino acid changes engineered into BAY 86–6150. Since the development of antibody in the BAY 86–6150 trial was detected in only 10% of individuals and only after multiple exposures, we believe the potential for the development of antibodies to CT‐001 is remote because it is fast‐clearing and intended for isolated emergency acute bleeding settings. Consequently, while FVIIa protein engineering can lead to an unwanted immunogenicity, it is important to note that CT‐001 is not envisioned for chronic treatment, thus eliminating the multiple exposures that have been documented as necessary for antibody formation.
Currently, there is no clinically approved safe and effective hemostatic therapy for acute bleeding. WT FVIIa has been used in acute bleeding indications with some successes but carries thrombotic risk. A recombinant FVIIa with lower thrombogenicity would be a breakthrough treatment for acute bleeding. Using a novel desialylation approach that exploits the body’s natural clearance mechanism, together with the Gla domain engineering for enhanced activity, CT‐001 demonstrates enhanced efficacy and faster clearance in vitro. Further in vivo studies are warranted to understand if CT‐001 can fulfill the unmet medical need of a safe prohemostatic agent and serve as a stand‐alone or complementing therapy to current treatments of acute bleeding.
RELATIONSHIP DISCLOSURE
DSS, CM, JT, and TWH are employees of CoagIulant Therapeutics Corporation. MB is a consultant to Coagulant Therapeutics Corporation. MB and TWH are inventors and patent holders of CT‐001 and dWT FVIIa.
AUTHOR CONTRIBUTIONS
DSS, CM, JT, RIF, MB, and TWH designed and performed experiments and analyzed data; DSS, CM, JT, RF, MB, TWH contributed to writing the manuscript.
Supporting information
Fig S1‐S2
ACKNOWLEDGMENTS
The authors thank Bayer HealthCare and Maxygen for their early contributions.
Sim DS, Mallari CR, Teare JM, Feldman RI, Bauzon M, Hermiston TW. In vitro characterization of CT‐001—a short‐acting factor VIIa with enhanced prohemostatic activity. Res Pract Thromb Haemost. 2021;5:e12530. 10.1002/rth2.12530
Handling Editor: Dr Johnny Mahlangu.
Funding information
This study was funded by Coagulant Therapeutics Corporation
REFERENCES
- 1. Tisherman SA, Schmicker RH, Brasel KJ et al. Detailed description of all deaths in both the shock and traumatic brain injury hypertonic saline trials of the resuscitation outcomes consortium. Ann Surg. 2015;261:586‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mayer SA, Brun NC, Begtrup K et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. New Engl J Med. 2008;358:2127‐2137. [DOI] [PubMed] [Google Scholar]
- 3. Mayer SA, Brun NC, Begtrup K et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2005;352:777‐785. [DOI] [PubMed] [Google Scholar]
- 4. Hauser CJ, Boffard K, Dutton R et al. Results of the CONTROL Trial: efficacy and safety of recombinant activated factor VII in the management of refractory traumatic hemorrhage. J Trauma. 2010;69:489‐500. [DOI] [PubMed] [Google Scholar]
- 5. Dutton RP, Parr M, Tortella BJ et al. Recombinant activated factor VII safety in trauma patients: results from the CONTROL trial. J Trauma. 2011;71:12‐19. [DOI] [PubMed] [Google Scholar]
- 6. Gill R, Herbertson M, Vuylsteke A et al. Safety and efficacy of recombinant activated factor VII. Circulation. 2009;120:21‐27. [DOI] [PubMed] [Google Scholar]
- 7. Narayan RK, Maas AIR, Marshall LF, Servadei F, Skolnick BE, Tillinger MN. Recombinant factor VIIa in traumatic intracerebral hemorrhage: results of a dose‐escalation clinical trial. Neurosurgery. 2008;62:776‐788. [DOI] [PubMed] [Google Scholar]
- 8. Lodge JPA, Jonas S, Oussoultzoglou E et al. Recombinant coagulation factor VIIa in major liver resection. Anesthesiology. 2005;102:269‐275. [DOI] [PubMed] [Google Scholar]
- 9. Lodge JPA, Jonas S, Jones RM et al. Efficacy and safety of repeated perioperative doses of recombinant factor VIIa in liver transplantation. Liver Transplant. 2005;11:973‐979. [DOI] [PubMed] [Google Scholar]
- 10. Planinsic RM, van der Meer J , Testa G et al. Safety and efficacy of a single bolus administration of recombinant factor VIIa in liver transplantation due to chronic liver disease. Liver Transplant. 2005;11:895‐900. [DOI] [PubMed] [Google Scholar]
- 11. Bosch J, Thabut D, Bendtsen F et al. Recombinant factor VIIa for upper gastrointestinal bleeding in patients with cirrhosis: a randomized, double‐blind trial. Gastroenterology. 2004;127:1123‐1130. [DOI] [PubMed] [Google Scholar]
- 12. Salama A, Rieke M, Kiesewetter H, Depka M. Experiences with recombinant FVIIa in the emergency treatment of patients with autoimmune thrombocytopenia: a review of the literature. Ann Hematol. 2009;88:11‐15. [DOI] [PubMed] [Google Scholar]
- 13. Sachs B, Delacy D, Green J et al. Recombinant activated factor VII in spinal surgery: a multicenter, randomized, double‐blind, placebo‐controlled dose‐escalation trial. Spine. 2007;32:2285‐2293. [DOI] [PubMed] [Google Scholar]
- 14. Lavigne‐Lissalde G, Aya AG, Mercier FJ et al. Recombinant human FVIIa for reducing the need for invasive second‐line therapies in severe refractory postpartum hemorrhage: a multicenter, randomized, open controlled trial. J Thromb Haemost. 2015;13:520‐529. [DOI] [PubMed] [Google Scholar]
- 15. Johansson PI, Eriksen K, Nielsen SL, Rojkjaer R, Alsbjørn B. Recombinant FVIIa decreases perioperative blood transfusion requirement in burn patients undergoing excision and skin grafting—results of a single centre pilot study. Burns. 2007;33:435‐440. [DOI] [PubMed] [Google Scholar]
- 16. Pihusch M, Bacigalupo A, Szer J et al. Recombinant activated factor VII in treatment of bleeding complications following hematopoietic stem cell transplantation. J Thromb Haemost. 2005;3:1935‐1944. [DOI] [PubMed] [Google Scholar]
- 17. Shenoy A, Savani BN, Barrett AJ. Recombinant factor VIIa to treat diffuse alveolar hemorrhage following allogeneic stem cell transplantation. Biol Blood Marrow Tr. 2007;13:622‐623. [DOI] [PubMed] [Google Scholar]
- 18. Heslet L, Nielsen JD, Levi M, Sengeløv H, Johansson PI. Successful pulmonary administration of activated recombinant factor VII in diffuse alveolar hemorrhage. Crit Care. 2006;10:R177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shakur H, Roberts I, Bautista R et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH‐2): a randomised, placebo‐controlled trial. Lancet. 2010;376:23‐32. [DOI] [PubMed] [Google Scholar]
- 20. Shakur H, Roberts I, Fawole B et al. Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities in women with post‐partum haemorrhage (WOMAN): an international, randomised, double‐blind, placebo‐controlled trial. Lancet. 2017;389:2105‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kreuz W, Meili E, Peter‐Salonen K et al. Efficacy and tolerability of a pasteurised human fibrinogen concentrate in patients with congenital fibrinogen deficiency. Transfus Apher Sci. 2005;32:247‐253. [DOI] [PubMed] [Google Scholar]
- 22. Collins PW, Cannings‐John R, Bruynseels D et al. Viscoelastometric‐guided early fibrinogen concentrate replacement during postpartum haemorrhage: OBS2, a double‐blind randomized controlled trial. Brit J Anaesth. 2017;119:411‐421. [DOI] [PubMed] [Google Scholar]
- 23. Shaw JR, Siegal DM. Pharmacological reversal of the direct oral anticoagulants—a comprehensive review of the literature. Res Pract Thrombosis Haemostasis. 2018;2:251‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lusher R, Davignon J, Smith S et al. A randomized, double‐blind comparison of two dosage levels of recombinant factor VIIa in the treatment of joint, muscle and mucocutaneous haemorrhages in persons with haemophilia A and B, with and without inhibitors. Haemophilia. 1998;4:790‐798. [DOI] [PubMed] [Google Scholar]
- 25. Tiede A, Worster A. Lessons from a systematic literature review of the effectiveness of recombinant factor VIIa in acquired haemophilia. Ann Hematol. 2018;97:1889‐1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brenner B, Wiis J. Experience with recombinant‐activated factor VII in 30 patients with congenital factor VII deficiency. Hematology Amsterdam Neth. 2007;12:55‐62. [DOI] [PubMed] [Google Scholar]
- 27. Mariani G, Konkle B, Ingerslev J. Congenital factor VII deficiency: therapy with recombinant activated factor VII—a critical appraisal. Haemophilia. 2006;12:19‐27. [DOI] [PubMed] [Google Scholar]
- 28. Poon M‐C, D’Oiron R, Depka MV et al. Prophylactic and therapeutic recombinant factor VIIa administration to patients with Glanzmann’s thrombasthenia: results of an international survey: rFVIIa and Glanzmann’s thrombasthenia. J Thromb Haemost. 2004;2:1096‐1103. [DOI] [PubMed] [Google Scholar]
- 29. Goodnough LT, Levy JH. The judicious use of recombinant factor VIIa. Semin Thromb Hemost. 2016;42:125‐132. [DOI] [PubMed] [Google Scholar]
- 30. Levy JH, Fingerhut A, Brott T, Langbakke IH, Erhardtsen E, Porte RJ. Recombinant factor VIIa in patients with coagulopathy secondary to anticoagulant therapy, cirrhosis, or severe traumatic injury: review of safety profile. Transfusion. 2006;46:919‐933. [DOI] [PubMed] [Google Scholar]
- 31. Thomas GOR, Dutton RP, Hemlock B et al. Thromboembolic complications associated with factor VIIa administration. J Trauma Inj Infect Critical Care. 2007;62:564‐569. [DOI] [PubMed] [Google Scholar]
- 32. O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA. 2006;295:293‐298. [DOI] [PubMed] [Google Scholar]
- 33. Levi M, Levy JH, Andersen HF, Truloff D. Safety of recombinant activated factor VII in randomized clinical trials. N Engl J Med. 2010;363:1791‐1800. [DOI] [PubMed] [Google Scholar]
- 34. Nordisk Novo . NovoSeven (recombinant) prescribing information. HYPERLINK "sps:urlprefix::https" https://www.novo‐pi.com/novosevenrt.pdf. Accessed June 21, 2021
- 35. Hagen FS, Gray CL, O’Hara P et al. Characterization of a cDNA coding for human factor VII. Proc National Acad Sci. 1986;83:2412‐2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O’Hara PJ, Grant FJ, Haldeman BA et al. Nucleotide sequence of the gene coding for human factor VII, a vitamin K‐dependent protein participating in blood coagulation. Proc National Acad Sci. 1987;84:5158‐5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cazenave J‐P, Ohlmann P, Cassel D, Eckly A, Hechler B, Gachet C. Platelets and megakaryocytes. Methods Mol Biol. 2004;272:013‐028. [DOI] [PubMed] [Google Scholar]
- 38. Blasko E, Brooks AR, Ho E, Wu JM, Zhao X‐Y, Subramanyam B. Hepatocyte clearance and pharmacokinetics of recombinant factor IX glycosylation variants. Biochem Bioph Res Co. 2013;440:485‐489. [DOI] [PubMed] [Google Scholar]
- 39. Nelsestuen GL, Stone M, Martinez MB, Harvey SB, Foster D, Kisiel W. Elevated function of blood clotting factor VIIa mutants that have enhanced affinity for membranes: behavior in a diffusion‐limited reaction. J Biol Chem. 2001;276:39825‐39831. [DOI] [PubMed] [Google Scholar]
- 40. Gavel Y, von Heijne G . Sequence differences between glycosylated and non‐glycosylated Asn‐X‐Thr/Ser acceptor sites: implications for protein engineering. Protein Eng Des Sel. 1990;3:433‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nilsson I, von Heijne G . Glycosylation efficiency of Asn‐Xaa‐Thr sequons depends both on the distance from the C terminus and on the presence of a downstream transmembrane segment. J Biol Chem. 2000;275:17338‐17343. [DOI] [PubMed] [Google Scholar]
- 42. Shakin‐Eshleman SH, Spitalnik SL, Kasturi L. The amino acid at the X Position of an Asn‐X‐Ser Sequon is an important determinant of N‐Linked core‐glycosylation efficiency. J Biol Chem. 1996;271:6363‐6366. [DOI] [PubMed] [Google Scholar]
- 43. Gupta SK, Shukla P. Glycosylation control technologies for recombinant therapeutic proteins. Appl Microbiol Biot. 2018;102:10457‐10468. [DOI] [PubMed] [Google Scholar]
- 44. Zhou Q, Qiu H. The mechanistic Impact of N‐glycosylation on stability, pharmacokinetics and immunogenicity of therapeutic proteins. J Pharm Sci. 2018;108:1366‐1377. [DOI] [PubMed] [Google Scholar]
- 45. Agersø H, Brophy DF, Pelzer H et al. Recombinant human factor VIIa (rFVIIa) cleared principally by antithrombin following intravenous administration in hemophilia patients. J Thromb Haemost. 2011;9:333‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morell AG, Gregoriadis G, Scheinberg IH, Hickman J, Ashwell G. The role of sialic acid in determining the survival of glycoproteins in the circulation. J Biol Chem. 1971;246:1461‐1467. [PubMed] [Google Scholar]
- 47. Petersen LC, Elm T, Ezban M et al. Plasma elimination kinetics for factor VII are independent of its activation to factor VIIa and complex formation with plasma inhibitors. Thromb Haemost. 2009;101:818‐826. [PubMed] [Google Scholar]
- 48. Lopez‐Sagaseta J, Montes R, Puy C, Diez N, Fukudome K, Hermida J. Binding of factor VIIa to the endothelial cell protein C receptor reduces its coagulant activity. J Thromb Haemost. 2007;5:1817‐1824. [DOI] [PubMed] [Google Scholar]
- 49. Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, Rao LVM. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007;282:11849‐11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Preston RJS, Ajzner E, Razzari C et al. Multifunctional specificity of the protein C/activated protein C Gla domain. J Biol Chem. 2006;281:28850‐28857. [DOI] [PubMed] [Google Scholar]
- 51. Fazavana JG, Muczynski V, Proulle V et al. LDL receptor‐related protein 1 contributes to the clearance of the activated factor VII–antithrombin complex. J Thromb Haemost. 2016;14:2458‐2470. [DOI] [PubMed] [Google Scholar]
- 52. Seested T, Appa RS, Jacobsen C, Christensen EI. Recombinant activated factor VII is reabsorbed in renal proximal tubules and is a ligand to megalin and cubilin. Nephron Exp Nephrol. 2011;117:e82‐e92. [DOI] [PubMed] [Google Scholar]
- 53. Sutkeviciute I, Mistiniene E, Sereikaite J, Bumelis VA. The influence of different glycosylation patterns on factor VII biological activity. Biochimie. 2009;91:1123‐1130. [DOI] [PubMed] [Google Scholar]
- 54. Böhm E, Seyfried BK, Dockal M et al. Differences in N‐glycosylation of recombinant human coagulation factor VII derived from BHK, CHO, and HEK293 cells. Bmc Biotechnol. 2015;15:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Patten P, Schellekens H. The immunogenicity of biopharmaceuticals. Lessons learned and consequences for protein drug development. Dev Biologicals. 2002;112:81‐97. [PubMed] [Google Scholar]
- 56. Rosenberg AS, Sauna ZE. Immunogenicity assessment during the development of protein therapeutics. J Pharm Pharmacol. 2018;70:584‐594. [DOI] [PubMed] [Google Scholar]
- 57. Lentz SR, Ehrenforth S, Karim FA et al. Recombinant factor VIIa analog in the management of hemophilia with inhibitors: results from a multicenter, randomized, controlled trial of vatreptacog alfa. J Thromb Haemost. 2014;12:1244‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mahlangu JN, Weldingh KN, Lentz SR et al. Changes in the amino acid sequence of the recombinant human factor VIIa analog, vatreptacog alfa, are associated with clinical immunogenicity. J Thromb Haemost. 2015;13:1989‐1998. [DOI] [PubMed] [Google Scholar]
- 59. Mahlangu JN, Coetzee MJ, Laffan M et al. Phase I, randomized, double‐blind, placebo‐controlled, single‐dose escalation study of the recombinant factor VIIa variant BAY 86–6150 in hemophilia. J Thromb Haemost. 2012;10:773‐780. [DOI] [PubMed] [Google Scholar]
- 60. Persson E, Olsen O, Bjorn S, Ezban M. Vatreptacog alfa from conception to clinical proof of concept. Semin Thromb Hemost. 2012;38:274‐281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2