

ABSTRACT
Subcutaneous injection of a low volume (<2 mL) high concentration (>100 mg/mL) formulation is an attractive administration strategy for monoclonal antibodies (mAbs) and other biopharmaceutical proteins. Using concentrated solutions may also be beneficial at various stages of bioprocessing. However, concentrating proteins by conventional techniques, such as ultrafiltration, can be time consuming and challenging. Isolation of the dense fraction produced by macroscopic liquid–liquid phase separation (LLPS) has been suggested as a means to produce high-concentration solutions, but practicality of this method, and the stability of the resulting protein solution have not previously been demonstrated. In this proof-of-concept study, we demonstrate that LLPS can be used to concentrate a mAb solution to >170 mg/mL. We show that the structure of the mAb is not altered by LLPS, and unperturbed mAb is recoverable following dilution of the dense fraction, as judged by 1H nuclear magnetic resonance spectroscopy. Finally, we show that the physical properties and stability of a model high concentration protein formulation obtained from the dense fraction can be improved, for example through the addition of the excipient arginine·glutamate. This results in a stable high-concentration protein formulation with reduced viscosity and no further macroscopic LLPS. Concentrating mAb solutions by LLPS represents a simple and effective technique to progress toward producing high-concentration protein formulations for bioprocessing or administration.
Abbreviations
Arginine·glutamate (Arg·Glu), Carr-Purcell-Meiboom-Gill (CPMG), critical temperature (TC), high-performance size-exclusion chromatography (HPSEC), liquid–liquid phase separation (LLPS), monoclonal antibody (mAb), nuclear magnetic resonance (NMR), transverse relaxation rate (R2)
KEYWORDS: Aggregation, bioprocessing, liquid-liquid phase separation, mAb stability, NMR spectroscopy
Introduction
Since the approval of the first recombinant protein therapeutic in 1982, biopharmaceutical proteins, including monoclonal antibodies (mAbs), have developed into critical treatments for a wide range of diseases.1–3 For the prolonged treatment of chronic conditions, such as arthritis and other autoimmune diseases, subcutaneous injection by the patient represents an attractive administration strategy for mAbs.4,5 Due to the limited volume (<2 mL) possible for injection into subcutaneous tissue,6 such strategies require high-concentration protein formulations, with protein concentrations typically greater than 100 mg/mL.7 Using high-concentrations solutions may also be beneficial during bioprocessing and manufacturing. However, achieving such high concentrations and stabilizing the final formulation against degradation remains challenging.8
During bioprocessing, biopharmaceutical protein solutions are typically concentrated using ultrafiltration techniques involving membranes, such as tangential-flow filtration.9 However, achieving high-protein concentrations can be challenging due to concentration-induced viscosity, self-association, and aggregation potentially resulting in significant back pressure, membrane fouling and reduced filtration rates.10–12 Alternatively, lyophilization followed by reconstitution in a reduced diluent volume may be used, but this may require substantial time for freeze-drying, plus additional reconstitution times,13 or may generate physical instabilities due to the stresses of freeze-drying and reconstitution.14 Therefore, other methods to concentrate biopharmaceutical proteins are required.
Liquid–liquid phase separation (LLPS) has been suggested as a novel technique to concentrate biopharmaceutical solutions.15–17 During macroscopic LLPS, a homogenous medium-concentration protein solution spontaneously separates into an upper low-concentration lean layer and a lower high-concentration dense layer. Whilst LLPS is typically considered an unwanted physical instability in biopharmaceutical solutions, 18,19 selective triggering of LLPS, for example, through addition of salts, or changes in temperature, may be used to concentrate a protein solution. Aqueous two-phase extraction, a similar approach involving polymers to trigger phase partitioning, is already widely used during purification in biotechnology.20–22
Despite the potential of LLPS as a method to concentrate mAb solutions, phase separation is still largely viewed as an undesired physical instability, and questions about the practicality and suitability of its use remain. For example, due to the attractive protein–protein interactions required for LLPS, there may be concerns about the promotion of aggregation in the high-concentration dense fraction.23,24 Additionally, the dense fraction may be excessively viscous 18,25 and potentially difficult to handle during bioprocessing or administration. Such concerns are common in the development of any highly concentrated biopharmaceutical protein formulation, where they are typically alleviated through optimization of solution conditions, including buffer, pH, ionic strength, and addition of excipients.10,26,27 However, to our knowledge, such a formulation approach has not been trialed for high-concentration dense fractions produced by LLPS.
In this study, we explore how the properties and stability of a high-concentration mAb solution produced by LLPS can be improved through the addition of an excipient, arginine·glutamate (Arg·Glu),27,28 to the isolated dense fraction. We demonstrate that LLPS can be used to concentrate a mAb from 80 mg/mL to >170 mg/mL, and show, using 1H nuclear magnetic resonance (NMR) spectroscopy, that structurally unperturbed mAb is recoverable following dilution of the dense fraction. Viscosity measurements and high-performance size-exclusion chromatography (HPSEC) analysis show that Arg·Glu improves the physical properties and stability of a model high-concentration formulation produced from the dense fraction. Controlled LLPS and subsequent addition of excipients offers a simple and effective method to produce stable high-concentration antibody formulations.
Results
Using LLPS to concentrate mAb solutions
To demonstrate the use of LLPS to concentrate a mAb, LLPS was triggered in 80 mg/mL COE-13 in 20 mM acetate buffer, pH 5.5, by addition of 75 mM NaCl and incubation at 4°C (Figure 1). The initially opalescent solutions underwent rapid macroscopic LLPS, with two layers visible after 20 min, and the boundary between these layers sharpening over time. After 24 hours, the upper lean and lower dense fractions were isolated. Protein concentration measurements showed that LLPS resulted in a > 2-fold increase in concentration in the dense layer (171 mg/mL) compared to the initial solution, while the lean fraction concentration was 10 mg/mL.
Figure 1.
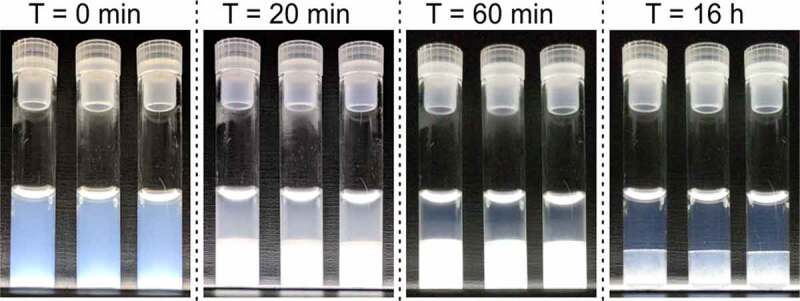
LLPS of COE-13 triggered by incubation at 4°C. Triplicate solutions illuminated from underneath to visualize light scattering and opalescence. Images taken at 4°C after the incubation times indicated. Beyond 20 minutes, the visible bottom layer corresponds to the dense fraction, and upper layer to the lean fraction
To investigate whether LLPS resulted in any aberrant irreversible changes in mAb structure and behavior, 1H NMR spectra and transverse relaxation rate (R2) profiles were recorded for the lean fraction, and the dense fraction before and after dilution with buffer (containing 75 mM NaCl to maintain ionic strength). Additionally, spectra were recorded for a dilute COE-13 solution that had not undergone LLPS. All samples were in equivalent buffers (20 mM acetate, pH 5.5, with 75 mM NaCl), with 10 mg/mL COE-13, apart from the intact dense fraction, where protein concentration was significantly higher (171 mg/mL). 1H NMR spectral fingerprints of the characteristic methyl region report on the apparent structure and colloidal behavior of mAbs.29 The lean fraction (green) and the diluted dense fraction (purple) exhibited very similar NMR spectra to the COE-13 solution that had not undergone LLPS (blue, Figure 2a), indicating that the protein in these solutions has very similar higher-order structure. NMR spectra of the neat dense fraction had significantly lower signal intensity than the other solutions (red, Figure 2a, and Figure S1), despite a 17-fold higher protein concentration, in agreement with our previous observations.19
Figure 2.
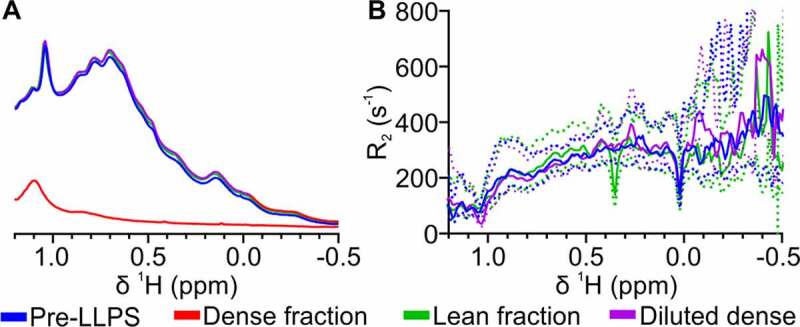
NMR comparison of mAb structure and behavior before and after LLPS. (a) 1D 1H NMR spectra and (b) R2 profiles comparing a non-phase separated protein solution, the intact lean fraction, and diluted dense fraction (all 10 mg/mL), with the intact dense fraction collected after LLPS (171 mg/mL). All solutions were in 20 mM acetate buffer, pH 5.5, with 75 mM NaCl. Dense fraction not shown on R2 profile due to rapid R2 rate and poor signal in CPMG experiments. Dotted lines indicate the respective 95% confidence bounds of the fitted relaxation rates
NMR transverse relaxation rates (R2) are coupled to molecular motions and apparent molecular size through rotational correlation time, and so report on apparent intermolecular interactions. The similar R2 spectral profiles (Figure 2b) between the three 10 mg/mL solutions show that protein–protein interactions and colloidal behavior of COE-13 are not perturbed irreversibly by LLPS. Conversely, R2 was markedly faster in the intact dense fraction, such that COE-13 relaxation rates were essentially unmeasurable in this fraction. Along with the significantly reduced signal, this suggests significant attractive protein–protein interactions and the occurrence of protein self-association and high viscosity in the highly concentrated dense fraction. On the other hand, the NMR observations indicate that unperturbed COE-13 is recoverable from the lean fraction and after dilution of the dense fraction, suggesting that the self-association observed in the dense fraction is fully reversible.
Production of model high-concentration formulations from the phase separated dense fraction
Whilst the neat dense fraction produced by LLPS may in principle have a satisfactorily high-protein concentration, its properties, such as viscosity, self-association, and protein stability, may render it unsuitable for bioprocessing, storage and administration. In our NMR assessments (Figure 2), we observed clear evidence of significant protein self-association, likely as a result of attractive protein–protein interactions. To understand if the self-association can be reduced while maintaining this high concentration, we next investigated whether the addition of an excipient, arginine·glutamate (Arg·Glu), could improve the properties of the mAb solution obtained from the dense fraction and shift the self-association equilibrium toward a monomeric, non-associated state. Two model high-concentration protein formulations were generated from the isolated dense fractions: 1) a formulation with 100 mM Arg·Glu (final concentration), and 2) a reference formulation without Arg·Glu. Final protein concentration in both formulations was 148 mg/mL, while final buffer and NaCl concentrations were identical to the starting solutions, at 20 and 75 mM, respectively. These model formulations were used for further comparative analysis.
Arg·Glu improves the stability of model formulations derived from the dense fraction
LLPS is typically considered an unwanted physical instability, and there may be concerns that proteins that have undergone LLPS may be inherently prone to further aggregation and degradation. To investigate this behavior, the physical and chemical stability of the model high-concentration formulations generated from the phase-separated dense fraction was examined by storage at standard refrigerated conditions (4°C) or under stressed degradation conditions (40°C) for 4 weeks.
The behavior of the two model COE-13 formulations during storage was markedly different (Figure 3). The reference formulation underwent LLPS again following storage at 4°C (Figure 3, right-hand side), with reestablishment of the LLPS equilibrium concentrations. Conversely, the formulation with 100 mM Arg·Glu remained as a single homogenous solution, with Arg·Glu preventing further LLPS at 4°C, although with mild opalescence occurring (Figure S2). Both formulations exhibited no LLPS when incubated at 40°C, above the apparent critical temperature (TC) for this system (∼13°C).
Figure 3.

Behavior of the model formulations after 24-hour incubation at 4 or 40°C. Image taken at ambient room temperature immediately after removal from incubation. Macroscopic LLPS present in the reference formulation stored at 4°C (right-hand side)
Antibody degradation by fragmentation and aggregation during storage was assessed using HPSEC (Figure 4, with example chromatograms in Figure S3). When stored at 4°C, both model high-concentration formulations exhibited no additional degradation after 28 days storage. This demonstrates that the protein solutions concentrated by LLPS are not inherently unstable or prone to degradation during storage at lower temperature. Meanwhile, model formulations stored under stressed degradation conditions at 40°C exhibited both aggregation and fragmentation (Figure 4a-c). However, the formulation with 100 mM Arg·Glu was more stable than the reference formulation, with Arg·Glu reducing the rate of antibody aggregation 2-fold, whilst also marginally reducing the rate of fragmentation (Table 1). This illustrates that excipients, such as Arg·Glu, can improve the stability of high concentration protein solutions generated by LLPS against degradation.
Figure 4.
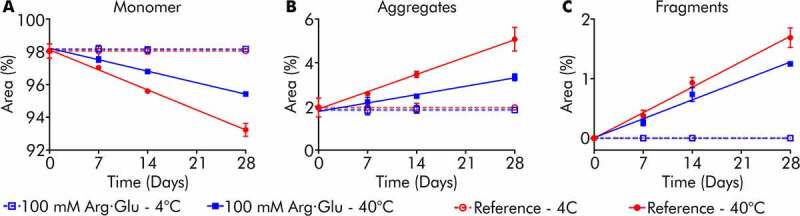
Monomer loss, aggregation, and fragmentation of model formulations following storage at 4 or 40°C. Monomer (a), aggregate (b), and fragment (c) content for COE-13 formulations over 4-week storage
Table 1.
Rates of degradation (± standard errors) for the model formulations stored at 40°C. Rates based on linear regression of HPLC data
| Model formulation | Monomer loss (%/month) | Aggregation (%/month) | Fragmentation (%/month) |
| Reference | 5.31 ± 0.18 | 3.45 ± 0.12 | 1.86 ± 0.10 |
| With 100 mM Arg·Glu | 3.04 ± 0.05 | 1.67 ± 0.18 | 1.38 ± 0.23 |
Next, the temperature dependence of the dynamic viscosities of the model high-concentration formulations produced by LLPS was explored (Figure 5). High viscosity resulting from protein self-association at high-protein concentration (e.g., 148 mg/mL here) may pose handling challenges during biopharmaceutical fill-finish and administration, and therefore formulation viscosity should be monitored and minimized. The reference model formulation was particularly viscous, with its viscosity exceeding 200 mPa·s at temperatures below 25°C. Such a high viscosity for a mAb formulation renders it largely unsuitable for processing and syringing, i.e., for subcutaneous administration.
Figure 5.
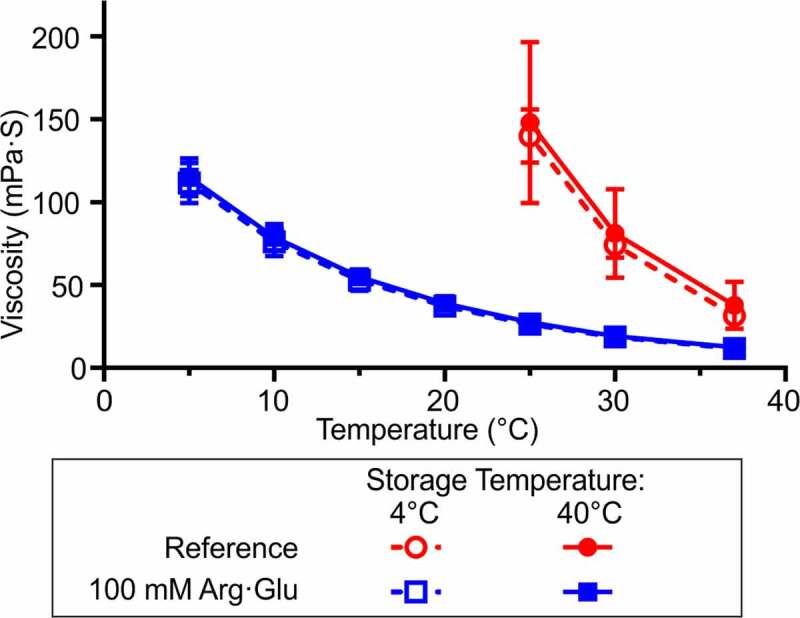
Viscosity of the model high-concentration formulations across a range of temperatures after 28 days storage at either 4°C or 40°C. Mean ± standard deviation of three replicate measurements. For the reference formulation, the viscosity of solutions below 25°C exceeded the measurement capacity of the specific VROC Initium chip (∼200 mPa·s)
Conversely, the model formulation with 100 mM Arg·Glu exhibited significantly lower viscosity, ranging from 12 to 113 mPa·s across the explored temperature window (37°C to 5°C). At ambient room temperature and above, this formulation may be syringeable, with viscosity beneath the widely accepted threshold of syringeability (∼25 mPa·s).30 Viscosities of the samples stored at 4°C and 40°C for 28 days were not significantly different, indicating that the aggregation and fragmentation observed following storage at 40°C (Figure 4) did not have a significant effect on the viscosity of either formulation. Both model formulations follow the expected exponential temperature-dependent viscosity relationship, with viscosity reduced at higher temperatures. In conclusion, this study shows that LLPS can yield high-concentration mAb formulation with satisfactory stability, viscosity and syringeability properties, by adding a suitable excipient to the dense fraction.
Discussion
Obtaining stable high-concentration antibody formulations is critical to the development of subcutaneous injections as a viable means of biopharmaceutical administration. However, concentrating some biopharmaceutical protein solutions beyond >100 mg/mL and stabilizing these solutions against self-association and degradation may be challenging. In this proof-of-principle study, we demonstrate that LLPS can be harnessed to generate high-concentration antibody solutions without adversely affecting higher-order protein structure, and that the addition of excipients improves the properties (e.g., viscosity, aggregation state and storage stability) of model formulations generated from the resulting dense fraction.
Here, LLPS was triggered in 80 mg/mL COE-13 in a formulation containing 75 mM NaCl by incubation at 4°C (Figure 1). This lead to a greater than twofold spontaneous increase in protein concentration to 175 mg/mL in the dense fraction, with such a concentration, in principle, suitable for subcutaneous administration.7 Although LLPS was relatively fast, the speed of macroscopic layer separation following LLPS, and thus the speed of concentrating of the protein solution, may be improved further through centrifugation of the solution during this process.31
NMR spectroscopy is increasingly used as a fingerprinting tool to assess the higher-order structure of biopharmaceutical proteins.32–34 Here, 1H NMR spectra showed that COE-13 in both the dense and lean fractions is not irreversibly affected by the process of LLPS, with the lean fraction and diluted dense fraction displaying similar fingerprint 1D spectra and transverse relaxation rate profiles to protein that had not undergone LLPS. Additionally, as protein in the lean fraction was shown to be structurally unperturbed following LLPS, this suggests that this extracted fraction may be recycled through conventional concentrating techniques, and then phase separated to produce further dense fraction, thus increasing yield and reducing waste.
While LLPS may be used to concentrate protein solutions, the attractive protein interactions that drive phase separation may in principle result in solutions with properties unsuitable for further handling during processing and administration. Moreover, recovery and dilution of this dense fraction may trigger further LLPS, which is undesirable in any final formulation. Here, we show that addition of an excipient, such as Arg·Glu, to the isolated dense fraction improves the properties of model high-concentration formulations generated from the dense fraction. As previously observed with other protein solutions,35,36 Arg·Glu reduced the viscosity of the model formulation (Figure 4), although further reductions in viscosity may be required to ensure syringeability as a final formulation. Importantly, Arg·Glu also prevented further LLPS of the model formulation (Figure 3). Furthermore, the high concentration antibody solutions generated here by LLPS were not inherently unstable when stored under typical refrigerated conditions, with Arg·Glu reducing both aggregation and fragmentation of the model formulations stored at 40°C to a level typical for other mAb formulations.27,37
Due to its spontaneous and self-driven nature, it may be tempting to incorporate LLPS in bioprocessing pipelines as a means of concentrating biopharmaceutical mAbs. To achieve that conditions under which LLPS occurs must first be identified for each individual mAb. While the primary sequence of some mAbs may render them inherently more prone to self-association and LLPS,25,38 a wide variety of additives, such as salts, macromolecular crowders and polyvalent ions,39–41 can be used alongside temperature and pH to induce LLPS. Selection of these additives and conditions could be achieved using conventional high-throughput screening platforms, for example, through detection of opalescence by light scattering techniques. To recover the favorable properties of solutions concentrated by LLPS, conventional formulation screening42,43 could be used to establish which excipients should be added to the separated dense fraction to prevent further phase separation, reduce self-association and solution viscosity, and achieve long-term storage stability. The practical issues regarding the scalability of this approach to the larger volumes necessary during industrial-scale bioprocessing require further investigation, and most likely would be mAb- and process-dependent. We suggest that in some cases concentrating biopharmaceutical proteins by LLPS may be more efficient than tangential flow ultrafiltration, given the cost of materials, energy and issues with membrane fouling.
In conclusion, the model system used in this study showed that LLPS is capable of concentrating mAb solutions to >150 mg/mL, with the LLPS ‘history’ of the concentrated sample not resulting in any additional instabilities. Applying a typical formulation optimization approach to the separated dense fraction should yield a stable high concentration protein formulation.
Materials and methods
Sample preparation
A full-length human IgG1κ mAb, COE-13 (MW 149 kDa, pI 8.1–8.6), known to be prone to LLPS under specific conditions19 was supplied by AstraZeneca. 20 mM acetate buffer (sodium acetate trihydrate (Sigma-Aldrich) and glacial acetic acid (Fisher Chemical)), pH 5.5, was prepared fresh as required. Concentrated NaCl (Fisher) was also prepared in buffer. Solutions were filtered with 0.2 µm syringe filters (Minisart SFCA, Sartorius) or 0.22 µm membrane filters (GSWP, Merck Millipore). Protein concentrations were determined by absorbance at 280 nm using a NanoDrop One (Thermo Scientific) in triplicates with dilution in buffer as required.
Stock solutions of COE-13 (46 mg/mL) were dialyzed in acetate buffer, using GeBAflex-Maxi dialysis tubes (3 mL, MWCO 30 kDa, Generon) or Slide-A-Lyzer MINI dialysis devices (0.5 mL, MWCO 20 kDa, Thermo Scientific) for large and small volumes, respectively. Extensive buffer exchange was conducted over five days. Samples not to undergo LLPS were subsequently prepared by dilution with additional buffer and concentrated NaCl to lean phase protein (10 mg/mL) and NaCl concentration (75 mM). Samples to undergo LLPS were initially concentrated using a centrifugal concentrator (30 kDa MWCO, Amicon) to >90 mg/mL, with subsequent dilution with buffer and concentrated NaCl to 80 mg/mL protein concentration and 75 mM NaCl. Macroscopic LLPS was induced by incubation at 4°C for 24 hours.
Following LLPS, the fractions were separated by removal of the lean fraction (Table 2). To generate two model high concentration formulations, the remaining dense fraction (with a small residual amount of lean fraction) was brought to 20°C, remixed and supplemented with 0.02% (w/v) sodium azide (Sigma-Aldrich) to inhibit bacterial growth. A 100 mM Arg·Glu (final concentration) formulation was generated by addition of concentrated Arg·Glu (equimolar L-arginine and L-glutamic acid mixture, both Sigma-Aldrich) dissolved in buffer supplemented with 75 mM NaCl, while a reference formulation was generated by addition of an equivalent volume of buffer with 75 mM NaCl. Aliquots of the model formulations were then stored under either refrigerated (4°C) or stressed stability (40°C) conditions. The sample aliquots were frozen after 0, 7, 14, or 28 days for further analysis.
Table 2.
Volumes and protein concentrations of the various solutions obtained during LLPS experiments. Measured concentrations determined by absorbance at 280 nm following appropriate dilution. High variability in measured dense fraction concentration caused by difficulties in handling such a viscous solution. Calculated concentrations were determined based on measured solution volumes (second column), and the measured concentration of the lean fraction
| Solution | Volume (µL) | Concentration (mg/mL) | |
| Measured | Calculated | ||
| Before LLPS | 545 | 80 ± 0.9 | - |
| Extracted lean fraction | 300 | 10 ± 0.3 | - |
| Dense layer with residual lean fraction | 245 | - | 165 ± 0.4 |
| Dense fraction | 230 | 171 ± 55 | 175 ± 5 |
NMR spectroscopy
NMR experiments were acquired using a Bruker 800 MHz Avance III spectrometer equipped with 5 mm TCI cryoprobe and variable temperature control unit. Sample temperature was calibrated against a standard methanol sample and verified with an external thermocouple placed in a sample tube in the probe. NMR samples (400 µL) were prepared in 5 mm NMR tubes (541-PP-7, Wilmad) with a coaxial insert (50 mm stem, Wilmad) filled with 2H2O to provide for external lock without sample adulteration. For variable temperature experiments, samples were left to equilibrate for 30 minutes upon reaching the desired temperate, with automated tuning, shimming and pulse calibration conducted following equilibration. For NMR analysis of mAbs after LLPS, lean fractions were assessed neat, whilst dense fractions were assessed neat or after dilution to lean phase concentration (10 mg/mL) with buffer containing 75 mM NaCl.
1D 1H spectra were recorded using excitation sculpting water suppression (Bruker pulse program zgesgp). Apparent transverse relaxation rates (R2) were measured using a Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence, with temperature compensation and a fixed echo time of 2.8 ms. Spectra were processed and analyzed with TopSpin 4.0 (Bruker). Apparent R2 at spectral points (0.05 ppm intervals) across the spectral width were calculated in Dynamics Center 2.6.1 (Bruker). Processed data were plotted in GraphPad Prism 8.0.
High-performance size-exclusion chromatography
Analysis of mAb monomeric, aggregate, and fragment species was performed using HPSEC. Analysis was performed using an Agilent 1200 series HPLC system with a TSKgel SWXL column (30 cm x 7.8 mm, 5 µm particle size, Tosoh Bioscience). Model formulations were diluted to 10 mg/mL and 0.45 µm filtered prior to analysis, Ultrafree-MC-HV, Merck Millipore). A 25 µL injection volume was used, with the system run at 1.0 mL/min with a mobile phase of 0.1 M Na2HPO4, 0.1 M Na2SO4, pH 6.8. Absorbance wavelength for detection was set at 280 nm. Chromatograms were analyzed in ChemStation (Agilent).
Viscosity measurements
Dynamic viscosities of the model formulations were measured at different temperatures using a VROC initium (Rheosense) with a B05 VROC cell and Peltier temperature control. Viscosities were measured according to the manufacturer’s instructions at a controlled shear rate of 345 s−1, with three repeated measurements per temperature. Samples were equilibrated for 2 min prior to analysis. Cleaning was conducted between samples with 1% Aquet detergent (Bel-Art), isopropyl alcohol, and filtered air.
Supplementary Material
Acknowledgments
J.E.B was supported by CASE DTP PhD studentship BB/M011208/1 from the UK Biotechnology and Biological Sciences Research Council (BBSRC) in partnership with AstraZeneca UK. We are grateful to Matthew Cliff for NMR facility management, to Grace Haagensen for assistance with HPSEC experiments, and to Leanne Amery for assistance with viscosity measurements.
Funding Statement
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) [BB/M011208/1].
Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Walsh G. Biopharmaceutical benchmarks 2018. Nat Biotechnol. 2018;36(12):1136–7. doi: 10.1038/nbt.4305. [DOI] [PubMed] [Google Scholar]
- 2.Cui Y, Cui P, Chen B, Li S, Guan H. Monoclonal antibodies: formulations of marketed products and recent advances in novel delivery system. Drug Dev Ind Pharm. 2017;43:519–30. [DOI] [PubMed] [Google Scholar]
- 3.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nature Rev. Cancer . 2012;12:278–87. doi: 10.1038/nrc3236 [DOI] [PubMed] [Google Scholar]
- 4.Viola M, Sequeira J, Seica R, Veiga F, Serra J, Santos AC, Ribeiro AJ. Subcutaneous delivery of monoclonal antibodies: how do we get there? J Control Release. 2018;286(301–314):301–14. doi: 10.1016/j.jconrel.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Tetteh E, Morris S. Systematic review of drug administration costs and implications for biopharmaceutical manufacturing. Appl Health Econ Health Policy. 2013;11(5):445–56. doi: 10.1007/s40258-013-0045-x. [DOI] [PubMed] [Google Scholar]
- 6.Mathaes R, Koulov A, Joerg S, Mahler HC. Subcutaneous injection volume of biopharmaceuticals-pushing the boundaries. J Pharm Sci. 2016;105(8):2255–59. doi: 10.1016/j.xphs.2016.05.029. [DOI] [PubMed] [Google Scholar]
- 7.Garidel P, Kuhn AB, Schafer LV, Karow-Zwick AR, Blech M. High-concentration protein formulations: how high is high? Eur J Pharm Biopharm. 2017;119(353–360):353–60. doi: 10.1016/j.ejpb.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 8.Shire SJ, Shahrokh Z, Liu J. (2010) Challenges in the Development of High Protein Concentration Formulations. In: Shire S., Gombotz W., Bechtold-Peters K., Andya J. editor(s). Current Trends in Monoclonal Antibody Development and Manufacturing. Biotechnology: Pharmaceutical Aspects, , New York: Springer. p 131–147. 10.1007/978-0-387-76643-0_9 [DOI] [Google Scholar]
- 9.Van Reis R, Zydney A. Membrane separations in biotechnology. Curr Opin Biotechnol. 2001;12(2):208–11. doi: 10.1016/S0958-1669(00)00201-9. [DOI] [PubMed] [Google Scholar]
- 10.Shire SJ. Formulation and manufacturability of biologics. Curr Opin Biotechnol. 2009;20(6):708–14. doi: 10.1016/j.copbio.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Allmendinger A, Mueller R, Huwyler J, Mahler HC, Fischer S. Sterile filtration of highly concentrated protein formulations: impact of protein concentration, formulation composition, and filter material. J Pharm Sci. 2015;104(10):3319–29. doi: 10.1002/jps.24561. [DOI] [PubMed] [Google Scholar]
- 12.Arakawa T, Ejima D, Akuta T. Protein aggregation under high concentration/density state during chromatographic and ultrafiltration processes. Int J Biol Macromol. 2017;95(1153–1158):1153–58. doi: 10.1016/j.ijbiomac.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Cao W, Krishnan S, Ricci MS, Shih LY, Liu D, Gu JH, Jameel F. Rational design of lyophilized high concentration protein formulations-mitigating the challenge of slow reconstitution with multidisciplinary strategies. Eur J Pharm Biopharm. 2013;85(2):287–93. doi: 10.1016/j.ejpb.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Bhatnagar BS, Bogner RH, Pikal MJ. Protein stability during freezing: separation of stresses and mechanisms of protein stabilization. Pharm Dev Technol. 2007;12(5):505–23. doi: 10.1080/10837450701481157. [DOI] [PubMed] [Google Scholar]
- 15.Johnson HR, Lenhoff AM. Characterization and suitability of therapeutic antibody dense phases for subcutaneous delivery. Mol Pharm. 2013;10(10):3582–91. doi: 10.1021/mp400006g. [DOI] [PubMed] [Google Scholar]
- 16.Raut AS, Kalonia DS. Pharmaceutical perspective on opalescence and liquid-liquid phase separation in protein solutions. Mol Pharm. 2016;13(5):1431–44. doi: 10.1021/acs.molpharmaceut.5b00937. [DOI] [PubMed] [Google Scholar]
- 17.Matsuda A, Mimura M, Maruyama T, Kurinomaru T, Shiuhei M, Shiraki K. Liquid droplet of protein-polyelectrolyte complex for high-concentration formulations. J Pharm Sci. 2018;107(10):2713–19. doi: 10.1016/j.xphs.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 18.Nishi H, Miyajima M, Nakagami H, Noda M, Uchiyama S, Fukui K. Phase separation of an igg1 antibody solution under a low ionic strength condition. Pharm Res. 2010;27(7):1348–60. doi: 10.1007/s11095-010-0125-7. [DOI] [PubMed] [Google Scholar]
- 19.Kheddo P, Bramham JE, Dearman RJ, Uddin S, Van Der Walle CF, Golovanov AP. Investigating liquid-liquid phase separation of a monoclonal antibody using solution-state nmr spectroscopy: effect of arg.Glu and arg.Hcl. Mol Pharm. 2017;14(8):2852–60. doi: 10.1021/acs.molpharmaceut.7b00418. [DOI] [PubMed] [Google Scholar]
- 20.Asenjo JA, Andrews BA. Aqueous two-phase systems for protein separation: a perspective. J Chromatogr A. 2011;1218(49):8826–35. doi: 10.1016/j.chroma.2011.06.051. [DOI] [PubMed] [Google Scholar]
- 21.Mao LN, Rogers JK, Westoby M, Conley L, Pieracci J. Downstream antibody purification using aqueous two-phase extraction. Biotechnol Prog. 2010;26(6):1662–70. doi: 10.1002/btpr.477. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt A, Richter M, Rudolph F, Strube J. Integration of aqueous two-phase extraction as cell harvest and capture operation in the manufacturing process of monoclonal antibodies. Antibodies (Basel). 2017;6(4):21. doi: 10.3390/antib6040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joubert MK, Luo Q, Nashed-Samuel Y, Wypych J, Narhi LO. Classification and characterization of therapeutic antibody aggregates. J Biol Chem. 2011;286(28):25118–33. doi: 10.1074/jbc.M110.160457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saluja A, Kalonia DS. Nature and consequences of protein-protein interactions in high protein concentration solutions. Int J Pharm. 2008;358(1–2):1–15. doi: 10.1016/j.ijpharm.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 25.Chow CK, Allan BW, Chai Q, Atwell S, Lu J. Therapeutic antibody engineering to improve viscosity and phase separation guided by crystal structure. Mol Pharm. 2016;13(3):915–23. doi: 10.1021/acs.molpharmaceut.5b00817. [DOI] [PubMed] [Google Scholar]
- 26.Bye JW, Platts L, Falconer RJ. Biopharmaceutical liquid formulation: a review of the science of protein stability and solubility in aqueous environments. Biotechnol Lett. 2014;36(5):869–75. doi: 10.1007/s10529-013-1445-6. [DOI] [PubMed] [Google Scholar]
- 27.Kheddo P, Tracka M, Armer J, Dearman RJ, Uddin S, Van Der Walle CF, Golovanov AP. The effect of arginine glutamate on the stability of monoclonal antibodies in solution. Int J Pharm. 2014;473(1–2):126–33. doi: 10.1016/j.ijpharm.2014.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golovanov AP, Hautbergue GM, Wilson SA, Lian LY. A simple method for improving protein solubility and long-term stability. J Am Chem Soc. 2004;126(29):8933–39. doi: 10.1021/ja049297h. [DOI] [PubMed] [Google Scholar]
- 29.Kheddo P, Cliff MJ, Uddin S, Van Der Walle CF, Golovanov AP. Characterizing monoclonal antibody formulations in arginine glutamate solutions using (1)h nmr spectroscopy. MAbs. 2016;8(7):1245–58. doi: 10.1080/19420862.2016.1214786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Liu Y. Recent progresses of understanding the viscosity of concentrated protein solutions. Curr Opin Chem Eng. 2017;16:48–55. doi: 10.1016/j.coche.2017.04.001. [DOI] [Google Scholar]
- 31.Wang Z, Zhang G, Zhang H. Protocol for analyzing protein liquid–liquid phase separation. Biophys Rep. 2018;5(1):1–9. doi: 10.1007/s41048-018-0078-7. [DOI] [Google Scholar]
- 32.Poppe L, Jordan JB, Lawson K, Jerums M, Apostol I, Schnier PD. Profiling formulated monoclonal antibodies by (1)h nmr spectroscopy. Anal Chem. 2013;85(20):9623–29. doi: 10.1021/ac401867f. [DOI] [PubMed] [Google Scholar]
- 33.Arbogast LW, Brinson RG, Marino JP. Mapping monoclonal antibody structure by 2d 13c nmr at natural abundance. Anal Chem. 2015;87(7):3556–61. doi: 10.1021/ac504804m. [DOI] [PubMed] [Google Scholar]
- 34.Brinson RG, Marino JP, Delaglio F, Arbogast LW, Evans RM, Kearsley A, Gingras G, Ghasriani H, Aubin Y, Pierens GK, et al. Enabling adoption of 2d-nmr for the higher order structure assessment of monoclonal antibody therapeutics. MAbs. 2019;11(1):94–105. doi: 10.1080/19420862.2018.1544454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borwankar AU, Dear BJ, Twu A, Hung JJ, Dinin AK, Wilson BK, Yue J, Maynard JA, Truskett TM, Johnston KP. Viscosity reduction of a concentrated monoclonal antibody with arginine·hcl and arginine·glutamate. Ind Eng Chem Res. 2016;55(43):11225–34. doi: 10.1021/acs.iecr.6b02042. [DOI] [Google Scholar]
- 36.Inoue N, Takai E, Arakawa T, Shiraki K. Specific decrease in solution viscosity of antibodies by arginine for therapeutic formulations. Mol Pharm. 2014;11(6):1889–96. doi: 10.1021/mp5000218. [DOI] [PubMed] [Google Scholar]
- 37.Bramham JE, Podmore A, Davies SA, Golovanov AP. Comprehensive assessment of protein and excipient stability in biopharmaceutical formulations using (1)h nmr spectroscopy. ACS Pharmacol Transl Sci. 2021;4(1):288–95. doi: 10.1021/acsptsci.0c00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du Q, Damschroder M, Pabst TM, Hunter AK, Wang WK, Luo H. Process optimization and protein engineering mitigated manufacturing challenges of a monoclonal antibody with liquid-liquid phase separation issue by disrupting inter-molecule electrostatic interactions. MAbs. 2019;11(4):789–802. doi: 10.1080/19420862.2019.1599634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Latypov RF, Lomakin A, Meyer JA, Kerwin BA, Vunnum S, Benedek GB. Quantitative evaluation of colloidal stability of antibody solutions using peg-induced liquid-liquid phase separation. Mol Pharm. 2014;11(5):1391–402. doi: 10.1021/mp400521b. [DOI] [PubMed] [Google Scholar]
- 40.Dumetz AC, Lewus RA, Lenhoff AM, Kaler EW. Effects of ammonium sulfate and sodium chloride concentration on peg/protein liquid-liquid phase separation. Langmuir. 2008;24(18):10345–51. doi: 10.1021/la801180n. [DOI] [PubMed] [Google Scholar]
- 41.Matsarskaia O, Roosen-Runge F, Lotze G, Moller J, Mariani A, Zhang F, Schreiber F. Tuning phase transitions of aqueous protein solutions by multivalent cations. Phys Chem Chem Phys. 2018;20(42):27214–25. doi: 10.1039/C8CP05884A. [DOI] [PubMed] [Google Scholar]
- 42.He F, Woods CE, Trilisky E, Bower KM, Litowski JR, Kerwin BA, Becker GW, Narhi LO, Razinkov VI. Screening of monoclonal antibody formulations based on high-throughput thermostability and viscosity measurements: design of experiment and statistical analysis. J Pharm Sci. 2011;100(4):1330–40. doi: 10.1002/jps.22384. [DOI] [PubMed] [Google Scholar]
- 43.Goldberg DS, Lewus RA, Esfandiary R, Farkas DC, Mody N, Day KJ, Mallik P, Tracka MB, Sealey SK, Samra HS. Utility of high throughput screening techniques to predict stability of monoclonal antibody formulations during early stage development. J Pharm Sci. 2017;106(8):1971–77. doi: 10.1016/j.xphs.2017.04.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.