

Abstract
Systemic treatment with resiniferatoxin (RTX) induces small-fiber sensory neuropathy by damaging TRPV1-expressing primary sensory neurons and causes distinct thermal sensory impairment and tactile allodynia, which resemble the unique clinical features of postherpetic neuralgia. However, the synaptic plasticity associated with RTX-induced tactile allodynia remains unknown. In this study, we found that RTX-induced neuropathy is associated with α2δ-1 upregulation in the dorsal root ganglion (DRG) and increased physical interaction between α2δ-1 and GluN1 in the spinal cord synaptosomes. RNAscope in situ hybridization showed that RTX treatment significantly increased α2δ-1 expression in DRG neurons labeled with calcitonin gene-related peptide, isolectin B4, NF200, and tyrosine hydroxylase. Electrophysiological recordings revealed that RTX treatment augmented the frequency of miniature excitatory postsynaptic currents (mEPSCs) and the amplitude of evoked EPSCs in spinal dorsal horn neurons, and these effects were reversed by blocking NMDA receptors with AP-5. Inhibiting α2δ-1 with gabapentin, genetically ablating α2δ-1, or targeting α2δ-1–bound NMDA receptors with α2δ-1Tat peptide largely normalized the baseline frequency of mEPSCs and the amplitude of evoked EPSCs potentiated by RTX treatment. Furthermore, systemic treatment with memantine or gabapentin and intrathecal injection of AP-5 or Tat-fused α2δ-1 C terminus peptide reversed allodynia in RTX-treated rats and mice. In addition, RTX-induced tactile allodynia was attenuated in α2δ-1 knock-out mice and in mice in which GluN1 was conditionally knocked out in DRG neurons. Collectively, our findings indicate that α2δ-1–bound NMDA receptors at presynaptic terminals of sprouting myelinated afferent nerves contribute to RTX-induced potentiation of nociceptive input to the spinal cord and tactile allodynia.
SIGNIFICANCE STATEMENT Postherpetic neuralgia (PHN), associated with shingles, is a distinct form of neuropathic pain commonly seen in elderly and immunocompromised patients. The synaptic plasticity underlying touch-induced pain hypersensitivity in PHN remains unclear. Using a nonviral animal model of PHN, we found that glutamatergic input from primary sensory nerves to the spinal cord is increased via tonic activation of glutamate NMDA receptors. Also, we showed that α2δ-1 (encoded by Cacna2d1), originally considered a calcium channel subunit, serves as an auxiliary protein that promotes activation of presynaptic NMDA receptors and pain hypersensitivity. This new information advances our understanding of the molecular mechanism underlying PHN and suggests new strategies for treating this painful condition.
Keywords: dorsal root ganglion, gabapentinoid, pregabalin, neuropathic pain, NMDA receptor, spinal cord, synaptic plasticity
Introduction
Postherpetic neuralgia (PHN) is a painful condition that occurs most often in elderly and in immunocompromised individuals and can last for months or years (Baron and Saguer, 1993; Rowbotham and Fields, 1996; Truini et al., 2008). PHN results from reactivation of the varicella-zoster virus, which predominantly infects and damages peripheral sensory neurons (Head et al., 1997; Johnson and Rice, 2014). Because of small-fiber deafferentation in many PHN patients (Rowbotham et al., 1996), temperature sensation is profoundly impaired, but light touch triggers severe pain in the same affected dermatomes (Fields et al., 1998; Pappagallo et al., 2000). Thus, tactile allodynia (cutaneous hypersensitivity) is a dominant clinical feature of PHN and often disrupts the lives of otherwise healthy individuals (Nurmikko and Bowsher, 1990). In PHN, tactile allodynia may be initiated and maintained by damaged myelinated nerves and/or aberrant afferent synaptic reorganization at the spinal cord level (Baron and Saguer, 1993). Current treatments for PHN remain unsatisfactory, and the synaptic mechanism involved in the distinct tactile allodynia in PHN remains unclear.
Our understanding of the mechanisms of neuropathic pain relies mostly on information obtained from animal models using traumatic nerve injury and chemotherapy, which generally increase both thermal and mechanical sensitivity. Although PHN is a common cause of neuropathic pain in patients, the mechanisms of PHN are rarely studied because viral-infection-induced animal models of PHN exhibit inconsistent sensory deficit, tissue lesions, and paralysis because of virus spread in the CNS (Takasaki et al., 2000; Dalziel et al., 2004). Furthermore, the varicella-zoster virus is highly contagious and must be handled in a biological-safety facility. Resiniferatoxin (RTX) is an ultrapotent agonist of transient receptor potential vanilloid subtype 1 (TRPV1; Szallasi and Blumberg, 1989). Systemic treatment with RTX diminishes thermal sensitivity by depletion of TRPV1-expressing primary sensory neurons, which results in a substantial degeneration of C-fiber afferent terminals in the spinal lamina I/II (Pan et al., 2003; Chen and Pan, 2005). Systemic RTX also induces aberrant sprouting of myelinated afferent terminals onto lamina I/II neurons (Pan et al., 2003; Wu et al., 2016). As a result, RTX treatment induces paradoxical thermal sensory impairment and tactile allodynia, which largely recapitulate the unique clinical features of PHN. Although RTX-induced structural changes in the peripheral nerve and spinal dorsal horn have been documented (Pan et al., 2003; Hsieh et al., 2012), little is known about how RTX-induced myelinated afferent nerve sprouting in the superficial dorsal horn initiates and sustains tactile allodynia.
Increased N-methyl-d-aspartate receptor (NMDAR) activity at the spinal cord level plays a crucial role in neuropathic pain caused by traumatic nerve injury and chemotherapy (Chen et al., 2014b; Xie et al., 2016). Although the NMDAR antagonist ketamine reduces pain hypersensitivity in PHN patients (Eide et al., 1994), nothing is known about whether and how synaptic NMDARs are activated in the spinal cord in PHN. Furthermore, α2δ-1, originally considered a Ca2+ channel subunit, is expressed in the dorsal root ganglion (DRG) and spinal dorsal horn neurons (Newton et al., 2001; Cole et al., 2005). Through its C-terminal domain, independent of its conventional role as a Ca2+ channel subunit, α2δ-1 forms a protein complex with NMDARs to promote synaptic trafficking of NMDARs in neuropathic pain (Chen et al., 2018; 2019). However, it is unclear whether α2δ-1 is involved in nociceptive transmission in PHN. In the present study, using RTX-induced neuropathy as a nonviral PHN model, we tested the hypothesis that RTX treatment causes tactile allodynia by augmenting glutamatergic input to spinal dorsal horn neurons via α2δ-1–dependent potentiation of NMDAR activity. Findings from our study advance a mechanistic understanding of synaptic plasticity in PHN and suggest new strategies for the treatment of this debilitating, painful condition.
Materials and Methods
Animal models
The experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas MD Anderson Cancer Center and conformed to guidelines from the National Institutes of Health on the ethical use of animals. Male Sprague Dawley rats (200–250 g; Harlan) were used in this study. All rats were housed in groups of 2–3 per cage at 22°C on a standard 12:12 h light-dark cycle and had access to food and water ad libitum. RTX was administered in rats as described in our previous studies (Pan et al., 2003; Chen and Pan, 2005), except that a lower dose was used to improve recovery and survival after RTX injection. RTX was dissolved in a solution of 10% ethanol and 10% tris-buffered saline (Tween) 80 in normal saline to a final concentration of 150 μg/ml. Rats received a single intraperitoneal administration of RTX (150 μg/kg; LC Laboratories) under 2% isoflurane-induced anesthesia. Rats in the control group were injected with the same volume of the vehicle.
We purchased Grin1flox/flox mice from The Jackson Laboratory (stock #005246). AdvillinCre/+ mice were kindly provided by Fan Wang (Duke University; da Silva et al., 2011). AdvillinCre/+::Grin1flox/+ mice were obtained by breeding the male AdvillinCre/+ mice with female Grin1flox/flox mice. We crossed male AdvillinCre/+::Grin1flox/+ mice with female Grin1flox/flox mice to generate AdvillinCre/+::Grin1flox/flox mice, referred to as Grin1-conditional knock-out (Grin1-cKO) mice. Cre-negative littermates (AdvillinCre/-::Grin1flox/flox) were used as wild-type (WT) control mice. The α2δ-1 (encoded by the Cacna2d1 gene) KO (Cacna2d1−/−) mice were originally obtained from the Medical Research Council (stock #6900) and were generated as described previously (Fuller-Bicer et al., 2009). The WT (Cacna2d1+/+) littermates were used as controls and obtained by breeding the Cacna2d1+/− heterozygous mice. Mice were earmarked 3 weeks after birth, and ear biopsies were used for genotyping. All the mice had a C57BL/6 genetic background and were housed no more than 5 per cage. Equal numbers of male and female adult (8–12 weeks old) mice were used for final experiments. The data were pooled because RTX produced similar effects in male and female mice. We intraperitoneally injected mice with 25 μg/kg of RTX because the RTX dose at or higher than 50 μg/kg unexpectedly killed the mice in our pilot experiments. Mice in the control group were injected with the same volume of the vehicle solution.
Drug administration
Gabapentin (Tocris Bioscience), memantine (Tocris Bioscience), 2-amino-5-phosphonopentanoic acid (AP-5; Hello Bio), Tat-fused α2δ-1 C terminus peptide (VSGLNPSLWSIFGLQFILLWLVSGSRHYLW), and Tat-fused scrambled control peptide (FGLGWQPWSLSFYLVWSGLILSVLHLIRSN; Bio Basic) were diluted with normal saline and injected intraperitoneally or intrathecally. Intrathecal injection was performed using a lumbar puncture technique, as previously described (Sun et al., 2019). Briefly, rats were anesthetized with 2% isoflurane and placed in a prone position with a tube under the abdomen to extend the lumbar vertebral space between L5 and L6. Lumbar puncture was performed using a small needle (30 gauge) connected to a 50-μl Hamilton syringe. Successful intrathecal injection (10 μl of drug solution) was indicated by brisk tail movement.
Nociceptive behavioral tests
To measure cutaneous hypersensitivity, we detected the paw withdrawal threshold of the animals with von Frey filaments, as described previously (Chen and Pan, 2005). A series of calibrated von Frey filaments were applied perpendicular to the plantar surface of the hindpaw with sufficient force to bend the filament for 6 s. The tactile stimulus producing a 50% likelihood of withdrawal was determined using the up-down method as described previously (Chaplan et al., 1994). To quantify the mechanical nociceptive threshold, we tested paw withdrawal thresholds using a Randall-Selitto analgesiometer (Ugo Basile), as described previously (Pan et al., 2003; Chen and Pan, 2005). A constantly increasing force was applied to the hindpaw. When the animal displayed a withdrawal response, the device was immediately stopped, and the nociceptive threshold was registered. In addition, to assess cutaneous thermal sensitivity, we measured the paw withdrawal latency using a thermal testing apparatus (IITC Life Science), as described previously (Pan et al., 2003; Chen and Pan, 2005). Animals were placed individually on a glass surface maintained at 30°C. A mobile radiant heat stimulus was applied to the plantar surface of the hindpaw until the animal displayed a withdrawal response or paw licking. All behavioral tests were done between 9:00 A.M. and 6:00 P.M.
Quantitative PCR
The DRG and dorsal spinal cord tissues were rapidly removed from rats deeply anesthetized with isoflurane. Total RNA was extracted from the tissue using Trizol/chloroform, and cDNA was prepared by using a RevertAid RT Reverse Transcription Kit (catalog #K1691, Thermo Fisher Scientific). Quantitative PCR was performed using SYBR Green PCR Master Mix (catalog #4913914001, Applied Biosystems) with QuantStudio 7-Flex Real-Time PCR System software (Thermo Fisher Scientific). The thermal cycling conditions used were the following: 1 cycle at 95°C for 5 min, 40 cycles at 95°C for 15 s, and 60°C for 45 s. The following primers were used: Cacna2d1 forward, GGACCTATTCAGTGGATGGCTTG; Cacna2d1 reverse, CCATTGGTCTTCCCAGAACATCTAGA; Grin1 forward, GGATAAGACATGGGTTCGGTATCAGG; Grin1 reverse, TGTGTCGCTTGTAGGCGATCTC; Gapdh forward, GACATGCCGCCTGGAGAAAC; Gapdh reverse, AGCCCAGGATGCCCTTTAGT; NeuN (encoded by Rbfox3), forward, CACCACTCTCTTGTCCGTTTGC; NeuN reverse, GGCTGAGCATATCTGTAAGCTGC. The relative mRNA amount of target gene in each sample was calculated using the 2−ΔΔCT method, normalized to the level of Gapdh or NeuN, and then normalized to its expression level in vehicle-treated rats.
Spinal cord synaptosome preparation
The dorsal spinal cord was harvested and homogenized in 10 volumes of ice-cold HEPES-buffered sucrose (0.32 m sucrose; 4 mm HEPES; and 1 mm EGTA, pH 7.4) in the present of the protease inhibitor cocktail (Sigma). The homogenate was centrifuged at 1000 × g at 4°C for 10 min to remove the sediment, and the supernatant was centrifuged again at 12 000 × g for 15 min to obtain the crude synaptosomal fraction. The crude synaptosomes were incubated in RIPA lysis buffer with a protease inhibitor cocktail for 1 h on ice and then centrifuged at 13,000 rpm for 15 min to obtain the synaptosomal fraction (Zhou et al., 2009; Xie et al., 2016; Chen et al., 2018; 2019). After the protein concentration was measured, 30 μg of protein was used for immunoblotting. Postsynaptic density protein 95 (PSD95), a known 95 kDa synaptic protein, was used as an internal loading control. The following antibodies were used for immunoblotting: mouse anti–α2δ-1 (1:1000; catalog #SC-271697, Santa Cruz Biotechnology), rabbit anti-GluN1 (1:1000; catalog #G8913, Sigma), and mouse anti-PSD95 (1:1000; catalog #75–348, NeuroMab). The amount of α2δ-1 and GluN1 in the spinal cord synaptosomes was normalized to that of PSD-95 on the same gels.
Immunoblotting
Proteins were extracted from the DRG and dorsal spinal cord of rats using RIPA lysis buffer (Thermo Fisher Scientific) in the presence of a mixture of proteinase inhibitors (Sigma). The samples with lysis buffer were placed on ice for 30 min after sonication. Lysates were centrifuged at 12 000 × g for 15 min at 4°C, and the supernatant was carefully collected. A total of 30 μg of protein was loaded and separated by SDS-PAGE with 4%–12% gel (Invitrogen). The resolved proteins were transferred to a polyvinylidene difluoride membrane. The membrane was incubated with 5% nonfat dry milk in Tris-buffered saline–Tween 20 (TBST) at 25°C for 1 h and then incubated with primary antibodies diluted with TBST containing 3% bovine serum albumin overnight at 4°C. The primary antibodies included mouse anti-α2δ-1 (1:1000; catalog #SC-271697, Santa Cruz Biotechnology), rabbit anti-GluN1 (1:1000; catalog #G8913, Sigma), rabbit anti-GAPDH (1:7000; catalog #5174S, Cell Signaling Technology), mouse anti-PSD95 (1:1000; catalog #75-348, NeuroMab), rabbit anti-NeuN (1:1000; catalog #12 943, Cell Signaling Technology), and mouse anti-synaptophysin (1:1000; catalog #4329, Cell Signaling Technology). The membrane was washed three times and then incubated with horseradish peroxidase conjugated anti-rabbit IgG (1:10,000; catalog #7074S, Cell Signaling Technology) or anti-mouse IgG (1:10,000; catalog #7076S, Cell Signaling Technology) for 1 h at 25°C. The protein bands were revealed with an ECL detection kit (Thermo Fisher Scientific). The protein band intensity was visualized and quantified using an Odyssey Fc Imager (LI-COR Biosciences). The amount of target proteins in each sample was first normalized to the level of GAPDH, synaptophysin, or NeuN and then normalized to its expression level in vehicle-treated rats.
Coimmunoprecipitation
Dorsal spinal cord tissues were collected and homogenized using immunoprecipitation lysis buffer (Thermo Fisher Scientific) in the presence of a proteinase inhibitor cocktail (Sigma). The protein lysis was incubated at 4°C overnight with protein G beads (catalog #16–266, MilliporeSigma) prebound to rabbit anti-GluN1 antibodies (catalog #G8913, Sigma). Protein G beads prebound to IgG were used as a control. All samples were washed three times with immunoprecipitation buffer and then immunoblotted. The following antibodies were used for immunoblotting: mouse anti-α2δ-1 (1:1000; catalog #SC-271697, Santa Cruz Biotechnology) and rabbit anti-GluN1 (1:1000; catalog #G8913; Sigma). We used anti-mouse IgG (1:10,000; catalog #7076S, Cell Signaling Technology) and TrueBlot anti-rabbit IgG (1:10,000; catalog #18–8816-33, Rockland Immunochemicals) as the secondary antibody. The amount of the α2δ-1 protein was normalized to that of GluN1 on the same gel.
RNAscope in situ hybridization
Single-molecule RNA signals can be visualized in individual cells by the use of a unique probe design strategy and a hybridization-based signal amplification system to amplify signals and suppress backgrounds. In the RNAscope assay, conventional fluorescent dyes can be applied to tissue sections for multiplex analysis (Li and Kim, 2015). We thus performed RNAscope in situ hybridization and immunofluorescence labeling to detect α2δ-1 mRNA and NeuN (a neuronal marker) in the DRG from three vehicle-treated and three RTX-treated rats 5 weeks after treatment. Animals were deeply anesthetized with 5% isoflurane and transcardially perfused with sterile PBS and then 4% paraformaldehyde. Lumbar DRGs were removed and postfixed in 4% paraformaldehyde overnight and then treated with 30% sucrose in PBS until the tissue settled at the base of the tube. Tissue was then embedded in M-1 embedding matrix (catalog #501951, Thermo Fisher Scientific) and kept at −80°C. The tissues were cut to 15 µm sections using a cryostat (catalog #CM1860, Leica Biosystems), mounted directly onto Superfrost Plus slides (catalog #22–178-277, Thermo Fisher Scientific), and kept frozen until the RNA in situ hybridization assay. The RNAscope Intro Pack for Multiplex Fluorescent Reagent Kit (catalog #323137, Advanced Cell Diagnostics) was applied to the slides. The RNAscope target-specific oligonucleotide probes for α2δ-1 were designed by Advanced Cell Diagnostics (catalog #818271). Opal 620 (catalog #FP1495001KT, Akoya Biosciences) was used to develop the horseradish peroxidase-C1 signal. The sections were then rinsed with Tris-buffered saline and incubated with Alexa Fluor 488 conjugated to isolectin B4 (IB4; (1:100; catalog #121411, Thermo Fisher Scientific) for 2 h at 22°C or the primary antibodies, including rabbit anti-NeuN (1:200; catalog #ab177487, Abcam), mouse anti-neurofilament (NF)200 (1:100; catalog #N0142, Sigma), goat anti-calcitonin gene-related peptide (CGRP; 1:100; catalog #ab36001, Abcam), and rabbit anti-tyrosine hydroxylase (1:200; catalog #NB300-10, Novus Biologicals), diluted in PBS solution containing 3% bovine serum albumin and 0.3% Triton X-100 overnight at 4°C. Subsequently, sections were rinsed in 0.1 m PBS and incubated with Alexa Fluor 488 conjugated to the secondary antibodies, including Alexa Fluor 488 conjugated to donkey anti-rabbit IgG (catalog #A-21 206, Thermo Fisher Scientific), goat anti-mouse IgG (catalog #A28175, Thermo Fisher Scientific), and donkey anti-goat IgG (catalog #ab150129, Abcam), at 1:200 concentrations for 1 h. The sections were then rinsed, mounted on slides, dried, and covered with coverslips.
Images were examined on a fluorescence microscope and acquired with a confocal laser-scanning microscope (Zeiss) with a 40× objective. For imaging analysis, we randomly selected two section images (40×) from each DRG and 4–5 DRGs from each group. RNAscope signal was identified as punctate staining with little or no background. We first counted the number of α2δ-1 mRNA punctate signals for each image and then compared the difference between vehicle- and RTX-treated groups. We classified neurons into small (<30 μm), medium (30–50 μm), and large (>50 μm) groups based on diameter and quantified the α2δ-1 mRNA punctate signals per cell in each group. The quantitative analysis was performed using the National Institutes of Health ImageJ Cell Counter Plugin.
Whole-cell patch-clamp recordings in spinal cord tissue slices
All electrophysiological recordings in spinal cord slices were conducted 5–6 weeks after treatment with RTX or vehicle. The lumbar spinal cord was obtained through laminectomy in animals anesthetized with 3% isoflurane, and transverse slices 400 μm thick were cut using a vibratome. To measure quantal glutamate release from presynaptic terminals, miniature excitatory postsynaptic currents (mEPSCs) were recorded from lamina II outer neurons in the presence of 0.5 μm tetrodotoxin at a holding potential of –60 mV using whole-cell voltage-clamp techniques, as we described previously (Li et al., 2002; Chen et al., 2014a). To measure glutamate release from primary afferent nerves, the dorsal root was electrically stimulated at 0.6 mA, 0.5 ms, and 0.1 Hz, and monosynaptic EPSCs were identified based on the constant latency and the absence of conduction failure of evoked EPSCs in response to a 20 Hz (lasting 1 s) electrical stimulation (Li et al., 2002; Zhou et al., 2010). For the electrical stimulation intensity used in our study, both A and C fibers can be activated (Kohno et al., 1999; Li et al., 2002; Zhou et al., 2010). To determine the paired-pulse ratio (PPR), two EPSCs were evoked by a pair of stimuli given at 50 ms intervals. The PPR was expressed as the ratio of the amplitude of the second synaptic response to the amplitude of the first synaptic response.
To record postsynaptic NMDAR and AMPAR currents, we puffed 100 μm NMDA or 20 μm AMPA directly onto the lamina II outer neuron using a positive pressure system (Toohey Company, Fairfield, NJ). The distance between the tip of the puff pipette and the recorded neuron was 150 μm (Chen et al., 2014a; Huang et al., 2019). The internal solution used for recording electrodes contained the following (in mm): 110 Cs2SO4, 2 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES, 0.3 Na2-GTP, 2 Mg-ATP, and 10 QX314 (280–300 mOsm, pH 7.2–7.4). To minimize the Mg2+ block of NMDARs at negative holding potentials, MgCl2 was replaced with CaCl2 in the extracellular solution during recording of puff NMDAR currents. Tetrodotoxin citrate and AP-5 were purchased from Hello Bio.
Study design and data analysis
We did not use any statistical means to predetermine sample sizes for each experiment, but our sample sizes were similar to those generally used in the field (Zhou et al., 2009; Xie et al., 2016; Chen et al., 2018). Only 1 neuron was recorded in each spinal cord slice, and four animals were used for each recording protocol. The amplitude and frequency of mEPSCs were analyzed using the MiniAnalysis peak detection program (Synaptosoft). The evoked EPSCs were analyzed using Clampfit 10.0 software (Molecular Devices), and the amplitude of currents was quantified by averaging six consecutive currents. Puff NMDA and AMPA currents were analyzed using Clampfit 10.0 software, and the amplitude of currents was quantified by averaging three consecutive currents. The investigators performing behavioral tests, biochemical assays, and electrophysiological recordings were blinded to the treatment and mouse genotypes. For the electrophysiological and biochemical data, a two-tailed Student's t test was used to compare two groups. One-way ANOVA followed by Tukey's post hoc test or repeated-measures ANOVA followed by Dunnett's post hoc test was used to compare more than two groups. For behavioral data analysis, two-way ANOVA followed by Tukey's post hoc test was used to compare between treatment groups or within subjects. Data are expressed as means ± SEM and were statistically analyzed using Prism software version 8 (GraphPad). Differences were considered significant when p < 0.05.
Results
RTX treatment leads to impaired thermal sensitivity and tactile allodynia
A single intraperitoneal administration of 150 μg/kg RTX caused a profound increase in the paw withdrawal latency in response to a noxious heat stimulus in all rats within 3 d of injection (p < 0.001, F(3,48) = 88.16, n = 9 rats; Fig. 1A). This diminished thermal sensitivity lasted for at least 5 weeks. In contrast, RTX treatment had no significant effect on the paw withdrawal threshold in response to a noxious pressure stimulus in these rats (p = 0.72, F(6,96) = 0.61; Fig. 1B). Intraperitoneal injection of the vehicle had no effect on the thermal or pressure withdrawal threshold in all rats tested (Fig. 1A,B).
Figure 1.
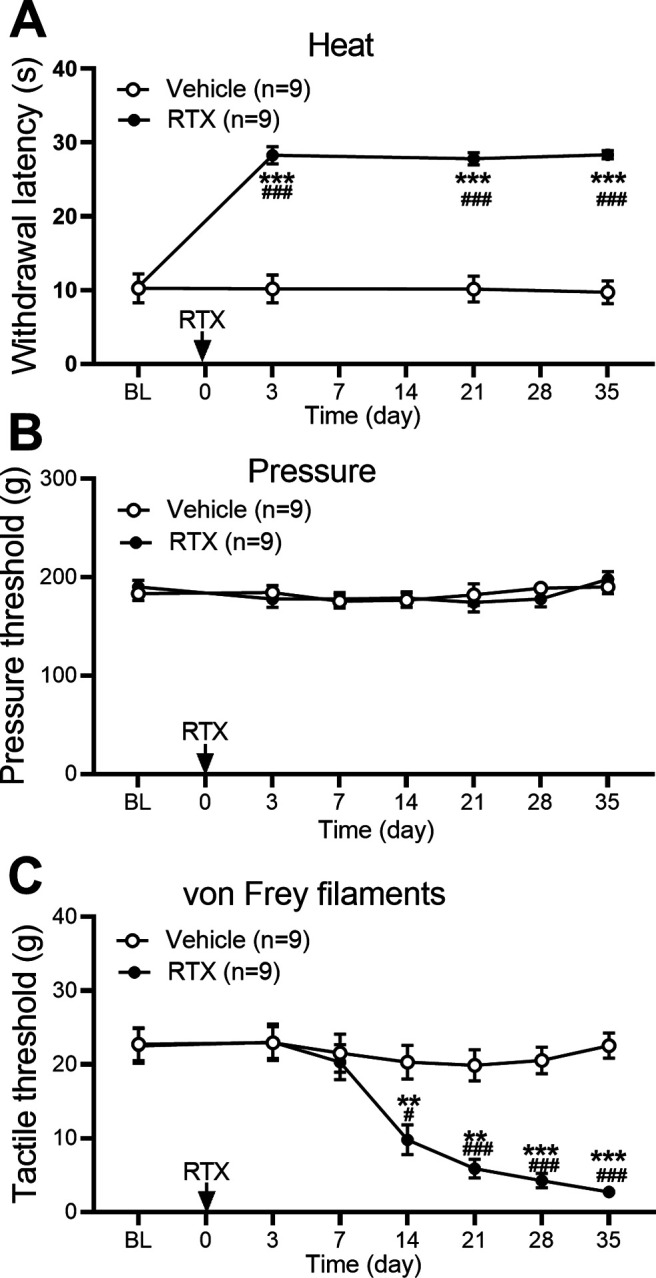
RTX treatment impairs thermal nociception but causes delayed and sustained tactile allodynia. A–C, Time course of the effect of RTX treatment on nociceptive responses to a heat stimulus (A), a pressure stimulus (B), and von Frey filaments (C) in rats. Data are expressed as means ± SEM (n = 9 rats per group). **p < 0.01, ***p < 0.001 compared with the baseline (BL) before RTX injection, #p < 0.05, ###p < 0.001 compared with the vehicle group at the same time point. Two-way ANOVA followed by Tukey's post hoc test.
Treatment with RTX, not vehicle, in rats caused a delayed and gradual reduction in the tactile withdrawal threshold in response to von Frey filaments applied to the hindpaw (Fig. 1C). The reduced tactile withdrawal threshold became evident 2 weeks after RTX injection and was progressively more pronounced for at least another 3 weeks (p < 0.001, F(6,96) = 23.19, n = 9 rats per group; Fig. 1C). The time course of reduced thermal sensitivity and tactile allodynia induced by 150 μg/kg of RTX was similar to that caused by 200 μg/kg of RTX reported previously (Pan et al., 2003).
RTX treatment increases α2δ-1, but not GluN1, expression levels in the DRG
Both peripheral nerve injury and paclitaxel treatment increase α2δ-1 expression levels in the DRG, which can physically interact with NMDARs (Chen et al., 2018; 2019). We used immunoblotting to determine how RTX treatment affects the protein levels of α2δ-1 and GluN1, an obligatory subunit of NMDARs, in the DRG and dorsal spinal cord. All tissues were obtained 5 weeks after treatment with RTX or vehicle. When normalized to GAPDH, the protein level of α2δ-1 was slightly increased in the DRG in the RTX group compared with the vehicle group (p = 0.027, t(16) = 2.44; Fig. 2A,B). Because RTX treatment kills TRPV1-expressing neurons in the DRG (Pan et al., 2003), using GAPDH for normalization likely underestimates RTX-treatment-induced changes in neuronal proteins in the DRG. We thus also normalized the α2δ-1 protein band to that of NeuN, a neuronal protein marker. Indeed, the α2δ-1 protein level in the DRG, when normalized to NeuN, showed a much larger increase in the RTX group than in the vehicle group (p < 0.001, t(16) = 5.12; Fig. 2C).
Figure 2.

RTX treatment potentiates the expression level of α2δ-1 in the DRG. A–C, Original gel images (A) and quantification of α2δ-1 and GluN1 protein levels (B, C; n = 9 rats per group) in the DRG after vehicle and RTX treatment. D–F, Original gel images (D) and quantification of α2δ-1 and GluN1 protein levels (E, F; n = 9 rats per group) in the dorsal spinal cord after vehicle and RTX treatment. G, H, Quantification of the mRNA level of α2δ-1 and GluN1 in the rat DRG (G) and dorsal spinal cord (H) after vehicle and RTX treatment (n = 9 rats per group). Data are shown as means ± SEM. GAPDH, synaptophysin (SYN), or NeuN was used as an endogenous loading control, and the mean value in the vehicle group was set to 1. *p < 0.05, ***p < 0.001 compared with the vehicle group. Two-tailed Student's t test.
The protein level of GluN1 in the DRG, when normalized to GAPDH, was significantly reduced in the RTX group compared with the vehicle group (p < 0.001, t(16) = 5.52; Fig. 2A,B). However, when normalized to NeuN, the protein level of GluN1 in the DRG showed no significant changes between the RTX and vehicle groups (p = 0.31, t(16) = 1.06; Fig. 2C).
In the dorsal spinal cord tissues, the protein levels of α2δ-1 and GluN1, when normalized to GAPDH or synaptophysin, a synaptic protein marker, did not differ significantly between the RTX and vehicle groups (n = 9 rats per group; Fig. 2D–F).
Furthermore, a quantitative PCR assay showed that the mRNA level of α2δ-1, but not GluN1, in the DRG was significantly greater in the RTX group than in the vehicle group (p < 0.001, t(16) = 5.24; Fig. 2G). However, the mRNA levels of α2δ-1 and GluN1 in the dorsal spinal cord did not differ significantly between the two groups (n = 9 rats per group; Fig. 2H). These data indicate that RTX treatment upregulates α2δ-1 expression in the DRG.
RTX treatment augments α2δ-1 expression in DRG neurons
Because RTX treatment increased the expression level of α2δ-1 in the DRG, we attempted to determine how RTX treatment affects the α2δ-1 expression in DRG neurons. However, all the α2δ-1 antibodies we tested showed nonspecific immunolabeling in the DRG from Cacna2d1-KO mice. We thus used RNAscope, a new in situ hybridization method, to determine expression patterns of α2δ-1 mRNA at the single-cell level in the DRG. In DRG tissues from vehicle-treated rats, the punctate signal of α2δ-1 mRNA was present in different sizes of DRG neurons labeled with NeuN (Fig. 3A–C). Because RTX treatment depletes TRPV1-expressing DRG neurons (Pan et al., 2003; Chen and Pan, 2006), the number of small- and medium-diameter neurons was reduced in RTX-treated rats compared with that in vehicle-treated rats. The proportion of NeuN-labeled DRG neurons expressing the α2δ-1 mRNA signal was much higher in RTX-treated rats (31.52 ± 2.92%) than in vehicle-treated rats (10.11 ± 1.64%; p < 0.001, t(18) = 6.396; Fig. 3B). The abundance of the α2δ-1 mRNA signal in small-, medium-, and large-diameter DRG neurons was also significantly greater in RTX-treated than in vehicle-treated rats (Fig. 3C).
Figure 3.
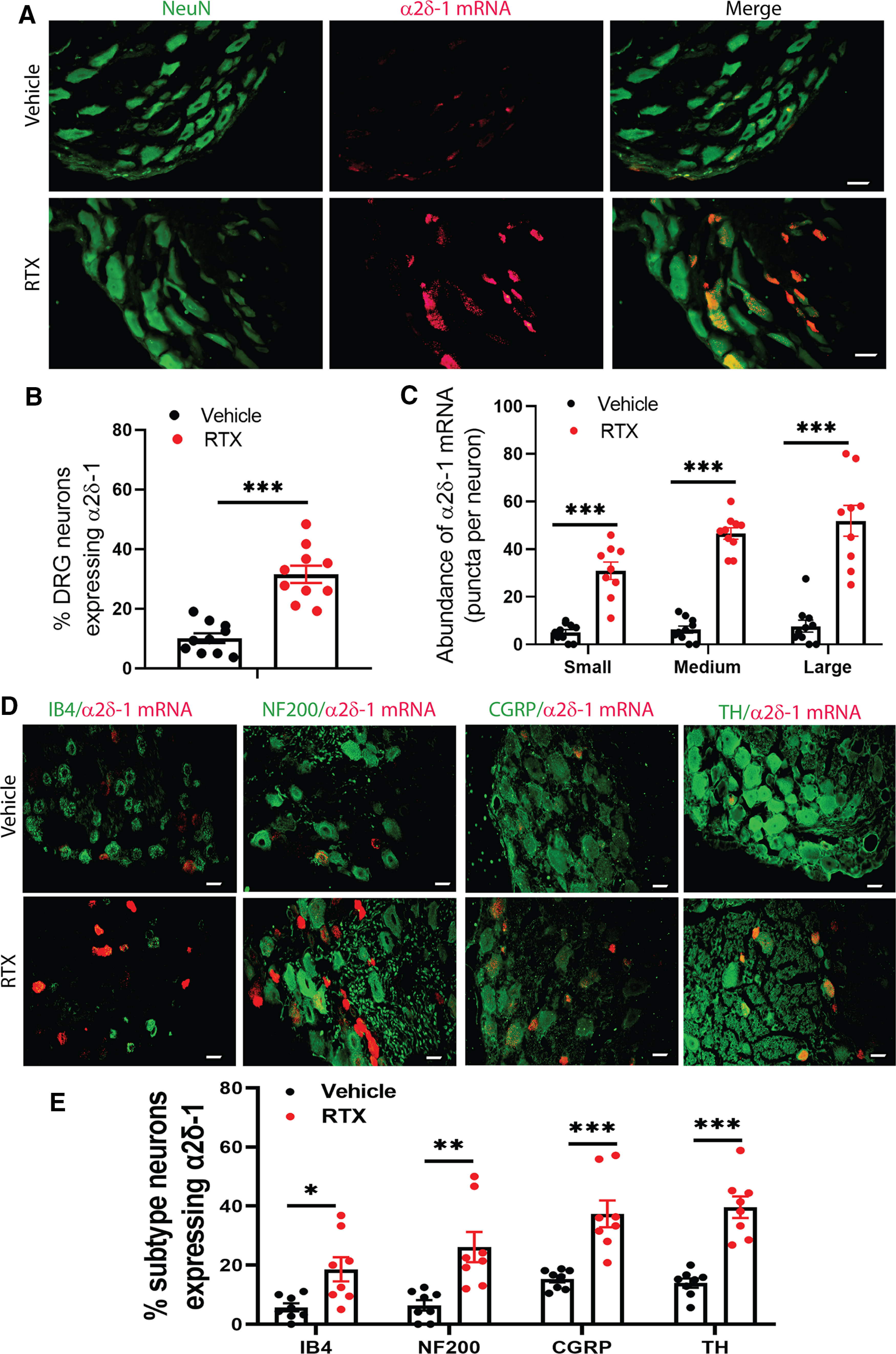
RTX treatment increases the α2δ-1 mRNA level in DRG neurons. A, Representative RNAscope images show colabeling of NeuN (green) and α2δ-1 mRNA (red) in DRG tissue sections from a vehicle-treated and an RTX-treated rat. B, C, Quantification of the proportion of total NeuN-labeled DRG neurons expressing α2δ-1 mRNA (B) and the number of α2δ-1 mRNA punctate signal present in small-, medium-, and large-diameter DRG neurons (C) in vehicle-treated and RTX-treated rats; n = 10 images per group. D, E, Representative overlay images (D) and quantification (E) show colabeling of α2δ-1 mRNA signal with IB4, NF200, CGRP, and tyrosine hydroxylase (TH) in DRG tissue sections from vehicle-treated and RTX-treated rats; n = 8 images per group. Scale bar, 20 µm. Data are shown as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the vehicle group. B, C, E, Two-tailed Student's t test (B) and one-way ANOVA followed by Tukey's post hoc test (C, E).
To further characterize the effect of RTX treatment on α2δ-1 upregulation in DRG neuronal subtypes, we conducted colabeling in DRG tissue sections with the α2δ-1 mRNA signal and one of the four molecular markers: IB4, NF200, CGRP, and tyrosine hydroxylase (Usoskin et al., 2015). Treatment with RTX significantly increased the proportion of α2δ-1-expressing DRG neurons labeled with IB4, NF200, CGRP, and tyrosine hydroxylase (Fig. 3D,E). Consistent with the above quantitative PCR and immunoblotting data, this complementary evidence indicates that RTX treatment causes α2δ-1 upregulation in primary sensory neurons.
RTX treatment increases the association of α2δ-1 with NMDARs and synaptic trafficking in the spinal cord
We next performed coimmunoprecipitation experiments to determine whether RTX treatment alters the physical interaction in the dorsal spinal cord between GluN1 and α2δ-1, a newly identified auxiliary protein for NMDARs in the spinal cord and brain (Chen et al., 2018; Zhou et al., 2018). In the samples immunoprecipitated with an anti-GluN1 antibody, immunoblotting with an α2δ-1 antibody indicated that the α2δ-1–GluN1 protein complex level was much greater in the RTX group than in the vehicle group (p < 0.001, t(16) = 4.97, n = 9 rats per group; Fig. 4A,B).
Figure 4.

RTX treatment potentiates the α2δ-1–NMDAR interaction and synaptic trafficking in the spinal cord. A, B, Original gel images (A) and quantification (B) show the interaction between α2δ-1 and GluN1 in the dorsal spinal cords from rats treated with vehicle (Veh) or RTX. Proteins were immunoprecipitated (IP) initially with a rabbit anti-GluN1 antibody or IgG. Immunoblotting was then performed using mouse anti–α2δ-1 or rabbit anti-GluN1 antibodies. IgG and input were used as negative and positive controls, respectively. The amount of α2δ-1 proteins was normalized to that of GluN1 on the same blot. C, D, Original gel images (C) and quantification (D) of the protein levels of GluN1 and α2δ-1 in synaptosomes isolated from the dorsal spinal cord from rats treated with vehicle or RTX. E, Immunoblotting images of α2δ-1 proteins in the dorsal spinal cord tissues from two WT mice and two α2δ-1 KO mice using the mouse anti-α2δ-1 antibody. Data are shown as means ± SEM (n = 9 rats per group). The synaptic proteins PSD95 and GAPDH were used as a loading control. **p < 0.01, ***p < 0.001 compared with the vehicle group. Two-tailed Student's t test.
To determine the effect of RTX treatment on the protein amounts of α2δ-1 and GluN1 in spinal cord synapses, we performed immunoblotting using isolated synaptosomes from the dorsal spinal cords. The protein levels of both GluN1 (p < 0.001, t(16) = 4.57, n = 9 rats per group) and α2δ-1 (p = 0.0012, t(16) = 3.92, n = 9 rats per group) in the spinal synaptosomes were significantly greater in RTX-treated rats than in vehicle-treated rats (Fig. 4C,D). The specificity of the mouse anti–α2δ-1 antibody was validated using spinal cord tissues from α2δ-1–KO mice (Fig. 4E). These findings suggest that RTX treatment potentiates the α2δ-1–NMDAR interaction and synaptic trafficking of α2δ-1–bound NMDARs in the spinal cord.
RTX treatment increases glutamatergic input to spinal dorsal horn neurons via α2δ-1 and endogenous activation of presynaptic NMDARs
Then, to determine whether RTX treatment alters glutamatergic input to spinal dorsal horn neurons, we recorded mEPSCs, which measure spontaneous quantal glutamate release from presynaptic terminals (Li et al., 2002; Chen et al., 2014a). The baseline frequency of mEPSCs in spinal lamina II neurons was much higher in RTX-treated rats than in vehicle-treated rats (p < 0.001, F(8,105) = 9.18, n = 13 neurons per group; Fig. 5A–C), whereas the amplitude of mEPSCs did not differ significantly between the two groups. Bath application of the specific NMDAR antagonist AP-5 (50 μm) rapidly decreased the frequency of mEPSCs in RTX-treated rats but had no such effect in vehicle-treated rats (Fig. 5A–C). These data suggest that RTX treatment augments glutamatergic input to spinal dorsal horn neurons via tonic activation of presynaptic NMDARs.
Figure 5.

RTX treatment increases glutamatergic input to spinal dorsal horn neurons via α2δ-1 and tonic activation of presynaptic NMDARs. A, B, Representative recording traces (A) and cumulative probability plots (B) show the effect of bath application of 50 μm AP-5 on mEPSCs of lamina II neurons from vehicle-treated rats, RTX-treated rats, and RTX-treated rats from which spinal cord slices were incubated with 100 μm gabapentin. C, Summary data show the effect of AP-5 on the frequency and amplitude of mEPSCs of lamina II neurons from vehicle-treated rats (n = 13 neurons), RTX-treated rats (n = 13 neurons), and RTX-treated rats from which spinal cord slices were incubated with GBP (n = 12 neurons). D, E, Representative current traces (D) and mean data (E) show currents elicited by puff application of 100 μm NMDA (n = 12 neurons in the vehicle group; n = 11 neurons in the RTX group) or 20 μm AMPA (n = 15 neurons per group) onto lamina II neurons from vehicle-treated rats and RTX-treated rats. Data are means ± SEM. ***p < 0.001 compared with baseline in RTX-treated rats. #p < 0.05 compared with baseline before AP-5 application. One-way ANOVA followed by Tukey's post hoc test.
To determine whether α2δ-1 plays a role in the increased presynaptic NMDAR activity caused by RTX treatment, we determined the effect of gabapentin (GBP), an α2δ-1 inhibitory ligand (Marais et al., 2001; Fuller-Bicer et al., 2009; Chen et al., 2018), on mEPSCs. Treatment with 100 μm gabapentin for 30 min significantly reduced the baseline frequency of mEPSCs of lamina II neurons in RTX-treated rats (p < 0.001, F(8,105) = 9.18, n = 12 neurons; Fig. 5A–C). In these spinal cord slices pretreated with gabapentin, bath application of AP-5 had no further effect on the frequency of mEPSCs (Fig. 5A–C).
We also determined whether RTX treatment alters postsynaptic NMDAR and AMPAR activity in the spinal dorsal horn. We recorded NMDAR and AMPAR activity by puff application of 100 μm NMDA and 20 μm AMPA, respectively, directly onto lamina II neurons (Chen et al., 2014a; Huang et al., 2019). The current amplitude elicited by puff NMDA (n = 12 neurons in the control group; n = 11 neurons in the RTX group) or AMPA (n = 15 neurons per group) in lamina II neurons did not differ significantly between RTX-treated and vehicle-treated rats (Fig. 5D,E).
To confirm the role of α2δ-1 in RTX-induced tonic activation of presynaptic NMDARs in the spinal cord, we compared RTX-treatment-induced changes in mEPSCs of spinal lamina II neurons in WT and Cacna2d1-KO mice. The baseline frequency of mEPSCs in spinal lamina II neurons was significantly higher in RTX-treated WT mice than in vehicle-treated WT mice (p < 0.001, F(8,111) = 7.28, n = 13 neurons per group), whereas the amplitude of mEPSCs did not differ significantly between the two groups (Fig. 6A–C). Bath application of AP-5 rapidly decreased the frequency of mEPSCs in RTX-treated WT mice but not in vehicle-treated WT mice (Fig. 6A–C). In contrast, in Cacna2d1-KO mice, RTX treatment had no significant effect on the baseline frequency or amplitude of mEPSCs in spinal lamina II neurons (n = 14 neurons). Also, bath application of AP-5 did not significantly alter the frequency or amplitude of mEPSCs in these neurons in RTX-treated Cacna2d1-KO mice (Fig. 6A–C).
Figure 6.

α2δ-1 is essential for RTX treatment-induced tonic activation of presynaptic NMDARs in the spinal dorsal horn. A, B, Representative recording traces (A) and cumulative probability plots (B) show the effect of bath application of 50 μm AP-5 on mEPSCs of lamina II neurons from vehicle-treated WT mice, RTX-treated WT mice, and RTX-treated Cacna2d1-KO mice. C, Summary data show the effect of AP-5 on the frequency and amplitude of mEPSCs of lamina II neurons from vehicle-treated WT mice (n = 13 neurons), RTX-treated WT mice (n = 13 neurons), and RTX-treated Cacna2d1-KO mice (n = 14 neurons). D, E, Representative current traces (D) and mean data (E) show currents elicited by puff application of 100 μm NMDA (n = 15 neurons per group) or 20 μm AMPA (n = 12 neurons from vehicle-treated WT mice; n = 16 neurons from RTX-treated WT mice; n = 16 neurons from RTX-treated Cacna2d1-KO mice) onto lamina II neurons from vehicle-treated WT mice, RTX-treated WT mice, and RTX-treated Cacna2d1-KO mice. Data are means ± SEM. ***p < 0.001 compared with baseline in RTX-treated WT mice. #p < 0.05 compared with baseline before AP-5 application. One-way ANOVA followed by Tukey's post hoc test.
In addition, the amplitude of currents elicited by puff application of 20 μm AMPA to lamina II neurons was similar between vehicle-treated WT (n = 12 neurons), RTX-treated WT (n = 16 neurons), and RTX-treated Cacna2d1-KO mice (n = 16 neurons; Fig. 6D,E). Also, puff application of 100 μm NMDA elicited similar increases in the current amplitudes of lamina II neurons in these three groups of mice (n = 16 neurons per group; Fig. 6D,E). Together, these results strongly suggest that α2δ-1 is required for RTX-induced tonic activation of presynaptic NMDARs in the spinal cord.
RTX treatment increases glutamatergic input to spinal dorsal horn neurons via α2δ-1 and NMDARs present at primary afferent terminals
To specifically determine the role of α2δ-1 in NMDAR-mediated synaptic glutamate release from primary afferent terminals, we examined the effect of gabapentin on the amplitude of EPSCs monosynaptically evoked from dorsal root stimulation. The baseline amplitude of evoked EPSCs was significantly higher in RTX-treated rats than in vehicle-treated rats (p = 0.04, F(8,102) = 5.21, n = 13 neurons per group; Fig. 7A,C). Bath application of 50 μm AP-5 for 6 min reversed the increased amplitude of evoked EPSCs of lamina II neurons in RTX-treated rats but had no inhibitory effect on evoked EPSCs in vehicle-treated rats (Fig. 7A,C). Furthermore, treatment with gabapentin (100 μm for 30 min) significantly reduced the baseline amplitude of evoked EPSCs of lamina II neurons from RTX-treated rats (n = 11 neurons; Fig. 7A,C). After gabapentin treatment, subsequent application of AP-5 had no effect on evoked EPSCs in these neurons (Fig. 7A,C).
Figure 7.

RTX treatment augments glutamatergic input to spinal dorsal horn neurons via α2δ-1 and NMDARs present at primary afferent terminals. A, B, Original recording traces show the effect of 50 μm AP-5 on evoked monosynaptic EPSCs (A) and PPR (B) in lamina II neurons from vehicle-treated rats, RTX-treated rats, and RTX-treated rats from which spinal cord slices were incubated with 100 μm GBP. C, Summary data show the effect of AP-5 on the amplitude of evoked EPSCs and PPR in lamina II neurons from vehicle-treated rats (n = 13 neurons), RTX-treated rats (n = 13 neurons), and RTX-treated rats from which spinal cord slices were incubated with gabapentin (n = 12 neurons). D, E, Representative recording traces show the effect of 50 μm AP-5 on evoked monosynaptic EPSCs (D) and paired-pulse evoked EPSCs (E) in lamina II neurons from vehicle-treated WT mice, RTX-treated WT mice, and RTX-treated Cacna2d1-KO mice. F, Mean data show the effect of AP-5 on the amplitude of evoked EPSCs and paired-pulse ratio in lamina II neurons from vehicle-treated WT mice (n = 13 neurons), RTX-treated WT mice (n = 14 neurons), and RTX-treated Cacna2d1-KO mice (n = 13 neurons). Data are means ± SEM. *p < 0.05, **p < 0.01 compared with baseline in RTX-treated rats or WT mice. ##p < 0.01, ###p < 0.001 compared with baseline before AP-5 application. One-way ANOVA followed by Tukey's post hoc test.
We also determined the effect of gabapentin on the paired-pulse ratio (PPR), a commonly used measure of the probability of neurotransmitter release from the presynaptic terminal based on responses to a pair of stimuli (Zhou et al., 2010; Xie et al., 2016). The baseline PPR of monosynaptically evoked EPSCs in lamina II neurons was significantly smaller in RTX-treated rats than in vehicle-treated rats (p = 0.007, F(5,70) = 4.74, n = 13 neurons per group; Fig. 7B,C). Bath application of 50 μm AP-5 inhibited the first evoked EPSCs more than the second evoked EPSCs, resulting in an increase in the PPR in RTX-treated rats. However, AP-5 had no significant effect on the PPR in vehicle-treated rats (Fig. 7B,C). Treatment with gabapentin (100 μm for 30 min) significantly increased the baseline PPR of evoked EPSCs in lamina II neurons from RTX treated rats (n = 12 neurons; Fig. 7B,C). After gabapentin treatment, subsequent AP-5 application had no significant effect on the PPR in these neurons.
To further substantiate the role of α2δ-1 in the RTX-induced potentiation of presynaptic NMDARs at primary afferent terminals, we recorded monosynaptically evoked EPSCs in RTX-treated WT and Cacna2d1-KO mice. The baseline amplitude of EPSCs in spinal lamina II neurons was significantly higher in RTX-treated WT mice (n = 13 neurons) than in vehicle-treated WT mice (p = 0.003, F(8,111) = 9.26, n = 14 neurons; Fig. 7D,F). Furthermore, the baseline PPR of evoked EPSCs in lamina II neurons was significantly lower in RTX-treated WT mice than in vehicle-treated WT mice (p = 0.002, F(5,74) = 7.35; Fig. 7E,F). Bath application of AP-5 readily decreased the amplitude of evoked EPSCs and increased the PPR in RTX-treated WT mice but had no such effects in vehicle-treated WT mice (Fig. 7D–F). However, treatment with RTX failed to significantly alter the baseline amplitude or PPR of evoked EPSCs in lamina II neurons in Cacna2d1-KO mice (n = 13 neurons; Fig. 7D–F). Also, AP-5 had no effect on the amplitude or the PPR of evoked EPSCs in RTX-treated Cacna2d1-KO mice (Fig. 7D–F). These data provide strong evidence that α2δ-1 functions as an auxiliary protein of NMDARs and is essential for RTX-induced tonic activation of NMDARs at primary afferent terminals in the spinal cord.
Presynaptic α2δ-1–bound NMDARs mediate RTX-induced potentiation of glutamatergic input to spinal dorsal horn neurons
To determine whether bound NMDARs are involved in RTX-treatment-induced potentiation of presynaptic NMDARs in the spinal dorsal horn, we examined the effect of α2δ-1Tat peptide, which specifically disrupts the α2δ-1–NMDAR interaction (Chen et al., 2018) on mEPSCs of lamina II neurons. Treatment with α2δ-1Tat peptide (1 μm; n = 11 neurons) for 30 min significantly reduced the baseline frequency of mEPSCs of lamina II neurons in RTX-treated rats (p < 0.001, F(5,63) = 10.23; Fig. 8A–C). After treatment with α2δ-1Tat peptide, application of AP-5 had no effect on the frequency of mEPSCs (Fig. 8A–C). In contrast, treatment with 1 μm Tat-fused scrambled control peptide did not reduce the baseline mEPSC frequency of lamina II neurons from RTX-treated rats, and AP-5 still significantly reduced the mEPSC frequency in these neurons (n = 12 neurons; Fig. 8A–C). These data suggest that α2δ-1–bound NMDARs play a key role in RTX-induced tonic activation of presynaptic NMDARs in the spinal cord.
Figure 8.

Presynaptic α2δ-1–bound NMDARs mediate RTX-induced potentiation of glutamatergic input to spinal dorsal horn neurons. A, B, Original recording traces (A) and cumulative probability plots (B) show the effect of bath application of 50 μm AP-5 on mEPSCs of lamina II neurons from RTX-treated rats in which spinal cord slices were incubated with 1 μm control peptide or 1 μm α2δ-1Tat peptide. C, Mean data show the effect of AP-5 on the frequency and amplitude of mEPSCs of lamina II neurons from RTX-treated rats in which spinal cord slices were incubated with control peptide (n = 12 neurons) or α2δ-1Tat peptide (n = 11 neurons). D, E, Original recording traces show the effect of 50 μm AP-5 on evoked monosynaptic EPSCs (D) and paired-pulse evoked EPSCs (E) in lamina II neurons from RTX-treated rats from which spinal cord slices were incubated with 1 μm control peptide or 1 μm α2δ-1Tat peptide. F, Summary data show the effect of AP-5 on the amplitude of evoked EPSCs and the paired-pulse ratio in lamina II neurons from RTX-treated rats in which spinal cord slices were incubated with control peptide or α2δ-1Tat peptide (n = 12 neurons per group). Data are means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with baseline in the control peptide group. #p < 0.05, ##p < 0.01 compared with baseline before AP-5 application. One-way ANOVA followed by Tukey's post hoc test.
RTX treatment potentiates glutamatergic input to spinal dorsal horn neurons via α2δ-1–bound NMDARs at primary afferent terminals
We next determined the role of α2δ-1–bound NMDARs in synaptic glutamate release from primary afferent terminals to lamina II neurons. Treatment with α2δ-1Tat peptide (1 μm for 30 min) significantly reduced the baseline amplitude of monosynaptic EPSCs evoked from the dorsal root in lamina II neurons from RTX-treated rats (p = 0.008, F(5,66) = 6.14, n = 12 neurons; Fig. 8D,F). Subsequent application of AP-5 had no further effect on the amplitude of evoked EPSCs in lamina II neurons treated with α2δ-1Tat peptide. In contrast, treatment with the control peptide had no effect on the baseline amplitude of evoked EPSCs in lamina II neurons from RTX-treated rats (n = 12 neurons). Also, after treatment with the control peptide, bath application of AP-5 still significantly reduced the amplitude of evoked EPSCs in lamina II neurons from RTX-treated rats (Fig. 8D,F).
Furthermore, treatment with α2δ-1Tat peptide significantly reduced the baseline PPR of lamina II neurons from RTX-treated rats (p = 0.039, F(3,42) = 4.71, n = 12 neurons; Fig. 8E,F). After α2δ-1Tat peptide incubation, bath application of AP-5 had no effect on the PPR in these neurons. However, treatment with the control peptide failed to alter the baseline PPR in lamina II neurons from RTX-treated rats, and AP-5 application significantly increased the PPR in these neurons (Fig. 8E,F). These findings suggest that α2δ-1–bound NMDARs at primary afferent terminals mediate the RTX-treatment-induced increase in glutamatergic input to spinal dorsal horn neurons.
α2δ-1–bound NMDARs at the spinal cord level contribute to RTX-induced tactile allodynia
To determine whether NMDARs play a role in RTX-treatment-induced tactile allodynia, we tested the effect of memantine, an orally active NMDAR antagonist used clinically, on tactile allodynia in rats 5–6 weeks after RTX treatment. Intraperitoneal injection of memantine (10 mg/kg; Chen et al., 2014a; Xie et al., 2016) caused a large increase in the tactile withdrawal threshold within 30 min in all RTX-treated rats tested (p < 0.001, F(6,84) = 68.61, n = 8 rats per group; Fig. 9A).
Figure 9.
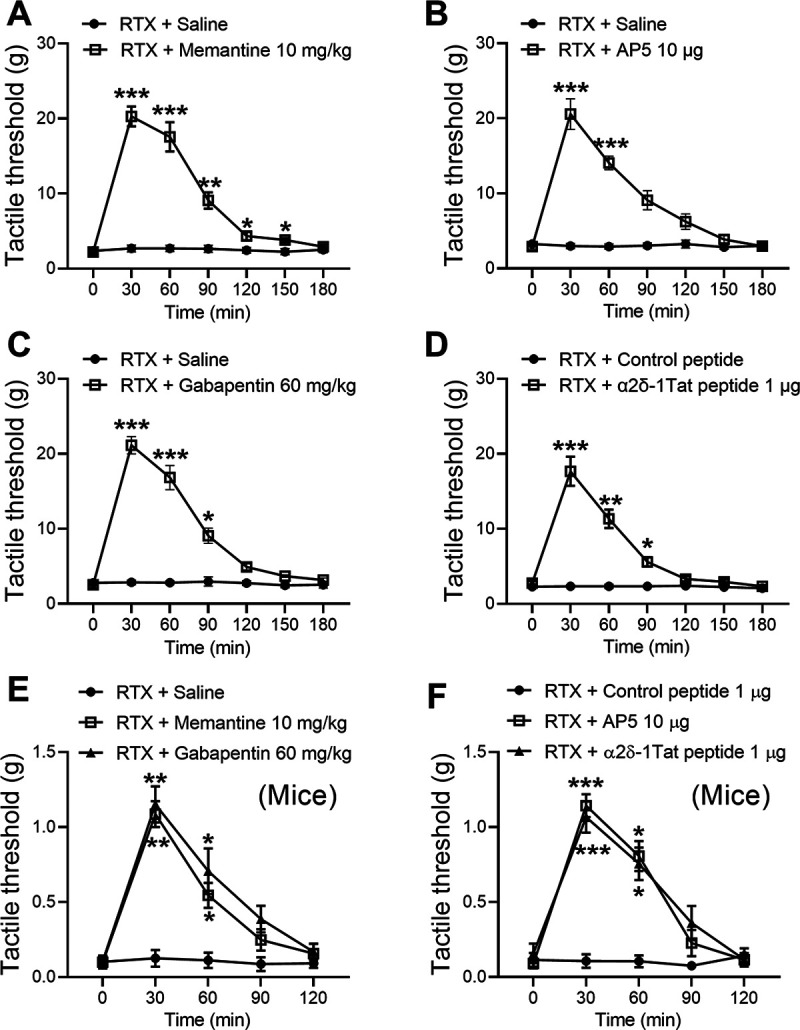
Blocking NMDARs or targeting α2δ-1–NMDARs reverses RTX-induced tactile allodynia. A, B, Time course of the effects of intraperitoneal injection of 10 mg/kg memantine (A; n = 8 rats) or intrathecal injection of 10 μg of AP-5 (B; n = 9 rats) on the tactile withdrawal threshold tested with von Frey filaments in RTX-treated rats (n = 8 rats per group). C, D, Time course of the effects of intraperitoneal injection of 10 μg of gabapentin (C; n = 8 rats) or intrathecal injection 1 μg of α2δ-1Tat peptide or 1 μg of control peptide (D; n = 9 rats) on the tactile withdrawal threshold in RTX-treated rats. E, F, Time course of the effects of intraperitoneal injection of memantine or gabapentin (E; n = 6 mice per group) and intrathecal injection of AP-5 or α2δ-1Tat peptide (F; n = 6 mice per group) on the tactile withdrawal threshold in RTX-treated mice. Data are expressed as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the baseline immediately before drug injection (time 0 min). Repeated-measures ANOVA followed by Dunnett's post hoc test.
Also, we determined whether NMDARs at the spinal cord level mediate RTX-induced tactile allodynia in additional rats. A single intrathecal injection of AP-5 (10 µg; Chen et al., 2014a; Xie et al., 2016) also significantly increased the tactile withdrawal threshold that had been reduced by RTX treatment (p < 0.001, F(6,96) = 35.54, n = 9 rats per group; Fig. 9B). Intraperitoneal or intrathecal injection of saline had no effect on the tactile withdrawal threshold in RTX-treated rats.
We next used gabapentin to determine whether α2δ-1 mediates RTX-treatment-induced tactile allodynia. Intraperitoneal injection of gabapentin (60 mg/kg; Pan et al., 1999; Chen and Pan, 2005) significantly increased the tactile withdrawal threshold in rats 5 weeks after RTX treatment (p < 0.001, F(6,84) = 70.27; n = 8 rats per group. Fig. 9C). We then determined the role of the α2δ-1–NMDAR interaction at the spinal cord level in RTX-induced tactile allodynia by testing the effect of intrathecal injection of α2δ-1Tat peptide (1 µg; Chen et al., 2018) in RTX-treated rats. Treatment with α2δ-1Tat peptide increased the tactile withdrawal threshold that had been reduced by RTX treatment within 30 min, and this effect lasted for ∼90 min (p < 0.001, F(6,96) = 58.99, n = 9 rats per group). However, intrathecal injection of the Tat-fused scrambled control peptide (1 µg) had no effect on the baseline tactile withdrawal threshold in RTX-treated rats (Fig. 9D).
Furthermore, we tested the effects of intraperitoneal administration of memantine or gabapentin as well as intrathecal injection of AP-5 or α2δ-1Tat peptide on tactile allodynia in WT mice 3–4 weeks after RTX treatment (25 µg/kg, see below). Intraperitoneal injection of memantine (10 mg/kg) or gabapentin (60 mg/kg) caused a large increase in the tactile withdrawal threshold in RTX-treated mice (n = 6 mice in each group, Fig. 9E). Similarly, intrathecal injection of AP-5 (10 µg) or α2δ-1Tat peptide (1 µg), but not control peptide (1 µg), readily reversed tactile allodynia (n = 6 mice in each group, Fig. 9F). Together, these results suggest that RTX-induced tactile allodynia is sustained by α2δ-1–bound NMDARs at the spinal cord level.
α2δ-1 and NMDARs in primary sensory neurons are involved in RTX-induced tactile allodynia
In addition, we used Cacna2d1-KO mice to validate the role of α2δ-1 in the development of tactile allodynia after RTX treatment. The baseline thermal and mechanical withdrawal thresholds did not differ significantly between WT and Cacna2d1-KO mice before RTX treatment. Intraperitoneal injection of 25 µg/kg of RTX in WT mice markedly increased the paw withdrawal latency in response to a noxious heat stimulus but had no effect on the withdrawal threshold tested with a noxious pressure stimulus (n = 8 mice per group; Fig. 10A,B). Unlike in rats (Fig. 1), RTX treatment caused a rapid reduction in the tactile withdrawal threshold in response to von Frey filaments in WT mice; tactile allodynia appeared 3 d after RTX injection and lasted for at least another 4 weeks (Fig. 10C). In Cacna2d1-KO mice, however, the reduction in the tactile withdrawal threshold induced by RTX was significantly attenuated (n = 8 mice per group; Fig. 10C). These data provide further evidence that α2δ-1 is involved in the development of tactile allodynia caused by RTX.
Figure 10.

NMDARs in primary sensory neurons and α2δ-1 contribute to RTX-induced tactile allodynia. A–C, Time course of changes in the paw-withdrawal threshold tested with a heat stimulus (A), a pressure stimulus (B), and von Frey filaments (C) in WT, Grin1-cKO, and Cacna2d1-KO mice treated with RTX (injected at time 0). RTX injections are indicated by arrows. Data are expressed as means ± SEM (n = 8 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001 compared with the baseline (BL) before drug injection, #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the corresponding value at the same time point in RTX-treated WT mice. Two-way ANOVA followed by Tukey's post hoc test.
Because our electrophysiological data suggest that NMDARs expressed at primary afferent central terminals mediate the increased glutamatergic input caused by RTX treatment, we used Grin1-cKO mice to determine the role of NMDARs expressed in primary sensory neurons in the development of tactile allodynia caused by RTX. We crossed AdvillinCre/+ mice with Grin1flox/flox mice to genetically ablate Grin1 in DRG neurons, which has been validated previously (Huang et al., 2020). The baseline thermal and mechanical withdrawal thresholds did not differ significantly between WT and Grin1-cKO mice. Treatment with 25 µg/kg RTX caused a similar increase in the thermal withdrawal latency, but it had no effect on the pressure withdrawal threshold in Grin1-cKO mice or WT mice (n = 8 mice per group; Fig. 10A,B). Finally, the reduction in the tactile withdrawal threshold induced by RTX was significantly attenuated in Grin1-cKO mice compared with RTX-treated WT mice (n = 8 mice per group; Fig. 10C). These findings strongly suggest that NMDARs present in primary sensory neurons contribute to the development of tactile allodynia induced by RTX.
Discussion
Our study provides the first direct evidence that nociceptive input from primary afferent nerves to spinal dorsal horn neurons is augmented via endogenously activated NMDARs in an animal model of PHN. Because of the loss of synaptic contacts typically made by nociceptive C-fiber afferents onto lamina I/II neurons, the central terminals of myelinated afferent nerves that normally terminate in deeper laminae can sprout into superficial laminae after the depletion of TRPV1-expressing DRG neurons by RTX (Pan et al., 2003). We demonstrated that RTX treatment markedly increased the glutamatergic input to lamina II neurons, which was largely reversed by the NMDAR antagonist AP-5. Also, systemically administered memantine or the use of AP-5 to block NMDARs at the spinal cord level reversed RTX-treatment-induced tactile allodynia. Thus, increased NMDAR activity at the spinal cord level likely plays a prominent role in transforming innocuous touch stimuli to nociceptive sensory input in the spinal dorsal horn in PHN.
Our study reveals that presynaptic NMDARs mediate spinal nociceptive transmission and cutaneous hypersensitivity in RTX-induced neuropathy. Although conventionally postsynaptic, NMDARs are also expressed in primary sensory neurons and central terminals in the spinal dorsal horn (Liu et al., 1994). These presynaptic NMDARs in the spinal dorsal horn are normally nonfunctional, and genetically ablating NMDARs in DRG neurons has no effect on normal nociception (McRoberts et al., 2011; Huang et al., 2020). Also, blocking NMDARs at the spinal cord level does not affect nociception under normal conditions (Zahn and Brennan, 1998; Chen et al., 2014a; Xie et al., 2016). Traumatic nerve injury increases the activity of both presynaptic and postsynaptic NMDARs in the spinal dorsal horn (Chen et al., 2014b; Li et al., 2016; Chen et al., 2018), whereas chemotherapy-induced neuropathic pain primarily augments the activity of presynaptic NMDARs (Xie et al., 2016; 2017; Chen et al., 2019). Remarkably, despite the denervation of unmyelinated C-fiber afferent terminals in the laminae II by RTX treatment, the frequency of mEPSCs did not decrease but instead increased in RTX-treated rats and mice. This finding suggests that sprouting myelinated afferent nerves form new synapses with lamina II neurons and/or display increased presynaptic NMDAR activity. Our complementary recording data documenting the effects of AP-5 on mEPSCs, EPSCs evoked from the dorsal root, and PPR strongly suggest that RTX treatment causes tonic activation of presynaptic NMDARs expressed at sprouting primary afferent terminals to potentiate nociceptive input to the spinal dorsal horn. Interestingly, RTX treatment did not alter postsynaptic NMDAR activity in spinal dorsal horn neurons, which is consistent with the finding that RTX treatment does not affect TRPV1-expressing neurons in the spinal dorsal horn (Zhou et al., 2009).
Another major finding of our study is that α2δ-1-dependent potentiation of NMDAR activity mediates augmented glutamatergic input and allodynia in RTX-induced neuropathy. We found that depletion of TRPV1-expressing neurons by RTX treatment increased α2δ-1 expression in different sizes of DRG neurons. Furthermore, RTX treatment not only increased the α2δ-1 expression level in the DRG but also potentiated the physical interaction between α2δ-1 and NMDARs at the spinal cord level. α2δ-1 is a highly glycosylated protein (Tétreault et al., 2016), and the increased expression of α2δ-1 and coupling to NMDARs promote trafficking from DRG neurons to the central terminals in the spinal dorsal horn, where they are activated by endogenous glutamate to augment nociceptive input (Chen et al., 2018). We showed that inhibiting α2δ-1 with gabapentin, disrupting the α2δ-1–NMDAR interaction with α2δ-1Tat peptide, or genetically ablating α2δ-1 all reversed the RTX-induced increase in glutamatergic input to dorsal horn neurons. We found that gabapentin had no effect on evoked EPSCs (Chen et al., 2018; 2019), a measure of synaptic glutamate release triggered mostly by activating voltage-gated Ca2+ channels, in control animals. Also, recent studies reported no or weak interactions between α2δ-1 and Ca2+ channel α1 subunits in neural tissues (Müller et al., 2010; Held et al., 2020). Thus, under neuropathic pain conditions, α2δ-1 regulates nociceptive transmission mainly through interacting with NMDARs, but not voltage-gated Ca2+ channels.
The relative importance of presynaptic NMDARs in neuropathic pain has not been previously examined specifically. We demonstrated that RTX-induced tactile allodynia was similarly attenuated in Cacna2d1-KO and Grin1-cKO mice. These findings consistently suggest an important role of α2δ-1–bound presynaptic NMDARs in the spinal cord in the development of tactile allodynia in this PHN model. The α2δ-1 inhibitory ligands, including gabapentin and pregabalin, are effective for PHN in patients (Rowbotham et al., 1998; Irving et al., 2009). We showed that NMDAR antagonists, gabapentin, and α2δ-1Tat peptide potently reversed RTX-induced tactile allodynia in both rats and WT mice. However, this RTX-induced pain hypersensitivity was only partially attenuated in the Cacna2d1-KO and Grin1-cKO mice used in our study. The limited effect of genetic knockout on RTX-induced allodynia may be because of the long-term loss of Cacna2d1 and Grin1 in mice. In the absence of α2δ-1 and presynaptic NMDARs, development of tactile allodynia may be initiated and/or maintained mainly by ectopic discharges from damaged afferent nerves and DRG neurons via other receptors and ion channels, such as voltage-gated sodium channels (Wasner et al., 2005; Yatziv and Devor, 2019).
In conclusion, our study reveals that in an animal model of PHN, tactile allodynia is mediated by α2δ-1–bound presynaptic NMDARs, which increases excitatory glutamatergic input from myelinated afferent nerves to the spinal dorsal horn. This information is important for understanding the mechanisms of synaptic plasticity underlying PHN and for the design of new treatment strategies. Although NMDAR antagonists can generally reduce neuropathic pain, these drugs indiscriminately inhibit all NMDARs and inevitably produce adverse effects in the CNS. Because gabapentinoids and α2δ-1Tat peptide target α2δ-1–bound NMDARs without affecting α2δ-1-free NMDARs (Chen et al., 2018; Huang et al., 2020), this selectivity represents a major advantage over general NMDAR antagonists for treating PHN. In addition, gabapentinoids inhibit both α2δ-1 and α2δ-2 (Marais et al., 2001; Fuller-Bicer et al., 2009), and the latter is highly expressed in the cerebellum (Cole et al., 2005) and likely mediates dizziness and ataxia associated with gabapentinoid actions. Therefore, selectively disrupting the α2δ-1–NMDAR interaction with α2δ-1Tat peptide could alleviate pain hypersensitivity in PHN patients and minimize the adverse effects associated with gabapentinoids.
Footnotes
This study was supported by Grants NS101880 and GM120844 from the National Institutes of Health and by the N. G. and Helen T. Hawkins Endowment. We thank Sarah Bronson in the Department of Scientific Publications, MD Anderson Cancer Center for proofreading the final version of the paper.
The authors declare no competing financial interests.
References
- Baron R, Saguer M (1993) Postherpetic neuralgia. Are C-nociceptors involved in signalling and maintenance of tactile allodynia? Brain 116: 1477–1496. 10.1093/brain/116.6.1477 [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chen J, Li L, Chen SR, Chen H, Xie JD, Sirrieh RE, MacLean DM, Zhang Y, Zhou MH, Jayaraman V, Pan HL (2018) The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep 22:2307–2321. 10.1016/j.celrep.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Pan HL (2005) Effect of systemic and intrathecal gabapentin on allodynia in a new rat model of postherpetic neuralgia. Brain Res 1042:108–113. 10.1016/j.brainres.2005.02.024 [DOI] [PubMed] [Google Scholar]
- Chen SR, Pan HL (2006) Loss of TRPV1-expressing sensory neurons reduces spinal mu opioid receptors but paradoxically potentiates opioid analgesia. J Neurophysiol 95:3086–3096. 10.1152/jn.01343.2005 [DOI] [PubMed] [Google Scholar]
- Chen SR, Hu YM, Chen H, Pan HL (2014a) Calcineurin inhibitor induces pain hypersensitivity by potentiating pre- and postsynaptic NMDA receptor activity in spinal cords. J Physiol 592:215–227. 10.1113/jphysiol.2013.263814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SR, Zhou HY, Byun HS, Chen H, Pan HL (2014b) Casein kinase II regulates N-methyl-D-aspartate receptor activity in spinal cords and pain hypersensitivity induced by nerve injury. J Pharmacol Exp Ther 350:301–312. 10.1124/jpet.114.215855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Chen SR, Chen H, Zhang J, Pan HL (2019) Increased α2δ-1-NMDA receptor coupling potentiates glutamatergic input to spinal dorsal horn neurons in chemotherapy-induced neuropathic pain. J Neurochem 148:252–274. 10.1111/jnc.14627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, Gu G (2005) Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491:246–269. 10.1002/cne.20693 [DOI] [PubMed] [Google Scholar]
- da Silva S, Hasegawa H, Scott A, Zhou X, Wagner AK, Han BX, Wang F (2011) Proper formation of whisker barrelettes requires periphery-derived Smad4-dependent TGF-beta signaling. Proc Natl Acad Sci U S A 108:3395–3400. 10.1073/pnas.1014411108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel RG, Bingham S, Sutton D, Grant D, Champion JM, Dennis SA, Quinn JP, Bountra C, Mark MA (2004) Allodynia in rats infected with varicella zoster virus—a small animal model for post-herpetic neuralgia. Brain Res Brain Res Rev 46:234–242. 10.1016/j.brainresrev.2004.07.008 [DOI] [PubMed] [Google Scholar]
- Eide PK, Jørum E, Stubhaug A, Bremnes J, Breivik H (1994) Relief of post-herpetic neuralgia with the N-methyl-D-aspartic acid receptor antagonist ketamine: a double-blind, cross-over comparison with morphine and placebo. Pain 58:347–354. 10.1016/0304-3959(94)90129-5 [DOI] [PubMed] [Google Scholar]
- Fields HL, Rowbotham M, Baron R (1998) Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiol Dis 5:209–227. 10.1006/nbdi.1998.0204 [DOI] [PubMed] [Google Scholar]
- Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang SP, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK, Schwartz A (2009) Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. Am J Physiol Heart Circ Physiol 297:H117–H124. 10.1152/ajpheart.00122.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head H, Campbell AW, Kennedy PG (1997) The pathology of Herpes Zoster and its bearing on sensory localisation. Rev Med Virol 7:131–143. [DOI] [PubMed] [Google Scholar]
- Held RG, Liu C, Ma K, Ramsey AM, Tarr TB, De Nola G, Wang SSH, Wang J, van den Maagdenberg A, Schneider T, Sun J, Blanpied TA, Kaeser PS (2020) Synapse and active zone assembly in the absence of presynaptic Ca(2+) channels and Ca(2+) entry. Neuron 107:667–683.e9. 10.1016/j.neuron.2020.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YL, Chiang H, Lue JH, Hsieh ST (2012) P2X3-mediated peripheral sensitization of neuropathic pain in resiniferatoxin-induced neuropathy. Exp Neurol 235:316–325. 10.1016/j.expneurol.2012.02.013 [DOI] [PubMed] [Google Scholar]
- Huang Y, Chen SR, Chen H, Pan HL (2019) Endogenous transient receptor potential ankyrin 1 and vanilloid 1 activity potentiates glutamatergic input to spinal lamina I neurons in inflammatory pain. J Neurochem 149:381–398. 10.1111/jnc.14677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Chen SR, Chen H, Luo Y, Pan HL (2020) Calcineurin inhibition causes α2δ-1-mediated tonic activation of synaptic NMDA receptors and pain hypersensitivity. J Neurosci 40:3707–3719. 10.1523/JNEUROSCI.0282-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving G, Jensen M, Cramer M, Wu J, Chiang YK, Tark M, Wallace M (2009) Efficacy and tolerability of gastric-retentive gabapentin for the treatment of postherpetic neuralgia: results of a double-blind, randomized, placebo-controlled clinical trial. Clin J Pain 25:185–192. 10.1097/AJP.0b013e3181934276 [DOI] [PubMed] [Google Scholar]
- Johnson RW, Rice AS (2014) Clinical practice. Postherpetic neuralgia. N Engl J Med 371:1526–1533. 10.1056/NEJMcp1403062 [DOI] [PubMed] [Google Scholar]
- Kohno T, Kumamoto E, Higashi H, Shimoji K, Yoshimura M (1999) Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J Physiol 518: 803–813. 10.1111/j.1469-7793.1999.0803p.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan YZ, Levey AI, Pan HL (2002) Role of presynaptic muscarinic and GABA(B) receptors in spinal glutamate release and cholinergic analgesia in rats. J Physiol 543:807–818. 10.1113/jphysiol.2002.020644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chen SR, Chen H, Wen L, Hittelman WN, Xie JD, Pan HL (2016) Chloride homeostasis critically regulates synaptic NMDA receptor activity in neuropathic pain. Cell Rep 15:1376–1383. 10.1016/j.celrep.2016.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kim J (2015) Neuronal expression of CB2 cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience 311:253–267. 10.1016/j.neuroscience.2015.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI (1994) Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci U S A 91:8383–8387. 10.1073/pnas.91.18.8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais E, Klugbauer N, Hofmann F (2001) Calcium channel alpha(2)delta subunits-structure and gabapentin binding. Mol Pharmacol 59:1243–1248. 10.1124/mol.59.5.1243 [DOI] [PubMed] [Google Scholar]
- McRoberts JA, Ennes HS, Marvizón JC, Fanselow MS, Mayer EA, Vissel B (2011) Selective knockdown of NMDA receptors in primary afferent neurons decreases pain during phase 2 of the formalin test. Neuroscience 172:474–482. 10.1016/j.neuroscience.2010.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, Schulte U (2010) Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 107:14950–14957. 10.1073/pnas.1005940107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RA, Bingham S, Case PC, Sanger GJ, Lawson SN (2001) Dorsal root ganglion neurons show increased expression of the calcium channel α2δ-1 subunit following partial sciatic nerve injury. Brain Res Mol Brain Res 95:1–8. 10.1016/S0169-328X(01)00188-7 [DOI] [PubMed] [Google Scholar]
- Nurmikko T, Bowsher D (1990) Somatosensory findings in postherpetic neuralgia. J Neurol Neurosurg Psychiatry 53:135–141. 10.1136/jnnp.53.2.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan HL, Eisenach JC, Chen SR (1999) Gabapentin suppresses ectopic nerve discharges and reverses allodynia in neuropathic rats. J Pharmacol Exp Ther 288:1026–1030. [PubMed] [Google Scholar]
- Pan HL, Khan GM, Alloway KD, Chen SR (2003) Resiniferatoxin induces paradoxical changes in thermal and mechanical sensitivities in rats: mechanism of action. J Neurosci 23:2911–2919. 10.1523/JNEUROSCI.23-07-02911.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappagallo M, Oaklander AL, Quatrano-Piacentini AL, Clark MR, Raja SN (2000) Heterogenous patterns of sensory dysfunction in postherpetic neuralgia suggest multiple pathophysiologic mechanisms. Anesthesiology 92:691–698. 10.1097/00000542-200003000-00013 [DOI] [PubMed] [Google Scholar]
- Rowbotham MC, Fields HL (1996) The relationship of pain, allodynia and thermal sensation in post-herpetic neuralgia. Brain 119:347–354. 10.1093/brain/119.2.347 [DOI] [PubMed] [Google Scholar]
- Rowbotham M, Harden N, Stacey B, Bernstein P, Magnus-Miller L (1998) Gabapentin for the treatment of postherpetic neuralgia: a randomized controlled trial. JAMA 280:1837–1842. 10.1001/jama.280.21.1837 [DOI] [PubMed] [Google Scholar]
- Rowbotham MC, Yosipovitch G, Connolly MK, Finlay D, Forde G, Fields HL (1996) Cutaneous innervation density in the allodynic form of postherpetic neuralgia. Neurobiol Dis 3:205–214. 10.1006/nbdi.1996.0021 [DOI] [PubMed] [Google Scholar]
- Sun J, Chen SR, Chen H, Pan HL (2019) μ-Opioid receptors in primary sensory neurons are essential for opioid analgesic effect on acute and inflammatory pain and opioid-induced hyperalgesia. J Physiol 597:1661–1675. 10.1113/JP277428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM (1989) Resiniferatoxin, a phorbol-related diterpene, acts as an ultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience 30:515–520. 10.1016/0306-4522(89)90269-8 [DOI] [PubMed] [Google Scholar]
- Takasaki I, Andoh T, Shiraki K, Kuraishi Y (2000) Allodynia and hyperalgesia induced by herpes simplex virus type-1 infection in mice. Pain 86:95–101. 10.1016/s0304-3959(00)00240-2 [DOI] [PubMed] [Google Scholar]
- Tétreault MP, Bourdin B, Briot J, Segura E, Lesage S, Fiset C, Parent L (2016) Identification of glycosylation sites essential for surface expression of the CaVα2δ1 subunit and modulation of the cardiac CaV1.2 channel activity. J Biol Chem 291:4826–4843. 10.1074/jbc.M115.692178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truini A, Galeotti F, Haanpaa M, Zucchi R, Albanesi A, Biasiotta A, Gatti A, Cruccu G (2008) Pathophysiology of pain in postherpetic neuralgia: a clinical and neurophysiological study. Pain 140:405–410. 10.1016/j.pain.2008.08.018 [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P (2015) Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18:145–153. 10.1038/nn.3881 [DOI] [PubMed] [Google Scholar]
- Wasner G, Kleinert A, Binder A, Schattschneider J, Baron R (2005) Postherpetic neuralgia: topical lidocaine is effective in nociceptor-deprived skin. J Neurol 252:677–686. 10.1007/s00415-005-0717-z [DOI] [PubMed] [Google Scholar]
- Wu CH, Yuan XC, Gao F, Li HP, Cao J, Liu YS, Yu W, Tian B, Meng XF, Shi J, Pan HL, Li M (2016) Netrin-1 contributes to myelinated afferent fiber sprouting and neuropathic pain. Mol Neurobiol 53:5640–5651. 10.1007/s12035-015-9482-x [DOI] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Chen H, Zeng WA, Pan HL (2016) Presynaptic N-Methyl-d-aspartate (NMDA) receptor activity is increased through protein kinase C in paclitaxel-induced neuropathic pain. J Biol Chem 291:19364–19373. 10.1074/jbc.M116.732347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Chen H, Pan HL (2017) Bortezomib induces neuropathic pain through protein kinase C-mediated activation of presynaptic NMDA receptors in the spinal cord. Neuropharmacology 123:477–487. 10.1016/j.neuropharm.2017.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatziv SL, Devor M (2019) Suppression of neuropathic pain by selective silencing of dorsal root ganglion ectopia using nonblocking concentrations of lidocaine. Pain 160:2105–2114. 10.1097/j.pain.0000000000001602 [DOI] [PubMed] [Google Scholar]
- Zahn PK, Brennan TJ (1998) Lack of effect of intrathecally administered N-methyl-D-aspartate receptor antagonists in a rat model for postoperative pain. Anesthesiology 88:143–156. 10.1097/00000542-199801000-00022 [DOI] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL (2009) The glutamatergic nature of TRPV1-expressing neurons in the spinal dorsal horn. J Neurochem 108:305–318. 10.1111/j.1471-4159.2008.05772.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL (2010) Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci 30:4460–4466. 10.1523/JNEUROSCI.5857-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JJ, Li DP, Chen SR, Luo Y, Pan HL (2018) The α2δ-1-NMDA receptor coupling is essential for corticostriatal long-term potentiation and is involved in learning and memory. J Biol Chem 293:19354–19364. 10.1074/jbc.RA118.003977 [DOI] [PMC free article] [PubMed] [Google Scholar]