

Abstract
Pantetheine is ubiquitous in nature in various forms of pantetheine-containing ligands (PCLs), including coenzyme A and phosphopantetheine. Lack of scalable force field libraries for PCLs has hampered the computational studies of biological macromolecules containing PCLs. We describe here the development of the first generation Pantetheine Force Field (PFF) library that is compatible with Amber force fields; parameterized using Gasteiger, AM1-BCC, or RESP charging methods combined with gaff 2 and ff14SB parameter sets. In addition, a “plug-and-play” strategy was employed to enable the systematic charging of computationally expensive molecules sharing common substructural motifs. The validation studies performed on the PFF library showed promising performance where molecular dynamics (MD) simulations results were compared with experimental data of three representative systems. The PFF library represents the first force field library capable of modeling systems containing PCLs in silico and will aid in various applications including protein engineering and drug discovery.
Graphical Abstract
INTRODUCTION
Pantetheine is the cysteamine amide analog of pantothenic acid (vitamin B5), which is ubiquitous in nature in various forms of pantetheine-containing ligands (PCLs). Playing a central role in energy metabolism, coenzyme A (CoA) is arguably one of the most important universal PCLs. It is present in all known organisms with genomes sequenced to date, and roughly 4% of known enzymes use either CoA or CoA thioesters as substrates.1 Coenzyme A is important as it plays two major roles in metabolism:2–5 (l) energy production, by participating in two key steps of the citric acid cycle in the form of acetyl-CoA and succinyl-CoA; and (2) fatty acid synthesis, by acting as an acyl group carrier that assists in transferring fatty acid from cytosol to mitochondria during fatty acid oxidation and from mitochondria to cytosol during fatty acid synthesis. Coenzyme A synthesis from pantothenate requires the following five steps:6,7 (l) Pantothenate phosphorylation to phsphopantothenate by pantothenate kinase; (2) cysteinylation to phospho-N-pantothenoylcysteine (PPC) by phosphopantothenoylcysteine synthetase; (3) PPC decarboxylation to phosphopantetheine (Ppant) by phosphopantothenoylcysteine decarboxylase; (4) Ppant adenylation to dephospho-CoA by phosphopantetheine adenylyltransferase (PPAT); (5) dephospho-CoA phosphorylation to form CoA by dephosphocoenzyme A kinase.
Another important PCL is phosphopantetheine (Ppant), which usually functions as a prosthetic group by covalently linking to carrier proteins (CPs), such as acyl carrier proteins (ACPs) for fatty acid synthases (FASs) or polyketide synthases (PKSs), and peptidyl carrier proteins or aryl carrier proteins for nonribosomal peptide synthetases (NRPSs).8–11 The Ppant moiety is post-translationally transferred from CoA to a conserved serine residue on CPs by the action of phosphopantetheinyl transferase.10 By forming an energy-rich thioester linkage with intermediates of fatty acids, polyketides, or nonribosomal peptides in their biosynthetic pathways, Ppant fulfills the demand of providing flexibility and relatively sufficient length (approximately 2 nm) that allows the covalently tethered intermediates to navigate and access spatially distinct and structurally diverse enzyme active sites.
Both CoA and Ppant play central roles in carrier protein-based biosynthesis of fatty acids, polyketides, and nonribosomal peptides, ultimately providing a wide array of complex, bioactive natural products including valuable pharmaceuticals and precious commodity chemicals. For fatty acid synthesis, the simplest model system available is the type II FAS in Escherichia coli. In this system, an ACP interacts with more than 10 different catalytic partners, catalyzing the formation of long fatty acid chains from malonyl-CoA with high efficiency and fidelity.12 For polyketide synthesis, besides a similar mechanism for polyketide chain elongation with the participation of an ACP and malonyl- CoA, nature has co-opted the assembly line strategy to produce macrocyclic polyketide natural products by utilizing additional tailoring domains for increased chemical diversity and biological function.13 Similarly, NRPSs utilize the carrier protein machinery with elongation by amino acids instead of acyl groups.14 Recent efforts have been made to engineer these systems to expand their product diversity as well as to optimize systems for expression in heterologous hosts.15,16 A major hurdle that remains is our poor understanding of the transient substrate—protein interactions between the CPs with their Ppant-bound intermediates, as well as protein-protein interactions between CPs and their catalytic partner domains. Molecular dynamics (MD) and other computational techniques can be used to provide models of these transient interactions that are difficult to capture experimentally, thus providing an additional tool to increase yields and expand product diversity for the biosynthesis of “unnatural” natural products.17–21
The reliability of MD simulations depends on the availability and quality of molecular mechanics force fields, including both the functional form and parameter sets. Current Amber force fields provide parameter sets to support modeling standard amino acids, nucleic acids, sugars, lipids, and other relatively common moieties.22–26 At present, no scalable force field parameter set exists for PCLs. Performing MD simulations on systems containing PCLs require extra parameterization works each time, thus reducing the computational accessibility to potentially critical information on protein—protein and protein—substrate interactions. In addition, nonstandard residues, such as a phosphopantetheinyl-serine (Ppant-Ser) covalently embedded in a protein, require more efforts in parameterization. Furthermore, the size of CoA, Ppant, and Ppant-Ser “apo” ligands and their corresponding thioesters are at least 80, 43, and 52 atoms, respectively, making their parameterization processes computationally expensive and time consuming. At the time of this writing, a search on Protein Data Bank (PDB) database returns about 1700 entries containing CoA, CoA thioesters, Ppant, or Ppant thioesters, the majority of which contain CoA (603 entries) and acetyl-CoA (222 entries).27,28 However, a search on PubMed for keywords “molecular dynamics” with “coenzyme A” or “pantetheine” reveals only 141 or 9 publications, respectively. The limited literature for MD studies of PCLs is directly linked to the lack of pantetheine force field (PFF) parameters. The availability of a PFF library would allow researchers to model these enzymes for engineering efforts and provide medicinal chemists better models for drug design efforts.
Here we report a PFF library built specifically to model and simulate systems containing PCLs compatible with standard Amber force fields,22 including 12 standalone CoA or CoA-thioesters, 9 standalone Ppant or Ppant-thioesters, and 9 covalently linked Ppant-Ser or Ppant-Ser-thioesters with compatible nomenclatures with the Protein Data Bank. The atomic partial charge parameters were calculated by one of three charging algorithms, including Gasteiger,29 AM1-BCC,30,31 and restrained electrostatic potential (RESP), matching similar techniques employed in current Amber force fields.32,33 Inspired by the development of the LIPID11 force field,34 a “plug-and-play” parameterization scheme utilizing modular splitting was employed to simplify the computational complexity of using the RESP algorithm, resulting in a fragmentation strategy that allows for systematic charging of large molecules sharing common substructural motifs. The remaining parameters, such as those for bond terms, angle terms, and dihedral angle terms, were adopted from either ff14SB23 or gaff 2 force fields.35 This library is expected to have a significant impact on researchers who wish to conduct MD simulations of any system that requires PCLs as either substrates or cofactors.
METHODS
Structural Preparation.
Structural files of PCLs in the CIF (Crystallographic Information File) format containing observed and idealized structures (calculated by software such as OpenEye’s Omega based on the known covalent geometry) were obtained from the RCSB Protein Data Bank.28,36 The original hydrogen atoms on each PCL structural file were removed and the Amber/Reduce program was used to add hydrogen atoms matching its physiological protonation state.37 A “plug-and-play” fragmentation scheme was employed for the computationally expensive RESP charging method, which splits CoA, Ppant, and Ppant-Ser into a pool of eight fragments: (1) methylphosphate, (2) adenosine, (3) dimethyldiphosphate, (4) pantoic acid, (5) beta-alanine, (6) cysteamine, (7) serine dipeptide, and (8) dimethylphosphate. Fragments 1—6 were obtained from the structural file of CoA (PDB ID: COA); fragments 7 and 8 were obtained from the structural file of phosphoserine (PDB ID: SEP); extending fragments for each selected PCL was obtained from the corresponding structural file directly. Fragments were capped with acetyl, methylamide, methyl, and/or hydroxyl groups using the Build Structure feature of UCSF Chimera.38
PFF Parameterization.
The RESP ESP charge Derive (R.E.D.)-III.5 tools were used for RESP charge fitting for each “plug and play” fragment.39 Gaussian 09 was used to optimize the geometry of each fragment at B3LYP/6–31G* level of quantum mechanical (QM) theory and to derive molecular electrostatic potential at HF/6–31G* level of QM theory.40 Extra care was taken during the optimization of the serine dipeptide fragment (fragment 7), where ϕ and ѱ angles were constrained at —60.70° and —31.32°, respectively. A four-step RESP fitting strategy was employed to derive final RESP partial charges, including (1) charge fitting for each fragment independently, (2) pairwise charge fitting for each pair of connecting fragments, with intermolecular charge constraints applied on corresponding caps whose net charge was constrained to 0, (3) fragment merging by averaging the two different charges of each atom derived at step (2), and (4) charge scaling to ensure integer total charges of intact molecules with the following equation:
where Ci is the partial charge of the ith atom of the molecule before normalization, and Ctot is the total integer charge of the molecule. To reduce the charging error, Rigid-Body Reorientation Algorithm (RBRA) embedded in R.E.D.-III.5 was applied in step (l).39 The Amber/Antechamber program was used to conduct the Gasteiger and AM1-BCC charge fitting procedures.41
Non-charge parameters include those for bond, angle, dihedral angle, and van der Waals terms. For covalent Ppant- Ser PCLs, these parameters were first derived from ff14SB force fields where possible.23 Missing parameters were adopted from gaff2.35 For standalone CoA and Ppant PCLs, non-charge parameters were derived from the gaff2 force field directly. The parameterization process was handled by the parmchk2 program to obtain parameter modification (frcmod) files. Finally, the Amber/tleap program was used to generate OFF library (lib) files.42
Structural and Normal Mode Analyses of Fragments.
Fragment geometries were sequentially minimized using increasing levels of QM theories in the order of B3LYP/6–311+G(2d,p), MP2/aug-cc-pVDZ, and MP2/aug-cc-pVTZ using Gaussian 09, after which the QM normal mode frequencies were obtained.40 Scaling factors of 0.967, 0.959, and 0.953 were applied to B3LYP/6–311+G(2d,p), MP2/aug-cc-pVDZ, and MP2/aug-cc-pVTZ calculated normal mode frequencies, respectively, as suggested by precomputed scaling factors of Computational Chemistry Comparison and Benchmark DataBase (CCCBDB).43 For molecular mechanical minimization with PFF and OL3 force fields, the Amber/ pmemd program was used.42,44 PFF normal mode analysis was then performed using the nmode function of the Nucleic Acid Builder (NAB) language.45
Structural alignment and RMSD calculation between QM minimized structures and PFF or OL3 minimized structures were conducted using the match command of UCSF Chimera.38
MD Preparation.
Three PCL containing systems were selected for validation purposes, including phosphopantetheine adenylyltransferase-phosphopantetheine (PPAT-Ppant, PDB ID: 1OD6),46 3-hydroxy-3-methylglutaryl synthase/acyl carrier protein complex (HGMS/ACP-Ppant-Ser, PDB ID: 5KP7),47 and diaminobutyrate acetyltransferase-coenzyme A (EctA-CoA, PDB ID: 6SK1).48 Missing residues in PPAT and HGMS/ACP were added using modeller.49 Topology and coordinate files were prepared using the Amber/tleap program, with standard residues parameterized by the ff14SB force field and PCLs parameterized by the PFF library.23,42 Following parameterization, each system was solvated in an octahedral box of TIP3P water molecules with thickness extending 10 Å from the protein surface.50 Complexes were neutralized by adding counter ions with opposite charges (sodium or chloride), and extra sodium—chloride ion pairs were added to match reported experimental salt concentrations.
MD Simulations.
The Amber/pmemd.cuda program was used for all MD simulations.42,44 A 10 Å cutoff was set for nonbonded interactions and short-range electrostatic corrections. The SHAKE algorithm was used to constrain the hydrogen atom bond lengths,51,52 and the particle mesh Ewald (PME) method was used to handle long-range electrostatic interactions.53,54 Energy minimization was performed to relieve any possible atomic spatial conflicts in two stages. The first stage was used to relax only water molecules and ions, while the second stage was used to relax the whole system. Langevin dynamics with a 1 ps−1 collision frequency were used to gradually increase the system temperature from 0 K to reported experimental temperatures over 200 ps.55 The systems were first equilibrated for 100 ns under constant pressure and temperature (NPT) to adjust the system density; then, 100 ns production simulations were performed under constant volume and temperature (NVT) conditions. Both equilibration and production phases employed a 2 fs integration time step and 200 ps interval for simulation snapshot extraction. Each simulation was repeated in triplicates with different random seeds, starting from identical minimized structures.
MD Analysis.
MD simulation results were analyzed using three metrics: comparisons of RMSD between simulated and experimental conformations, comparisons of simulated and experimental B-factors, and our previously developed binding stability scoring.56 All metrics of each simulation were calculated using the Amber/cpptraj program, employing commands rmsd, atomicfluct, and nativecontacts, respectively.57 Simulated B-factor calculations only included snapshots of the last 10 ns. Both experimental and simulated B-factors were standardized using the following equation:
where μ and σ are the mean value and standard deviations of all B-factors, respectively. The stability score (SS) was developed to determine the binding stability of the receptor—ligand pair during simulation.56 The native atom pairs are defined as the heavy atom pairs that are within the distance of 7 Å in the crystal structure, and the stability score is calculated using the following equation:
where the stability score of the ith frame SSi is the fraction of the amount of these pairs that remain within 7 Å of each other. fstart and fend are the start and end frame numbers, respectively. In this work, fstart was set as 101 and fend was set as 200 to include the trajectory snapshots of the last 100 ns. Gaussian kernel density estimation (KDE) plots for RMSD and scatterplots for standardized B-factors were generated by the Matplotlib package of Python. B-factor visualizations were generated using the Render by Attribute feature of UCSF Chimera.38 Statistical analyses of stability scores were conducted by using the R statistical package.
RESULTS AND DISCUSSION
PFF Library Design.
The current pantetheine force field (PFF) library includes parameters for 30 PCLs available in the Protein Data Bank (Table 1). Besides “apo” CoA, Ppant, and Ppant-Ser, the PFF library contains thioesters of CoA, Ppant, and Ppant-Ser with extending units from saturated fatty acids, whose lengths range from 3 to 16 carbons, or the intermediates of fatty acid synthesis, including acetyl-, malonyl-, acetoacetyl- CoA; acetyl-Ppant; and acetyl-Ppant-Ser. All PCLs included in the CoA library and the Ppant library are standalone ligands, and all PCLs included in the phosphopantetheinyl-serine (Ppant- Ser) library are non-standard residues covalently linked to proteins. The URL links to the individual page of each PCL is also shown in Table 1.
Table 1.
Pantetheine-Containing Ligands Included in the Pantetheine Force Field Library
The functional form of a typical force field includes terms responsible for bond stretching, angle bending, dihedral angle torsion, van der Waals, and electrostatic interactions. Forexample, the additive Amber force field functional form for the total potential energy (Etotal) is
In this equation, ε is the dielectric constant, which has a default value of 1 in Amber and thus can be omitted. A parameter set including the following parameters has to be provided to perform tasks, such as minimization and molecular dynamics simulations:
bond parameters: kb, r0
angle parameters: kө,Ө0
torsional angle parameters: Vn, γ
van der Waals parameters: AijBij,
charge parameters: qi, qj
For Ppant-Ser PCLs, both covalent parameters (bond, angle, and torsional angle) and noncovalent van der Waals parameters were first derived from ff14SB where possible, to ensure compatibility with parameters for standard amino acid residues;23 missing parameters were then obtained from the gaff 2 force field, which were designed for general organic molecules.35 For standalone CoA and Ppant PCLs, these parameters were directly derived from the gaff 2 force field. Charge parameters have to be treated separately, since individual partial charge has to be assigned to each atom for widely used point-charge electrostatic models. In the PFF library, three common charging algorithms were applied, including Gasteiger,29 AM1-BCC,30,31and RESP.32,33
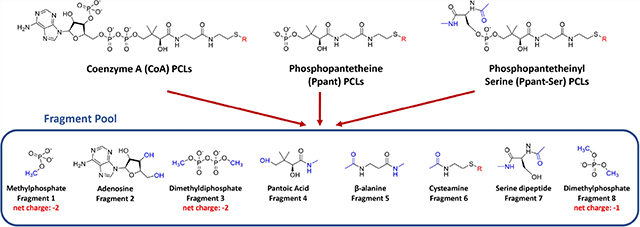
The RESP charges depend on molecular geometries provided as input. However, large, flexible molecules tend to form intramolecular interactions such as hydrogen bonds during the geometry optimization step, introducing a bias in fitted charges. Moreover, the CPU time of geometry optimization is positively correlated with molecular sizes. Therefore, a “plug-and-play” fragmentation approach was employed serving as a consistent charging scheme for the PFF library development, which splits common substructures of PCLs (CoA, Ppant, and Ppant-Ser) into a fragment pool including eight components: (1) methylphosphate, (2) adenosine, (3) dimethyldiphosphate, (4) pantoic acid, (5) beta-alanine, (6) cysteamine, (7) serine dipeptide, and (8) dimethylphosphate, as shown in Figure 1. Fragments were capped with acetyl, methylamide, methyl, and/ or hydroxyl groups mimicking the natural chemical environments of the fragments, and these caps were constrained to 0 net charge and removed during the fragment merging process. This approach was deemed necessary due primarily to the flexibility and relatively large size of the pantetheine moiety itself. Indeed, it is common for primed CoA and Ppant-Ser thioesters to achieve sizes greater than 200 atoms.58 During the geometry optimization step, extra care was taken for the serine dipeptide fragment (fragment 7), where Φ and Ψ angles were constrained at −60.70° and - 31.32° respectively, according to the analysis of Φ and Ψ angle distributions of 320 Ppant-Ser conformations from the Protein Data Bank (Figure S1). In contrast, Gasteiger charges and AM1-BCC charges were obtained with the whole molecule strategy, i.e., the structural files of the intact molecule of each PCL were used as inputs, because of the much higher efficiency of the two charging algorithms than that of the RESP charging method.
Figure 1.

“Plug-and-play” fragmentation strategy of PFF library development. Coenzyme A (CoA) PCLs, phosphopantetheine (Ppant) PCLs, and phosphopantetheinyl-serine (Ppant-Ser) PCLs can be fragmented into a fragment pool consisting of eight components: (1) methylphosphate, (2) adenosine, (3) dimethyldiphosphate, (4) pantoic acid, (5) beta-alanine, (6) cysteamine, (7) serine dipeptide, and (8) dimethylphosphate. CoA PCLs can be reconstructed with fragments 1, 2, 3, 4, 5, and 6; Ppant PCLs can be reconstructed with fragments 1, 4, 5, and 6; Ppant-Ser PCLs can be reconstructed with fragments 4, 5, 6, 7, and 8. Various extending units that form thioester bonds with CoA, Ppant, or Ppant-Ser are labeled with “R” in red. Acetyl, methylamide, methyl, and hydroxyl caps that were constrained to 0 net charge and removed during the fragment merging process are depicted in blue.
A caveat during the PFF library development is that the atomic nomenclatures of common substructures between different PCLs are inconsistent on the PDB. For example, the amine nitrogen atom of adenine of coenzyme A (PDB ID: COA), malonyl CoA (PDB ID: MLC) and propionyl CoA (PDB ID: 1VU) are named as N6A, N6, and N4, respectively. The nomenclature inconsistency prevents parameter transferability that is necessary for our fragmentation strategy. To address this problem, an atom renaming program called PyRenamer written in Python that enables converting atom names to corresponding atom names of a reference molecule was developed. The source code of PyRenamer can be obtained by contacting the authors.
Structural Comparisons of QM and PFF Optimized Fragments.
To validate PFF parameters, fragments were sequentially minimized with the increasing level of QM theories in the order of B3LYP/6–311+G(2d,p), MP2/aug-cc-pVDZ. and MP2/aug-cc-pVTZ as benchmarks. An acetyl cysteamine fragment was also tested as a representative thioester extending unit. Due to the increasing computational time complexity for larger fragments, the highest level of theory used for fragment 2 (adenosine) was B3LYP/6–311+G(2d,p). For fragments 3 (dimethyldiphosphate), 4 (pantoic acid), and 5 (beta-alanine) and the acetyl cysteamine fragment, the highest level of theory used was MP2/aug-cc-pVDZ. MP2/aug-cc-pVTZ was only applied to smaller fragments including fragments 1 (methylphosphate), 6 (cysteamine), and 8 (dimethylphosphate). The RMSD between QM and PFF optimized fragments ranged from 0.095 to 0.465 Å when RESP charges were used (denoted as PFF/RESP below) (Table 2 and Figure S2) In particular, since the structure of fragment 2 (adenosine) matches the adenosine (entry name: AN) available in the Amber/OL3 force field, the QM- and OL3-optimized fragments were also compared.59 The RMSD comparison shows that PFF/RESP (0.327 Å) has higher accuracy than OL3 (0.550 Å) for adenosine. (Figure S3) Additionally, PFF with Gasteiger (PFF/Gasteiger) and AM1- BCC (PFF/AM1-BCC) charges were also validated similarly. The RMSD between QM- and PFF/Gasteiger-optimized fragments ranged from 0.102 to 0.519 Å, and for PFF/AM1- BCC-optimized fragments, the RMSD ranged from 0.097 to 0.509 A (Table S1). Overall, PFF parameters with all three charging methods perform similarly in reproducing QM-optimized structures for the tested fragments.
Table 2.
RMSD between QM and PFF/RESP Optimized Fragments
| fragment no. | fragment name | highest level of theory | RMSD |
|---|---|---|---|
| 1 | methylphosphate | MP2/aug-cc-pVTZ | 0.095 |
| 2 | adenosine | B3LYP/6-311+G(2d,p) | 0.327 |
| 3 | dimethyldiphosphate | MP2/aug-cc-pVDZ | 0.465 |
| 4 | pantoic acid | MP2/aug-cc-pVDZ | 0.281 |
| 5 | beta-alanine | MP2/aug-cc-pVDZ | 0.386 |
| 6 | cysteamine | MP2/aug-cc-pVTZ | 0.098 |
| 8 | dimethylphosphate | MP2/aug-cc-pVTZ | 0.113 |
| - | acetyl-cysteamine | MP2/aug-cc-pVDZ | 0.401 |
| average | 0.271 |
Normal Mode Analysis of QM- and PFF-Optimized Fragments.
In order to gain further insights of the quality of PFF parameters, the QM normal mode frequencies of each fragment were obtained with the same level of theories described above. Due to the fact that ab initio calculated harmonic vibrational frequencies are typically larger than the experimental vibrational frequencies,60 scaling factors were applied to QM-calculated normal mode frequencies. Normal mode plots agreed well between QM calculations and PFF calculations, except for modes in the high frequency (above 2000 cm−1) region. (Figure 2 and Figures S4 and S5). For example, the frequencies observed in the 450–1100 cm−1 range include C—O and O—P bond stretching, O—P—O twisting, O—P—O wagging, and O—P—O scissoring. S—C bond stretching is observed at 645 and 749 cm−1, O—C—S bending is observed at 439 cm−1, and the characteristic intense carbonyl stretch for thioesters at 1720 cm−1 at the MP2/aug-cc-pVDZ level of theory. The PFF frequencies were in good agreement with QM frequencies for both cases.
Figure 2.

Comparison of normal mode frequencies of fragments calculated with PFF/RESP and B3LYP/6–311+G(2d,p), MP2/aug-cc-pVDZ, and MP2/aug-cc-pVTZ levels of theories. Scaling factors of 0.967, 0.959, and 0.953 were applied to B3LYP/6–311 + G(2d,p), MP2/aug-cc-pVDZ, and MP2/aug-cc-pVTZ-calculated normal mode frequencies, respectively.
Partial Charge Comparisons between Three Charge Fitting Methods.
Since the accuracy of PFF parameters for individual fragments has been validated, a four-step RESP fitting strategy was employed to derive final RESP partial charges as stated in the Methods section. Figure 3 shows the atom names and partial charges derived by RESP (fragmentation strategy), Gasteiger (whole molecule strategy), and AM1-BCC methods (whole molecule strategy), including their deviations from the unconstrained fragmental partial charges (the “differences” column) for standalone phosphopantetheine (PDB ID: PNS).
Figure 3.

Comparison of RESP charges, Gasteiger charges, and AM1-BCC charges with unconstrained fragmental partial charges for standalone phosphopantetheine. The “differences” column associated with each charging method shows the differences with unconstrained partial charges of corresponding atoms (the “No Constraints” column). Color schemes were applied to “differences” columns, where blue indicates negative differences and red indicates positive differences. The bottom row shows the sum of corresponding columns.
It can be observed that the greatest deviations are from charges derived by the Gasteiger method, where 17 atoms have differences above 0.15, including O27, P24, O23, O24, O25, C29, O33, H33, C34, O35, N36, H36, C39, O40, N41, H4, and S44. This is due to the fact that Gasteiger charges are not derived to reproduce electrostatic potentials (ESP) as the other two methods do.29 In contrast, ESP-based AM1-BCC30,31 and RESP32,33 charging methods produced only 7 or 1 atomic partial charges with differences above 0.15, respectively. It is reasonable to set 0.15 partial charge deviation as the “red line”, as indicated by LIPID11 force field development involving a similar fragmentation approach.34 Therefore, it is expected that AM1- BCC and RESP charges perform better than Gasteiger charges in subsequent validation tests.
Parameter Validations in MD Simulations.
Three representative systems containing PCLs with available experimental structures were used for validation purposes: phosphopantetheine adenylyltransferase-phosphopantetheine (PPAT-Ppant, PDB ID: 1OD6),46 3-hydroxy-3-methylglutaryl synthase/acyl carrier protein complex (HGMS/ACP-Ppant-Ser, PDB ID: 5KP7),47 and diaminobutyrate acetyltransferase-coenzyme A (EctA-CoA, PDB ID: 6SK1).48 It is notable that (l) Ppant is the substrate of PPAT in PPAT-Ppant; (2) Ppant-Ser is covalently linked to an ACP as a prosthetic group in HGMS/ACP-Ppant-Ser; (3) CoA is the cofactor of EctA in EctA-CoA. Since covalent bonds are typically stronger than noncovalent interactions and cofactors typically remain bound with proteins, it is reasonable to expect that their binding strengths increase in the order of PPAT-Ppant, EctA-CoA, and HGMS/ACP-Ppant-Ser.61 Each system was simulated under reported experimental temperatures and salt concentrations.
RMSD of simulation trajectories to the crystal structure is considered as an important validation metric of the quality of a force field, since it is reasonable to assume that protein crystal structures are typically close to the structures at the physiological condition.62 Therefore, the RMSD’s relative to crystal structures were computed for heavy atoms of both PCLs and protein residues in contact with PCLs (Figures S6–S8), and the probability density functions estimated were also analyzed via the Gaussian kernel density estimation (KDE), as shown in Figure 4. For contact residues, the parameter sets of all three charging methods show similar RMSD distribution patterns, as illustrated in the left panel of Figure 4. For Ppant-Ser and CoA PCLs, the PFF/AM1-BCC parameter set gave significantly lower RMSD than the other two charging methods (Figure 4D,F). However, for Ppant PCLs, the PFF/AM1-BCC parameter set and the PFF/RESP parameter set lead to higher RMSD than the PFF/Gasteiger parameter set, and the highest RMSD value observed reaches 7 Å (Figure 4B). Nevertheless, the PFF/AM1-BCC parameter set is the best in capturing the expected trend that PPAT-Ppant, EctA-CoA, and HGMS/ACP-Ppant-Ser are in the increasing order of binding strengths.
Figure 4.

Gaussian kernel density estimates (KDEs) of computed RMSD values of heavy atoms of contact residues (left panel) and PCLs (right panel) relative to the experimental structures. The diamond markers indicate the mean RMSD values.
The second quantitative validation of the PFF parameter set is the comparison of the experimental and simulated B-factors, or the temperature factor, reflecting the mobility or flexibility of various parts of the molecule caused by thermal motion. High B-factors indicate greater uncertainty about the actual atom position. Figure 5 displays the scatterplots of standardized B-factors simulated from three charging methods compared with experimental B-factors of the ligands and contact residues of the PPAT-Ppant system. The PFF/AM1-BCC parameter set resulted in highest correlation coefficients for both Ppant and contact residues, and next comes PFF/RESP and PFF/Gasteiger parameter sets. The visualization of standardized experimental B-factor and simulated B-factors of the PPAT-Ppant system illustrating the average structures of the last 10 ns are shown in Figure 6. PFF/AM1-BCC and PFF/RESP simulations yield similar agreement in Ppant conformations with respect to experimental structures (Figure 6C,D), although all three charging methods resulted in similar B-factor patterns for Ppant with the two ends of the linear structure having higher flexibility than the middle region. The corresponding scatterplots and structural visualizations of HGMS/ACP-Ppant-Ser and EctA-CoA systems are shown in Figures S9–S12. Highest correlation coefficients with experimental B-factors are always observed in PFF/AM1-BCC simulations. However, the simulated B-factors of CoA with all three charging methods failed to capture the trend that the phosphate group has higher flexibility than the rest of the molecules (Figures S11 and S12). A closer look into the X-ray structures of EctA-CoA revealed the existence of the cation—pi interaction between the adenine ring and Arg 99, which is difficult to be modeled in the current nonpolarizable Amber force field63 but is actively investigated in ongoing Amber polarizable force field developments.64–69
Figure 5.

Correlation analysis of standardized simulated and experimental B-factors of the contact residues (upper panel) and the PCLs (lower panel) for the PPAT-Ppant system. The residue names of contact residues or atom names of Ppant are annotated. R is the Pearson correlation coefficient.
Figure 6.

Visual comparison of standardized simulated and experimental B-factors for the PPAT-Ppant system. Ppant is depicted as ball-and-stick; contact residues are depicted as wire. Colors (red color indicates high B-factors, and blue color indicates low B-factors) and thickness of the protein backbone also indicate B-factor values; (A) the experimental structure, (B) the average structure of the last 10 ns trajectory with Gasteiger charges, (C) the average structure of the last 10 ns trajectory with AM1-BCC charges, (D) the average structure of the last 10 ns trajectory with RESP charges.
The last quantitative validation for MD simulations is our previously defined binding stability scoring, which reflects the binding stability between two molecules (ligands and proteins for example) by counting the native atomic contacts between the two molecules in each trajectory snapshots.56 Higher stability score indicates stronger binding. The last 50 ns of each trajectory was used for t test analysis of stability scores (Figures S13—S15). Consistent with previous expectations, the stability scores in PFF/AM1-BCC simulations are the highest among the three for HGMS/ACP-Ppant-Ser and EctA-CoA, while the lowest for PPAT-Ppant, reflecting the nature of their expected binding strengths (Figure 7).
Figure 7.

t test of binding stability scores of the last 50 ns trajectories of (A) PPAT-Ppant, (B) HGMS/ACP-Ppant-Ser, and (C) EctA-CoA. Significance levels: ***, p ≤ 0.001; ****, p ≤ 0.0001.
Pantetheine Force Field (PFF) Library Website Interface.
A website hosting the pantetheine force field library (http://rayluolab.org/pff-library/) has been developed. Published on the website are three libraries of force fields for CoA PCLs, Ppant PCLs, and Ppant-Ser PCLs. For each PCL, an OFF library (lib) file with all structures and charges and one or two parameter modification (frcmod) file with all missing non-charge parameters for each charging method are present. OFF library files contain the same atom names and coordinates as present in the Protein Data Bank for compatibility. Only one frcmod file is provided for CoA or Ppant PCL, since they are derived from only the gaff 2 force field, while two frcmod files are present for Ppant-Ser PCL, due to the fact that non-charge parameters of Ppant-Ser PCLs are first derived from the ff14SB force field then from the gaff 2 force field. Users of PFF files for Ppant-Ser PCLs are expected to load gaff 2 frcmod files first then ff14SB frcmod files to overwrite overlapping parameters. In addition, tutorials are present on the website to provide detailed protocols and input files on how to model and set up simulations containing PCLs. These structures can be used for minimizations, MD simulations, or as a part of docking studies.
CONCLUSIONS
In this paper, we present the first Amber-compatible force field library for various pantetheine-containing ligands. The PFF library was parameterized using Gasteiger, AM1-BCC, or RESP charging methods in combination with gaff2 parameters. Among three commonly used charging schemes, the PFF/ AM1-BCC parameter set shows better MD simulation performance than PFF/Gasteiger and PFF/RESP parameter sets, as indicated by MD validations. Furthermore, a “plug-and-play” fragmentation strategy was designed to enable systematic charge fitting for large molecules sharing common substructures. However, the parameter sets with the RESP charges derived from the fragmentation strategy does not perform better than that with the AM1-BCC charging method that can be applied to whole molecules in terms of MD simulations. In fact, the “plug-and-play” strategy applied in this study generating a fragment pool with extremely small fragments ranging from 9 atoms (methylphosphate) to 32 atoms (adenosine) has the following disadvantages: First, the increased amount of manual work overshadows the benefits of cheaper computational expenses during parameterization. Second, many charging errors were introduced due to the existence of too many merging interfaces between adjacent fragments. Therefore, a natural improvement of the “plug-and-play” strategy is to employ larger fragments. In a subsequent version of the PFF library, larger fragments will be explored to reduce errors in the RESP charging method.
This work paves the foundation for easy setup of MD simulations of biological systems containing PCLs in silico, and it is hoped to be applied in applications such as protein engineering for the production of novel compounds, or drug discovery for targeting certain PCL-containing proteins that play critical roles in diseases.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Fanglue Ni for making Figure 1. This work is supported in part by NIH (GM076330, GM093040 &GM100305). This material is based upon the work supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE-1321846.
ABBREVIATIONS
- ACP
acetyl carrier protein
- CoA
coenzyme A
- CP
carrier protein
- EctA
diaminobutyrate acetyltransferase
- FAS
fatty acid synthase
- HGMS/ACP
3-hydroxy-3-methylglutaryl synthase/ acyl carrier protein complex
- NRPS
nonribosomal peptide synthetases
- PCL
pantetheine-containing ligands
- PKS
polyketide synthase
- Ppant
phosphopantetheine
- Ppant-Ser
phosphopantetheinyl-serine
- PPAT
phosphopantetheine adenylyltransferase
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c01384.
(Figure S1) Ф/Ψ values of covalently bound phosphopantetheinyl-serine (Ppant-Ser) from the protein data bank (PDB ID: PNS) for a total of 320 data points; (Figures S2 and S3) Comparison of QM and PFF minimized structures of “plug and play” fragments; (Figures S4 and S5) Comparison of QM and PFF calculated normal mode frequencies of “plug and play” fragments; (Figures S6–S8) RMSD (angstrom) time series for simulation trajectories of representative PCL-containing systems; (Figures S9 and S10) Correlation analysis and visual analysis of standardized simulated and experimental B-factors of the HGMS/ACP-Ppant-Ser system; (Figures S11 and S12) Correlation analysis and visual analysis of standardized simulated and experimental B-factors of the EctA-CoA system; (Figures S13–S15) Stability score time series of simulation trajectories of representative PCL-containing systems; (Table S1) RMSD (angstrom) between QM and PFF/Gasteiger or PFF/AM1-BCC optimized “plug and play” fragments (PDF)
Contributor Information
Shiji Zhao, Department of Molecular Biology and Biochemistry, Department of Chemistry, Department of Chemical and Biomolecular Engineering, Department of Materials Science and Engineering, Department of Biomedical Engineering, and Department of Pharmaceutical Sciences, University of California, Irvine, Irvine, California 92697, United States.
Andrew J. Schaub, Department of Molecular Biology and Biochemistry, Department of Chemistry, Department of Chemical and Biomolecular Engineering, Department of Materials Science and Engineering, Department of Biomedical Engineering, and Department of Pharmaceutical Sciences, University of California, Irvine, Irvine, California 92697, United States.
Shiou-Chuan Tsai, Department of Molecular Biology and Biochemistry, Department of Chemistry, and Department of Pharmaceutical Sciences, University of California, Irvine, Irvine, California 92697, United States.
Ray Luo, Department of Molecular Biology and Biochemistry, Department of Chemical and Biomolecular Engineering, Department of Materials Science and Engineering, and Department of Biomedical Engineering, University of California, Irvine, Irvine, California 92697, United States.
REFERENCES
- (1).Mishra PK; Drueckhammer DG Coenzyme A analogues and derivatives: Synthesis and applications as mechanistic probes of coenzyme A ester-utilizing enzymes. Chem. Rev. 2000, 100, 3283–3310. [DOI] [PubMed] [Google Scholar]
- (2).Shi L; Tu BP Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hoagland MB; Novelli GD Biosynthesis of coenzyme A from phosphopantetheine and of pantetheine from pantothenate. J. biol. Chem 1954, 207, 767–773. [PubMed] [Google Scholar]
- (4).Harwood JL Fatty acid metabolism. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1988, 39, 101–138. [Google Scholar]
- (5).Daugherty M; Polanuyer B; Farrell M; Scholle M; Lykidis A; de Crécy-Lagard V; Osterman A Complete reconstitution of the human coenzyme A biosynthetic pathway via comparative genomics. J. Biol. Chem. 2002, 277, 21431–21439. [DOI] [PubMed] [Google Scholar]
- (6).Leonardi R; Jackowski S Biosynthesis of Pantothenic Acid and Coenzyme A. EcoSal Plus; 2007, 2, DOI: 10.1128/ecosalplus.3.6.3.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Leonardi R; Zhang Y-M; Rock CO; Jackowski S Coenzyme A: back in action. Prog. Lipid Res. 2005, 44, 125–153. [DOI] [PubMed] [Google Scholar]
- (8).Qiao C; Wilson DJ; Bennett EM; Aldrich CC A mechanism-based aryl carrier protein/thiolation domain affinity probe. J. Am. Chem. Soc. 2007, 129, 6350–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhou Z; Lai JR; Walsh CT Directed evolution of aryl carrier proteins in the enterobactin synthetase. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 11621–11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Elovson J; Vagelos PR Acyl carrier protein: X. Acyl carrier protein synthetase. J. Biol. Chem. 1968, 243, 3603–3611. [PubMed] [Google Scholar]
- (11).Byers DM; Gong H Acyl carrier protein: structure-function relationships in a conserved multifunctional protein family. Biochem. Cell Biol. 2007, 85, 649–662. [DOI] [PubMed] [Google Scholar]
- (12).White SW; Zheng J; Zhang Y-M; Rock CO The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 2005, 74, 791–831. [DOI] [PubMed] [Google Scholar]
- (13).Sattely ES; Fischbach MA; Walsh CT Total biosynthesis: in vitro reconstitution of polyketide and nonribosomal peptide pathways. Nat. Prod. Rep. 2008, 25, 757–793. [DOI] [PubMed] [Google Scholar]
- (14).Schwarzer D; Finking R; Marahiel MA Nonribosomal peptides: from genes to products. Nat. Prod. Rep. 2003, 20, 275–287. [DOI] [PubMed] [Google Scholar]
- (15).Nielsen J; Keasling JD Engineering cellular metabolism. Cell 2016, 164, 1185–1197. [DOI] [PubMed] [Google Scholar]
- (16).Yuzawa S; Kim W; Katz L; Keasling JD Heterologous production of polyketides by modular type I polyketide synthases in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 727–735. [DOI] [PubMed] [Google Scholar]
- (17).Nguyen C; Haushalter RW; Lee DJ; Markwick PRL; Bruegger J; Caldara-Festin G; Finzel K; Jackson DR; Ishikawa F; O’Dowd B; McCammon JA; Opella SJ; Tsai S-C; Burkart MD Trapping the dynamic acyl carrier protein in fatty acid biosynthesis. Nature 2014, 505, 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dowling DP; Kung Y; Croft AK; Taghizadeh K; Kelly WL; Walsh CT; Drennan CL Structural elements of an NRPS cyclization domain and its intermodule docking domain. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bravo-Rodriguez K; Klopries S; Koopmans KRM; Sundermann U; Yahiaoui S; Arens J; Kushnir S; Schulz F; Sanchez-Garcia E Substrate flexibility of a mutated acyltransferase domain and implications for polyketide biosynthesis. Chem. Biol. 2015, 22, 1425–1430. [DOI] [PubMed] [Google Scholar]
- (20).Barajas JF; Phelan RM; Schaub AJ; Kliewer JT; Kelly PJ; Jackson DR; Luo R; Keasling JD; Tsai S-C Comprehensive structural and biochemical analysis of the terminal myxalamid reductase domain for the engineered production of primary alcohols. Chem. Biol. 2015, 22, 1018–1029. [DOI] [PubMed] [Google Scholar]
- (21).Milligan JC; Lee DJ; Jackson DR; Schaub AJ; Beld J; Barajas JF; Hale JJ; Luo R; Burkart MD; Tsai S-C Molecular basis for interactions between an acyl carrier protein and a ketosynthase. Nat. Chem. Biol. 2019, 15, 669–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Salomon-Ferrer R; Case DA; Walker RC An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar]
- (23).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Galindo-Murillo R; Robertson JC; Zgarbová M; Šponer J; Otyepka M; Jurečka P; Cheatham TE III Assessing the current state of amber force field modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kirschner KN; Yongye AB; Tschampel SM; González- Outeiriño J; Daniels CR; Foley BL; Woods RJ GLYCAM06: a generalizable biomolecular force field Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Dickson CJ; Madej BD; Skjevik á. A.; Betz RM; Teigen K; Gould IR; Walker RC Lipid14: the amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Burley SK; Berman HM; Bhikadiya C; Bi C; Chen L; Di Costanzo L; Christie C; Dalenberg K; Duarte JM; Dutta S; Feng Z; Ghosh S; Goodsell DS; Green RK; Guranović V; Guzenko D; Hudson BP; Kalro T; Liang Y; Lowe R; Namkoong H; Peisach E; Periskova I; Prlić A; Randle C; Rose A; Rose P; Sala R; Sekharan M; Shao C; Tan L; Tao Y-P; Valasatava Y; Voigt M; Westbrook J; Woo J; Yang H; Young J; Zhuravleva M; Zardecki C RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gasteiger J; Marsili M Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar]
- (30).Jakalian A; Bush BL; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [DOI] [PubMed] [Google Scholar]
- (31).Jakalian A; Jack DB; Bayly CI Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- (32).Bayly CI; Cieplak P; Cornell W; Kollman PA A wellbehaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar]
- (33).Cieplak P; Cornell WD; Bayly C; Kollman PA Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. [Google Scholar]
- (34).Skjevik á. A.; Madej BD; Walker RC; Teigen K LIPID11: a modular framework for lipid simulations using amber. J. Phys. Chem. B 2012, 116, 11124–11136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (36).Hawkins PCD; Skillman AG; Warren GL; Ellingson BA; Stahl MT Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Word JM; Lovell SC; Richardson JS; Richardson DC Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [DOI] [PubMed] [Google Scholar]
- (38).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (39).Dupradeau F-Y; Pigache A; Zaffran T; Savineau C; Lelong R; Grivel N; Lelong D; Rosanski W; Cieplak P The R.E.D. Tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Frisch M; Trucks G; Schlegel H; Scuseria G; Robb M; Cheeseman J; Scalmani G; Barone V; Mennucci B; Petersson G, Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009, 32, 5648–5652. [Google Scholar]
- (41).Wang J; Wang W; Kollman PA; Case DA Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- (42).Case DA; Cheatham TE III; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).NIST Computational Chemistry Comparison and Benchmark Database; NIST Standard Reference Database Number 101, Release 20, August 2020. Editor: Johnson RD, III http://cccbdb.nist.gov/. [Google Scholar]
- (44).Le Grand S; Götz AW; Walker RC SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar]
- (45).Macke TJ; Case DA Modeling Unusual Nucleic Acid Structures. In Molecular Modeling of Nucleic Acids; American Chemical Society: 1997; Vol. 682, Chapter 24, pp. 379–393. [Google Scholar]
- (46).Takahashi H; Inagaki E; Fujimoto Y; Kuroishi C; Nodake Y; Nakamura Y; Arisaka F; Yutani K; Kuramitsu S; Yokoyama S; Yamamoto M; Miyano M; Tahirov TH Structure and implications for the thermal stability of phosphopantetheine adenylyltransferase from Thermus thermophilus. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 97–104. [DOI] [PubMed] [Google Scholar]
- (47).Maloney FP; Gerwick L; Gerwick WH; Sherman DH; Smith JL Anatomy of the β-branching enzyme of polyketide biosynthesis and its interaction with an acyl-ACP substrate. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 10316–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Richter AA; Kobus S; Czech L; Hoeppner A; Zarzycki J; Erb TJ; Lauterbach L; Dickschat JS; Bremer E; Smits SHJ The architecture of the diaminobutyrate acetyltransferase active site provides mechanistic insight into the biosynthesis of the chemical chaperone ectoine. J. Biol. Chem. 2020, 295, 2822–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Eswar N; Webb B; Marti-Renom MA; Madhusudhan MS; Eramian D; Shen M -y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinf. 2006, 15, 5.6. 1–5.6. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- (51).Miyamoto S; Kollman PA Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar]
- (52).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- (53).Darden T; York D; Pedersen L Particle mesh Ewald: An N. log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- (54).Crowley M; Darden T; Cheatham T III; Deerfield D II Adventures in improving the scaling and accuracy of a parallel molecular dynamics program. J. Supercomput. 1997, 11, 255–278. [Google Scholar]
- (55).Loncharich RJ; Brooks BR; Pastor RW Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers 1992, 32, 523–535. [DOI] [PubMed] [Google Scholar]
- (56).Zhao S; Ni F; Qiu T; Wolff JT; Tsai S-C; Luo R Molecular Basis for Polyketide Ketoreductase-Substrate Interactions. Int. J. Mol. Sci. 2020, 21, 7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Roe DR; Cheatham TE III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (58).Ray L; Valentic TR; Miyazawa T; Withall DM; Song L; Milligan JC; Osada H; Takahashi S; Tsai S-C; Challis GL A crotonyl-CoA reductase-carboxylase independent pathway for assembly of unusual alkylmalonyl-CoA polyketide synthase extender units. Nat. Commun. 2016, 7, 13609–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zgarbová M; Otyepka M; Šponer J; Mládek A. t.; Banáš P; Cheatham TE III; Jurečka P Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Scott AP; Radom L Harmonic vibrational frequencies: an evaluation of Hartree- Fock, Moller- Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem. 1996, 100, 16502–16513. [Google Scholar]
- (61).Hashim OH; Adnan NA Coenzyme, cofactor and prosthetic group: ambiguous biochemical jargon. Biochem. Educ. 1994, 22,93–94. [Google Scholar]
- (62).Wang L-P; McKiernan KA; Gomes J; Beauchamp KA; Head-Gordon T; Rice JE; Swope WC; Martínez TJ; Pande VS Building a more predictive protein force field: a systematic and reproducible route to AMBER-FB15. J. Phys. Chem. B 2017, 121, 4023–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Lamoureux G; Orabi EA Molecular modelling of cation-л interactions. Mol. Simul. 2012, 38, 704–722. [Google Scholar]
- (64).Wang J; Cieplak P; Li J; Hou T; Luo R; Duan Y Development of Polarizable Models for Molecular Mechanical Calculations I: Parameterization of Atomic Polarizability. J. Phys. Chem. B 2011, 115, 3091–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wang J; Cieplak P; Li J; Wang J; Cai Q; Hsieh M; Lei H; Luo R; Duan Y Development of Polarizable Models for Molecular Mechanical Calculations II: Induced Dipole Models Significantly Improve Accuracy of Intermolecular Interaction Energies. J. Phys. Chem. B 2011, 115, 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Wang J; Cieplak P; Cai Q; Hsieh MJ; Wang JM; Duan Y; Luo R Development of Polarizable Models for Molecular Mechanical Calculations. 3. Polarizable Water Models Conforming to Thole Polarization Screening Schemes. J. Phys. Chem. B 2012, 116, 79998008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Wang J; Cieplak P; Li J; Cai Q; Hsieh M-J; Luo R; Duan Y Development of Polarizable Models for Molecular Mechanical Calculations. 4. van der Waals Parametrization. J. Phys. Chem. B 2012, 116,7088–7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Wang J; Cieplak P; Luo R; Duan Y Development of Polarizable Gaussian Model for Molecular Mechanical Calculations I: Atomic Polarizability Parameterization To Reproduce ab Initio Anisotropy. J. Chem. Theory Comput. 2019, 15, 1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Wei H; Qi R; Wang J; Cieplak P; Duan Y; Luo R Efficient formulation of polarizable Gaussian multipole electrostatics for biomolecular simulations. J. Chem. Phys. 2020, 153, 114116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.