

Abstract
BACKGROUND:
To the authors’ knowledge, there are no approved therapies for recurrent, metastatic (R/M) salivary gland carcinoma (SGC), but molecularly targeted therapies warrant ongoing investigation. In the current study, the authors have reported on the efficacy of tipifarnib in patients with aggressive HRAS-mutant, R/M SGC.
METHODS:
The current prospective, nonrandomized, multicenter, international cohort study involved 8 centers and was conducted from May 2015 to June 2019. The median follow-up was 22 months (range, 6–55 months). Subjects with HRAS-mutant R/M SGC (any histology) and disease progression within the last 6 months were enrolled. Tipifarnib was dosed orally twice daily. The authors determined the objective response rate using Response Evaluation Criteria in Solid Tumors (version 1.1), duration of response, and molecular predictors of response.
RESULTS:
A total of 13 patients with R/M SGC were enrolled; all had received prior systemic therapy (1–3 regimens). One objective response was observed; an additional 7 of 12 evaluable patients (58%) had stable disease as their best response with a median duration of 9 months (range, 3–14 months). Five of 7 patients had >10% tumor regression and 6 of 7 had stable disease lasting >6 months. Q61R was the most frequent activating HRAS mutation noted (7 of 13 patients; 54%), but gene variant and allele frequency did not correlate with outcomes. The median progression-free survival was 7 months (95% confidence interval, 5.9–10.1 months), and the median overall survival was 18 months (95% confidence interval, 9.6–22.4 months) with approximately 58.6% of patients alive at 1 year. Survival was similar regardless of HRAS mutant variant or co-occurring PIK3CA alterations. No participant discontinued treatment because of toxicity.
CONCLUSIONS:
Tipifarnib resulted in modest clinical activity with a promising disease control rate among patients with HRAS-mutant, R/M SGC who developed disease progression within the last 6 months.
Keywords: HRAS, rare cancers, salivary cancer, targeted therapy, tipifarnib
INTRODUCTION
Salivary gland cancers (SGCs) comprise a rare group of head and neck malignancies with wide variations in histopathologic and molecular features, dictating a spectrum of clinical behavior from indolent to more aggressive.1 Although patients with localized disease often are treated with upfront surgery followed by radiation,2 to the best of our knowledge standard therapeutic approaches for those with recurrent, incurable, or metastatic disease are lacking. There is no clear evidence suggesting that cytotoxic chemotherapy improves survival in the advanced disease setting3 and early trials investigating immunotherapy have demonstrated low response rates.4 Therefore, there is a critical need to evaluate molecularly targeted approaches for SGC. Recent prospective trials in salivary duct carcinoma (SDC) have provided validation that targeting androgen receptor5 or HER2/neu amplification6,7 translates into high degrees of clinical efficacy, exemplifying the potential benefit of a personalized therapeutic approach for these rare cancers.8–12 The ongoing challenge is to translate findings from the next-generation sequencing efforts taking place in recent years into clinical trials that will identify more actionable therapeutic targets for patients with SGC.
HRAS is a member of the RAS proto-oncogene family, encoding for a GTPase (a hydrolase enzyme) that functions as a molecular switch to regulate cell survival and proliferation pathways in cancer.13 Sequencing efforts in SGCs have identified HRAS mutations in up to 20% of high-grade histologic subtypes, such as mucoepidermoid carcinoma, adenocarcinoma, and SDC.14,15 Targeting RAS remains an open challenge in cancer medicine, but preclinical data have demonstrated that the HRAS isoform is uniquely susceptible to farnesyltransferase inhibition. Farnesyltransferase inhibitors (FTIs) block HRAS localization to the plasma membrane, thereby abrogating downstream oncogenic signaling.16,17 Recent clinical data demonstrating a favorable response rate (8 of 15 patients; 53%) with the first-in-class FTI tipifarnib in patients with high variant allele frequency (VAF) HRAS-mutant head and neck squamous cell carcinoma (HNSCC) validated that inhibiting farnesyltransferase can successfully target HRAS activity in human cancers.18 In the current study, we have reported the efficacy of tipifarnib in a cohort of patients with HRAS-mutant, recurrent, or metastatic SGC.
MATERIALS AND METHODS
Patients and Design
The original study was an open-label, single-arm, phase 2 trial evaluating the safety and efficacy of tipifarnib in patients with unresectable or metastatic, HRAS-mutant thyroid cancer (cohort 1) and other HRAS-mutant solid tumors (cohort 2) (ClinicalTrials.gov identifier NCT02383927).19 Seven patients with SGC were enrolled in cohort 2, and an additional 6 treated patients received tipifarnib under individual patient US Food and Drug Administration Investigational New Drug Application (IND) or single patient expanded access requests (with sponsor permission) during the study period after written informed consent was obtained (total of 13 patients). Herein, we have reported the outcomes of these 13 molecularly and pathologically similar patients with SGC who were treated with tipifarnib.
For inclusion, patients had to have an HRAS mutation; cytologically confirmed, locoregionally advanced, unresectable or metastatic SGC (2 pathologists independently confirmed the SGC histology in each case); and measurable disease. Any number of prior lines of systemic therapy were permitted. The study was approved by the institutional review board at each participating institution. Tipifarnib for the other HRAS-mutant SGC cases was accessed with sponsor agreement (Kura Oncology, San Diego, California) through submission of a single-patient IND application to the US Food and Drug Administration or the equivalent regulatory agency for cases outside the United States, and treatment proceeded with local institutional review board approval.
Molecular Characterization
Nine of 13 patients (69%) were evaluated using in-house, next-generation targeted sequencing platforms unique to their academic institution to identify actionable mutations, performed at Clinical Laboratory Improvement Amendments–certified laboratories. The remaining 4 patients (31%) had FoundationOne CDx (Foundation Medicine Inc, Cambridge, Massachusetts) next-generation sequencing performed, which surveys 324 genes and selected rearrangements. The VAF (as a percentage) of the HRAS mutation present in each case was reported if available, along with other coexisting genetic alterations. Nine of 13 patient tumor samples (69%) were obtained at the time of disease recurrence, and thus most VAFs determined by next-generation sequencing were not based on biopsies at the time of initial diagnosis.
Treatment
Tipifarnib was dosed in the original study at 900 mg given orally twice daily on days 1 to 7 and days 15 to 21 of a 28-day cycle until progressive disease (PD), unacceptable toxicity, or death. The 6 patients being treated off protocol were dosed on a similar schedule with starting doses ranging from 600 mg to 900 mg twice daily, at the discretion of the treating physician. Dose adherence was monitored by pill diary in most cases. Adverse events were reported using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0). Patients who developed grade 3 or intolerable toxicities were instructed to withhold tipifarnib, with the option of resuming at a prespecified lower dose. Patients were evaluated for adverse events and laboratory abnormalities at least every 6 weeks while receiving treatment, regardless of whether they were being treated on or off protocol.
Assessments
Baseline computed tomography imaging was required prior to the initiation of therapy and performed at 8-week to 12-week intervals (depending on whether the patient was being treated on or off protocol) to evaluate treatment response. Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1)20 was used to define the best overall response in each on-protocol patient, and was reported only when independently verified (centrally reviewed by a board-certified radiologist blinded to the clinical data) in those patients treated off protocol. Patients were followed after discontinuation of study treatment by their treating physician.
Statistical Analysis
Descriptive statistics were used to summarize the study cohort. The Kaplan-Meier method was used to evaluate survival outcomes. Progression-free survival (PFS) was evaluated from the time of the initiation of study treatment to either PD or death, whichever occurred first; otherwise, the patient was censored at the time of last known follow-up. Overall survival (OS) was defined from the start of study treatment to death or the patient was censored at the time of last known follow-up. Binary logistic regression analysis was used to determine the impact of Q61R mutational status on the presence or absence of tumor regression. The Spearman rho (correlation coefficient) was used to compare VAFs (as a percentage) and tumor regression (as a percentage). Statistical significance was set at a 2-sided P < .05 (Stata/IC 14.2; StataCorp LLC, College Station, Texas).
RESULTS
Study Population
Between May 2015 and June 2019, a total of 7 patients were enrolled on protocol, as outlined above, while an additional 6 patients were treated with tipifarnib (for a total of 13 patients) through expanded access provided by Kura Oncology. All participating investigators confirmed that study participants had clinical or radiographic PD within 6 months prior to the initiation of tipifarnib. All patients received at least 1 dose of the study treatment. Baseline demographics and patient characteristics are summarized in Table 1. With a median age of 58 years (range, 34–75 years), the cohort was comprised mostly of men (11 of 13 patients; 85%). Nearly all patients had primary parotid SGC (12 of 13 patients; 92%) with 8 of 13 patients demonstrating 1 of 2 dominant histologies (4 with SDC and 4 with epithelial-myoepithelial carcinoma). The majority of patients had locoregionally advanced disease at the time of initial presentation (11 of 13 patients; 85%), and initial therapy included surgery followed by radiotherapy with or without chemotherapy in most cases (77%). Two patients (15%) received systemic therapy for their initial disease (which was metastatic at the time of presentation) (Table 2). The median time to first disease recurrence was 22 months (range, 4–81 months) among the 11 patients who had prior locoregional disease. Six of 13 patients (46%) received palliative cytotoxic chemotherapy agents (most often carboplatin or cisplatin with or without a taxane) prior to initiating study treatment, with all having had received at least 1 prior line of systemic therapy for advanced disease.
Table 1.
Demographics and clinicopathologic information among HRAS-mutant salivary cancer patients receiving tipifarnib
| Characteristic | All patients N = 13a |
|---|---|
| Age at diagnosis (median) | 58 (34–75) |
| Gender | |
| Female | 2 (15) |
| Male | 11 (85) |
| ECOG performance status | |
| 0–1 | 13 (100) |
| Ethnicity & Race | |
| White | 10 (77)b |
| Asian | 2 (15) |
| Black | 1 (8) |
| Site of Primary Disease | |
| Parotid gland | 12 (92) |
| Minor salivary gland | 1 (8) |
| Histologic subtype (high grade) | |
| Salivary duct carcinoma | 4 (31) |
| Myoepithelial-epithelial carcinoma | 4 (31) |
| Mucoepidermoid carcinoma | 1 (8) |
| Acinic cell carcinoma | 1 (8) |
| Oncocytic carcinoma | 1 (8) |
| Adenocarcinoma | 1 (8) |
| Carcinoma ex pleomorphic adenoma | 1 (8) |
| Initial Disease Stagingc | |
| Stage I, II | 2 (15) |
| Stage III, IVA-C | 11 (85)d |
| Initial or Primary Therapy | |
| Surgery alone | 1 (8) |
| Surgery + radiation (RT) | 6 (46) |
| Surgery + chemoradiation (CRT) | 4 (31) |
| Systemic therapy | 2 (15) |
| Median time to recurrence (in months) | 22.0 (4–81) |
| # of Lines of Prior Therapy for Advanced Disease | |
| 1–2 | 9 (69) |
| 3+ | 4 (31) |
parentheses indicate % except for age which represents range;
no patients identified as Hispanic;
American Joint Committee on Cancer (AJCC) 8th edition Staging (2017);
N=2 had metastatic disease at initial presentation; ECOG = Eastern Cooperative Oncology Group
Table 2.
HRAS-mutant salivary cancer patients treated with tipifarnib
| Subject | Age at Dx | Gender | Race | Date of initial Dx | Primary site | Histology | Initial Stage | ARa | HER2 statusb | Initial Tx | Completed Initial Tx | First recurrence | Prior Systemic Tx | Started tipifarnib | Starting dosec | Time on therapy (months)d | BOR | % Tumor changee |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 48 | M | W | 06/15/13 | PAR | AcCC | IVA | − | 0 | S+R | 04/15/14 | 09/23/14 | ICI | 10/25/18 | 600 mg | 14.0+ | PR | −31.4 |
| 2 | 75 | M | W | 05/12/17 | PAR | CexPA | IVA | + | 0 | S+R | 09/26/17 | 01/16/18 | ADT | 03/08/19 | 600 mg | 1.0 | PD | +15.6 |
| 3 | 61 | M | W | 05/21/15 | PAR | SDC | IVC | + | 1+ | C | -- | -- | ADT | 09/09/16 | 900 mg | 1.0 | PD | +6.7 |
| 4 | 69 | M | W | 12/03/10 | PAR | MyoEpC | IV | − | 0 | S+R | 02/28/11 | 11/22/13 | Phase I | 09/30/15 | 900 mg | 13.0 | SD | −27.5 |
| 5 | 34 | M | A | 08/23/11 | PAR | MyoEpC | IV | S | 10/11/11 | 01/14/14 | CT | 05/13/15 | 300 mg | 1.0 | PD | +20.6 | ||
| 6 | 58 | M | B | 09/26/14 | PAR | Adeno | IV | + | 0 | S+CRT | 01/16/15 | -- | Cisplatin | 01/14/16 | 900 mg | 3.0 | SD | −14.1 |
| 7 | 65 | M | W | 03/07/14 | PAR | SDC | IV | S+CRT | 06/30/14 | 03/07/15 | Cisplatin | 10/15/15 | 900 mg | 7.0 | SD | +6.9 | ||
| 8 | 53 | F | W | 12/09/11 | PAR | OncCA | IV | S+CRT | 03/26/12 | 09/04/15 | Cisplatin | 01/21/16 | 900 mg | 8.0 | SD | +3.1 | ||
| 9 | 73 | M | W | 08/16/17 | PNS | SGC | IV | C | -- | -- | CD | 02/20/18 | 900 mg | 10.0 | SD | −16.9 | ||
| 10 | 38 | M | A | 06/03/10 | PAR | SDC | IVA | + | 2+ | S+CRT | 09/09/10 | 02/15/12 | Alpelisib | 06/18/19 | 600 mg | 6.0+ | UE | -- |
| 11 | 56 | F | W | 08/25/11 | PAR | MyoEpC | I | + | 1+ | S+R | 11/16/11 | 10/25/16 | CT | 05/08/18 | 900 mg | 5.0 | SD | −14.2 |
| 12 | 59 | M | W | 06/15/08 | PAR | MyoEpC | II | S+R | 08/15/08 | 05/15/15 | ICI | 02/14/19 | 600 mg | 10.0+ | SD | −10.3 | ||
| 13 | 50 | M | W | 05/27/16 | PAR | MEC | IVA | + | 2+ | S+R | 08/15/16 | 11/28/17 | ADT | 05/20/19 | 600 mg | 2.0 | PD | +21.1 |
reported ‘+’ if greater than or equal to 1% expression by immunohistochemistry (IHC) was observed, blank spaces were not available for testing;
HER-2/neu status was defined by IHC using standard immune expression intensity scoring from 0 to 1–3+ (none had amplification), if available;
reported milligram (mg) doses were dosed by mouth twice daily;
a ‘+’ indicates that the patient remains on therapy at the date of analysis (1/6/20);
as determined by RECIST v1.1 with central review; Dx = diagnosis, AR = androgen receptor, Tx = treatment, BOR = best overall response per RECIST v1.1, PAR = parotid, PNS = paranasal sinuses, AcCC = acinic cell carcinoma, CexPA = carcinoma ex pleomorphic adenoma, SDC = salivary duct carcinoma, MyoEpC = myoepithelial-epithelial carcinoma, Adeno = adenocarcinoma, OncCA = oncocytic carcinoma, SGC = salivary gland cancer not otherwise specified, MEC = mucoepidermoid carcinoma, S = surgery, R = radiation, CRT = concurrent chemoradiation, ICI = immune checkpoint inhibitor, ADT = androgen deprivation therapy, CT = carboplatin/paclitaxel, CD = carboplatin/docetaxel, SD = stable disease, PD = progression of disease, UE = unevaluable.
Efficacy and Tolerability
Twelve of 13 patients (92%) were evaluable using RECIST (version 1.1) after independent verification by a central radiology reviewer. There was 1 partial response (PR) (duration of 14 months and ongoing at the time of last follow-up) for a best overall response rate of 8%, but it is interesting to note that an additional 7 of 12 patients (58%) demonstrated stable disease (SD) as a best response with a median duration of 9 months (range, 3–13 months) (Fig. 1). Five of 7 patients with SD had tumor regression of >10% (range, 10.3%−27.5%). Six of 7 patients (86%) demonstrated SD for ≥6 months, 1 of whom remained on therapy at the time of analysis (January 6, 2020). Four of 12 patients (33%) had confirmed PD (2 due to new sites of disease) as their best response when considering the entire cohort (1 case was not evaluable due to bone-only metastatic disease that precluded central imaging review using RECIST criteria).
FIGURE 1.
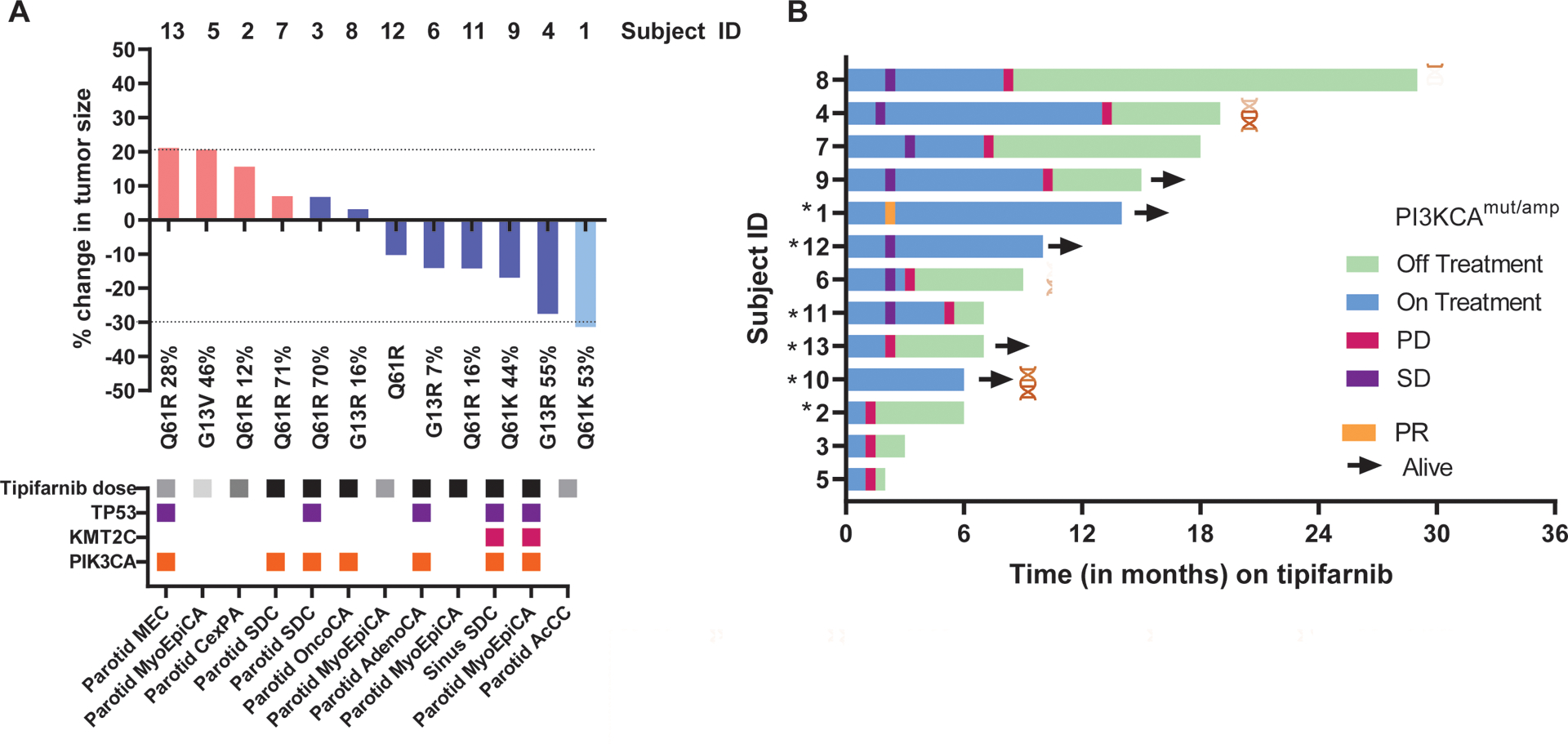
Response assessment and duration in patients with HRAS-mutant, advanced salivary gland cancer who are treated with tipifarnib. (A) Waterfall plot of maximum percentage change in tumor size from baseline as measured by Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1) (if evaluable; 12 of 13 patients). Subject identification (ID) is reported above each patient column. Below each patient column is the specific HRAS mutational variant (if known), and the variant allele frequency (VAF) expressed as a percentage of the altered genome. Below each column are matched clinical and mutational data, including the histologic subtype of disease, tipifarnib starting dose (black indicates 900 mg, gray indicates 600 mg, and light gray indicates 300 mg), and co-mutations with other selected genes (TP53, KMT2C, and PIK3CA). (B) Swimmer plot from the initiation of tipifarnib (13 patients) to the time of last tipifarnib dose and last known follow-up with each horizontal column representing a single patient. Colored ticks indicate response assessments and results, and coexisting PIK3CA mutational or amplification status is indicated. An asterisk indicates those patients who were treated as part of the expanded access program (off protocol). AcCC indicates acinic cell carcinoma; AdenoCA, adenocarcinoma; CexPA, carcinoma ex pleomorphic adenoma; EMC, epithelial-myoepithelial carcinoma; MEC, mucoepidermoid carcinoma; OncoCA, oncocytic carcinoma; PD, progressive disease; PR, partial response; SD, stable disease; SDC, salivary duct carcinoma.
Expected treatment-related adverse events included gastrointestinal upset (nausea, emesis, or dyspepsia) and moderate cytopenias (see Supporting Table 1). Six patients (46%) required a dose adjustment for toxicity: 3 patients underwent a single dose reduction from 900 to 600 mg (2 patients for grade 3 acute renal failure that was reversible and 1 patient for grade 3–4 cytopenias), 1 patient underwent a dose reduction from 900 mg to 600 mg and finally to 300 mg twice daily for grade 3 neutropenia, and 2 patients underwent a dose reduction (600 mg to 300 mg and 300 mg to 200 mg, respectively) for grade 3 to 4 cytopenias. All dose-adjusted patients but one (who remained on therapy at the time of last follow-up) later withdrew from treatment for PD. No patients discontinued treatment due to toxicity alone.
Survival
At a median follow-up of 22 months (range, 6–55 months), 8 patients had died. The median PFS for the entire cohort was 7 months (95% confidence interval [95% CI], 5.9–10.1 months), with 10 PD events observed at the time of last analysis (January 6, 2020); the remaining 3 patients remained on treatment at the time of last follow-up. The median OS was 18 months (95% CI, 9.6–22.4 months), with 58.6% and 19.5% of patients alive at 12 months and 24 months, respectively (Fig. 2).
FIGURE 2.
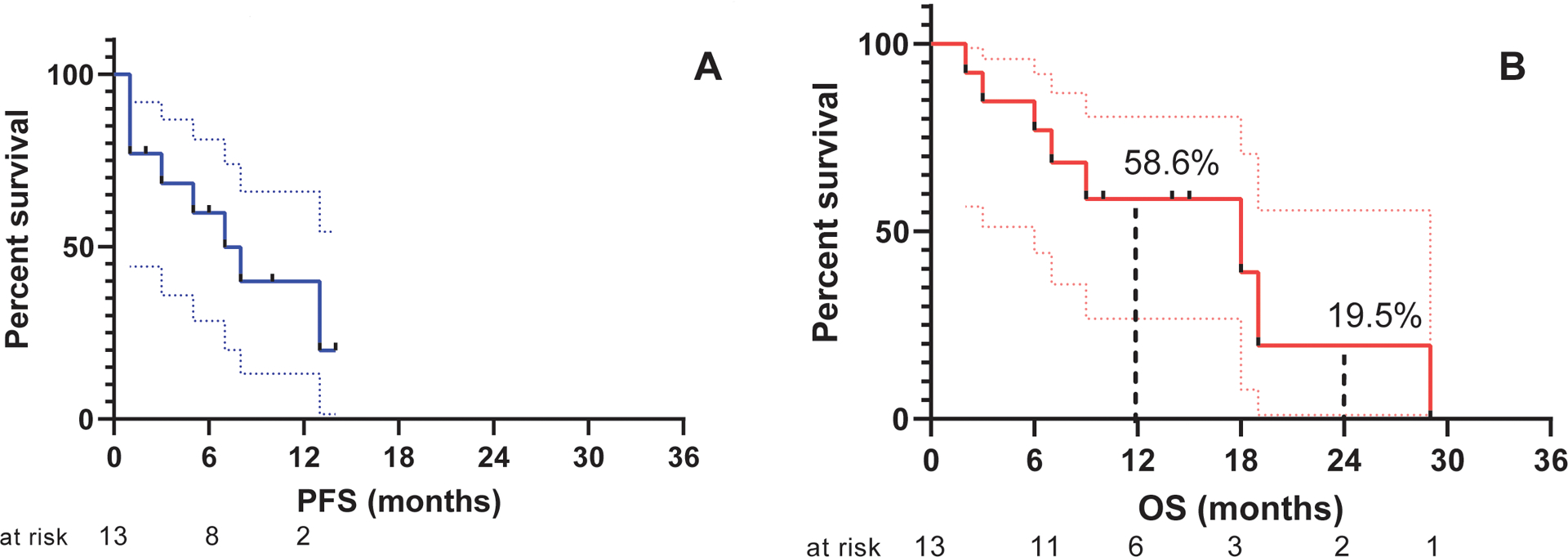
Progression-free survival (PFS) and overall survival (OS) outcomes for patients with HRAS-mutant, advanced salivary gland cancer who were treated with tipifarnib. Kaplan-Meier survival curves showing (A) PFS measured in months from the time of the initiation of tipifarnib dosing to first instance of disease progression or death (whichever occurred first) or censored at the time of last known follow-up and (B) OS measured in months from the time of the initiation of tipifarnib therapy to death or else censored at the time of last known follow-up. The number of subjects at risk are shown below each curve.
Molecular Findings
Q61R was the most commonly detected missense HRAS alteration (7 of 13 patients; 54% of known variants) among the entire cohort. When considering an individual’s gene variant leading to HRAS mutation, there was no clearly observed tendency for genomic subgroups to yield greater clinical benefit (odds ratio, 0.37; P = .12). However, both patients with a Q61K HRAS alteration (VAFs of 44%−53%) had >15% tumor reduction (Fig. 1). When available, the VAF was reported for each gene variant. There was no correlation between VAF (as a percentage) and change in tumor size (r = −0.08; P = .81) among the 11 patients with evaluable VAFs. When considering the most common gene variant among the cohort (HRAS Q61R missense mutation), there was no difference in OS (from the initial diagnosis of SGC) noted compared with other gene variants (hazard ratio, 0.62; P = .47) (Fig. 3). In addition, co-mutation or coamplification of PIK3CA with HRAS did not appear to have an effect on outcomes after treatment with tipifarnib (hazard ratio, 0.49; P = .28), and our single responding patient did not demonstrate androgen receptor or HER2 overexpression. It is interesting to note that no gene fusions or rearrangements of interest were observed among the cohort.
FIGURE 3.
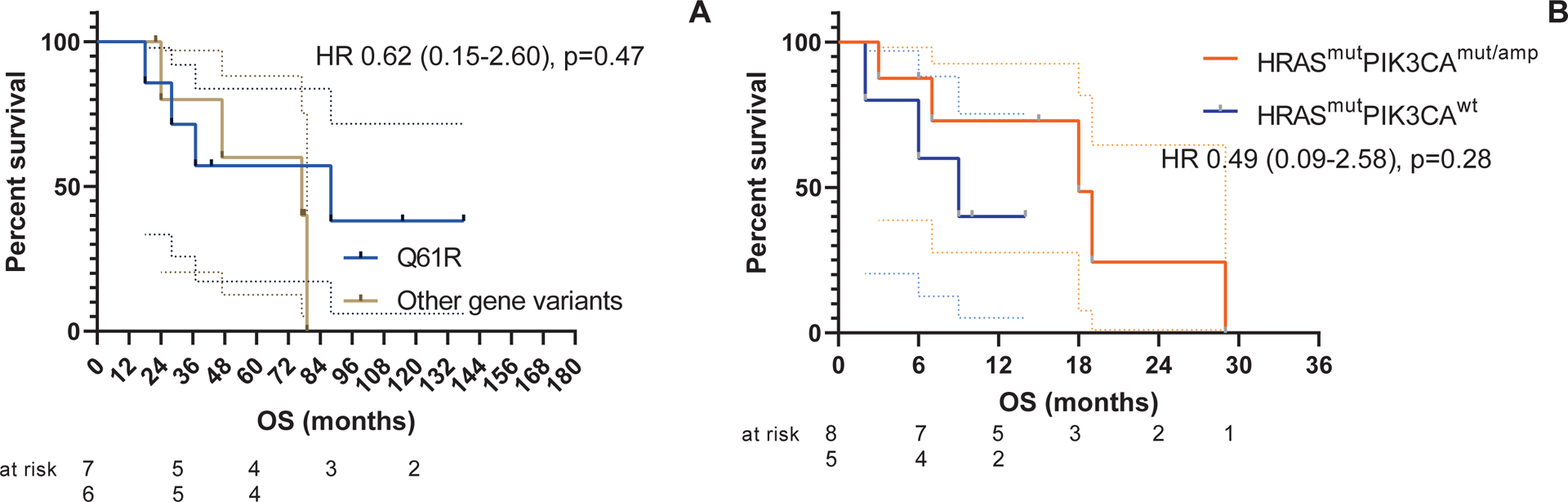
Survival outcomes for patients with HRAS-mutant, advanced salivary gland cancer who were treated with tipifarnib based on molecular findings. Kaplan-Meier survival curves showing (A) overall survival (OS) measured in months from the time of the initial diagnosis of salivary gland cancer to death or else censored at the time of last known follow-up for those subjects with a Q61R HRAS variant (7 patients) versus other gene variants in HRAS (6 patients) and (B) OS measured in months from the time of initiation of tipifarnib dosing to death or else censored at the time of last known follow-up between HRAS/PIK3CA co-mutated (mut) or coamplified (amp) subjects (8 patients vs 5 patients without co-mutation or coamplification). The number of subjects at risk is shown below each curve. HR indicates hazard ratio. *P < .05 using the log-rank test.
DISCUSSION
Next-generation sequencing efforts have demonstrated that HRAS mutations can be found in a significant number of aggressive histologic SGC subtypes (mucoepidermoid carcinoma, adenocarcinoma, and SDC),14,15 although to the best of our knowledge a targeted therapeutic approach has never been investigated among these patients. In the absence of a dedicated clinical trial for this rare orphan disease, we launched an international, multicenter effort to pool together an unprecedented number of patients with HRAS-mutant SGC who were treated with tipifarnib; to the best of our knowledge, the current study is the largest targeted therapy experience for this molecular subset performed to date.
The rationale for administering tipifarnib to target oncogenic RAS activity was based on the understanding that membrane localization is required for its downstream signaling. Subcellular trafficking to the membrane in turn is reliant on the posttranslational addition of hydrophobic groups to the C-terminal tail of RAS (known as “prenylation”). Because the predominant form of RAS prenylation is farnesylation (the addition of hydrophobic farnesyl groups) catalyzed by farnesyltransferase, FTIs were developed >10 years ago as a new class of cancer therapeutics that could abrogate mutant RAS signaling in human malignancies. Despite this hypothesis, initial trials of tipifarnib in patients with solid tumor subtypes enriched for RAS mutations failed to demonstrate significant clinical activity.21,22 This likely reflects the existence of redundant forms of prenylation that can facilitate membrane localization despite farnesyltransferase inhibition.17 However, HRAS differs from other N-RAS and K-RAS isoforms in that it is modified only by farnesylation, thereby predicting its unique susceptibility to FTIs.23 Based on this biologic rationale, a phase 2 tipifarnib basket study (ClinicalTrials.gov identifier NCT02383927)19 was developed to test the hypothesis that tipifarnib possesses activity against HRAS-mutant solid tumors, including SGCs. After that trial was no longer accruing patients with SGCs, subsequent patients were treated on single-patient INDs requested by individual investigators or other modes of single patient expanded access requests, with sponsor permission. Combined, this resulted in a cohort of 13 patients with SGC who were treated with tipifarnib reported herein.
The one major response observed in this SGC cohort occurred in a patient with Q61K HRAS-mutant acinic cell carcinoma who experienced marked tumor regression in the clivus and lungs that was ongoing 14 months into treatment (as of January 13, 2020) (Fig. 4). Five of 12 evaluable patients (42%) had >10% tumor regression (to a maximum of 27.5%). Approximately 86% of patients with SD (6 of 7 patients) had experienced SD for ≥6 months at the time of last follow-up (with 1 patient having maintained SD for >1 year). The overall clinical benefit rate (partial response plus SD) of 67%is posited to represent clinical activity with tipifarnib given the aggressive SGC histologic subtypes noted among the enrolled patients. Indeed, all patients had PD noted within 6 months prior to the initiation of FTI therapy. It also is worth noting that the one responding patient was treated with a dose of 600 mg twice daily, and 6 of 7 patients with SD (86%) were dosed at 900 mg twice daily. Beyond the modest activity achieved with tipifarnib, the tumor regressions observed represent critical supporting evidence that mutant HRAS is a potential oncogenic driver for SGCs, and that future therapeutic developments should focus on optimizing the inhibition of HRAS signaling alone or in combination to enhance therapeutic activity.
FIGURE 4.
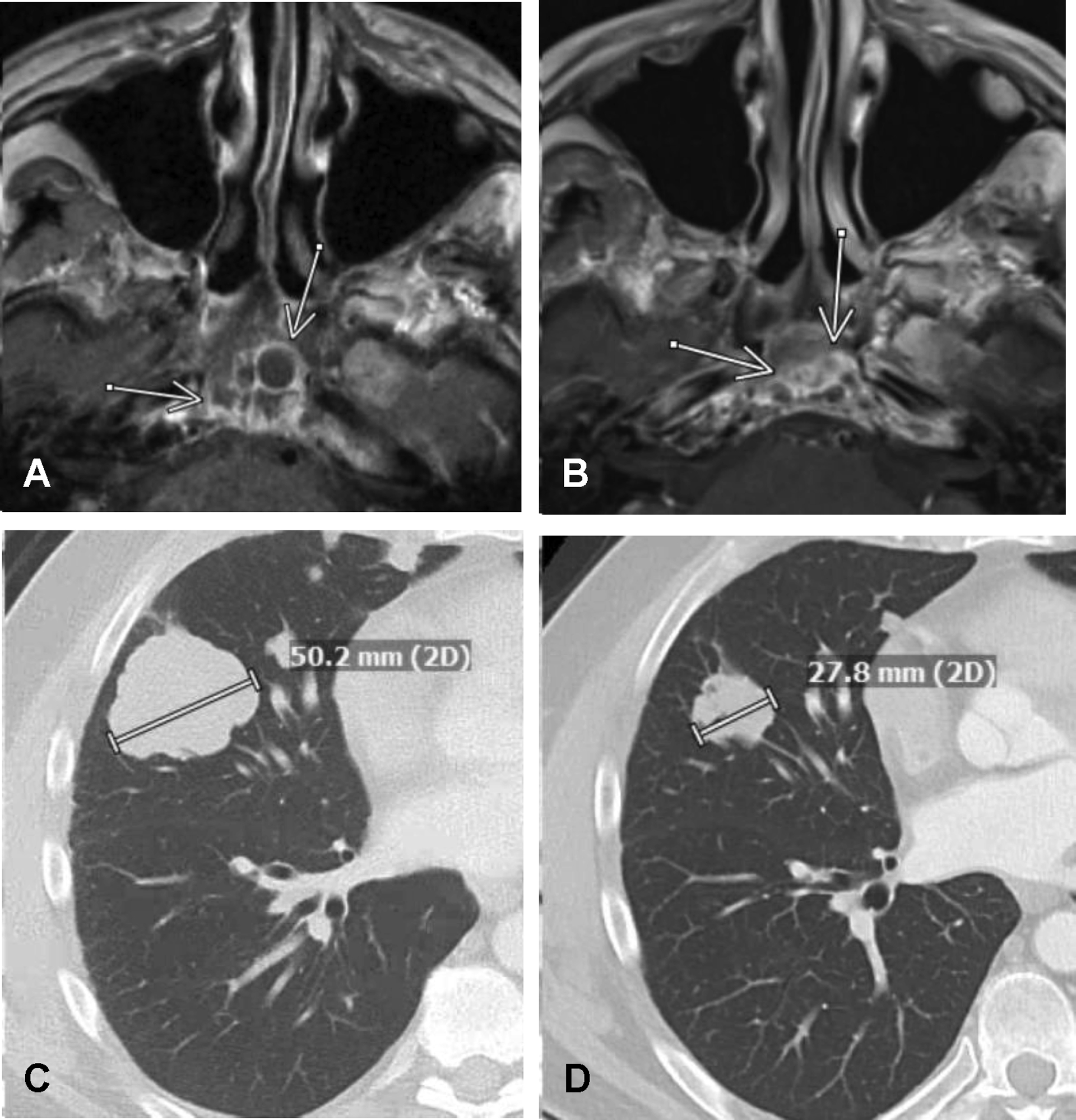
Response to tipifarnib in patients with Q61K HRAS-mutant, metastatic salivary cancer of the parotid gland. (A) Pretreatment, axial, contrast-enhanced, T1-weighted magnetic resonance imaging demonstrating a multilobulated and partially cystic mass (arrows) arising from the clivus prior to the initiation of tipifarnib and (B) a later marked reduction in the size and cystic nature of the mass after 8 months of tipifarnib therapy. (C) Axial contrast-enhanced computed tomography images in lung windows demonstrating a 50.2-mm, right middle lobe mass prior to the initiation of tipifarnib therapy and (D) a later reduction in the size of the mass to 27.8 mm after 5 months of tipifarnib therapy (patient 1).
These findings with tipifarnib compare favorably with other molecularly targeted therapies that have been investigated in trials of patients with advanced SGC that were performed without molecular selection. For example, agents such as lapatinib, targeting HER-2 and EGFR,24 and gefitinib25 have yielded disease stability rates of approximately 60% to 80% in patients with adenoid cystic carcinoma.3 Only more recently has molecular selection in SGC trials translated into greater efficacy with targeted approaches, such as strategies to inhibit the androgen receptor, overexpressed or amplified HER2, or NTRK3 rearrangements.12,26 These efforts have highlighted the intrinsic value of molecular selection and profiling even for rare diseases, not only to identify effective new therapies but also to delineate potential mechanisms of acquired resistance that can suggest new directions for clinical investigation.
Beyond HRAS-mutant SGC, we recently reported in abstract form a response rate of 56% (10 of 18 patients) with tipifarnib in heavily pretreated patients with high VAF (20% to ≥35%), HRAS-mutant HNSCC.18 The disparity in efficacy for patients with SGC versus those with HNSCC may reflect biologic distinctions between these tumor lineages that require further investigation. Although supporting preclinical and clinical evidence have suggested that HRAS is the primary target for tipifarnib in these tumors, the potential contribution of other farnesylated proteins to tipifarnib susceptibility or resistance cannot be ruled out.
Reviewing the molecular data from larger genomic characterization efforts in patients with SDC specifically, HRAS-activating mutations were reported to occur in approximately 23% of these tumors (7 of 31 tumors)27,28 and in 2% of sequenced adenoid cystic carcinoma cases.29 Among participants in the current study cohort for whom detailed molecular data were available, 9 of 13 patients (69%) had mutations at codon 61 (Q61R/K). Specifically, Q61R was the most commonly detected missense HRAS mutation (54% of known variants). The remainder of HRAS mutational events (31%) occurred at codon 13 (G13R/V). The specific HRAS-mutant single-nucleotide variant detected did not predict for clinical benefit at this cohort size, whereas a high VAF did appear to be associated with greater tipifarnib activity in patients with HNSCC. In the SGC cohort in the current study, the 6 patients with tumor regression had VAFs ranging from 7% to 55%, with 1 patient (patient 6) demonstrating 14.1% tumor regression as a best response with a G13R HRAS mutation at a VAF of only 7%. It is interesting to note that the VAF did not directly correlate with the degree of tumor regression achieved (P = .81) at this cohort size. In addition, 2 patients with a high VAF (patients 3 and 5) had PD as their best overall response. Although lowering the dose of tipifarnib to 200 mg twice daily may have contributed to the outcome noted in patient 5, patient 3, with a 70% VAF for Q61R, failed tipifarnib despite maintaining the high dose of 900 mg.
Other mechanisms of intrinsic resistance to inhibiting HRAS activity may be related to the disruption of a negative feedback loop upstream, leading to increased protein-kinase signaling through wild-type RAS (NRAS, KRAS). Acquired resistance also has been reported with the emergence of inactivating NF1 and activating GNAS mutations in thyroid cancer models.24,30 Co-occurring PIK3CA mutations have been hypothesized as activating a parallel biochemical pathway that could limit the efficacy of inhibiting RAS/mitogen-activated protein kinase (MAPK) signaling in malignancies, although to our knowledge the significance in patients with SGC has not been explored to date. Dalin et al previously observed 100% co-mutation with PIK3CA H1047R and G13R mutations in their molecularly defined SGC cohort.28 In our cohort, we observed that 3 of 3 patients with G13R mutations and 5 of 9 patients with Q61R/K mutations also had a co-occurring PIK3CA mutation, although we failed to find a clear association between the presence of hotspot PIK3CA mutations and poor outcomes in the current study. Further investigation with larger patient cohorts certainly is warranted.
Although the limitations of the current study included a small sample size, heterogeneous starting doses of tipifarnib, and some variation in imaging schedules, we speculated that the observed median PFS of 7 months (95% CI, 5.9–10.1 months), along with evidence of minor tumor regression, suggested meaningful clinical activity with tipifarnib. The median OS of 18 months (95% CI, 9.6–22.4 months) suggested a limited interval between PD while receiving the drug and death in this cohort, another illustration of the poor prognosis and limited treatment options for these patients. Patients in the current study with HRAS-mutant SGC experienced a 1-year OS rate of 58.6% compared with prior studies of subgroups of patients with aggressive SGC, which have reported 1-year OS rates of 66% to ≥80%.8,31
Conclusions
There are limitations to this report related to the rarity and orphan disease status of HRAS-mutant SGCs, including the small sample size and the inclusion of patients who were being treated outside of a single prospective trial. Nonetheless, to the best of our knowledge, the current study is the first report of its kind demonstrating modest, promising clinical activity with tipifarnib in a molecularly defined cohort of patients with a disease for which prospective trial data are completely lacking. It is important to note that this initial observation strongly recommended that novel approaches to optimizing the inhibition of HRAS signaling, including drug combinations with tipifarnib, could translate into even greater clinical activity for patients with HRAS-mutant SGC.
Supplementary Material
FUNDING SUPPORT
The study was also supported in part at Memorial Sloan Kettering Cancer Center through the NIH/NCI Cancer Center Support Grant/Core Grant P30 CA008748, the Geoffrey Beene Cancer Research Center and the Overman Fund.
CONFLICT OF INTEREST DISCLOSURES
Glenn J. Hanna has received grants and personal fees from Bristol-Myers Squibb, Regeneron, and Sanofi Genzyme; grants from GlaxoSmithKline, Kartos, Kite Pharma, NantKwest, Exicure, American Society of Clinical Oncology Conquer Cancer Foundation, V Foundation, and Gateway for Cancer Research; and personal fees from Maverick, Merck, Kura Oncology, Bio-Rad Laboratories, Bicara, and Prelude for work performed outside of the current study. Nicole G. Chau has received grants from Merck, GlaxoSmithKline, Pfizer, and Bristol-Myers Squibb for work performed outside of the current study. Cyrus M. Sayehli has received travel support from Celgene/Bristol-Myers Squibb. Deborah J. Wong received a grant from Kura Oncology for work performed as part of the current study and grants from Merck Sharpe and Dohme, Bristol-Myers Squibb, Astellas, FSTAR, Iovance, AstraZeneca, Regeneron, Roche/Genentech, Pfizer, Enzychem, and Lilly and grants and personal fees from Sanofi-Aventis for work performed outside of the current study. Mohammad Razaq has participated in a speaker panel for Merck. Elisabeth Pérez-Ruiz has received grants to her institution from Roche and Bristol-Myers-Squibb; has acted as a paid member of the Speaker’s Bureau for Bristol-Myers Squibb; has taken part in a training course for Novartis; and has received travel grants from Merck Sharpe and Dohme, Roche, and Bristol-Myers Squibb. Ezra E.W. Cohen has received research support and/or consulting fees from ALX Oncology, Ascendis Pharma, Bayer, Bioline Rx, Bristol-Myers Squibb, Debiopharm, Dynavax Technologies, Merck Sharpe and Dohme, Merck, Regeneron, and Sanofi. Rahul Aggarwal has received grants and personal fees from Janssen and Merck; grants from AstraZeneca, Novartis, and Xynomic Pharmaceuticals; and personal fees from Dendreon for work performed outside of the current study. Catherine Scholz and Antonio Gualberto are employed by and own stock in Kura Oncology. Alan L. Ho has acted as a paid member of the advisory board for Sanofi Genzyme (Aventis); has received clinical trial funding and personal fees and other fees from Novartis; has received clinical trial funding from, acted as a paid member of the advisory board for, and received travel expenses and conference fees from Kura Oncology; has received clinical trial funding from, acted as a paid member of the advisory board for, and received travel expenses from AstraZeneca; has received clinical trial funding from, acted as a paid member of the advisory board for, and received travel expenses from Merck; has received clinical trial funding from and acted as a paid member of the advisory board for Bristol-Myers Squibb; has received travel expenses and conference fees from Ignyta; has received clinical trial funding from and acted as a paid consultant for Genentech/Roche; has acted as a paid member of the advisory board for Sun Pharmaceuticals; has received clinical trial funding from Celldex (previously Koltan Pharmaceuticals), Bayer, and Lilly; has received clinical trial funding from, acted as a paid member of the advisory board for, and received travel expenses and conference fees from Ayala Pharmaceuticals; has acted as a paid member of the advisory board for Regeneron and TRM Oncology; has received clinical trial funding from Astellas and Daiichi Sankyo; has acted as a paid consultant for CureVac; has received travel expenses from Janssen; has received clinical trial support from and acted as a member of the advisory board for Eisai Pharmaceuticals; has received clinical trial funding from Allos Pharm and Pfizer; has acted as a paid speaker for Medscape; has received other fees from Klus Pharma; has received clinical trial funding from Elevar Therapeutics; has acted as a paid member of the advisory board for Prelude Therapeutics; has acted as a paid consultant for McGivney Global Advisors; has received honoraria from Leidos, ASTRO, and Shanghai Jia Tong University School of Medicine; has received travel expenses from RASopathy conference; and has received honoraria and travel expenses from Partners Health. The other authors made no disclosures.
Footnotes
Additional supporting information may be found in the online version of this article.
REFERENCES
- 1.Guzzo M, Locati LD, Prott FJ, Gatta G, McGurk M, Licitra L. Major and minor salivary gland tumors. Crit Rev Oncol Hematol. 2010;74:134–148. [DOI] [PubMed] [Google Scholar]
- 2.Amini A, Waxweiler TV, Brower JV, et al. Association of adjuvant chemoradiotherapy vs radiotherapy alone with survival in patients with resected major salivary gland carcinoma: data from the National Cancer Data Base. JAMA Otolaryngol Head Neck Surg. 2016;142:1100–1110. [DOI] [PubMed] [Google Scholar]
- 3.Laurie SA, Ho AL, Fury MG, Sherman E, Pfister DG. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: a systematic review. Lancet Oncol. 2011;12:815–824. [DOI] [PubMed] [Google Scholar]
- 4.Cohen RB, Delord JP, Doi T, et al. Pembrolizumab for the treatment of advanced salivary gland carcinoma: findings of the phase 1b KEYNOTE-028 Study. Am J Clin Oncol. 2018;41:1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu B, Dogan S, Haroon Al Rasheed MR, Ghossein R, Katabi N. Androgen receptor immunohistochemistry in salivary duct carcinoma: a retrospective study of 188 cases focusing on tumoral heterogeneity and temporal concordance. Hum Pathol. 2019;17:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Can NT, Lingen MW, Mashek H, et al. Expression of hormone receptors and HER-2 in benign and malignant salivary gland tumors. Head Neck Pathol. 2018;12:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clauditz TS, Reiff M, Gravert L, et al. Human epidermal growth factor receptor 2 (HER2) in salivary gland carcinomas. Pathology. 2011;43:459–464. [DOI] [PubMed] [Google Scholar]
- 8.Boon E, van Boxtel W, Buter J, et al. ; Nationwide Network and Registry of Histopathology and Cytopathology (PALGA) Group, van der Graaf WTA, van Herpen CML. Androgen deprivation therapy for androgen receptor–positive advanced salivary duct carcinoma: a nationwide case series of 35 patients in The Netherlands. Head Neck. 2018;40:605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor–positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haddad R, Colevas AD, Krane JF, et al. Herceptin in patients with advanced or metastatic salivary gland carcinomas. A phase II study. Oral Oncol. 2003;39:724–727. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi H, Tada Y, Saotome T, et al. Phase II trial of trastuzumab and docetaxel in patients with human epidermal growth factor receptor 2–positive salivary duct carcinoma. J Clin Oncol. 2019;37:125–134. [DOI] [PubMed] [Google Scholar]
- 12.Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross JS, Gay LM, Wang K, et al. Comprehensive genomic profiles of metastatic and relapsed salivary gland carcinomas are associated with tumor type and reveal new routes to targeted therapies. Ann Oncol. 2017;28:2539–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato S, Elkin SK, Schwaederle M, et al. Genomic landscape of salivary gland tumors. Oncotarget. 2015;6:25631–25645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Makarewicz JM, Knauf JA, Johnson LK, Fagin JA. Transformation by Hras(G12V) is consistently associated with mutant allele copy gains and is reversed by farnesyl transferase inhibition. Oncogene. 2014;33:5442–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whyte DB, Kirschmeier P, Hockenberry TN, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. [DOI] [PubMed] [Google Scholar]
- 18.Ho AL, Branna I, Haddad R, et al. Preliminary results from a phase 2 trial of tipifarnib in squamous cell carcinomas (SCCs) with HRAS mutations. American Association for Cancer Research–National Cancer Institute–European Organisation for Research and Treatment of Cancer International Conference on Molecular Targets and Cancer Therapeutics; October 26–30, 2019; Boston, MA. [Google Scholar]
- 19.Ho AL, Chau N, Bauman J, et al. Preliminary results from a phase 2 trial of tipifarnib in squamous cell carcinomas (SCCs) with HRAS mutations. ESMO 2018 Congress. Ann Oncol. 2018;29(suppl_8):viii372–viii399. [Google Scholar]
- 20.Eisenhauer EA, Therasse P, Bogaerts J, et al. New Response Evaluation Criteria in Solid Tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 21.Harousseau JL. Farnesyltransferase inihibitors in hematologic malignancies. Blood Rev. 2007;21:173–182. [DOI] [PubMed] [Google Scholar]
- 22.Ashar HR, James L, Gray K, et al. The farnesyl transferase inhibitor SCH 66336 induces a G2→M or G1 pause in sensitive human tumor cell lines. Exp Cell Res. 2001;262:17–27. [DOI] [PubMed] [Google Scholar]
- 23.Untch BR, Dos Anjos V, Garcia-Rendueles MER, et al. Tipifarnib inhibits HRAS-driven dedifferentiated thyroid cancers. Cancer Res. 2018;78:4642–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agulnik M, Cohen EWE, Cohen RB, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol. 2007;25:3978–3984. [DOI] [PubMed] [Google Scholar]
- 25.Glisson B, Blumenschein G, Francisco M, Erasmus J, Zinner R, Kies M. Phase II trial of gefitinib in patients with incurable salivary gland cancer [abstract]. Proc Am Soc Clin Oncol. 2005;23:Page. Abstract 5532. [Google Scholar]
- 26.Jakob JA, Kies MS, Glisson BS, et al. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head & Neck 2015;37:644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalin MG, Desrichard A, Katabi N, et al. Comprehensive molecular characterization of salivary duct carcinoma reveals actionable targets and similarity to apocrine breast cancer. Clin Cancer Res. 2016;22:4623–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013;3:112–123. [DOI] [PubMed] [Google Scholar]
- 31.Ho AL, Foster NR, Zoroufy AJ, et al. Alliance A091404: a phase II study of enzalutamide (NSC# 766085) for patients with androgen receptor–positive salivary cancers. J Clin Oncol. 2019;37(15 suppl): 6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.