


Abstract
Proper repair of damaged DNA is crucial for genetic integrity and organismal survival. As semi-autonomous organelles, plastids have their own genomes whose integrity must be preserved. Several factors have been shown to participate in plastid DNA damage repair; however, the underlying mechanism remains unclear. Here, we elucidate a mechanism of homologous recombination (HR) repair in chloroplasts that involves R-loops. We find that the recombinase RecA1 forms filaments in chloroplasts during HR repair, but aggregates as puncta when RNA:DNA hybrids accumulate. ssDNA-binding proteins WHY1/3 and chloroplast RNase H1 AtRNH1C are recruited to the same genomic sites to promote HR repair. Depletion of AtRNH1C or WHY1/3 significantly suppresses the binding of RNA polymerase to the damaged DNA, thus reducing HR repair and modulating microhomology-mediated double-strand break repair. Furthermore, we show that DNA polymerase IB works with AtRNH1C genetically to complete the DNA damage repair process. This study reveals the positive role of R-loops in facilitating the activities of WHY1/3 and RecA1, which in turn secures HR repair and organellar development.
INTRODUCTION
Double-strand breaks (DSBs) are one of the most toxic types of DNA damage. They are caused by both endogenous and exogenous agents, such as oxidative stress, ultraviolet light, and ionizing radiation, and can lead to genomic instability and even cell death (1,2). To prevent deleterious effects, organisms employ two major pathways to repair DSBs: homologous recombination (HR) and non-homologous end joining (NHEJ). HR is an error-free pathway in which identical DNA segments are used as templates. By contrast, NHEJ is an error-prone pathway and can be further divided into two sub-pathways: classical nonhomologous end joining (c-NHEJ) and alternative end joining (alt-EJ). In c-NHEJ, the DSB is repaired by end ligation independently of sequence homology, while alt-EJ requires microhomology recombination (3).
Mitochondria and plastids are the descendants of endosymbiotic bacteria and harbor their own genomes whose integrity must be preserved. It is believed that HR operates in organelles to repair DSBs, since counterparts of most of the HR-associated factors in bacteria have been identified in plant organelles (4,5). Recent studies have also found microhomology-mediated break-induced repair (MMBIR) in plant organelles (6). To date, no direct evidence indicates that c-NHEJ occurs in plant mitochondria or plastids (7–9).
Eubacterial recombinase RecA and its eukaryotic homolog RAD51 are key factors for accurate recombination during HR, assembling into filaments to promote DNA strand invasion and exchange (10). Homologs of RecA are present in the organelles of various plants (11). In Arabidopsis, there are three nuclear-encoded RecA homologs: RecA1 and RecA3 localize to chloroplasts and mitochondria, respectively, and RecA2 localizes to both (11,12). Mutation in RecA1 leads to increased single-stranded DNA (ssDNA) and reduced chloroplast DNA (cpDNA) copy number (13). Both RecA2 and RecA3 control mitochondrial DNA (mtDNA) rearrangement and repair, and loss of RecA2 is lethal (11,12). Several other factors facilitating organellar HR have been identified, including Whirly proteins (WHYs) and DNA polymerase IB (Pol IB). Members of WHYs are plant-specific ssDNA-binding proteins having no sequence-preference (14). Arabidopsis WHY1 and WHY3 participate in the maintenance of chloroplast genomes with functional redundancy, and the why1why3 (hereafter referred to as why1/3) double mutant accumulates rearranged cpDNA through MMBIR (15). A similar effect is observed in mitochondria when the why2 mutant is treated with DSB-inducer ciprofloxacin (CIP) (16). Thus, it was proposed that WHY proteins negatively regulate error-prone MMBIR and consequently facilitate high-fidelity HR-dependent repair (16). Genetic studies indicate that Pol IB synergistically interacts with WHY1 and WHY3 in chloroplasts, as why1/3/pollb triple mutants have a stronger developmental phenotype and more cpDNA rearrangements than why1/3 or pollb mutants (17). why1/3/reca1 triple mutant also shows an aggravated phenotype (18) and the quadruple mutant why1/3/reca1/pollb is unable to survive (18). Apparently, all these factors work in concert to protect the integrity of chloroplast genomes (Supplementary Figure S1). Nevertheless, their precise functions in HR are still obscure.
Over the last decade, unusual nucleic acid structures termed R-loops have been revealed as potent sources of genome instability (19–21). R-loops are formed when the nascent RNA hybridizes with the template DNA, generating an RNA:DNA hybrid and a displaced ssDNA (22). The exposed unpaired ssDNAs in R-loops are vulnerable to genotoxic agents or DNA-modifying enzymes, such as nucleotide excision repair (NER) endonucleases XPF and XPG, leading to DNA damage (23,24). R-loops may even act as obstacles to stall replication-fork progression, which ultimately gives rise to genetic instability (25,26). Conversely, recent studies have uncovered a positive role of R-loops in genome stability, as transient R-loops formed at DSB sites facilitate HR-mediated DNA repair (27–32). Genome-wide mapping shows that R-loops are present throughout genomes ranging from plants to humans (33–35). To prevent the deleterious effects of over-accumulated R-loops, cells possess several factors to suppress the formation of these structures, including RNA binding and processing factors, helicases, DNA replication- and repair-associated factors, and nucleases (36–40). RNase H family proteins are the most important players, specifically cleaving the RNA moiety of the RNA:DNA hybrid (40,41).
Recently, we identified three members of the RNase H1 family in the model plant Arabidopsis, namely AtRNase H1A (AtRNH1A), AtRNase H1B (AtRNH1B) and AtRNase H1C (AtRNH1C). AtRNH1C is mainly localized to chloroplasts and restricts chloroplast R-loop levels to prevent genome instability (42). Interactome analysis has identified some potential interactors of AtRNH1C in chloroplasts, including WHY1 and WHY3 (42). Given the roles of WHY1 and WHY3 in DNA repair (16), we speculated that AtRNH1C and R-loops may also be involved in cpDNA repair. Here, we tested this hypothesis and found that AtRNH1C works in concert with both RecA1 and WHY1/3 in maintaining chloroplast genome integrity. Loss of AtRNH1C suppresses the formation of RecA1 filaments and inhibits the accumulation of WHY1/3 at unstable cpDNA sites. The why1/3/atrnh1c and reca1/atrnh1c mutants show more severe phenotypes than atrnh1c. Remarkably, WHY1/3 are positive regulators of RNA:DNA hybrids through recruiting Plastid-Encoded RNA Polymerases (PEP). Additionally, we found DNA Pol IB also functions genetically with AtRNH1C in DNA repair. Overall, this study deepens our understanding of how R-loops promote HR-mediated DNA damage repair, especially in chloroplasts.
MATERIALS AND METHODS
Plant materials and growth conditions
All Arabidopsis thaliana plants used in this study are in the Columbia (Col-0) background. The why1/3, reca1, pollb, why1/3/reca1 T-DNA mutants have been described previously (13,15,17,18) and were kindly provided by Prof. Normand Brisson (Université de Montréal). OE-AtRNH1C-3xFLAG, OE-AtRNH1C-HA and proAtRNH1C:AtRNH1C3xFLAG/atrnh1c (herein named as CM-AtRNH1C-3xFLAG) were generated in previous work (42,43). All seeds were surface sterilized and sown onto 1/2 Murashige and Skoog (MS) medium plates supplemented with 0.8% phytagar. Plates were kept in the dark for 2 days at 4°C, then moved into growth chamber and cultivated under long-day conditions (16 h light/8 h dark, 22°C/20°C) with an average photon flux density of 120–150 μE m−2 s−1. The germinated seedlings were transferred onto fresh medium or soil for further growth after one week.
For CIP treatment, one-week-old seedlings were transferred onto fresh 1/2 MS medium supplemented with 1 μM CIP for 1 week, and 1/2 MS medium supplemented with DMSO was used as control. Plant materials were harvested and used immediately for further experiments.
Construction of transformation vectors and generation of transgenic plants
The why1/3/atrnh1c, reca1/atrnh1c, why1/3/reca1/atrnh1c and pollb/atrnh1c mutants were produced by CRISPR-Cas9-mediated deletion of the AtRNH1C gene in the corresponding mutant as described previously (42).
To generate transgenic plants expressing RecA1-GFP, the genomic sequence, plus promoter and terminator region, of RecA1 was amplified and cloned into the binary vector pCambia1300. Then the coding sequence of GFP was amplified and inserted into the intermediate construct before the stop code of RecA1. The resulting construct was transferred into Agrobacterium tumefaciens GV3101 and then transformed into Col-0, reca1, and atrnh1c using the floral dip method. Positive transformants were identified through selection for hygromycin resistance and verified by PCR and immunoblot. For RecA1-GFP/reca1/atrnh1c and RecA1-GFP/why1/3/reca1, the RecA1-GFP/reca1 plant was crossed with atrnh1c and why1/3/reca1, respectively. Segregating populations were screened based on hygromycin resistance, the pale-green phenotype of homozygous atrnh1c, and the genotype of reca1 to get homozygotes. The proWHY1:WHY1-3xFLAG/why1 (herein named as CM-WHY1-3xFLAG) complemental line was generated in the same way.
To generate CM-AtRNH1C-3xFLAG/RecA1-GFP/reca1 transgenic plants, the hygromycin-resistance gene in pCambia1300-proAtRNH1C:AtRNH1C-3xFLAG construct, produced previously (42), was replaced with kanamycin-resistance gene. The resulting construct was transformed into RecA1-GFP/reca1/atrnh1c. The T0 seeds were sown onto 1/2 MS medium with kanamycin and hygromycin antibiotics to select transformants.
To generate OE-WHY1-GFP transgenic plants, the coding sequence of WHY1 gene without the stop codon was cloned into binary vector pEarleyGate202, fused with GFP. Then the construct was transformed into Col-0, and transformants were selected in the presence of hygromycin antibiotics.
To generate OE-AtRNH1C-3xFLAG/RecA1-GFP/reca1 transgenic plants, the coding sequence of AtRNH1C without the stop codon was cloned into binary vector pEarleyGate202, fused with 3xFLAG, then the hygromycin-resistance gene was replaced with kanamycin-resistance gene, The resulting construct was transformed into RecA1-GFP/reca1. The T0 seeds were sown onto 1/2 MS medium with kanamycin and hygromycin to select transformants. All the constructs were produced based on the FastCloning method (44). The primers used for cloning are listed in Supplementary Table S1.
Chloroplast isolation and chloroplast DNA extraction
Three-week-old sterile-grown Arabidopsis leaf material was harvested and ground in cold chloroplast isolation buffer (CIB, 10 mM HEPES–KOH (pH 8.0), 150 mM sorbitol, 2.5 mM EDTA (pH 8.0), 2.5 mM EGTA (pH 8.0), 2.5 mM MgCl2, 5 mM NaHCO3, and 0.1% BSA). The homogenate was filtered through three layers of Miracloth (Millipore) and centrifuged at 200 g /4°C for 3 min to remove unwanted whole cells and cell wall debris. Then transferred the supernatant to fresh, chilled 50 ml tubes and centrifuged for 10 min at 1100 g /4°C. The chloroplasts were used for pulsed-field gel electrophoresis (PFGE), DNA:RNA Hybrid Immunoprecipitation (DRIP), and chromatin immunoprecipitation (ChIP).
For slot-blot assay, the crude chloroplasts were further loaded onto 40%/80% Percoll gradient and spun for 15 minutes at 3200 g /4°C with the brake off. Intact chloroplasts were collected from the interface between the 40% and 80% Percoll layers and washed twice in CIB buffer without BSA.
For comet assay, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, and immunofluorescence staining, the intact chloroplasts were extracted using MinuteTM Chloroplast Isolation Kit (Invent) following the manufacturer's instructions.
The chloroplast DNA extraction was performed as previously described (42).
Slot-blot hybridization analysis of chloroplast RNA:DNA hybrids
Chloroplast DNA was treated with 1 U of RNase III (New England Biolabs) per 5 μg of DNA at 37°C for 2 h, then purified with HiPure Gel Pure DNA Mini Kit (Magen). A certain amount of DNA was further treated with 1 U of commercial RNase H (New England Biolabs) per 1 μg of DNA at 37°C for 1 h, then purified again. Different amounts of DNA as indicated in the figure were slotted on to Hybond N+ membrane (GE Healthcare Life Science). Following UV crosslinking at 1200 μJ, the membrane was blocked in 5% milk/TBS-Tween (0.05% Tween20) and incubated overnight with 1 μg/mL S9.6 antibody (purified from HB-8730 (ATCC)). After washing three times for 10 min with TBS-Tween (0.05% Tween20), the membrane was incubated for 1 h at RT with anti-Mouse-Hrp (Easybio). The membrane was washed three times for 10 min with TBS-Tween (0.05% Tween20) then incubated with Super ECL Detection Reagent ECL (Yeasen). S9.6 signals were detected with ImageQuant LAS 4000 (GE Healthcare).
DNA:RNA hybrid immunoprecipitation (DRIP)
Chloroplast DNA was fragmented using 1 U of DdeI, MseI, RsaI and AluI (New England Biolabs) each per 2 μg of DNA at 37°C for 12 h. RNase H pretreated cpDNA of Col-0 was included as negative control. Then the DNA was purified with the phenol–chloroform method and precipitated with 1 volume of isopropyl alcohol at –20°C overnight. 2 μg of fragmented DNA was incubated with 10 μg S9.6 antibody overnight at 4°C. Samples were further incubated with 50 μl Protein G beads (Invitrogen) for 4 h at 4°C. DRIPed DNA was purified with phenol-chloroform as mentioned above. Primers used for qPCR are listed in Supplementary Table S1.
Comet assay
The comet assay was performed as previously described (42) using the Comet Assay kit (Trevigen). Briefly, 10 μL of chloroplast suspension (1 × 106/ml) was mixed with 90 μl of LMAgarose, and 50 μL of each sample was added onto the CometSlide. After solidification, the slides were incubated in lysis solution overnight at 4°C in the dark and then incubated in neutral electrophoresis buffer (50 mM Tris and 150 mM sodium acetate, pH 9.0) for 30 min. The slides were run at 1 V/cm for 20 min in neutral electrophoresis buffer and then incubated in DNA precipitation solution (1 M NH4Ac in 95% ethanol) for 30 min and 70% ethanol for another 30 min. After dried at 37°C, the slides were stained with SYBR Gold for 30 min followed by a wash of water. Samples were visualized by epifluorescence microscopy (Nikon) at 480 nm. The Casplab software (https://casplab.com/) was used for data analysis and quantification of the results.
TUNEL assay
TUNEL assay was performed using TUNEL Apoptosis Assay Kit-FITC (7sea Pharmatech Co. Ltd.) according to the manufacturer's instructions. Briefly, the fixed chloroplasts were adhered to PLL-coated slides and incubated with blocking solution (1% BSA and 0.3% Triton X-100 in 1× PBS) for 30 min at room temperature. The slides were rinsed three times with 1× PBS and then incubated in TdT reaction mixture for 1 h at 37°C in a humidified chamber. After three washes with 1× PBS, the slides were imaged under confocal laser-scanning microscopy Zeiss LSM880. The fluorescence intensity was quantified by using ImageJ software.
Protein coimmunoprecipitation analysis
Five gram of 3-week-old seedlings was used to isolated chloroplasts, which were then lysed with 5 ml buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM DTT, 1 mM PMSF, 1% Triton X-100, 10% glycerol, protease inhibitor (Roche)) for 30 min on ice. After centrifugation, the supernatant was added to prewashed beads (40 μl GFP-Trap magnetic beads (ChromoTek), anti-HA beads (Life Technologies), or 50 μk anti-FLAG M2 magnetic beads (Sigma-Aldrich)) and incubated for 3 h at 4°C with rotation. The beads were separated and washed at least three times with lysis buffer. SDS loading buffer was added to the beads followed by boiling at 100°C for 5 min to release the immunoprecipitated complex. The immunoprecipitated proteins were subjected to SDS-PAGE and immunoblot analyses. 5% or 1% input was loaded as control. The antibodies used for immunoblotting were diluted as follows: anti-HA antibody (Sigma-Aldrich) 1/5000, anti-GFP antibody (ABclonal) 1/5000, anti-FLAG (Sigma-Aldrich) 1/5000, anti-WHY1/3 (produced by our own) 1/5000, anti-Mouse-Hrp (Easybio) 1/10 000, and anti-Rabbit-Hrp (Easybio) 1/10 000. The nuclease sensitivity assay was performed as previously described (42).
Immunofluorescence staining
Intact chloroplasts were fixed in 3.7% formaldehyde for 10 min at room temperature, followed by three washes with 1× PBS. Then, the chloroplasts were resuspended in 8% paraformaldehyde and applied to a PLL-coated slide (Solarbio) for 20 min. After a gentle wash, the slide was covered by blocking solution (1% BSA and 0.1% TritonX-100 in 1× PBS) and incubated for 20 min at room temperature. Subsequently, the chloroplasts were incubated overnight at 4°C with primary antibodies (1:100 dilution in blocking solution). After three washes with 1× PBS, Alexa 594-labeled anti-rabbit (Abcam) and Alexa 488-labeled anti-mouse (Abcam) secondary antibodies were added to the chloroplasts and incubated for 1 h at room temperature. The nucleoid of chloroplast was stained with 4′,6-diamidino-2-phenylindole (DAPI) in mounting medium (SouthernBiotech). The observation was performed with the Zeiss LSM880.
PFGE
The chloroplasts were embedded in 0.5% low-melting-point agarose (Promega) and lysed in 1% sarkosyl, 0.45 M EDTA, 10 mM Tris-HCl (pH 8.0) and 2 mg/mL proteinase K at 48°C for 16 h with shaking. Agarose plugs were washed in TE buffer six times at 4°C, with the first two washes containing 1 mM PMSF, then filled into 1% agarose gel. The gel was subjected to electrophoresis in 0.5 × TBE buffer (45 mM Tris, 45 mM boric acid, 1 mM EDTA, pH 8.0) for 36 h at 14°C using a CHEF Mapper XA system (Bio-Rad), with switch times ramped from 5 to 120 s at 4.5 V/cm. After EtdBr staining and photography, the gel was blotted onto a Hybond N+ membrane (GE Healthcare Life Science). A 505-bp fragment of chloroplast rbcL gene (55677–56181) was labeled with [α-32P]dCTP using Random Primer DNA Labeling Kit Ver. 2 (Takara) and used as probe for hybridization.
Confocal laser-scanning microscopy
For colocalization of WHY1 and AtRNH1C in chloroplasts, the coding sequences of WHY1 and AtRNH1C were cloned into vector pUC19, fused with eGFP, and mCherry, respectively, for transient expression. Arabidopsis leaf mesophyll protoplast isolation and transfection were performed as previously described (45). The transformed protoplasts were cultured for 15 h at room temperature in the dark. The GFP and mCherry fluorescence and chlorophyll autofluorescence were visualized with the Zeiss LSM880.
For morphology observation of RecA1-GFP assemblies, leaves from transgenic lines were detached and mounted on slides with a drop of 1× PBS and observed immediately under Zeiss LSM880. To measure the length of filaments, a series of images were taken using the Z-stack function on the ZEN software and processed into 2D images by the maximum intensity projection method. Then the 2D images were analyzed via ImageJ software.
Chloroplast ChIP
Crude chloroplasts were crosslinked in 1% formaldehyde at 25°C for 10 min with rotation. Glycine was added to a final concentration of 0.125 M to quench the cross-linking. Then the chloroplasts were pelleted and washed twice with CIB, and lysed in buffer containing 50 mM Tris–HCl (pH 7.6), 0.15 M NaCl, 1 mM EDTA (pH 8.0),1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, and plant protease inhibitor cocktail (Sigma-Aldrich). Subsequently, chloroplast DNA was sheared by sonication into fragments of ∼500 bp. Rabbit anti-WHY1/3 antibody and mouse anti-RpoB (PhytoAB) antibody coupled Dynabeads Protein G (Invitrogen), anti-FLAG M2 magnetic beads (Sigma-Aldrich), and GFP-Trap magnetic beads (ChromoTek) were used in corresponding assays to capture the protein–DNA complex. Plants without targeted proteins or beads without antibodies were adopted as negative controls. qPCR was performed on the immunoprecipitated DNA and input DNA using primer sets corresponding to four rDNA regions. The ChIP-reChIP assay was performed according to the protocol reported previously (46), and the cpDNA was sheared into fragments of ∼200 bp. The primer sets are listed in Supplementary Table S1.
Blue native gel electrophoresis
The PEP complex was analyzed by blue-native polyacrylamide gel electrophoresis (BN-PAGE) as previously described (47,48). The three-week-old seedlings were ground in liquid nitrogen and resuspended in 3 volumes of BN-Lysis buffer (100 mM Tris–Cl, pH 7.2; 10 mM MgCl2; 25% glycerol; 1% Triton X-100; 10 mM NaF; 5 mM β-mercaptoethanol; 1× protease inhibitor cocktail). BNSample buffer (1× NativePAGE Sample Buffer, 50 mM 6-aminocaproic acid, 1% n-dodecyl β-d-maltoside, and 1 U/μl benzonase) was added and the samples were incubated for 60 min at room temperature. Samples were mixed with 0.25% NativePAGE Coomassie blue G-250 Sample Additive and centrifuged at 17 500 g for 10 min at 4°C. Proteins from the supernatant were separated on a 4–16% Native-PAGE Bis–Tris protein gel. NativeMark Unstained Protein Standard (Thermo Fisher Scientific) was used to determine protein size. Briefly, electrophoresis was performed at a constant 50 V at 4°C until the blue dye migrated through one-third of the gel. Blue cathode buffer was replaced with cathode buffer without dye, and electrophoresis continued at a constant 100 V overnight (12–14 h) at 4°C. After electrophoresis was complete, the gel was soaked in precooled (10°C) transfer buffer (0.5% SDS, 25 mM Tris–HCl (pH 7.6), 192 mM glycine, 10% methanol) for 30 min. The separated proteins were transferred onto a PVDF membrane using semi-dry blotting apparatus (Bio-Rad) at a constant 15 V for 3 h. After transfer, the membrane was destained with methanol for 3 min and rinsed with ddH2O, and washed with 1× TBST for 5 min, then immunoblotting was performed.
RNA preparation and qPCR
Total RNA was extracted from ∼100 mg of three-week-old plants with Trizol reagent and following the manufacturer's protocol (Invitrogen). The crude RNA extract was subject to DNase I treatment (Thermo Fisher Scientific), then proceeded to reverse transcription using RevertAid RT Reverse Transcription Kit (Thermo Fisher Scientific). The resulting cDNA was diluted 50-fold for qPCR reaction. ACTIN2 was used as reference gene. The primer sets are listed in Supplementary Table S1.
DNA preparation, sequencing and qPCR
Total DNA was extracted from ∼100 mg pools of three-week-old plants using the Hi-DNAsecure Plant Kit (Tiangen) according to the manufacturer's instructions. The DNA quality was assessed on a Nanodrop 2000 (Thermo Fisher Scientific) and the concentration was determined by Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). Five nanograms of DNA were used to prepare the library with TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme). The sequencing was performed on an Illumina HiSeq X Ten instrument. The chloroplast genome rearrangements analysis was conducted as previously reported (18) with some modifications and U-Turn-like rearrangements (<50 bp) were filtered out as they mainly resulted from replication fork collapse. Two biological replicates were performed.
To determine the cpDNA copy number, the isolated DNA was used for qPCR. The AtFTSZ2-1 in the nuclear genome was included as reference gene. The primer sets are listed in Supplementary Table S1.
qPCR analysis
qPCR reactions were performed on a LightCycler 480 Instrument II (Roche) using LightCycler 480 SYBR Green I Master (Roche) following the manufacturer's instructions. Quantitative data were obtained from at least three biological replicates and were analyzed using LightCycler 480 software version 1.5 (Roche).
RESULTS
Accumulated RNA:DNA hybrids disrupt filamentation of RecA1 recombinase
During HR, the double-strand broken DNA ends are processed to generate 3′-ssDNA overhangs for the filamentation of RecA and its homologs (10). Several recent studies show that in nucleus the 3′-ssDNA overhangs are pre-used as templates to produce RNA:DNA hybrids and it is important for the following repair process (27–31). Thus, it would be interesting to determine whether RNA:DNA hybrids are also involved in DSB repair in chloroplasts. As the RNA moiety in RNA:DNA hybrids need to be removed properly for the coating of ssDNA-binding protein RPA, which is then replaced by Rad51 to catalyze HR (27,29,49,50), we wanted to check the effect of accumulated RNA:DNA hybrids on the assembly of chloroplast RecA1 in Arabidopsis. We generated transgenic plants expressing RecA1-GFP under the native promoter (herein named as RecA1-GFP) in Col-0 and atrnh1c backgrounds. We observed the GFP signal in detached leaves from RecA1-GFP/Col-0 and RecA1-GFP/atrnh1c seedlings under confocal microscopy. In both cases, the GFP fluorescence was captured exclusively in chloroplasts (Supplementary Figure S2A). Surprisingly, in Col-0 background, RecA1-GFP mainly formed filaments, while in atrnh1c, it aggregated as puncta. Moreover, this difference was not due to the different expression levels (Supplementary Figure S2B), although the fluorescence intensity was positively related to RecA1-GFP protein levels. Note that the different expression levels under native promoter may be because of different insertion sites in the genome (51).
To rule out the potential influence of different insertion sites and endogenous RecA1 on the formation of filaments, we produced RecA1-GFP/reca1 complementation lines. We then crossed complemented lines with atrnh1c to generate RecA1-GFP/reca1/atrnh1c plants. After obtained the homozygotes, we found that, in RecA1-GFP/reca1, most RecA1-GFP proteins were assembled into filaments, while in RecA1-GFP/reca1/atrnh1c, the majority displayed as puncta (Figure 1A). Additionally, the granular structure did not result from chloroplast defects, since in normally developed chloroplasts, which account for about 28% in the atrnh1c mutant (42), RecA1-GFP also formed puncta (Supplementary Figure S2C). The filamentous structure was restored when the RecA1-GFP/reca1/atrnh1c line was complemented by AtRNH1C (Figure 1A).
Figure 1.

Depletion of AtRNH1C suppresses the assembly of RecA1 filaments. (A) Comparison of the structures of RecA1 in reca1, reca1/atrnh1c, why1/3/atrnh1c and CM-AtRNH1C-3xFLAG/reca1 backgrounds (left panel). For quantification, the lengths of at least 100 fluorescence signals were measured (right panel). Data are mean ± SD. Statistical testing was done using t-test. ****P < 0.0001; n.s, not significant. Bar = 5 μm. (B) ChIP analysis showed the enrichment of RecA1-GFP at rDNA regions. Col-0 was included as negative ChIP control. Four loci in rDNA regions were examined by qPCR. The mitochondrial gene Cox2 was used as negative control locus. The IP/Input ratios were normalized to Col-0. Three biological replicates were performed and indicated as open circles. Graphs represent mean ± SD. Statistical testing was done using multiple t-test. **P < 0.01; ***P < 0.001. (C) Slot-blot assay of the overall R-loops in chloroplast DNA of RecA1-GFP/reca1 and OE-AtRNH1C-3xFLAG/RecA1-GFP/reca1 treated with or without CIP. RNase H treated DNA was used as control. RNA:DNA hybrids were detected using S9.6 antibody (left panel). SYBR Gold staining indicated DNA loading (right panel). (D) The structure of RecA1 in seedlings treated with or without CIP (left panel). For quantification, the length of about 100 fluorescence signals were measured (right panel). Data are mean ± SD. Statistical testing was done using t-test. ****P < 0.0001; n.s, not significant. Bar = 5 μm. (E) PFGE detected the cpDNA damage in seedlings treated with or without CIP. The left panel shows the image of the gel after staining with ethidium bromide. The right panel is blot hybridization of the probe 55677–56181 (a 505-bp rbcL gene fragment). Arrowheads indicate the bands representing monomer and dimer cpDNA molecule.
Next, we compared the size/length of RecA1 filaments. Images of GFP fluorescence were taken in three dimensions and the lengths of signals were measured. The average length of RecA1-GFP filaments was around 3 μm in RecA1-GFP/reca1 but only 1 μm in RecA1-GFP/reca1/atrnh1c (Figure 1A, right panel). As WHY1/3 are believed to work cooperatively with RecA1 in cpDNA repair (18), we wanted to clarify their function on RecA1 assembly by generating RecA1-GFP/why1/3/reca1 transgenic line in the same as RecA1-GFP/reca1/atrnh1c. In fact, loss of WHY1/3 did not affect RecA1 filamentation (Figure 1A). Next, we wondered whether the failed filamentation in the absence of AtRNH1C represents inhibition of RecA1 binding to cpDNA. We performed ChIP-qPCR to examine the accumulation of RecA1 on four loci located in rDNA regions, which are highly unstable due to transcription-replication head-on conflicts (HO-TRCs) (42,43). The binding of RecA1-GFP to cpDNA in atrnh1c was less than 30% of that in complementation plants (Figure 1B).
To further confirm the adverse effect of RNA:DNA hybrids on the filamentation of RecA1, we treated the seedlings of RecA1-GFP/reca1 with ciprofloxacin (CIP), which induces R-loop accumulation and DNA damage in chloroplasts by disrupting the function of AtRNH1C and AtGyrases (42). Consistent with our previous report (42), after CIP treatment, R-loops in chloroplast DNA accumulated as determines by slot blot (Figure 1C),. As a result, the RecA1 structure was converted from filaments to puncta (Figure 1C), and the cpDNA was greatly broken as detected by PFGE (Figure 1E and Supplementary Figure S2E). Overexpressing AtRNH1C could dramatically alleviate R-loop accumulation and the structural conversion (Figure 1D and Supplementary Figure S2D). Moreover, cpDNA damage was also alleviated (Figure 1E and Supplementary Figure S2E). Together, our data suggest that accumulation of RNA:DNA hybrids inhibits RecA1 filamentation and promotes its improper aggregation and AtRNH1C might be required to restrict the hybrids around DSBs for efficient HR.
AtRNH1C works synergistically with WHY1/3
Then, we wondered whether disrupting the filamentous structure of RecA1 would impair its biological function in plant development. A previous study had shown that RecA1 works synergistically with WHY1/3 (18). Under normal growth conditions, both reca1 and why1/3 mutants have no apparent phenotype, while why1/3/reca1 displays strong leaf variegation (18). Thus, we were promoted to determine the effect of failed filamentation by generating why1/3/atrnh1c triple mutant. We introduced atrnh1c null mutation into both reca1 and why1/3 by CRISPR-Cas9-mediated deletion of the AtRNH1C gene (Supplementary Figure S3). As expected, reca1/atrnh1c displayed a similar phenotype as atrnh1c (Figure 2A and C). However, the combination of why1/3 and atrnh1c showed obvious aggravation in phenotype. While the leaf color of atrnh1c was pale-green, the color of why1/3/atrnh1c was nearly white (Figure 2A). The intensity of chlorophyll autofluorescence was further diminished in why1/3/atrnh1c compared to atrnh1c (Figure 2B). Moreover, the plant fresh weight of why1/3/atrnh1c was also dramatically reduced (Figure 2C). Cytological observations showed that the cells of why1/3/atrnh1c contained fewer and smaller chloroplasts than those of atrnh1c and reca1/atrnh1c (Figure 2A). The thylakoids in atrnh1c and reca1/arnh1c were similar, but less ordered and developed in why1/3/atrnh1c (Supplementary Figure S4A). We also tried to obtain the why1/3/reca1/atrnh1c quadruple mutant by knocking out AtRNH1C in the why1/3/reca1 background. The why1/3/reca1/atrnh1c homozygotes were isolated from why1/3/reca1/atrnh1c+/−, displaying pure white leaf color and significantly suppressed growth (Figure 2A). Nevertheless, the seedlings were unable to survive without MS media and sucrose support. These results indicate that the filamentous structure of RecA1 is important for plant development, and AtRNH1C works upstream to ensure its function. Moreover, the more severe phenotype of why1/3/atrnh1c than why1/3/reca1 might be because the function of RecA2 in chloroplast was also impaired (11,12).
Figure 2.
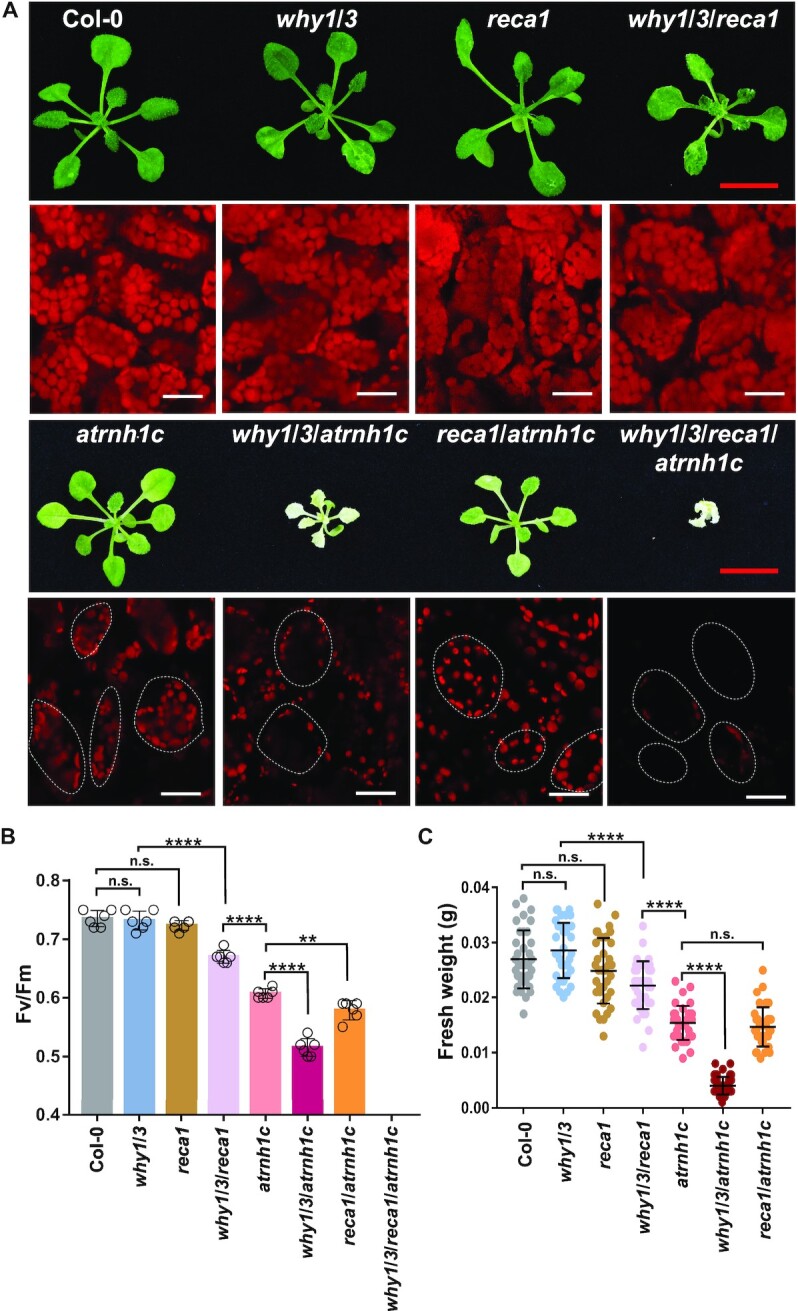
The why1/3/atrnh1c mutant shows severe developmental defects. (A) Phenotypes of three-week-old seedlings of Col-0, why1/3, reca1, why1/3/reca1, atrnh1c, why1/3/atrnh1c, reca1/atrnh1c, and why1/3/reca1/atrnh1c. Cytological observation of chloroplasts (chlorophyll autofluorescence) was conducted under confocal microscopy. Red scale bars, 1 cm. White scale bars 10 μm. (B) The maximum photochemical efficiency of photosystem II (Fv/Fm) of plants indicated in (A). Data shown are mean ± SD, n = 6 individuals. (C) Fresh weight collected from plants indicated in (A). Graphs represent mean ± SD, n ≥ 50 individuals. Statistical testing was done using t-test. **P < 0.01; ****P < 0.0001; n.s, not significant.
why1/3/atrnh1c mutant boosts DNA damage
To further investigate their relationship in chloroplast genome maintenance, we examined the cpDNA damage in these mutants. First, we performed neutral comet assays, which are typically used to detect double-stranded breaks. The DNA damage was represented by the percentage of DNA in the comet tail versus the entire nucleoid. In Col-0 and atrnh1c, the average ratios of tailed DNA were around 7% and 30%, respectively (Figure 3A), in agreement with previous reports (42,43). The why1/3 and reca1 mutants exhibited slightly higher levels of DNA breaks than Col-0, although they were indistinguishable in phenotype, and the level was further increased in why1/3/reca1. Consistent with the defective phenotype, the why1/3/atrnh1c mutant had the highest ratio of tailed DNA, up to about 40% (Figure 3A). We did not detect any significant difference between atrnh1c and reca1/atrnh1c. We obtained a similar cpDNA damage pattern by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Supplementary Figure S4B). Next, we determined the chloroplast genome integrity using PFGE. In Col-0, the cpDNA molecules mainly existed as monomers and oligomers, and these forms were mildly reduced in why1/3 and reca1 (Figure 3B). In why1/3/reca1, atrnh1c, why1/3/atrnh1c and reca1/atrnh1c mutants, large amounts of fragmented DNA were detected, indicating serious cpDNA breaks in these mutants. Strikingly, the lowest band in why1/3/atrnh1c was even smaller than in other mutants (Figure 3B).
Figure 3.

DNA damage in Col-0, why1/3, reca1, why1/3/reca1, atrnh1c, why1/3/atrnh1c and reca1/atrnh1c. (A) Comet assay of cpDNA integrity of each genotype. DNA damage was expressed as the ratio of the signal intensity of a comet tail versus that of the cpDNA. For each genotype, at least 100 chloroplasts were analyzed. Representative images of comets are shown on the left. Statistical testing was done using t-test. **P < 0.01; ****P < 0.0001; n.s, not significant. n.s., not significant. (B) PFGE assay of cpDNA from each genotype. The left panel shows ethidium bromide staining. The right panel shows the result of blot hybridization with the probe 55677–56181 (a 505-bp rbcL gene fragment). Arrowheads indicate the structures of cpDNA monomers and dimers. (C) Chloroplast DNA rearrangement analysis. The whole genomes of indicated genotypic lines were isolated for next-generation sequencing (NGS). The chloroplast rearrangement frequency per 10,000 genomes in each line was depicted in heatmaps. Each tile represents a region spanning 1 kb along each axis. The red bar represents the position of IR region. (D) Normalized number of rearrangements per plastid genome for each of the indicated genotypic lines in (C). Asterisk indicates a significant difference with P-value < 0.00001 using a χ2 test.
As RecA1 and WHY1/3 are also involved in cpDNA replication (18), the exacerbated growth defects in why1/3/atrnh1c and reca1/atrnh1c might be caused by enhanced replication stress. To rule out this possibility, we detected the copy number of cpDNA. We isolated whole genomes and subjected them to next-generation sequencing. The cpDNA sequencing coverage curves for mutants were normalized to Col-0, which indicated changes in the relative copy number of the different cpDNA regions. The results showed that the copy number was indeed changed in atrnh1c, with increase in large single-copy (LSC) region but reduction in inverted repeats (IRs) (Supplementary Figure S5A). Loss-of-function of WHY1/3 or RecA1 aggravated the reduction in IRs of atrnh1c but did not affect LSC region. These patterns were confirmed using quantitative PCR (Supplementary Figure S5B). The increasement of copy number in LSC indicates that the DNA replication is highly activated in atrnh1c, why1/3/atrnh1c, and reca1/atrnh1c. As the replication origins are located in IRs, the apparent reduction of copy number in these regions should be because of extensive DNA breaks and/or degradation, which could hinder DNA amplification. It is consistent with the observations that rDNA regions, located in IRs as well, are more unstable (42,43). These results indicate that the phenotypes are mainly caused by genome instability.
Chloroplast DNA breaks are always accompanied by the accumulation of cpDNA rearrangements, which is promoted by MMBIR, especially when Whirly proteins are depleted (15,18). To investigate the global impact of RecA1, WHY1/3 and/or AtRNH1C depletion on cpDNA rearrangements, we analyzed the above-mentioned next-generation sequencing data. Consistent with the previous results (18), both Col-0 and reca1 had few rearrangements in the whole chloroplast genome, while why1/3 and why1/3/reca1 displayed high frequencies of rearrangement (Figure 3C, D, Supplementary Figure S5C, D, Supplementary Tables S2 and S3). Even though the DNA damage in atrnh1c and reca1/atrnh1c was strikingly increased, the overall cpDNA rearrangements in these two mutants were comparable to Col-0. Moreover, the amount of rearrangement in why1/3/atrnh1c was higher than that in why1/3 but significantly lower than in why1/3/reca1 (Figure 3C D, Supplementary Figure S5C, D, Supplementary Tables S2 and S3). We hypothesized that in the absence of AtRNH1C, the transient RNA:DNA hybrids formed at DSB sites could not be removed effectively, thereby blocking the following DNA repair process both in HR and MMBIR. Taken together, these results suggested that WHY1/3 work synergistically with AtRNH1C in maintaining genome stability.
AtRNH1C and WHY1/3 co-localize on chloroplast DNA
In our previous work, we detected interaction between AtRNH1C and WHY1/WHY3 by Immunoprecipitation-Mass Spectrometry (IP-MS) (42). Here, we found that AtRNH1C works synergistically with WHY1/3, which drove us to further characterize the interaction. First, we performed coimmunoprecipitation (Co-IP) assay with transgenic plants co-expressing WHY1-GFP and AtRNH1C-HA under cauliflower mosaic virus 35S promoter. AtRNH1C-HA coimmunoprecipitated with WHY1-GFP (Figure 4A) and a consistent result was obtained in the reciprocal experiment (Figure 4B). We also conducted yeast two-hybrid and bimolecular fluorescence complementation assays to verify the physical interaction, but neither of them detected the interaction between AtRNH1C and WHY1/3 (data not shown), suggesting the interaction is most likely indirect. Given that the function of these proteins is related to DNA and RNA metabolism, the interaction might rely on different nucleic acid forms. To test this possibility, we repeated the Co-IP assay, with half the immunoprecipitated HA-beads treated with nucleases before immunodetection. After DNase I and/or RNase A treatment, the WHY1-GFP protein was completely lost from immunoprecipitated AtRNH1C-HA (Figure 4C–E). To further confirm the result, we intended to the interaction using endogenous proteins expressed under their own promoters. Thus, we produced an antibody against both WHY1 and WHY3 (Supplementary Figure S6A and S6B, hereafter referred to as anti-WHY1/3 antibody). The Co-IP assay was performed with CM-AtRNH1C-3xFLAG complemental line. The interaction was detected, although lower than that in the plants with AtRNH1C overexpression (Figure 4B and F). Moreover, RNase H treatment eliminated the interaction (Figure 4F). These results indicate that the interaction we detected between AtRNH1C and WHY1 was RNA:DNA hybrids dependent. Subsequently, we tested whether AtRNH1C and WHY1 co-localize in chloroplasts. We cloned the coding sequences of AtRNH1C and WHY1 into expression vectors in fusion with mCherry and eGFP, respectively, and co-transformed the vectors into Arabidopsis protoplasts. Results from fluorescence microscopy showed that both AtRNH1C and WHY1 were localized in chloroplasts, as in previous reports (42,52), and many of their fluorescence signals overlapped (Figure 4G). We sought to examine whether the co-localization of AtRNH1C and WHY1 occurred at the chloroplast nucleoid. We isolated the chloroplasts of transgenic plants overexpressing AtRNH1C-3xFLAG and subjected them to immunofluorescence (IF) assays with anti-WHY1/3 antibody and anti-FLAG antibody simultaneously. The signals of WHY1/3 significantly overlapped with those of AtRNH1C-3xFLAG and co-localized in nucleoids indicated by DAPI staining (Figure 4H). Moreover, we conducted ChIP-reChIP assay to examine the physical proximity of these two proteins. Chloroplast chromatin of OE-AtRNH1C-3xFLAG was sheared extensively into ∼200 bp (Figure 4I, right panel) and immunoprecipitated sequentially by anti-WHY1/3 antibody and anti-FLAG beads. The results showed that WHY1/3 and AtRNH1C co-occupy the same regions in cpDNA (Figure 4I, left panel). These data suggest that AtRNH1C and WHY1/3 are co-localized, and most possibly function at the same sites in chloroplast nucleoids for maintaining genome stability.
Figure 4.

AtRNH1C and WHY1/3 co-localize at the same sites in the chloroplast genome. (A, B) WHY1 coimmunoprecipitation with AtRNH1C. The three-week-old seedlings of OE-WHY1-GFP/OE-AtRNH1C-HA were used for immunoprecipitation with anti-GFP (A) and anti-HA (B) beads. Then the immunoprecipitated protein was detected with anti-HA and anti-GFP antibodies. (C–E) WHY1 co-immunoprecipitation with AtRNH1C depends on DNA/RNA. After immunoprecipitation with anti-HA beads as performed in (B), anti-GFP antibody was used to detect WHY1 protein. RT-PCR and digestion control were performed by amplifying an rbcL fragment from cDNA reverse transcribed from RNA or DNA isolated from beads with or without nuclease treatment, and primers are listed in Supplementary Table S1. (F) Co-IP assay with CM-AtRNH1C-3xFLAG complemental line. The anti-FLAG beads were used for immunoprecipitation. Col-0 was included to indicate the specificity of anti-FLAG beads. AtRNH1C-3xFLAG and WHY1/3 were detected with anti-FLAG and anti-WHY1/3 antibodies. RNase H treatment was performed to examine the dependency on RNA:DNA hybrids. (G) Fluorescence micrographs showing the co-localization of WHY1 with AtRNH1C. Plasmids encoding fusion proteins WHY1-mCherry and AtRNH1C-eGFP were transiently expressed in Arabidopsis protoplasts. Red indicates chloroplast autofluorescence; magenta indicates mCherry fluorescence; green indicates eGFP signals. (H) Immunofluorescence analysis of the co-localization of WHY1/3 and AtRNH1C. The chloroplasts from OE-AtRNH1C-3xFLAG plants were used for detection. Red indicates chloroplast autofluorescence; yellow indicates WHY1/3 proteins detected by anti-WHY1/3 antibody; green indicates AtRNH1C-3xFLAG proteins detected by anti-FLAG antibody; blue indicates the nucleoid detected by DAPI staining. (I) ChIP-reChIP analysis of AtRNH1C and WHY1/3 co-occupation at chloroplast genome. The chloroplast chromatin of OE-AtRNH1C-3xFLAG was first immunoprecipitated by anti-WHY1/3 polyclonal antibodies and then by anti-FLAG antibodies. Protein G was used as a negative control. Four loci indicated in Figure 1B were used for qPCR. The left panel shows mean ± SD (n = 3 biologically independent samples, indicated as open circles). The right panel shows one representative gel image of sonicated cpDNA. M, Marker; S, Sonicated.
WHY1/3 work in concert with AtRNH1C to regulate RNA:DNA hybrids
Whirly proteins can nonspecifically bind to ssDNA overhangs to suppress error-prone MMBIR (16). We wondered whether the almost unaffected rearrangement in the atrnh1c mutant despite remarkable DNA damage is due to enhanced binding of WHY1/3 to cpDNA. To test this possibility, we first analyzed the protein levels of WHY1/3 in Col-0 and mutant lines. In reca1, WHY1/3 protein level was comparable to Col-0, while in both atrnh1c and reca1/atrnh1c, the protein levels were significantly increased (Supplementary Figure S7A). Next, we compared the enrichment of WHY1/3 on chloroplast genomes among Col-0, reca1 and atrnh1c by performing ChIP-qPCR at the rDNA regions. Contrary to our assumption, the WHY1/3 enrichment was significantly lower in atrnh1c (Figure 5A). However, the WHY1/3 enrichment was obviously increased at these loci in the reca1 mutant.
Figure 5.

WHY1/3 works in concert with AtRNH1C to regulate R-loops. (A) ChIP analysis showed the enrichment of WHY1/3 at rDNA regions. The why1/3 mutant line was included as negative ChIP control. The IP/Input ratios were normalized to why1/3. (B) DRIP was performed with cpDNA isolated from Col-0, why1/3, reca1, why1/3/reca1, atrnh1c, why1/3/atrnh1c and reca1/atrnh1c. cpDNA of Col-0 pretreated with RNase H was included as negative control. The DRIP/Input ratios were normalized to Col-0. (C) Immunoblots showing the level of the PEP complex (BN-PAGE) as well as the total amount of RpoB (SDS-PAGE) in Col-0, why1/3, reca1, atrnh1c, why1/3/reca1, why1/3/atrnh1c and reca1/atrnh1c. Actin was used as loading control. The relative protein level of RpoB was quantified with ImageJ and indicated at the bottom. (D) ChIP analysis showed the enrichment of RpoB at rDNA regions. The Col-0 incubated with Protein G was used as negative ChIP control. (E) ChIP analysis showed the enrichment of AtRNH1C-3xFLAG at rDNA regions. The Col-0 was used as a negative control. The primers used in Figure 1B were adapted for qPCR in these experiments. The IP/Input ratios were normalized to negative ChIP control. Three biological replicates were performed. Graphs represent mean ± SD. The significance was calculated by t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Recently, several studies have demonstrated that the transient RNA:DNA hybrids induced around DSB sites could promote HR, thus minimizing illegitimate recombination (29,31,32). Therefore, we determined the levels of RNA:DNA hybrids in cpDNA with DRIP-qPCR. The results showed that RNA:DNA hybrids were accumulated in atrnh1c but decreased in the why1/3 mutant (Figure 5B). why1/3/reca1 also had fewer RNA:DNA hybrids than Col-0. Interestingly, the level of RNA:DNA hybrids in why1/3/atrnh1c were slightly lower than that in atrnh1c, while they were even higher in some regions (5S, 16S) in reca1/atrnh1c. These results suggest that WHY1/3 are important in suppressing MMBIR; however, this suppression role is less important in RNA:DNA hybrids accumulated mutant atrnh1c than that in reca1. This agrees with the results of cpDNA rearrangements (Figure 3C, D, Supplementary Figure S5C and D).
The formation of RNA:DNA hybrids around DSBs requires the recruitment of different RNA polymerases (27,29,32). Thus, it is reasonable to check the binding of RNA polymerase to cpDNA. In chloroplasts, there are two types of RNA polymerase: nuclear-encoded RNA polymerase (NEP) and plastid-encoded RNA polymerase (PEP), both of which are active at all stages of development. PEP plays a major role in rRNA synthesis (53) and is involved in R-loop formation in some conditions (43). Thus, we determined the loading of PEP on cpDNA. The expression levels of the four core subunits of PEP were similar in Col-0, why1/3, and reca1 and slightly upregulated in why1/3/reca1; however, they were significantly increased in atrnh1c, why1/3/atrnh1c and reca1/atrnh1c (Supplementary Figure S8A). We resolved the PEP complex using blue native gel electrophoresis and monitored the level by immunoblotting using antibodies against RpoB. In line with the expression level, the PEP complex level was unaffected in Col-0, why1/3 and reca1, but increased in atrnh1c, why1/3/atrnh1c and reca1/atrnh1c (Figure 5C). Then, we performed ChIP assay with a RpoB-specific antibody to examine the loading of the PEP complex on cpDNA. The binding of RpoB to cpDNA was significantly reduced in both atrnh1c and why1/3, and further reduced in why1/3/atrnh1c in the regions we tested (Figure 5D). Similar to the changes of R-loop levels (Figure 5B), the cpDNA affinity of RpoB in reca1 was indistinguishable from that in Col-0. Nevertheless, the expression of most PEP-dependent genes was not significantly affected in why1/3 and even increased in atrnh1c (Supplementary Figure S8B), indicating that the declined recruitment of PEP to rDNA regions was not because of reduction in protein abundance or transcription activity. Moreover, loss of WHY1/3 also reduced the binding of AtRNH1C to these sites (Figure 5E and Supplementary Figure S7B). Taken together, our results suggest that, in chloroplasts, RNA:DNA hybrids could prevent the broken DNA from error-prone repair, as shown in other organisms (30,31), and ssDNA binding proteins WHY1/3 could facilitate RNA:DNA hybrids generation by recruiting RNA polymerase to the damaged sites, successively, the hybrids are resolved by AtRNH1C for the following repair.
Combination of atrnh1c and polIb aggravated the chloroplast genome instability
Because Arabidopsis DNA Pol IB has been shown to maintain the integrity of chloroplast DNA, mostly in the last step of synthesis to complete the gapped genome (17,54,55), we were prompted to test the relationship between AtRNH1C and Pol IB. To address this question, we established a polIb/atrnh1c double mutant by CRISPR-Cas9-mediated deletion of the AtRNH1C gene in polIb (Supplementary Figure S3). First, we checked the growth phenotype of plants. Although polIb plants looked similar to Col-0, and atrnh1c exhibited the pale-green phenotype as described previously (42,43), the polIb/atrnh1c double mutant clearly showed more severe growth defects than atrnh1c (Figure 6A). The intensity of chlorophyll autofluorescence was further reduced in polIb/atrnh1c (Supplementary Figure S9) and chloroplast development was seriously inhibited (Figure 6A). Next, we examined the integrity of cpDNA by PFGE and detected more fragmented DNA in polIb/atrnh1c than in other plants (Figure 6B). These results suggest that Pol IB and AtRNH1C may work concertedly in maintaining genome integrity.
Figure 6.

Combination of atrnh1c and polIb enhanced the chloroplast genome instability. (A) Phenotypes of three-week-old seedlings of Col-0, pollb, atrnh1c, and pollb/atrnh1c. Cytological observation of chloroplasts (chlorophyll autofluorescence) was conducted under confocal microscopy. Red scale bars, 1 cm. White scale bars, 10 μm. (B) PFGE assay of cpDNA from plants indicated. The assay was performed as described in Figure 1E. Arrowheads indicate the bands representing monomer, dimer, and trimer cpDNA molecule. (C) During DSBR, WHY1/3 protect ssDNA overhang from MMBIR and recruit PEP to generate RNA:DNA hybrids at DSB ends. Then the hybrids are resolved by AtRNH1C, allowing RecA1 filamentation for succeeding HR.
DISCUSSION
It has been proposed that Whirly proteins maintain genome stability in organelles by favoring accurate repair of DNA DSBs over an error-prone MMBIR pathway (15,16). The binding of Whirly to ssDNA generated during DNA damage is believed to be important, but the mechanism remains mysterious. Here, we revealed that deletion of WHY1/3 reduced PEP enrichment and thus decreased the RNA:DNA hybrid levels in the rDNA regions of cpDNA (Figure 5B and D). There is a growing body of evidence indicating that RNA:DNA hybrids specifically accumulate at DSB sites and contribute to HR-mediated DNA repair by recruiting repair-associated factors (27–32). Thus, our data suggest that WHY1/3 could fulfill this function through promoting the generation of RNA:DNA hybrids by RNA polymerase. In our previous work, we characterized another nuclear ssDNA-binding protein, NDX1, that can stabilize RNA:DNA hybrids in the COOLAIR promoter region (56). WHY1/3 may also protect RNA:DNA hybrids from rapid removal to facilitate HR.
Although RNA:DNA hybrids are required for efficient DSB repair, they must be properly removed for the subsequent processes (27,29,31). In Schizosaccharomyces pombe, RNase H and senataxin are required to limit RNA:DNA hybrids at DSBs. Losing either of them suppresses HR-mediated DSB repair (27,31). Moreover, RNase H2 is recruited to DSBs to resolve RNA:DNA hybrids in HeLa cells (29). In our study, deletion of AtRNH1C led to the accumulation of broken DNA in chloroplasts (Figure 3A and B). However, error-prone repair-mediated rearrangements were not significantly accumulated (Figure 3C, D, Supplementary Figure S5C and D). We propose that AtRNH1C is required to remove RNA:DNA hybrids generated at DSBs in chloroplasts. In the absence of AtRNH1C, MMBIR was suppressed because of the presence of Whirly proteins WHY1/3, and HR-mediated repair was suppressed because the accumulation of RNA:DNA hybrids transformed RecA1 filamentation into puncta and thus inhibited downstream HR processes (Figure 6C).
RecA is a defined member of a class of proteins that are essential for HR across all forms of life (57). Three RecA-like proteins with organelle localization had been identified in Arabidopsis. In our study, we explored the role of RecA1 in cpDNA damage response, as RecA1 is solely targeted to chloroplasts, whereas RecA2 is localized to both chloroplasts and mitochondria (12). Additionally, RecA1 is suggested to work in concert with Whirly to maintain chloroplast genome stability (18). To function, RecA should nucleate and assemble into filaments. While previous reports only determined the subcellular localization of RecA1 (12), we clearly observed the filamentous structure of RecA1 (Figure 1A). Strikingly, the filaments were widely present in chloroplasts in the Col-0 background, suggesting that HR-mediated repair is extensively taking place. This might be the reason that the cpDNA suffers DNA damage all the time, but the genome is even more stable than the nuclear genome (58,59). The genome stability assays in this study also detected DNA damaged even in Col-0 (Figures 3 and 6B). Different from Escherichia coli RecA, which fulfills the function of recombination mainly during cell proliferation when the homologous sequence is available (10), RecA1 may be able to work beyond the division period, as chloroplasts have more than one copy of the genome (60). Interestingly, in most cases, there is only one filament in each chloroplast. We speculate that, in order to efficiently and accurately search for the homologous sequence, the vast majority of RecA1 is recruited to one place in the chloroplast for proper filamentation. A similar phenomenon has been observed in Escherichia coli (61).
In atrnh1c or after CIP treatment, RecA1 mainly aggregated as puncta instead of filaments when R-loops accumulated (Figure 1A and C). We further demonstrated that this is not due to different protein levels or transformants (Supplementary Figure S2A and S2B). Moreover, the size of puncta is indeed significantly correlated with the RecA1 protein levels (Supplementary Figure S2A). This is reminiscent of the feature that RecA1 can self-assemble into puncta as storage structures, with the size proportional to the protein level (62). However, in atrnh1c, the DNA damage was severe, thus one can infer that the deficiency of AtRNH1C represses the DNA-damage response of RecA1, which is in line with previous studies (27,31). Recently, it was reported that DSB-induced RNA synthesis stimulates liquid-liquid phase separation (LLPS) of DNA-damage-response factor (63). Moreover, the Escherichia coli single-stranded DNA-binding proteins (SSB) could form LLPS condensates (64). Therefore, RecA1-GFP puncta could be self-assembled storage structures or LLPS condensates. It is also possible that, in absence of AtRNH1C, there are still very short ssDNAs exposed at DSB ends in where RecA1 might be aggregated.
The addition of why1/3 null mutation aggravated the DNA damage in atrnh1c (Figures 2 and 3), indicating that, in absence of AtRNH1C, WHY1/3 still execute its function. Recently, we characterized an R-loop helicase RHON1 in chloroplast, which works in parallel with AtRNH1C to resolve RNA:DNA hybrids (43). Thus, WHY1/3 may co-operate with RHON1 to promote HR in the same way as with AtRNH1C. Besides, the reca1/atrnh1c double mutant also exhibited a slightly worse phenotype comparing to atrnh1c (Figure 2A and B). These results indicate that RecA1 may participate in other pathways in maintaining chloroplast genome stability as well. It will be interesting to explore the relationship of RHON1 to WHY1/3 and RecA1. Moreover, the potential impairment of DNA replication may also partially account for aggravated phenotype, as unrepaired damage could induce DNA replication collapse and vice versa (65).
In summary, we propose a model (Figure 6C) in which WHY1/3 protect 3′-ssDNA overhangs generated by DNA-end resection from MMBIR and promote the recruitment of PEP RNA polymerase. Then RNA:DNA hybrids were produced through transcription, probably to activate unknown repair-associated factors and/or prevent degradation of ssDNA. Subsequently, the RNA:DNA hybrids are resolved by AtRNH1C to expose the ssDNA for RecA1 filamentation, thus ensuring efficiently HR-mediated repair. If the RecA1 cannot bind in time, the ssDNA may need to be protected by WHY1/3 again. As WHY1/3 and RecA1 are also supposed to secure DNA replication (18), we think our model should apply to the single-end DSB generated as replication fork collapse as well.
DATA AVAILABILITY
All newly generated sequencing data have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number PRJNA698745.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Core Facility of the Center of Biomedical Analysis, Tsinghua University, for assistance with Confocal Microscopy analysis, and Prof. Normand Brisson (Université de Montréal) for why1/3, reca1, pollb and why1/3/reca1 T-DNA mutants.
Authors contributions: Q. Sun and W. Wang conceived and designed the experiments. K. Li analyzed the NGS data (Figure 3C and Supplementary Figure S5C). Z. Yang and W. Wang did the DRIP-qPCR and ChIP-qPCR (Figure 5B and D). Q. Hou did the Co-IP assay (Figure 4A-E) and helped W. Wang and Wei W Zhao to generate the plant materials, and W. Wang performed all the remaining experiments. W. Wang and Q. Sun wrote the manuscript, and all authors read and approved the final manuscript.
Contributor Information
Wenjie Wang, Center for Plant Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Beijing 100084, China.
Kuan Li, Center for Plant Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Beijing 100084, China.
Zhuo Yang, Center for Plant Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Quancan Hou, Center for Plant Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China.
Wei W Zhao, Center for Plant Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Beijing 100084, China.
Qianwen Sun, Center for Plant Biology, School of Life Sciences, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Beijing 100084, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by grants from the Ministry of Science and Technology of China [2016YFA0500800] and National Natural Science Foundation of China [91940306, 91740105, 31822028]. The Sun Lab is supported by Tsinghua-Peking Center for Life Sciences. W. Wang is supported by a postdoctoral fellowship from Tsinghua-Peking Center for Life Sciences. Funding for open access charge: Ministry of Science and Technology of China [2016YFA0500800]; National Natural Science Foundation of China [91940306, 91740105, 31822028].
Conflict of interest statement. None declared.
REFERENCES
- 1. Manova V., Gruszka D.. DNA damage and repair in plants - from models to crops. Front. Plant Sci. 2015; 6:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scully R., Panday A., Elango R., Willis N.A.. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019; 20:698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang H.H.Y., Pannunzio N.R., Adachi N., Lieber M.R.. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017; 18:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marechal A., Brisson N.. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010; 186:299–317. [DOI] [PubMed] [Google Scholar]
- 5. Gualberto J.M., Newton K.J.. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 2017; 68:225–252. [DOI] [PubMed] [Google Scholar]
- 6. Kwon T., Huq E., Herrin D.L.. Microhomology-mediated and nonhomologous repair of a double-strand break in the chloroplast genome of Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:13954–13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kohl S., Bock R.. Transposition of a bacterial insertion sequence in chloroplasts. Plant J. 2009; 58:423–436. [DOI] [PubMed] [Google Scholar]
- 8. Kazama T., Okuno M., Watari Y., Yanase S., Koizuka C., Tsuruta Y., Sugaya H., Toyoda A., Itoh T., Tsutsumi N.et al.. Curing cytoplasmic male sterility via TALEN-mediated mitochondrial genome editing. Nat Plants. 2019; 5:722–730. [DOI] [PubMed] [Google Scholar]
- 9. Chevigny N., Schatz-Daas D., Lotfi F., Gualberto J.M.. DNA repair and the stability of the plant mitochondrial genome. Int. J. Mol. Sci. 2020; 21:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bell J.C., Kowalczykowski S.C.. RecA: Regulation and Mechanism of a Molecular Search Engine. Trends Biochem. Sci. 2016; 41:491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller-Messmer M., Kuhn K., Bichara M., Le Ret M., Imbault P., Gualberto J.M.. RecA-dependent DNA repair results in increased heteroplasmy of the Arabidopsis mitochondrial genome. Plant Physiol. 2012; 159:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shedge V., Arrieta-Montiel M., Christensen A.C., Mackenzie S.A.. Plant mitochondrial recombination surveillance requires unusual RecA and MutS homologs. Plant Cell. 2007; 19:1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rowan B.A., Oldenburg D.J., Bendich A.J.. RecA maintains the integrity of chloroplast DNA molecules in Arabidopsis. J. Exp. Bot. 2010; 61:2575–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prikryl J., Watkins K.P., Friso G., van Wijk K.J., Barkan A.. A member of the Whirly family is a multifunctional RNA- and DNA-binding protein that is essential for chloroplast biogenesis. Nucleic Acids Res. 2008; 36:5152–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marechal A., Parent J.S., Veronneau-Lafortune F., Joyeux A., Lang B.F., Brisson N.. Whirly proteins maintain plastid genome stability in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:14693–14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cappadocia L., Marechal A., Parent J.S., Lepage E., Sygusch J., Brisson N.. Crystal structures of DNA-Whirly complexes and their role in Arabidopsis organelle genome repair. Plant Cell. 2010; 22:1849–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parent J.S., Lepage E., Brisson N.. Divergent roles for the two PolI-like organelle DNA polymerases of Arabidopsis. Plant Physiol. 2011; 156:254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zampini E., Lepage E., Tremblay-Belzile S., Truche S., Brisson N.. Organelle DNA rearrangement mapping reveals U-turn-like inversions as a major source of genomic instability in Arabidopsis and humans. Genome Res. 2015; 25:645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aguilera A., Garcia-Muse T.. R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 2012; 46:115–124. [DOI] [PubMed] [Google Scholar]
- 20. García-Muse T., Aguilera A.. R Loops: from Physiological to Pathological Roles. Cell. 2019; 179:604–618. [DOI] [PubMed] [Google Scholar]
- 21. Crossley M.P., Bocek M., Cimprich K.A.. R-Loops as cellular regulators and genomic threats. Mol. Cell. 2019; 73:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santos-Pereira J.M., Aguilera A.. R loops: new modulators of genome dynamics and function. Nat. Rev. Genet. 2015; 16:583–597. [DOI] [PubMed] [Google Scholar]
- 23. Sollier J., Stork C.T., Garcia-Rubio M.L., Paulsen R.D., Aguilera A., Cimprich K.A.. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell. 2014; 56:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sollier J., Cimprich K.A.. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015; 25:514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamperl S., Bocek M.J., Saldivar J.C., Swigut T., Cimprich K.A.. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017; 170:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gan W., Guan Z., Liu J., Gui T., Shen K., Manley J.L., Li X.. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011; 25:2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ohle C., Tesorero R., Schermann G., Dobrev N., Sinning I., Fischer T.. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell. 2016; 167:1001–1013. [DOI] [PubMed] [Google Scholar]
- 28. Yasuhara T., Kato R., Hagiwara Y., Shiotani B., Yamauchi M., Nakada S., Shibata A., Miyagawa K.. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell. 2018; 175:558–570. [DOI] [PubMed] [Google Scholar]
- 29. D’Alessandro G., Whelan D.R., Howard S.M., Vitelli V., Renaudin X., Adamowicz M., Iannelli F., Jones-Weinert C.W., Lee M., Matti V.et al.. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun. 2018; 9:5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu W.T., Hawley B.R., Skalka G.L., Baldock R.A., Smith E.M., Bader A.S., Malewicz M., Watts F.Z., Wilczynska A., Bushell M.. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018; 9:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen S., Puget N., Lin Y.L., Clouaire T., Aguirrebengoa M., Rocher V., Pasero P., Canitrot Y., Legube G.. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018; 9:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu S., Hua Y., Wang J., Li L., Yuan J., Zhang B., Wang Z., Ji J., Kong D.. RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell. 2021; 184:1314–1329. [DOI] [PubMed] [Google Scholar]
- 33. Costantino L., Koshland D.. Genome-wide map of R-loop-induced damage reveals how a subset of R-loops contributes to genomic instability. Mol. Cell. 2018; 71:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu W., Xu H., Li K., Fan Y., Liu Y., Yang X., Sun Q.. The R-loop is a common chromatin feature of the Arabidopsis genome. Nat Plants. 2017; 3:704–714. [DOI] [PubMed] [Google Scholar]
- 35. Chan Y.A., Aristizabal M.J., Lu P.Y., Luo Z., Hamza A., Kobor M.S., Stirling P.C., Hieter P.. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 2014; 10:e1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tuduri S., Crabbe L., Conti C., Tourriere H., Holtgreve-Grez H., Jauch A., Pantesco V., De Vos J., Thomas A., Theillet C.et al.. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009; 11:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dominguez-Sanchez M.S., Barroso S., Gomez-Gonzalez B., Luna R., Aguilera A.. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011; 7:e1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mischo H.E., Gomez-Gonzalez B., Grzechnik P., Rondon A.G., Wei W., Steinmetz L., Aguilera A., Proudfoot N.J.. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell. 2011; 41:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bhatia V., Barroso S.I., Garcia-Rubio M.L., Tumini E., Herrera-Moyano E., Aguilera A.. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature. 2014; 511:362–365. [DOI] [PubMed] [Google Scholar]
- 40. Wahba L., Amon J.D., Koshland D., Vuica-Ross M.. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell. 2011; 44:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reijns M.A., Rabe B., Rigby R.E., Mill P., Astell K.R., Lettice L.A., Boyle S., Leitch A., Keighren M., Kilanowski F.et al.. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012; 149:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Z., Hou Q., Cheng L., Xu W., Hong Y., Li S., Sun Q.. RNase H1 cooperates with DNA gyrases to restrict R-loops and maintain genome integrity in Arabidopsis chloroplasts. Plant Cell. 2017; 29:2478–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang Z., Li M., Sun Q.. RHON1 co-transcriptionally resolves R-loops for Arabidopsis chloroplast genome maintenance. Cell Rep. 2020; 30:243–256. [DOI] [PubMed] [Google Scholar]
- 44. Li C., Wen A., Shen B., Lu J., Huang Y., Chang Y.. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotech. 2011; 11:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yoo S.D., Cho Y.H., Sheen J.. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2007; 2:1565–1572. [DOI] [PubMed] [Google Scholar]
- 46. Furlan-Magaril M., Rincón-Arano H., Recillas-Targa F.. Leblanc B., Moss T.. Sequential chromatin immunoprecipitation protocol: ChIP-reChIP. DNA-Protein Interactions: Principles and Protocols. 2009; 3rd ednTotowa, NJ: Humana Press; 253–266. [DOI] [PubMed] [Google Scholar]
- 47. Yang E.J., Yoo C.Y., Liu J., Wang H., Cao J., Li F.W., Pryer K.M., Sun T.P., Weigel D., Zhou P.et al.. NCP activates chloroplast transcription by controlling phytochrome-dependent dual nuclear and plastidial switches. Nat. Commun. 2019; 10:2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pfalz J. Investigating the assembly status of the plastid encoded polymerase using BN-PAGE and sucrose gradient centrifugation. Bio-protocol. 2016; 6:e1873. [Google Scholar]
- 49. Sessa G., Gomez-Gonzalez B., Silva S., Perez-Calero C., Beaurepere R., Barroso S., Martineau S., Martin C., Ehlen A., Martinez J.S.et al.. BRCA2 promotes R-loop resolution by DDX5 helicase at DNA breaks to facilitate their repair by homologous recombination. EMBO J. 2021; 40:e106018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. San Filippo J., Sung P., Klein H.. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008; 77:229–257. [DOI] [PubMed] [Google Scholar]
- 51. Shapiguzov A., Vainonen J.P., Hunter K., Tossavainen H., Tiwari A., Jarvi S., Hellman M., Aarabi F., Alseekh S., Wybouw B.et al.. Arabidopsis RCD1 coordinates chloroplast and mitochondrial functions through interaction with ANAC transcription factors. Elife. 2019; 8:e43284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krause K., Kilbienski I., Mulisch M., Rodiger A., Schafer A., Krupinska K.. DNA-binding proteins of the Whirly family in Arabidopsis thaliana are targeted to the organelles. FEBS Lett. 2005; 579:3707–3712. [DOI] [PubMed] [Google Scholar]
- 53. Borner T., Aleynikova A.Y., Zubo Y.O., Kusnetsov V.V.. Chloroplast RNA polymerases: role in chloroplast biogenesis. Biochim. Biophys. Acta. 2015; 1847:761–769. [DOI] [PubMed] [Google Scholar]
- 54. Baruch-Torres N., Brieba L.G.. Plant organellar DNA polymerases are replicative and translesion DNA synthesis polymerases. Nucleic Acids Res. 2017; 45:10751–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Garcia-Medel P.L., Baruch-Torres N., Peralta-Castro A., Trasvina-Arenas C.H., Torres-Larios A., Brieba L.G.. Plant organellar DNA polymerases repair double-stranded breaks by microhomology-mediated end-joining. Nucleic Acids Res. 2019; 47:3028–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun Q., Csorba T., Skourti-Stathaki K., Proudfoot N.J., Dean C.. R-Loop stabilization represses antisense transcription at the Arabidopsis FLC locus. Science. 2013; 340:619–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin Z., Kong H., Nei M., Ma H.. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. USA. 2006; 103:10328–10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Green B.R. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011; 66:34–44. [DOI] [PubMed] [Google Scholar]
- 59. Wolfe K.H., Li W.H., Sharp P.M.. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jarvis P., Lopez-Juez E.. Biogenesis and homeostasis of chloroplasts and other plastids. Nat. Rev. Mol. Cell Biol. 2013; 14:787–802. [DOI] [PubMed] [Google Scholar]
- 61. Lesterlin C., Ball G., Schermelleh L., Sherratt D.J.. RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature. 2014; 506:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ghodke H., Paudel B.P., Lewis J.S., Jergic S., Gopal K., Romero Z.J., Wood E.A., Woodgate R., Cox M.M., van Oijen A.M.. Spatial and temporal organization of RecA in the Escherichia coli DNA-damage response. Elife. 2019; 8:e42761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pessina F., Giavazzi F., Yin Y., Gioia U., Vitelli V., Galbiati A., Barozzi S., Garre M., Oldani A., Flaus A.et al.. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 2019; 21:1286–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Harami G.M., Kovacs Z.J., Pancsa R., Palinkas J., Barath V., Tarnok K., Malnasi-Csizmadia A., Kovacs M.. Phase separation by ssDNA binding protein controlled via protein-protein and protein-DNA interactions. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:26206–26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Branzei D., Foiani M.. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010; 11:208–219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All newly generated sequencing data have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number PRJNA698745.