


Abstract
The INO80 chromatin remodeler is involved in many chromatin-dependent cellular functions. However, its role in pluripotency and cell fate transition is not fully defined. We examined the impact of Ino80 deletion in the naïve and primed pluripotent stem cells. We found that Ino80 deletion had minimal effect on self-renewal and gene expression in the naïve state, but led to cellular differentiation and de-repression of developmental genes in the transition toward and maintenance of the primed state. In the naïve state, INO80 pre-marked gene promoters that would adopt bivalent histone modifications by H3K4me3 and H3K27me3 upon transition into the primed state. In the primed state, in contrast to its known role in H2A.Z exchange, INO80 promoted H2A.Z occupancy at these bivalent promoters and facilitated H3K27me3 installation and maintenance as well as downstream gene repression. Together, our results identified an unexpected function of INO80 in H2A.Z deposition and gene regulation. We showed that INO80-dependent H2A.Z occupancy is a critical licensing step for the bivalent domains, and thereby uncovered an epigenetic mechanism by which chromatin remodeling, histone variant deposition and histone modification coordinately control cell fate.
Graphical Abstract
Graphical Abstract.
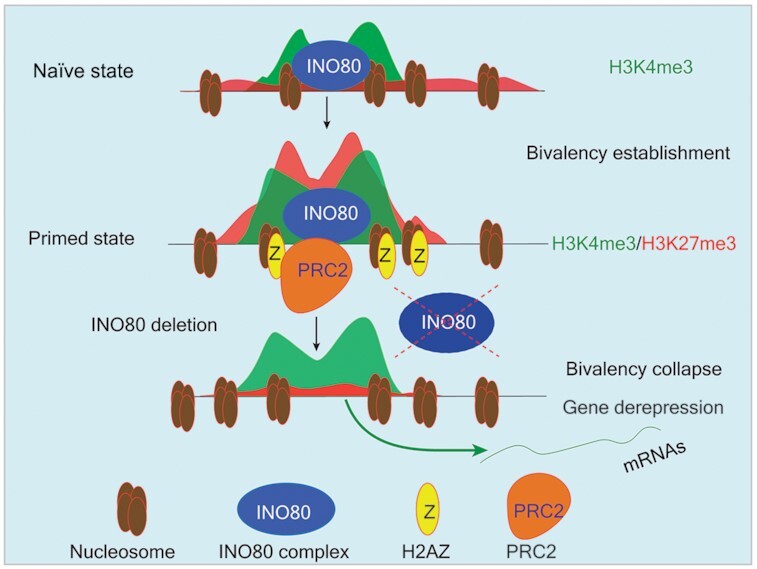
INO80-dependent H2A.Z occupancy licenses the establishment of the bivalent domains and maintains developmental genes in a repressed state.
INTRODUCTION
Pluripotent stem cells can provide important insights to both stem cell and developmental biology. Recent studies showed that there are different pluripotent states that correspond to the sequential developmental stages in embryos (1,2). These states can be captured in vitro using defined culture conditions, and they are characterized by distinct developmental potential, gene expression profile, and epigenetic status (3–12). Among them, the naïve pluripotent state is the earliest stage in pluripotency during embryonic development. It is represented by mouse embryonic stem cells (ESCs) cultured in medium containing MEK, GSK3 inhibitors and leukemia inhibitory factor (LIF) (2iL) (3). The primed pluripotent state is a later stage, and is represented by mouse epiblast stem cells (EpiSCs) cultured in medium containing FGF2 and Activin A (FA) (13,14) or FGF2, Activin A and the tankyrase inhibitor XAV939 (FAX) (15,16). These pluripotent stem cell models are valuable tools to investigate the molecular events that regulate pluripotency and early development.
The chromatin of pluripotent stem cells is characterized by unique epigenetic features that contribute to their special developmental potentials. Of particular interest, the pluripotent chromatin contains regions that are decorated by both the active histone H3 lysine 4 trimethylation (H3K4me3) and repressive H3 lysine 27 trimethylation marks (H3K27me3) (17–20). These bivalent chromatin domains are enriched at the promoters of key developmental genes (17), and they facilitate the formation of specific 3D chromatin conformations (21). The presence of the two apparently contradictory histone marks is thought to set downstream developmental genes in a state that allows for either rapid activation or stable repression upon differentiation, which contribute to the intricate balance between self-renewal and differentiation (22,23). Importantly, bivalent domains were also found in other cell types including tissue stem cells (24,25) and cancer (26,27), as well as in other organisms (28,29), suggesting that they play critical roles in development and disease. However, how the bivalent domains are formed and regulated remain incompletely understood.
The Polycomb Repressive Complex 2 (PRC2) catalyzes H3K27 methylation and is essential for bivalency (30). Intriguingly, H3K27me3 can be accurately established de novo at bivalent domains when PRC2 activity is restored in PRC2-deficient ESCs (31). Similarly, the bivalent modifications are initially absent from developmental gene promoters in mouse pre-implantation embryos but soon established after implantation (32,33). Thus, there likely exist additional factors that facilitate the establishment and maintenance of the bivalent domains. Indeed, one such factor is the histone variant H2A.Z. H2A.Z is highly conserved across eukaryotes and plays important roles in gene regulation (reviewed in (34,35)). Its incorporation into and removal from the nucleosomes is mediated by ATP-dependent chromatin remodeling complexes. In yeast, H2A.Z loading is carried out by the SWR1 complex (36) and its eviction by the INO80 complex (37). In ESCs, H2A.Z is enriched at bivalent promoters (38–40), and it promotes H3K27me3 occupancy by promoting PRC2 expression (41), recruitment (42) and enzymatic activity (43). Together, these findings suggested that chromatin remodelers may participate in the regulation of the bivalent chromatin via H2A.Z.
Because of the distinctive chromatin characteristics, it is not surprising that pluripotent stem cells are highly dependent on chromatin regulators such as the chromatin remodeling enzymes (44). The chromatin remodelers use the energy of ATP hydrolysis to regulate chromatin structure and dynamics. They can change nucleosome compositions, or remove, exchange and slide nucleosomes, and thereby play critical roles in a wide range of chromatin-dependent functions (45). The INO80 chromatin remodeling complex belongs to the INO80 sub-family of chromatin remodelers (46,47). It has DNA-dependent ATPase activity and catalyzes ATP-dependent nucleosome sliding and spacing (46,48), as well as the exchange of the histone variant H2A.Z (37,49). INO80 has been implicated in many cellular and physiological processes such as stem cell maintenance, embryogenesis, germ cell development, neurological diseases, and cancer (50–54). In mouse ESCs cultured in serum and LIF, INO80 occupies pluripotency gene promoters and facilitates downstream gene activation (51). In mice, INO80 promotes proximal-distal axis establishment and its deletion led to early embryonic lethality (50,53). Therefore, INO80 plays important roles in pluripotency and early development.
As earlier studies relied on ESCs cultured in heterogenous conditions, the role of INO80 in defined pluripotent states has not been carefully investigated. Here, we set out to examine the impact of Ino80 deletion on the naïve and primed pluripotent state. We found that INO80 is selectively required in the primed but not the naïve state. It promotes H2A.Z occupancy to facilitate the establishment and maintenance of the bivalent domains at developmental genes, keeping them in a repressed state. Our findings uncovered a critical step in the formation and regulation of the bivalent chromatin structure in pluripotent stem cells. As both INO80 and H2A.Z are heavily involved in development and disease, we propose that the INO80-H2A.Z axis may have similar functions during other cell fate transitions.
MATERIALS AND METHODS
Cell culture
For routine culture, mouse ESCs were maintained in the ESGRO medium (Millipore) on tissue culture plates pretreated with 0.1% gelatin. For the naïve pluripotent state, mouse ESCs were cultured in 2iL medium containing N2B27 medium supplemented with PD0325901 (1 mM, Selleck), CHIR99021 (3 mM, Selleck) and 1000 units/ml recombinant murine LIF (Millipore) on gelatin-coated plates (2iL). For the primed pluripotent state, mouse ESCs were cultured in FA or FAX medium containing N2B27 supplemented with FGF2 (Peprotech, 12 ng/ml), Activin A (Peprotech, 20 ng/ml) and XAV-939 (2 μM Selleck) on human fibronectin-coated plates (Millipore). The N2B27 medium contains (1000 ml): 500 ml DMEM/F12 (Invitrogen; 11320), 500 ml Neurobasal (Invitrogen; 21103), 5 ml N2 supplement (Invitrogen; 17502048), 10 ml B27 supplement (Invitrogen; 17504044) and 0.1% bovine serum albumin fraction V (Thermofisher).
Mouse embryonic stem cell lines
The E14Tg2a ESCs were obtained from MMRRC. The Ino80 conditional deletion ESC line (Ino80flox/flox;UBC-CreERT2) was derived from E3.5 blastocysts from the breeding of Ino80flox/+;UBC-CreErt2 and Ino80flox/+ in the 129S1/SvImJ background. The Ino80flox/flox mouse strain was obtained from blastocyst injection of the Ino80flox/flox ESCs ordered from EuMMCR. ESC liness expressing the INO80 ATPase-dead mutants were generated by transfecting the Ino80 conditional deletion ESCs with PiggyBac vectors (kindly provided by Dr Joanna Wysocka, Stanford University) containing the corresponding Ino80 mutant cDNAs and the PB transposase (System Biosciences). The INO80-HA and INO80 ATPase-dead knock-in ESCs were generated by CRISPR-mediated genome editing. Briefly, sgRNAs targeting the desired genomic loci were cloned into pX330 (addgene #42230). Single-stranded oligo DNA donor (ssODN) homologous recombination templates were synthesized by IDT. sgRNAs and ssODN templates were co-transfected into E14Tg2a cells, and correctly targeted clones were identified by PCR genotyping and confirmed with secondary assays. The H2az1 flox/flox; H2az2 Δ/Δ; Rosa26-CreERT2 (H2az1-cKO/H2az2-KO) ESC line was generated by CRISPR-mediated genome targeting in the Rosa26:CreERT2 ESCs (kindly provided by Dr. Shaun Cowley, University of Leicester). To induce gene deletion, 0.1 mM 4-OHT (Selleck) was added to culture medium for two days.
Embryoid body formation assay
For embryoid body (EB) formation, ESCs were induced to primed state in FA for two days. Simultaneously, cells were treated with DMSO or 0.1 mM 4-OHT. Then 500 000 cells/ml single cell suspension in DMEM medium containing with 10% FBS were added to ultra-low attachment plate (Corning) in a 37°C, 5% CO2 incubator to allow them to form EBs. Medium were refreshed every other day. EBs were collected at indicated days.
Protein extract and Western blotting
Cells were lysed in RIPA buffer (ThermoFisher) containing protease inhibitor cocktail (PIC, Roche) and PMSF, and total protein concentration was determined using the BCA kit (Pierce). Cell lysate was loaded into a NuPAGE® Bis–Tris gels and transferred onto nitrocellulose membrane. The blot was blocked with 5% non-fat milk at room temperature for 30 min, followed by incubation with primary antibodies at 4°C overnight. The blot was subsequently incubated with either horse-radish peroxidase (HRP)-conjugated anti-mouse or rabbit IgG. Signal was detected using ChemiDoc Imaging system (Bio-Rad).Antibodies against specific antigens are provided in Supplementary Table S1. ImageJ (NIH) was used to quantify blot band intensity.
RNA isolation, quantitative real-time PCR and RNA-seq
Total RNA was extracted using the GeneJET RNA purification kit (ThermoScientific). RNA was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad). Quantitative real-time PCR (qPCR) were performed with the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) on the CFX384 real-time PCR detection system. All experiments were carried out with at least two biological replicates. For RNA-seq with spike-in controls, 10 μl of 1:100 dilution of the ERCC Spike-In Mix #1 was added to the lysate of 1 million cells before RNA extraction. Total RNA was then extracted as described above. For RNA-seq library preparation, 1 μg total RNA was used to prepare the RNA-seq library with the Truseq RNA Library Prep Kit V2 with Ribo-Zero (Illumina). Primers used in this study are provided in Supplementary Table S2.
ATAC-seq, MNase ChIP-seq, MNase-seq and ChIP-qPCR
Omni-ATAC was performed with Nextera DNA library prep kit (Illumina) as described (55). Then tagmentated DNA was purified using DNA Clean & Concentrator-5 Kit (Zymo). ATAC-seq library was generated by PCR amplification and purified with AMPure XP beads (Beckman Coulter).
For ChIP-seq, mouse ESCs were digested by trypsin (Naïve state) or Accutase (Primed state) to single cells and fixed in 1% formaldehyde for 10 min at room temperature. Crosslinking was quenched by the addition of glycine to the final concentration of 0.25 M and the incubation at room temperature for 5 min. Cells were washed twice with ice-cold PBS containing the complete protease inhibitor cocktail (PIC, Roche). Cell pellet was resuspended and permeabilized with hypotonic buffer (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1% Triton X-100, and freshly add 1 mM DTT, 0.5% N-P40 and Protease inhibitors) on ice for 10 min. Cells were pelleted by centrifugation at 1350 × g, washed once with the hypotonic buffer, and again with the MNase buffer (20 mM Tris×HCl pH 7.5, Sucrose 0.34 M, KCl 60 mM, NaCl 15 mM and 1 mM CaCl2). Cell pellet was resuspended in the MNase buffer and digested with MNase (100–200 units per 10 M cells) at 37 °C for 12 min on a shaker. Digestion was stopped by the addition of 10% volume of 0.5 M EDTA and mixed with one volume of 2× lysis buffer (20 mM Tris–HCl pH8.0, 300 mM NaCl, 2 mM EDTA, 20% Glycerol, 1% Triton X-100, 2× PIC, 0.2 mM PMSF). Cells were sonicated by 4 cycles of 30 s ON and 90 s OFF on the Misonix S3000 sonicator with 30-W power output, and insoluble materials were removed by centrifugation at 21 000 × g at 4°C for 20 min. 25 μg chromatin was used for each ChIP experiment, and was incubated with the primary antibody (Supplementary Table S2) and the protein A/G Dynabeads (ThermoFisher) overnight at 4°C. The beads were washed with low salt buffer (20 mM Tris–Cl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100), high salt buffer (20 mM Tris–Cl (pH 8.0), 250 mM LiCl, 1 mM EDTA, 1% Triton X-100) and lithium chloride buffer (20 mM Tris–Cl (pH 8.0), 500 mM NaCl, 1 mM EDTA, 1% Triton X-100 and 1% sodium deoxycholate), and then washed twice with the TE buffer (10 mM Tris–HCl, 1mM EDTA). Immunoprecipitated DNA-protein complex was eluted with the elution buffer (50 mM Tris–Cl (pH 7.5), 10 mM EDTA, 1% sodium dodecyl sulphate) at 65°C for 15 min. The elution was collected and reverse-crosslinked at 65°C overnight, diluted with 1 volume of TE buffer and treated with RNase A at 37°C for 30 min followed by 0.2 μg/ml Proteinase K at 55°C for 60 min. The immunoprecipitated DNA was purified from the elution by the Zymo DNA Clean & Concentrator Kit. For ChIP-seq, 2 ng ChIP DNA or input was used to generate the sequencing library using the Nextflex Rapid DNA-seq Kit (PerkinEImer) or Nextflex DNA and the resulting libraries were sequenced on the Illumina platform. Two biological replicates were carried out for each experiment, and combined reads were used for further analyses.
For MNase-seq, fixed cells were treated as described above for titrated MNase digestion. Either 4, 16, 64 or 256 U of MNase (NEB) were added to per 2 M pre-warmed cells and incubated at 37°C for 10 min. Digestion was halted by addition of MNase stop buffer (20 mM EDTA, 20 mM EGTA, 0.4% SDS and 0.5 mg/ml Proteinase K). The mixtures were incubated at 65°C for 6 hours to reverse crosslinks. DNA was cleaned up by Gel Purification Column (Qiagen). Fragment size were estimated by Bioanalyzer 2000 (Agilent). 100 ng DNA from each digestion were used to prepare DNA libraries as described in the Nextflex Rapid DNA-seq Kit (PerkinEImer). Size selection was performed after adaptor ligation with Agencourt AMPure XP beads (Beckman Coulter). 8 cycles of PCR were used to amplify the products.
For ChIP-qPCR, either 0.1 ng ChIP DNA or 0.1 ng Input DNA was used as template, and fold-enrichments were determined by the 2−ΔCT method. Primers used in this study are provided in Supplementary Table S2.
Data processing and analyses
For all sequencing runs, only reads with Phred quality score ≥20 and length >15 were kept for analysis. For RNA-seq, reads were first trimmed with cutadapt (v1.9), and filtered reads were aligned to mm9 reference with STAR (v2.5.2b) with parameters ‘–outFilterMismatchNoverLmax 0.04’. Mapped reads were annotated to gene exons to generate a count matrix on gene level with featureCounts (v1.5.1). Differential expressed genes (DEGs) were identified with the R package DESeq2, and DEGs were selected as |log2FC| > 1 and adjusted P value <0.05. For RNA-seq with ERCC spike-ins, the ERCC reads were aligned separately to the ERCC92 reference. Gene count matrix was generated with the same pipeline as standard RNA-seq. To adjust the RNA-seq result using the ERCC, estimateSizeFactors function in DESeq2 was used with the control genes set to ERCCs. To normalize RPKM of genes to the ERCC spike-in, loess normalization function from R affy package was used to adjust the values. RPKM data for genes were log2 transformed (after adding 1). Gene ontology enrichment of was analyzed by GO Consortium powered by PANTHER.
For ChIP-seq analysis, reads were aligned to mm9 reference using bowtie (v1.1.2) with the following parameters ‘-v 2 -m 1 –best –strata -I 15 -X 1000’ after filtering as described above. PCR duplicates were removed after alignment. For peak calling, SICER (v1.1) was used with window size 200 and gap 600. Peaks were cleaned-up by FDR <0.0001 and excluded signals from the Encode mm9 blacklist for further analyses. Two-fold enrichment against input was applied to high confidence H3K27me3 peaks. For visualization, Bedtools (v2.21.0) genomeCoverageBed and UCSC utility bedGraphToBigWig were used to build the tracks of the samples. The coverage of the samples was then normalized to the total reads per 10 million for between sample comparison. Deeptools (v3.3.0) was used for spearman correlation, heatmap and metagene (density) plot. Peaks were annotated to nearest TSS with Homer. Promoter was defined as regions of 500 bp around TSS. Promoter number was consolidated into H3K4me3 peaks in promoter region. Bivalent promoters were defined by promoters with overlapped H3K4me3 and H3K27me3 peaks.
For MNase-seq, reads were aligned using bowtie (v1.1.2) with the same parameters as in ChIP-seq. The reads fragments between 100 and 200 bp were selected for further analyses. For visualization of the nucleosome position pattern in metagene profiles, mid-point of each fragment was selected and followed by median normalization (56). Gaussian smooth was used to smooth the curves in the final plot (57).
For ATAC-seq, same filtering pipeline was used as described in ChIP-seq. The first 9 bp from the end of the aligned fragments were counted as open chromatin reads and normalized to library depth. Reads from chrM were removed.
Software and algorithms used for data analysis were listed in Supplementary Table S3.
Statistical analysis
RT-qPCR and ChIP-qPCR were analyzed with student t-test. RNA-seq, ATAC-seq or ChIP-seq signals were analyzed with Wilcoxon rank sum test (when unpaired) or Wilcoxon signed rank test (when paired).
RESULTS
INO80 is selectively required for the maintenance of the primed pluripotent state
To investigate the role of INO80 in pluripotent stem cells, we generated an inducible deletion mouse ESC line for the core ATPase INO80 (Ino80-cKO ESCs, Supplementary Figure S1A). The Ino80-cKO cells have a normal karyotype (Supplementary Figure S1B) and show efficient deletion of Ino80 at both the mRNA and protein levels upon 4-hydroxytamoxifen (4-OHT) treatment (Supplementary Figure S1C-D). Moreover, chromatin immunoprecipitation followed by high throughput sequencing (ChIP-seq) using an antibody that recognizes the C-terminus of INO80 confirmed the loss of INO80 genomic occupancy in the deletion cells (Supplementary Figure S1E).
In order to study the naïve and primed pluripotent states, we cultured ESCs in the 2i/LIF (2iL) or FGF2/Activin-A/XAV-939 (FAX) medium as previously described (3,4,8,9,16). We were also able to induce the transition from the naïve to the primed state by switching the cells from the 2iL to FGF2/Activin-A (FA) medium (Figure 1A) (58). With these in vitro culture models, we set out to systematically dissect the molecular function of INO80 in different pluripotent states.
Figure 1.

INO80 is selectively required in the primed pluripotent state. (A) Culture model for the naïve and primed pluripotent state. Mouse ESCs were cultured in 2iL for the naïve state, switched to FA to induce the transition toward the primed state, and maintained in FAX for the primed state. Scale bar = 100 μm. (B) INO80 expression in different pluripotent states. Ino80-HA knock-in ESCs were cultured in 2iL, FA, or FAX, and INO80 expression was detected by western blot using the HA antibody. Histone H3 was blotted as the loading control. The experiment was repeated three times and one representative result was shown. (C, D) Ino80 deletion in the naïve state. Ino80-cKO ESCs were cultured in 2iL and treated with DMSO or 4-OHT for 2 days to induce Ino80 deletion. Cell morphology (C) and lineage marker expression (D) were examined by imaging and RT-qPCR. Scale bar = 100 μm. Relative mRNA levels between wild-type and Ino80 KO cells were first normalized by Gapdh and then normalized to wild-type cells. Results from three independent experiments were plotted as mean ± SEM. p-values were calculated by Student's t-test: * <0.05, ** <0.01, *** <0.001. (E, F) Ino80 deletion in the primed state. Ino80-cKO ESCs were cultured in FAX and treated with DMSO or 4-OHT for 2 days. Cell morphology (E) and lineage marker expression (F) were examined by imaging and RT-qPCR. Results from three independent experiments were plotted as mean (G) ± SEM. p-values (H) were calculated by Student's t-test: * <0.05, ** <0.01, *** <0.001. (G, H) Gene expression changes after Ino80 deletion in the naïve and primed state. Differentially expressed genes (DEGs) were determined by RNA-seq from two replicate experiments and highlighted in the volcano plots. Numbers of DEGs were also listed. Blue: down-regulated genes; Red: up-regulated genes.
The INO80 protein was expressed at comparable levels in the naïve state and during the naïve-to-primed transition, but became slightly reduced in the primed state (Figure 1B). Surprisingly, Ino80 deletion had no effect on cell morphology (Figure 1C) and minimal impact on the expression of the pluripotency and differentiation markers in the naïve state (Figure 1D). Furthermore, Ino80-null ESCs can be stably maintained for more than five passages in 2iL. In the primed state, however, Ino80 deletion led to differentiation and cell death (Figure 1E, Supplementary Figure S1F). In addition, the deletion resulted in reduced expression of pluripotency markers such as Nanog and Esrrb, and increased expression of lineage markers such as Eomes, Gata6, and T (Figure 1F). Together, these results indicated that INO80 is dispensable for the naïve state but required for the primed pluripotent state.
To understand why INO80 is selectively required in the primed state, we carried out RNA-seq to examine the impact of Ino80 deletion on gene expression. We found that Ino80 deletion in the naïve state only led to modest changes in the transcriptome (Figure 1G), consistent with the cellular phenotype. However, its deletion in the primed state resulted in much more prominent perturbations, as shown by both the number and magnitude of differentially expressed genes (Figure 1H). It also led to a strong bias in gene expression changes, with 3822 up-regulated but only 550 down-regulated genes (Figure 1H). This is consistent with the phenotypic observation (Figure 1E) and further suggested that INO80 primarily represses gene expression in the primed state.
INO80 occupies bivalent gene promoters
To investigate how INO80 regulates gene expression, we examined its genomic occupancy by ChIP-seq. We made the following improvements to the INO80 ChIP-seq strategy. First, we generated a knock-in ESC line by inserting the HA-tag in the C-terminus of the endogenous Ino80 and carried out INO80 ChIP-seq with the HA antibody (Supplementary Figure S2A). The HA-antibody ChIP produced a larger number of peaks (Supplementary Figure S2B), and improved signal-to-noise ratio (Supplementary Figure S2C) over the INO80-antibody ChIP used in previous works (51,52). Second, we used the MNase digestion instead of sonication for chromatin fragmentation. In addition to the peaks identified by sonication, MNase ChIP-seq captured many more INO80-bound peaks (Supplementary Figure S2D-E). It is worth noting that these additional peaks were located in regions with lower H3K4me3 but higher H3K27me3 signals (Supplementary Figure S2F), suggesting that they may be less accessible and less efficiently recovered in sonication-based ChIP-seq.
With the optimized ChIP-seq procedure, we determined the genomic occupancy of INO80 in both the naïve and primed pluripotent state. We found that INO80 occupies a large number of genomic regions in both states, and preferentially binds near the transcription start sites (TSSs) in the primed state (Supplementary Figure S2G). When intercepted with the RNA-seq data from the Ino80 deletion cells, INO80 genomic occupancy was found near many of the differentially expressed genes (Supplementary Figure S2H), suggesting that INO80 may directly regulate their expression. In agreement with previous reports (51,52), many INO80-bound TSSs were marked by the active histone mark H3K4me3 (Figure 2A). To our surprise, however, a significant fraction of INO80-bound TSSs were bivalently marked by both H3K4me3 and H3K27me3, especially in the primed state (Figure 2A). In fact, INO80 occupied the majority of the bivalent promoters in the primed state (Figure 2B), suggesting that it may play an important role in their regulation.
Figure 2.

INO80 occupies bivalent gene promoters. (A) H3K4me3, H3K27me3 and bivalent domains in INO80 occupied promoters in the naïve and primed state. Number of total INO80 peaks (bottom) and INO80 peaks with bivalent marks (top) in the promoter region were listed. (B) Percentage of bivalent promoters occupied by INO80 in the naïve and primed state. Number of total bivalent promoters were listed at the bottom. (C) Gene Ontology analysis of INO80 bound bivalent genes in primed state. Heatmap shows the Author: –log10(FDR) values of selected top enriched categories. The complete list of enriched categories can be found in Supplementary Figure S2I. (D) Box plot to show the relative expression of all genes, active genes (top 25%), INO80-bound bivalent genes, and lowly or not-expressed genes (bottom 25%) in the primed state. Gene expression was determined by RNA-seq and calculated as log2(RPKM + 1). (E) Violin plot of normalized ATAC-seq signals to show the chromatin accessibility of all genes, active genes (top 25%), INO80-bound bivalent genes, and lowly or not-expressed genes (bottom 25%) in the primed state. (F) Box plots to show relative gene expression changes of active and INO80-bound bivalent genes as in D during mouse embryonic development from E6.5-E8.5. Gene expression changes between Ectoderm, Mesoderm, or Endoderm cells and E6.5 epiblast cells were calculated as log2(RPKM + 1) from published single cell RNA-seq data (E-MTAB-6967).
INO80 is required for the establishment and maintenance of the bivalent gene promoters
To test whether and how INO80 regulates bivalent genes, we examined the behavior of the INO80-bound bivalent genes in the presence and absence of Ino80 in the primed state. We found that the INO80-bound bivalent genes were highly enriched for those that are involved in development (Figure 2C and Supplementary Figure S2I). They were expressed at low to moderate levels in comparison to active and lowly/un-expressed genes (Figure 2D), and their promoters displayed low to moderate chromatin accessibilities (Figure 2E). In addition, on average they were up-regulated during mouse embryonic development from the epiblast cells at E6.5 to the three main germ layers at E8.5 (Figure 2F). Thus, some of the INO80-bound bivalent genes appear to be repressed in pluripotent cells and activated at the onset of differentiation and lineage commitment. Consistent with this notion, a significant fraction of the INO80-bound bivalent genes were up-regulated upon Ino80 deletion in the primed state (Figure 3A, B), and some of them also showed premature or excessive activation during embryoid body differentiation (Figure 3C). Therefore, INO80 is required for the repression of bivalent genes and prevents them from precocious activation.
Figure 3.

INO80 is required for H3K27me3 occupancy at the bivalent promoters. (A) Gene expression changes in INO80-bound bivalent genes in the primed state. (B) Box plot to show changes in the expression of INO80-bound bivalent genes. Gene expression changes were determined by RNA-seq and plotted in log2 fold-change of (RPKM+1). p-value was calculated by the Wilcoxon signed rank test. (C) Expression of selected INO80-bound bivalent genes during EB differentiation. ESCs were cultured in FA and treated with DMSO or 4-OHT to induce Ino80 deletion for two days, and then aggregated to form EBs for 2 days to initiate differentiation. Relative mRNA levels in wild-type and Ino80 KO EBs were determined by RT-qPCR, normalized by Gapdh, normalized to wild-type ESCs, and log2 transformed. Scattered dots represent three independent experiments. p-values were calculated by student t-test: * <0.05, ** <0.01, *** <0.001. (D, E) H3K27me3 occupancy at INO80-bound bivalent TSSs in WT and Ino80 deletion cells in the primed state. ESCs were cultured in FAX and treated with DMSO or 4-OHT to induce Ino80 deletion for two days. H3K27me3 occupancy was determined by ChIP-seq, and normalized ChIP-seq signal was used for metagene (D) and box (E) plots. (F) Genome browser view of INO80, H3K27me3, H3K4me3, and H3K27ac occupancy near T in WT and Ino80 deletion cells in the primed state. (G) H3K27me3 occupancy at representative bivalent gene promoters as determined by ChIP-qPCR. Fold-enrichment was plotted as mean ± SEM from three replicates. p-values were calculated by student t-test: * <0.05, ** <0.01, *** <0.001. (H, I) H3K4me3 occupancy at INO80-bound bivalent TSSs in WT and Ino80 deletion cells in the primed state. Normalized ChIP-seq signal was used for metagene (H) and box (I) plots.
To understand the underlying mechanism, we carried out ChIP-seq for H3K4me3 and H3K27me3. We found that Ino80 deletion led to dramatic reductions in H3K27me3 occupancy at INO80-bound bivalent gene promoters (Figure 3D–F) in the primed state, and we validated such changes by ChIP-qPCR (Figure 3G). Similarly, Ino80 deletion led to reduced H3K27me3 occupancy at INO80-bound H3K27me3-only promoters as well (Supplementary Figure S3A). But because the number of H3K27me3-only promoters is small and the overall H3K27me3 signal at those promoters is weak, we focused on the role of INO80 at bivalent promoters. In contrast to H3K27me3, Ino80 deletion only slightly affected H3K4me3 occupancy (Figure 3H, I). Together, our data strongly suggested that INO80 is required for the maintenance of H3K27me3 at bivalent promoters for the repression of downstream genes in the primed state. Interestingly, in comparison to the primed state, Ino80 deletion only had very subtle effects on H3K27me3 or H3K4me3 occupancy in the naïve state (Supplementary Figure S3B, C), which may explain the muted gene expression changes (Supplementary Figure S3D, S1G).
During the transition from the naïve to the primed state, the number of bivalent promoters increases significantly (Figure 4A) and the increase can be mainly attributed to the gradual gain of H3K27me3 (Figure 4B, Supplementary Figure S3E, F). This is largely similar to what was observed during mouse embryonic development (32,33). We found that the majority of the INO80-bound bivalent genes in the primed state, such as T, were already occupied by INO80 in the naïve state (Figure 4C–E, Supplementary Figure S3G), suggesting that INO80 may be involved in the establishment of H3K27me3 at these regions. To test this, we carried out H3K27me3 ChIP-seq during the naïve to primed transition in wild-type and Ino80 deletion cells. We found that Ino80 deletion led to significant cell loss (Figure 4F, Supplementary Figure S3H). Furthermore, it resulted in impaired H3K27me3 deposition at INO80-bound bivalent promoters (Figure 4G–I), as well as the de-repression of bivalent genes (Figure 4J). Compared to wild type cells in which 55% bivalent promoters were established after 2 days of culture in FA, Ino80 KO cells had only 21% detectable bivalent promoters (Figure 4K). Therefore, INO80 is required not only for the maintenance of H3K27me3 at bivalent promoters in the primed state, but also for the proper establishment of H3K27me3 and bivalency at key developmental genes during the naïve to primed transition.
Figure 4.

INO80 is required for the establishment of bivalency. (A) Venn diagram to show the overlap of bivalent promoters between the naïve and primed state. (B) Violin plots to show normalized H3K4me3 and H3K27me3 ChIP-seq signal intensity at bivalent promoters during the naïve to primed transition. (C) Violin plot to show normalized INO80 ChIP-seq signal intensity at bivalent promoters during the naïve to primed transition. (D) Pie chart to show INO80 occupancy in the naïve state at the primed bivalent promoters. (E) Genome browser view of INO80, H3K4me3, H3K27me3 occupancy at T in the naïve and primed state. (F–K)Ino80 deletion during the naïve to primed transition. ESCs were cultured in 2iL and treated with 4-OHT to induce Ino80 deletion for two days. Cells were then transferred to the FA medium for another 2 days, imaged (F), and collected for ChIP-seq and RT-qPCR (G–K). Normalized H3K27me3 ChIP-seq signal at INO80-bound primed bivalent gene promoters was examined by metagene (G) and box (H) plots. INO80 and H3K27me3 occupancy near T was shown by the genome browser view (I). The relative expression of selected primed bivalent genes in wild-type and Ino80 deletion cells was determined by RT-qPCR, first normalized to Gapdh and then normalized to wild-type cells, and plotted as mean ± SEM from three independent experiments. p-values were calculated by Student's t-test: * <0.05, ** <0.01, *** <0.001 (J). The numbers of successfully established bivalent promoters during the naïve to primed transition in WT or Ino80 deletion cells were shown by the pie chart (K).
INO80 promotes H2A.Z occupancy as a chromatin remodeler
Next, we wanted to investigate how INO80 regulates H3K27me3 occupancy. Because INO80 is a chromatin remodeler, we hypothesized that it regulates H3K27me3 via its chromatin remodeling function. To test the hypothesis, we introduced dox-inducible wild-type or ATPase-dead INO80 mutants (K551A or E665Q) (59,60) into the Ino80-cKO ESCs. As expected, the expression of wild-type INO80 rescued the differentiation and cell death caused by Ino80 deletion when switched into the primed state (Figure 5A, Supplementary Figure S4A, B). However, the expression of ATPase-dead mutants, albeit at similar levels (Supplementary Figure S4A), was not able to maintain the cells under the same condition (Figure 5A, Supplementary Figure S4A, B). In addition, we also mutated the endogenous Ino80 gene and generated an Ino80 ATPase-dead knock-in ESC line (the K551A/E665Q double mutant, or Ino80-KA/ED) by CRISPR-mediated genome editing. The Ino80 ATPase-dead knock-in did not drastically affect the INO80 protein level (Supplementary Figure S4C). However, similar to Ino80 deletion, it led to cell loss (Supplementary Figure S4D) and elevated expression of bivalent genes during transition toward the primed state (Supplementary Figure S4E). Finally, we examined H3K27me3 occupancy at selected bivalent promoters by ChIP-qPCR in Ino80 KO cells expressing wild-type or ATPase-dead INO80. We found that H3K27me3 occupancy was significantly reduced in the ATPase-dead mutants (Figure 5B). Together, the above results indicated that the chromatin remodeling activity of INO80 is essential for cells during the transition toward the primed state, and it is also required for the repression of bivalent genes and the deposition of H3K27me3 at their promoters. It is worth noting that in both the Ino80 deletion and ATPase-dead mutant cells, the down-regulation of naïve markers Tfcp2l1 and Esrrb was similar to that in the wild-type cells during the naïve to primed transition, suggesting that Ino80 deletion or mutation does not compromise the exit of the naïve state (Supplementary Figure S4F, G).
Figure 5.

INO80 promotes H2A.Z occupancy. (A) Rescue of Ino80 deletion phenotype by WT INO80 or INO80-ATPase-dead mutants. Ino80 deletion ESCs were transfected with the piggyBac vectors expressing Dox-inducible WT or ATPase-dead Ino80 (K551A, KA or E665Q, EQ). Ino80 deletion was induced by 4-OHT treatment and exogenous Ino80 expression was induced by Dox treatment simultaneously in serum/LIF. Cell morphology was imaged after one passage. Scale bar = 100 μm. (B) ChIP-qPCR of H3K27me3 occupancy at selected bivalent promoters in Ino80 deletion cells expressing WT or ATPase-dead mutants during naïve to primed transition. Fold-enrichment was plotted as mean ± SEM. The experiment was repeated twice and shown as exp1 and exp2. (C, D) Normalized H2A.Z ChIP-seq signal at the INO80-bound bivalent promoters was examined by metagene (C) and violin (D) plots. Ino80 deletion ESCs were cultured in FAX and treated with DMSO or 4-OHT for 2 days and collected for ChIP-seq. (E) Genome browser view of INO80 and H2A.Z occupancy near T. (F) ChIP-qPCR to show H2A.Z occupancy at representative bivalent gene promoters in WT and Ino80 deletion cells. Fold-enrichment was plotted as mean ± SEM from three replicates. p-values were calculated by Student's t-test: * <0.05, ** <0.01, *** <0.001.
As a remodeler, INO80 can regulate nucleosome positioning and composition. To test which of these functions is involved, we first examined the role of INO80 in nucleosome occupancy by MNase titration followed by high throughput sequencing (61) (Supplementary Figure S4H). To our surprise, Ino80 deletion only had very limited impact on nucleosome positioning and density (Supplementary Figure S4I). Moreover, the small changes in nucleosome signals after Ino80 deletion were similar in both the naïve and primed state and showed no clear correlation with gene expression changes. Thus, changes in the nucleosomes could not explain the Ino80 deletion phenotypes.
Next, we examined the role of INO80 in the histone variant exchange, as INO80 has been reported to control the eviction of H2A.Z (37,49). Using public ChIP-seq data in mouse ESCs (62), we found that INO80 and H2A.Z showed the highest correlation in genomic occupancy among all the examined chromatin remodelers including BRG1, MBD3, EP400, TIP60, CHD1, CHD2, CHD4, CHD6, CHD7, CHD8 and CHD9 (Supplementary Figure S5A). This co-localization is consistent with the notion that INO80 may be an important regulator for H2A.Z. We then carried out H2A.Z ChIP-seq in wild-type and Ino80 deletion cells. In wild-type cells, a sizable portion of H2A.Z localized at TSSs (Supplementary Figure S5B) and its signal intensity was much higher in the primed state compared to the naïve state (Supplementary Figure S5C). In Ino80 deletion cells in the primed state, although the H2A.Z protein level did not change (Supplementary Figure S5D, E), H2A.Z occupancy at INO80-bound TSSs was significantly reduced (Figure 5C-F). Similarly, H2A.Z occupancy was also reduced at INO80-bound active promoters and enhancers (Supplementary Figure S5F). Therefore, INO80 is required for the maintenance of H2A.Z genomic occupancy in the primed state. Interestingly, reminiscent of what was observed for H3K27me3 (Supplementary Figure S3B), Ino80 deletion had minimal impact on H2A.Z occupancy in the naïve state (Supplementary Figure S5G, H), possibly due to the already low basal H2A.Z occupancy (Supplementary Figure S5C).
Because our results showed an opposite role of INO80 in H2A.Z occupancy compared to earlier studies (37,49), we wondered whether our observation was specific to pluripotent stem cells. We thus tested the impact of Ino80 deletion in mouse embryonic fibroblasts (MEFs, Supplementary Figure S5I). Similar to the above results, Ino80 deletion led to significant reduction in H2A.Z signals in MEFs as well (Supplementary Figure S5J). Therefore, the requirement for INO80 in H2A.Z genomic occupancy appears to be conserved among different cellular states in mammalian cells.
INO80-dependent H2A.Z occupancy licenses the establishment of bivalency
Based on the above data, we hypothesized that INO80-dependent H2A.Z occupancy is a determining factor in bivalent promoter regulation. Consistent the notion, INO80 pre-occupied 80.9% of all bivalent promoters in the primed state (Figure 4D). Moreover, almost all the INO80-bound bivalent promoters were co-occupied by H2A.Z (Supplementary Figure S6A) and showed stronger H2A.Z signal (Supplementary Figure S6B). To test the hypothesis, we generated H2A.Z conditional deletion ESCs, in which the isoform H2az2 was constitutively deleted and H2az1 could be deleted upon 4-OHT treatment (Supplementary Figure S6C–E). Similar to Ino80 deletion, H2az1/H2az2 double deletion had limited impact on the naïve state but led to severe cellular differentiation and cell loss in the transition to primed state (Figure 6A). Importantly, in the primed state, the double deletion resulted in impaired H3K27me3 occupancy at bivalent promoters (Figure 6B–D) and the impairment was more obvious at regions with stronger H3K27me3 signal (Supplementary Figure S6F). Consistently, H2az1/H2az2 deletion caused de-repression of bivalent genes in the primed state (Figure 6E), as well as premature activation of developmental genes during subsequent embryoid body differentiation (Figure 6F), phenocopying Ino80 deletion (Figure 1F, 3C). Therefore, H2A.Z is required for the maintenance of H3K27me3 and the repression of bivalent genes.
Figure 6.

INO80-dependent H2A.Z deposition licenses the establishment of bivalency. (A) H2az1/H2az2 deletion in the naïve state and during the naïve to primed transition. H2az1-cKO/H2az2-KO ESCs were cultured in the naïve state in 2iL and treated with DMSO or 4-OHT for 2 days. Cells were then maintained in the naïve state in 2iL or transitioned into FA for another 2 days to initiate the primed state. Cells were imaged and collected for ChIP-seq and RT-qPCR experiments. Scale bars = 100 μm. (B, C) H3K27me3 occupancy at INO80-bound bivalent TSSs in WT and H2az1/H2az2 deletion cells. Metagene and box plots were generated using normalized H3K27me3 ChIP-seq signals. (D) Genome browser view of H2A.Z, H3K4me3, H3K27me3, PRC2 component SUZ12 and EZH2 near T. (E) Expression of representative bivalent genes in WT and H2az1/H2az2 deletion cells. Relative mRNA levels were determined by RT-qPCR, first normalized to Gapdh and then normalized to wild-type cells, and log2 transformed. Scattered dots represent three independent experiments. p-values were calculated by Student's t-test: * <0.05, ** <0.01, *** <0.001. (F) Expression of representative bivalent genes in WT and H2az1/H2az2 deletion cells during EB formation. WT or H2az1/H2az2 deletion cells were induced to transition toward the primed state as described in A. Cells were aggregated to form EB for 2 and 4 days and collected for RT-qPCR. Expression of representative bivalent genes in wild-type and H2az1/H2az2 deletion ESCs and EBs was determined by RT-qPCR, first normalized to Gapdh and then normalized to wild-type EBs, and log2 transformed. Scattered dots represent three independent experiments. p-values were calculated by Student t-test: * <0.05, ** <0.01, *** <0.001.
To understand how INO80 and H2A.Z regulates H3K27me3, we examined key components of the PRC2 complex in wild-type, Ino80 deletion, and H2az1/H2az2 deletion cells in the primed state. We found that Ino80 deletion did not affect the expression of PRC2 components (Supplementary Figure S7A), and H2az1/H2az2 deletion led to reductions in EZH2 and SUZ12 proteins (Supplementary Figure S7B). Importantly, both Ino80 and H2az1/H2az2 deletion impaired the genomic occupancy of the PRC2 structural component SUZ12 (Figure 7A–E). In addition, H2az1/H2az2 deletion also impaired the occupancy of the PRC2 enzymatic subunit EZH2 (Figure 7F, G). We validated these results by ChIP-qPCR (Figure 7H). In contrast, H2A.Z occupancy was not significantly affected by the deletion of PRC2 components EZH2 or EED (Supplementary Figure S7C–E). These results suggested that INO80 and H2A.Z are required for the recruitment of a fully-assembled PRC2 for H3K27me3 deposition at bivalent promoters.
Figure 7.

INO80-dependent H2A.Z occupancy is required for PRC2 recruitment. (A, B) SUZ12 occupancy at INO80-bound bivalent TSSs in the primed state in WT and Ino80 deletion cells. Ino80-cKO ESCs were cultured in FAX and treated with 4-OHT for 2 days. Cells were cultured for 2 additional days and collected for ChIP-seq. SUZ12 occupancy was shown by metagene (A) and box (B) plots using normalized ChIP-seq signals. (C) Genome browser track to show SUZ12 occupancy near T in WT and Ino80 deletion cells. (D–G) SUZ12 and EZH2 occupancy at INO80-bound bivalent TSSs in the primed state in WT and H2az1/H2az2 deletion cells. H2az1-cKO/H2az2-KO ESCs were cultured in the primed state in FAX and treated with DMSO or 4-OHT for 2 days. Cells were cultured for another 2 days and collected for ChIP-seq. SUZ12 and EZH2 occupancy was shown by metagene (D, F) and box (E, G) plots using normalized ChIP-seq signals. (H) ChIP-qPCR to show SUZ12 (upper) and EZH2 (lower) at representative bivalent gene promoters in WT and Ino80 deletion cells. Fold enrichment was plotted as mean ± SEM from three replicates and p-values were calculated by Student's t-test: * <0.05, ** <0.01, *** <0.001. (I) Spearman correlation of genomic occupancy among chromatin factors. Except for that of INO80 and H2AZ, genomic occupancy of all other chromatin factors was based on public datasets (GSE64825).
To add further support, we also examined H2A.Z and SUZ12 occupancy at selected bivalent promoters in Ino80 KO cells expressing wild-type or ATPase-dead INO80 proteins. In comparison to cells expressing the wild-type INO80, cells expressing the ATPase-dead mutants had reduced H2A.Z and SUZ12 occupancy (Supplementary Figure S7F, G), consistent with the above findings. Finally, we analyzed the genomic occupancy by chromatin factors in ESCs using public data (62,63). We found that INO80 closely clustered with H2A.Z and PRC2 at bivalent promoters among major chromatin remodelers (Figure 7I). Besides INO80, the NuRD complex has been shown to repress bivalent genes via H3K27ac deacetylation (64). As expected, the core NuRD components CHD4 and MBD3 also showed strong co-localization with PRC2, supporting the validity of this analysis.
Taken together, we propose the following model (graphical abstract): During the naïve to primed transition, INO80 occupancy at TSSs near developmental genes promotes the deposition of H2A.Z. In turn, increased H2A.Z binding facilitates the recruitment of PRC2 and methylation of H3K27 to establish bivalent domains. The bivalent histone modifications maintain the developmental genes in repressed state for activation during further differentiation and development. Our model strongly argues that INO80-dependent H2A.Z deposition is a critical licensing step in bivalent chromatin establishment and maintenance during cell fate transitions.
DISCUSSION
ATP‐dependent chromatin remodeling complexes play important roles in pluripotent stem cells. While many interact with and target specific nucleosomes (62), we showed that INO80 does not appear to drastically change global nucleosome positioning or density. Instead, it promotes H2A.Z occupancy and licenses the formation and maintenance of bivalent domains at developmental gene promoters. Our findings support the notion that different remodelers use different mechanisms and act on different genes to maintain the transcription program in pluripotent stem cells.
Our findings are also consistent with the mouse phenotypes. Specifically, Ino80 deletion led to post-implantation lethality and the embryos failed to develop beyond the egg cylinder stage (50,53). Deletion of H2A.Z and PRC2 components resulted in developmental arrest around gastrulation (65–67). The similarities in the developmental defects and timing in the animal models support the idea that INO80, H2A.Z and PRC2 coordinately regulate gene expression in post-implantation embryonic development. They also support our result that Ino80 is required in the primed pluripotent state and bivalent gene regulation. Interestingly, Ino80 is dispensable for the naïve pluripotent state, but is necessary to maintain ESC self-renewal in the serum/LIF condition (51). The differences may likely lie in the specific cellular state, as ESCs cultured in serum/LIF are heterogenous and known to require many more factors that are otherwise expendable in the naïve state (3,68). In contrast, the naïve state is thought to be the ground state that is more robust and stable (69). It is characterized with an epigenome that has low levels of DNA and repressive histone methylations (5,6), and it can be maintained without many of the repressive epigenetic mechanisms (70). Consistently, we found that H2A.Z genomic occupancy is low in the naïve state and Ino80 deletion did not further reduce its level significantly, which may explain why Ino80 is not required under this condition.
INO80 has been shown to facilitate H2A.Z eviction in vitro, though the impact on H2A.Z levels in vivo in yeast appears to be variable (37,49,71,72). In mouse ESCs, we found that INO80 showed the highest co-localization with H2A.Z in the genome among major ATP-dependent chromatin remodelers. Furthermore, it is required for the maintenance of H2A.Z occupancy in the primed state. Finally, Ino80 deletion led to reduced H2A.Z occupancy in MEFs as well, suggesting that its role in H2A.Z deposition and maintenance is not unique to ESCs. Consistent with our data, a recent study showed that Ino80 mutant caused decreased H2A.Z binding at key flowering genes in Arabidopsis (73). Thus, INO80 likely plays a complex role in H2A.Z regulation especially in higher eukaryotes. Intriguingly, our results showed that INO80 pre-marks bivalent promoters in the naïve state before significant levels of H2A.Z are established. Upon transition into the primed state, H2A.Z occupancy at bivalent promoters is greatly increased and becomes dependent on INO80. These results suggest that INO80 alone is insufficient for H2A.Z deposition. Instead, it likely facilitates H2A.Z deposition by other chromatin remodelers in the establishment and maintenance of the bivalent domains. In support of this view, the mammalian SWR1 homologues TIP60/EP400 and SRCAP can both catalyze H2A.Z incorporation within the chromatin (74,75), while INO80 has no such activity. Furthermore, TIP60/EP400 was found to bind many bivalent promoters and repress developmental gene expression in ESCs (76). Gas41, a subunit found in both TIP60/EP400 and SRCAP, is required for H2A.Z occupancy at bivalent domains in ESCs (77). It will be interesting to further investigate the roles of INO80, TIP60/EP400, and SRCAP in H2A.Z deposition in the naïve and primed state.
The histone variant H2A.Z can activate or repress downstream genes in a context dependent manner and is involved in many developmental and disease processes (35). In serum/LIF cultured ESCs, H2A.Z is enriched at gene promoters and PRC2 target genes, and it facilitates both positive and negative regulatory complexes to access chromatin and regulate genes important for ESC self-renewal and differentiation (38,39,42). Consistently, we showed that H2A.Z binds both active and repressed gene promoters in the naïve and primed pluripotent state. However, we found that H2A.Z shows stronger signals at the bivalent gene promoters, and it is selectively required in the primed but not the naïve state. This is consistent with the phenotype of H2A.Z deletion in mice, in which the null embryos were normal up to E4.5 and started to show defects after implantation (65). In addition, we identified H2A.Z as crucial prerequisite in H3K27me3 deposition and bivalent domain formation, adding further support to earlier reports on the interplay between H2A.Z and H3K27me3 (38,42,43). Finally, our results provided a possible mechanism to explain the de novo formation of bivalent domains after H3K27me3 erasure (31), in that H2A.Z may serve as a bookmark in the absence of H3K27me3. The bivalent modifications by H3K4me3 and H3K27me3 at developmental gene promoters are thought to keep downstream genes in a state for rapid activation or silencing upon further differentiation (22,23). In agreement with this notion, we showed that reduced H3K27me3 occupancy at bivalent promoters in Ino80 or H2az1/H2az2 deletion cells led to de-repression of the downstream genes and disruption of the primed pluripotent state. Our data suggests that the bivalent domains may play an important role in maintaining an intricate balance between self-renewal and differentiation in pluripotency and early development. Indeed, recent reports showed that bivalent modifications are largely absent from developmental gene promoters in mouse pre-implantation embryos but become enriched soon after implantation, and a strong bivalent chromatin is a hallmark for the primed pluripotent state (32,33). In summary, we identified INO80 as an essential regulator for bivalent domains and primed pluripotency via H2A.Z. Because INO80 and H2A.Z have been heavily implicated in development and disease, it is conceivable that the INO80-H2A.Z-H3K27me3 axis may play important roles in a similar fashion during other cell fate transitions.
DATA AVAILABILITY
Genomic data generated for this study have been deposited in the GEO repository with the accession number GSE158545. We used the following published datasets for analysis: ChIP-seq: ATPase chromatin remodelers (GSE64825), SUZ12 and EZH2 (GSE49431), H3K4me3 and H3K27me3 (GSE117896); scRNA-seq: E-MTAB-6967.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Michael Stadler and Dr Dirk Schübeler (FMI science) for the help with MNase-seq data normalization. We thank Dr Joanna Wysocka (Stanford University) for the PiggyBac expression vectors and Dr Shaun Cowley (University of Leicester) for the Rosa26-CreERT2 ESCs. We thank Dr Raja Jothi and Dr Dhirendra Kumar (NIEHS) for scientific discussions on bivalency. We thank the NIEHS Epigenomics and DNA Sequencing core, Imaging and Animal facilities for assistance with various experiments.
Author contributions: H.Y. and G.H. conceived the study. H.Y., B.L. and G.H. carried out the experiments. J.W., H.Y. and B.B. analyzed the data. H.Y., J.W. B.L., B.B., J.L. and G.H. interpreted the data. H.Y. and G.H. wrote the manuscript.
Contributor Information
Hongyao Yu, Epigenetics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709, USA.
Jiajia Wang, Epigenetics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709, USA.
Brad Lackford, Epigenetics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709, USA.
Brian Bennett, Integrative Bioinformatics Support Group, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709, USA.
Jian-liang Li, Integrative Bioinformatics Support Group, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709, USA.
Guang Hu, Epigenetics and Stem Cell Biology Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the NIH, National Institute of Environmental Health Sciences [Z01ES102745 to G.H., in part]. Funding for open access charge: NIEHS [Z01ES102745].
Conflict of interest statement. None declared.
REFERENCES
- 1. Nichols J., Smith A.. Naïve and primed pluripotent states. Cell Stem Cell. 2009; 4:487–492. [DOI] [PubMed] [Google Scholar]
- 2. Kinoshita M., Smith A.. Pluripotency deconstructed. Dev. Growth Differ. 2018; 60:44–52. [DOI] [PubMed] [Google Scholar]
- 3. Ying Q.L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A.. The ground state of embryonic stem cell self-renewal. Nature. 2008; 453:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guo G., Yang J., Nichols J., Hall J.S., Eyres I., Mansfield W., Smith A.. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development. 2009; 136:1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marks H., Kalkan T., Menafra R., Denissov S., Jones K., Hofemeister H., Nichols J., Kranz A., Stewart A.F., Smith A.et al.. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012; 149:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Habibi E., Brinkman A.B., Arand J., Kroeze L.I., Kerstens H.H., Matarese F., Lepikhov K., Gut M., Brun-Heath I., Hubner N.C.et al.. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013; 13:360–369. [DOI] [PubMed] [Google Scholar]
- 7. Hackett J.A., Dietmann S., Murakami K., Down T.A., Leitch H.G., Surani M.A.. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Rep. 2013; 1:518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buecker C., Srinivasan R., Wu Z., Calo E., Acampora D., Faial T., Simeone A., Tan M., Swigut T., Wysocka J.. Reorganization of enhancer patterns in transition from naïve to primed pluripotency. Cell Stem Cell. 2014; 14:838–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Factor D.C., Corradin O., Zentner G.E., Saiakhova A., Song L., Chenoweth J.G., McKay R.D., Crawford G.E., Scacheri P.C., Tesar P.J.. Epigenomic comparison reveals activation of “seed" enhancers during transition from naïve to primed pluripotency. Cell Stem Cell. 2014; 14:854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kalkan T., Olova N., Roode M., Mulas C., Lee H.J., Nett I., Marks H., Walker R., Stunnenberg H.G., Lilley K.S.et al.. Tracking the embryonic stem cell transition from ground state pluripotency. Development. 2017; 144:1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Mierlo G., Dirks R.A.M., De Clerck L., Brinkman A.B., Huth M., Kloet S.L., Saksouk N., Kroeze L.I., Willems S., Farlik M.et al.. Integrative proteomic profiling reveals PRC2-dependent epigenetic crosstalk maintains ground-state pluripotency. Cell Stem Cell. 2019; 24:123–137. [DOI] [PubMed] [Google Scholar]
- 12. Yang P., Humphrey S.J., Cinghu S., Pathania R., Oldfield A.J., Kumar D., Perera D., Yang J.Y.H., James D.E., Mann M.et al.. Multi-omic profiling reveals dynamics of the phased progression of pluripotency. Cell Syst. 2019; 8:427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brons I.G., Smithers L.E., Trotter M.W., Rugg-Gunn P., Sun B., Chuva de Sousa Lopes S.M., Howlett S.K., Clarkson A., Ahrlund-Richter L., Pedersen R.A.et al.. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007; 448:191–195. [DOI] [PubMed] [Google Scholar]
- 14. Tesar P.J., Chenoweth J.G., Brook F.A., Davies T.J., Evans E.P., Mack D.L., Gardner R.L., McKay R.D.. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007; 448:196–199. [DOI] [PubMed] [Google Scholar]
- 15. Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R.. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013; 153:910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalkan T., Bornelov S., Mulas C., Diamanti E., Lohoff T., Ralser M., Middelkamp S., Lombard P., Nichols J., Smith A.. Complementary activity of ETV5, RBPJ, and TCF3 drives formative transition from naïve pluripotency. Cell Stem Cell. 2019; 24:785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K.et al.. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006; 125:315–326. [DOI] [PubMed] [Google Scholar]
- 18. Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P.et al.. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007; 448:553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pan G., Tian S., Nie J., Yang C., Ruotti V., Wei H., Jonsdottir G.A., Stewart R., Thomson J.A.. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007; 1:299–312. [DOI] [PubMed] [Google Scholar]
- 20. Zhao X.D., Han X., Chew J.L., Liu J., Chiu K.P., Choo A., Orlov Y.L., Sung W.K., Shahab A., Kuznetsov V.A.et al.. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007; 1:286–298. [DOI] [PubMed] [Google Scholar]
- 21. Denholtz M., Bonora G., Chronis C., Splinter E., de Laat W., Ernst J., Pellegrini M., Plath K.. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 2013; 13:602–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Voigt P., Tee W.W., Reinberg D.. A double take on bivalent promoters. Genes Dev. 2013; 27:1318–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harikumar A., Meshorer E.. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 2015; 16:1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mohn F., Weber M., Rebhan M., Roloff T.C., Richter J., Stadler M.B., Bibel M., Schubeler D.. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008; 30:755–766. [DOI] [PubMed] [Google Scholar]
- 25. Cui K., Zang C., Roh T.Y., Schones D.E., Childs R.W., Peng W., Zhao K.. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009; 4:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McGarvey K.M., Van Neste L., Cope L., Ohm J.E., Herman J.G., Van Criekinge W., Schuebel K.E., Baylin S.B.. Defining a chromatin pattern that characterizes DNA-hypermethylated genes in colon cancer cells. Cancer Res. 2008; 68:5753–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodriguez F.J., Scheithauer B.W., Giannini C., Bryant S.C., Jenkins R.B.. Epithelial and pseudoepithelial differentiation in glioblastoma and gliosarcoma: a comparative morphologic and molecular genetic study. Cancer. 2008; 113:2779–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vastenhouw N.L., Zhang Y., Woods I.G., Imam F., Regev A., Liu X.S., Rinn J., Schier A.F.. Chromatin signature of embryonic pluripotency is established during genome activation. Nature. 2010; 464:922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lesch B.J., Silber S.J., McCarrey J.R., Page D.C.. Parallel evolution of male germline epigenetic poising and somatic development in animals. Nat. Genet. 2016; 48:888–894. [DOI] [PubMed] [Google Scholar]
- 30. Kuroda M.I., Kang H., De S., Kassis J.A.. Dynamic competition of polycomb and trithorax in transcriptional programming. Annu. Rev. Biochem. 2020; 89:235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hojfeldt J.W., Laugesen A., Willumsen B.M., Damhofer H., Hedehus L., Tvardovskiy A., Mohammad F., Jensen O.N., Helin K.. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018; 25:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng H., Huang B., Zhang B., Xiang Y., Du Z., Xu Q., Li Y., Wang Q., Ma J., Peng X.et al.. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol. Cell. 2016; 63:1066–1079. [DOI] [PubMed] [Google Scholar]
- 33. Xiang Y., Zhang Y., Xu Q., Zhou C., Liu B., Du Z., Zhang K., Zhang B., Wang X., Gayen S.et al.. Epigenomic analysis of gastrulation identifies a unique chromatin state for primed pluripotency. Nat. Genet. 2020; 52:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scacchetti A., Becker P.B.. Variation on a theme: Evolutionary strategies for H2A.Z exchange by SWR1-type remodelers. Curr. Opin. Cell Biol. 2020; 70:1–9. [DOI] [PubMed] [Google Scholar]
- 35. Giaimo B.D., Ferrante F., Herchenrother A., Hake S.B., Borggrefe T.. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin. 2019; 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mizuguchi G., Shen X., Landry J., Wu W.H., Sen S., Wu C.. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004; 303:343–348. [DOI] [PubMed] [Google Scholar]
- 37. Papamichos-Chronakis M., Watanabe S., Rando O.J., Peterson C.L.. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell. 2011; 144:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Creyghton M.P., Markoulaki S., Levine S.S., Hanna J., Lodato M.A., Sha K., Young R.A., Jaenisch R., Boyer L.A.. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell. 2008; 135:649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ku M., Jaffe J.D., Koche R.P., Rheinbay E., Endoh M., Koseki H., Carr S.A., Bernstein B.E.. H2A.Z landscapes and dual modifications in pluripotent and multipotent stem cells underlie complex genome regulatory functions. Genome Biol. 2012; 13:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Subramanian V., Mazumder A., Surface L.E., Butty V.L., Fields P.A., Alwan A., Torrey L., Thai K.K., Levine S.S., Bathe M.et al.. H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLos Genet. 2013; 9:e1003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Courtney A.J., Kamei M., Ferraro A.R., Gai K., He Q., Honda S., Lewis Z.A.. Normal patterns of histone H3K27 methylation require the histone variant H2A.Z in neurospora crassa. Genetics. 2020; 216:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu G., Cui K., Northrup D., Liu C., Wang C., Tang Q., Ge K., Levens D., Crane-Robinson C., Zhao K.. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2013; 12:180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y., Long H., Yu J., Dong L., Wassef M., Zhuo B., Li X., Zhao J., Wang M., Liu C.et al.. Histone variants H2A.Z and H3.3 coordinately regulate PRC2-dependent H3K27me3 deposition and gene expression regulation in mES cells. BMC Biol. 2018; 16:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hota S.K., Bruneau B.G.. ATP-dependent chromatin remodeling during mammalian development. Development. 2016; 143:2882–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clapier C.R., Iwasa J., Cairns B.R., Peterson C.L.. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017; 18:407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shen X., Mizuguchi G., Hamiche A., Wu C.. A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000; 406:541–544. [DOI] [PubMed] [Google Scholar]
- 47. Gerhold C.B., Gasser S.M.. INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends Cell Biol. 2014; 24:619–631. [DOI] [PubMed] [Google Scholar]
- 48. Yen K., Vinayachandran V., Pugh B.F.. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell. 2013; 154:1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brahma S., Udugama M.I., Kim J., Hada A., Bhardwaj S.K., Hailu S.G., Lee T.H., Bartholomew B.. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nat. Commun. 2017; 8:15616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Min J.N., Tian Y., Xiao Y., Wu L., Li L., Chang S.. The mINO80 chromatin remodeling complex is required for efficient telomere replication and maintenance of genome stability. Cell Res. 2013; 23:1396–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang L., Du Y., Ward J.M., Shimbo T., Lackford B., Zheng X., Miao Y.L., Zhou B., Han L., Fargo D.C.et al.. INO80 facilitates pluripotency gene activation in embryonic stem cell self-renewal, reprogramming, and blastocyst development. Cell Stem Cell. 2014; 14:575–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou B., Wang L., Zhang S., Bennett B.D., He F., Zhang Y., Xiong C., Han L., Diao L., Li P.et al.. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 2016; 30:1440–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qiu Z., Elsayed Z., Peterkin V., Alkatib S., Bennett D., Landry J.W.. Ino80 is essential for proximal-distal axis asymmetry in part by regulating Bmp4 expression. BMC Biol. 2016; 14:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Serber D.W., Runge J.S., Menon D.U., Magnuson T.. The mouse INO80 chromatin-remodeling complex is an essential meiotic factor for spermatogenesis. Biol. Reprod. 2016; 94:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B.et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barisic D., Stadler M.B., Iurlaro M., Schubeler D.. Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature. 2019; 569:136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maehara K., Ohkawa Y.. Exploration of nucleosome positioning patterns in transcription factor function. Sci. Rep. 2016; 6:19620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hayashi K., Ohta H., Kurimoto K., Aramaki S., Saitou M.. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell. 2011; 146:519–532. [DOI] [PubMed] [Google Scholar]
- 59. Chen L., Cai Y., Jin J., Florens L., Swanson S.K., Washburn M.P., Conaway J.W., Conaway R.C.. Subunit organization of the human INO80 chromatin remodeling complex: an evolutionarily conserved core complex catalyzes ATP-dependent nucleosome remodeling. J. Biol. Chem. 2011; 286:11283–11289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kapoor P., Bao Y., Xiao J., Espejo A., Yang L., Bedford M.T., Peng G., Shen X.. Phosphorylation-dependent enhancement of Rad53 kinase activity through the INO80 chromatin remodeling complex. Mol. Cell. 2015; 58:863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mieczkowski J., Cook A., Bowman S.K., Mueller B., Alver B.H., Kundu S., Deaton A.M., Urban J.A., Larschan E., Park P.J.et al.. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016; 7:11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Dieuleveult M., Yen K., Hmitou I., Depaux A., Boussouar F., Bou Dargham D., Jounier S., Humbertclaude H., Ribierre F., Baulard C.et al.. Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature. 2016; 530:113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kaneko S., Son J., Shen S.S., Reinberg D., Bonasio R.. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat. Struct. Mol. Biol. 2013; 20:1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reynolds N., Salmon-Divon M., Dvinge H., Hynes-Allen A., Balasooriya G., Leaford D., Behrens A., Bertone P., Hendrich B.. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J. 2012; 31:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Faast R., Thonglairoam V., Schulz T.C., Beall J., Wells J.R., Taylor H., Matthaei K., Rathjen P.D., Tremethick D.J., Lyons I.. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 2001; 11:1183–1187. [DOI] [PubMed] [Google Scholar]
- 66. Pasini D., Bracken A.P., Jensen M.R., Lazzerini Denchi E., Helin K.. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004; 23:4061–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. O’Carroll D., Erhardt S., Pagani M., Barton S.C., Surani M.A., Jenuwein T.. The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 2001; 21:4330–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yamaji M., Ueda J., Hayashi K., Ohta H., Yabuta Y., Kurimoto K., Nakato R., Yamada Y., Shirahige K., Saitou M.. PRDM14 ensures naïve pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell. 2013; 12:368–382. [DOI] [PubMed] [Google Scholar]
- 69. Halley J.D., Smith-Miles K., Winkler D.A., Kalkan T., Huang S., Smith A.. Self-organizing circuitry and emergent computation in mouse embryonic stem cells. Stem Cell Res. 2012; 8:324–333. [DOI] [PubMed] [Google Scholar]
- 70. Shukla R., Mjoseng H.K., Thomson J.P., Kling S., Sproul D., Dunican D.S., Ramsahoye B., Wongtawan T., Treindl F., Templin M.F.et al.. Activation of transcription factor circuity in 2i-induced ground state pluripotency is independent of repressive global epigenetic landscapes. Nucleic. Acids. Res. 2020; 48:7748–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jeronimo C., Watanabe S., Kaplan C.D., Peterson C.L., Robert F.. The histone chaperones FACT and Spt6 restrict H2A.Z from intragenic locations. Mol. Cell. 2015; 58:1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tramantano M., Sun L., Au C., Labuz D., Liu Z., Chou M., Shen C., Luk E.. Constitutive turnover of histone H2A.Z at yeast promoters requires the preinitiation complex. Elife. 2016; 5:e14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang C., Cao L., Rong L., An Z., Zhou W., Ma J., Shen W.H., Zhu Y., Dong A.. The chromatin-remodeling factor AtINO80 plays crucial roles in genome stability maintenance and in plant development. Plant J. 2015; 82:655–668. [DOI] [PubMed] [Google Scholar]
- 74. Ruhl D.D., Jin J., Cai Y., Swanson S., Florens L., Washburn M.P., Conaway R.C., Conaway J.W., Chrivia J.C.. Purification of a human SRCAP complex that remodels chromatin by incorporating the histone variant H2A.Z into nucleosomes. Biochemistry. 2006; 45:5671–5677. [DOI] [PubMed] [Google Scholar]
- 75. Gevry N., Chan H.M., Laflamme L., Livingston D.M., Gaudreau L.. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007; 21:1869–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fazzio T.G., Huff J.T., Panning B.. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008; 134:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hsu C.C., Zhao D., Shi J., Peng D., Guan H., Li Y., Huang Y., Wen H., Li W., Li H.et al.. Gas41 links histone acetylation to H2A.Z deposition and maintenance of embryonic stem cell identity. Cell Discov. 2018; 4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic data generated for this study have been deposited in the GEO repository with the accession number GSE158545. We used the following published datasets for analysis: ChIP-seq: ATPase chromatin remodelers (GSE64825), SUZ12 and EZH2 (GSE49431), H3K4me3 and H3K27me3 (GSE117896); scRNA-seq: E-MTAB-6967.