

Abstract
Male infertility impacts millions of couples yet, the etiology of primary infertility remains largely unknown. A critical element of successful spermatogenesis is maintenance of genome integrity. Here, we present a genomic study of spermatogenic failure (SPGF). Our initial analysis (n = 176) did not reveal known gene-candidates but identified a potentially significant single-nucleotide variant (SNV) in X-linked germ-cell nuclear antigen (GCNA). Together with a larger follow-up study (n = 2049), 7 likely clinically relevant GCNA variants were identified. GCNA is critical for genome integrity in male meiosis and knockout models exhibit impaired spermatogenesis and infertility. Single-cell RNA-seq and immunohistochemistry confirm human GCNA expression from spermatogonia to elongated spermatids. Five identified SNVs were located in key functional regions, including N-terminal SUMO-interacting motif and C-terminal Spartan-like protease domain. Notably, variant p.Ala115ProfsTer7 results in an early frameshift, while Spartan-like domain missense variants p.Ser659Trp and p.Arg664Cys change conserved residues, likely affecting 3D structure. For variants within GCNA’s intrinsically disordered region, we performed computational modeling for consensus motifs. Two SNVs were predicted to impact the structure of these consensus motifs. All identified variants have an extremely low minor allele frequency in the general population and 6 of 7 were not detected in > 5000 biological fathers. Considering evidence from animal models, germ-cell-specific expression, 3D modeling, and computational predictions for SNVs, we propose that identified GCNA variants disrupt structure and function of the respective protein domains, ultimately arresting germ-cell division. To our knowledge, this is the first study implicating GCNA, a key genome integrity factor, in human male infertility.
Introduction
Male infertility is a significant health problem that impacts millions of couples trying to conceive worldwide. The etiology of primary infertility, a condition directly affecting the reproductive system and germ cell production, is not well understood, with nearly 50% of male cases described as unexplained (Agarwal et al. 2021). The consensus view is that male infertility is a complex condition with a significant genetic component (Krausz and Riera-Escamilla 2018). As such, current genetic standards for diagnosis of severe male infertility include testing for numerical and structural chromosomal aberrations and deletions of the AZF regions on the Y chromosome. These approaches, however, yield low diagnosis rates of between 10 and 20% (Tüttelmann et al. 2018). Massive research efforts in animal reproductive biology have identified hundreds of infertility gene candidates, suggesting that disruption of their human orthologs would also result in male infertility (Cooke and Saunders 2002; Schultz et al. 2003; Singh and Schimenti 2015). However, progress in integrating diagnostic testing for variants in these genes into human reproductive medicine remains modest, with only a handful readily translated to the clinic. Recent genomic analyses of complex disorders have improved the perspective of conclusive diagnoses, successfully elucidating underlying variants for many disorders with a strong genetic component, including male infertility. Translational studies have identified several clinically recognized male fertility gene markers (e.g., CFAP65, M1AP, SOLHLH1, SYCP2, SYCP3, and TEX11) (Miyamoto et al. 2003; Choi et al. 2010; Yang et al. 2015; Yatsenko et al. 2015; Wang et al. 2019; Schilit et al. 2020, Wyrwoll et al. 2020). Still, overall progress in detection of causal variants in primary spermatogenic failure (SPGF) is limited with only a fraction of the genes responsible for non-obstructive azoospermia, oligozoospermia, teratozoospermia, and asthenozoospermia uncovered (Oud et al. 2019).
A notable clinical challenge within the broad spectrum of male infertility is defining the etiology of men who present with severe germ-cell arrest at the mitotic or meiotic phases of spermatogenesis. These individuals have no prospect for natural conception and assisted reproductive technology methods often involve highly invasive techniques such as microdissection testicular sperm extraction (mTESE). Several key biological processes in germ cell division include cross-over exchange of genetic material and subsequent maintenance of genome stability yet, it remains unclear whether aberrant germ cell-specific genome integrity factors contribute to male infertility. To date, only a few genome integrity genes with pathogenic variants responsible for some forms of human infertility (e.g., MCM8 and MCM9) have been reported (Wood-Trageser et al. 2014; AlAsiri et al. 2015; Desai et al. 2017).
X-linked, germ cell nuclear antigen (GCNA), previously known as acidic repeat containing (ACRC), encodes an ancient germ cell-specific protein with multiple highly conserved functional domains, including an N-terminus intrinsically disordered region (IDR) and C-terminus Spartan (SprT)-like and zinc finger domains in humans (Carmell et al. 2016). GCNA is a crucial regulator of germ cell division and development in multiple eukaryotes. Recent animal studies suggest a major role for GCNA in DNA repair and genome maintenance during chromosomal crossing over as well as chromatin condensation during sperm maturation (Bhargava et al. 2020; Dokshin et al. 2020). Furthermore, knockout male mice along with other transgenic animal models present with infertility marked by abnormal chromosome segregation and meiotic germ cell loss. Thus, we hypothesize that selected variants disrupt critical GCNA domains and contribute to male infertility in humans. Here, using whole exome sequencing (WES), we identified variants in the germ-cell-specific genome integrity factor, GCNA, associated with spermatogenic failure and human male infertility.
Materials and methods
Patients and controls
Participants were recruited from University of Pittsburgh, United States (n = 120), Polish Academy of Sciences, Poland (n = 56), Münster University, Germany (n = 1100), GEMINI Consortium (Weill Cornell Medicine, United States and University of Tartu, Estonia) (n = 924), and Monash University, Australia (n = 25). Patients were diagnosed with non-obstructive azoospermia (no sperm in the ejaculate due to impaired sperm production) or cryptozoospermia (no sperm in fresh ejaculate without evidence of partial reproductive tract obstruction, but with rare sperm seen in centrifuged pellet) according to American Urology Association and American Society for Reproductive Medicine guidelines (Schlegel et al. 2021). Briefly, male infertility patients were evaluated with reproductive history and physical examination based on AUA and ASRM guidelines. For these men, evaluation also included semen analysis, serum follicle stimulating hormone, luteinizing hormone, testosterone, and prolactin levels, testicular biopsy, and ultrasound (where appropriate). Individuals with a history of non-genetic causes for SPGF (e.g., trauma, surgery, or medication), obstructive azoospermia (e.g., CBAVD), and with abnormal sex chromosome evaluation on karyotype or Y chromosome micro-deletions were excluded from the study.
Whole exome sequencing and data analysis
DNA from whole blood collected by venipuncture was isolated using standard methods. For samples processed in Pittsburgh, sequencing libraries were constructed using the SureSelect XT All Exon V4 + UTRs capture kit and SureSelect XT Target Enrichment System (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s protocol. Captured libraries were sequenced using 100 bp paired-end sequencing with TruSeq PE Cluster Kit v3 cBot HS and 4 × 50 TruSeq SBS V3 HS sequencing kit (Illumina, San Diego, CA) on the HiSeq2000 (Illumina, San Diego, CA). Approximately 6 Gb of sequencing data per sample was generated with average read coverage of 62–100X. For the Australian cohort, whole exome target capture was achieved using Twist Bioscience’s Twist Human Core Exome Kit (Twist Bioscience, San Francisco, CA) according to the manufacturer’s protocol. Sample libraries were dual-indexed using primers of the Twist Universal Adapter System. Pooled libraries were sequenced on the NovaSeq 6000 platform (Illumina, San Diego, CA). 150 bp paired-end sequencing was carried out at an average sequencing depth of 90 × per sample.
Sequencing reads were aligned to human reference genome hg19 with the Burrows-Wheeler Aligner (Li and Durbin 2010). Variant calling and quality control were carried out with the Genome Analysis Toolkit (McKenna et al. 2010). Variants were annotated using ANNOVAR or Variant Effect Predictor (Wang et al. 2010; McLaren et al. 2016). For WES analysis, we evaluated coding, non-synonymous, single nucleotide variants (SNVs) with minor allele frequency (MAF) < 1% in males according to GnomAD v2.1.1 (https://gnomad.broadinstitute.org/). SNVs were cross referenced against reported monogenic gene candidates with definitive association with human male infertility (Oud et al. 2019; Alhathal et al. 2020). Variant disease association was determined using public and private databases dbSNP (https://www.ncbi.nlm.nih.gov/snp/), OMIM (https://www.ncbi.nlm.nih.gov/omim/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and HGMD (Qiagen, Redwood City, CA). SNV pathogenicity was assessed on the basis of American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015).
Missense mutation predicted effect was derived from computational tools, MutationT@ster (Schwarz et al. 2010), SIFT (Ng and Henikoff 2001), Polyphen (Adzhubei et al. 2010), CADD (Rentzsch et al. 2019), MetaDome (Wiel et al. 2019) and Revel (Ioannidis et al. 2016). Conservation was determined via PhyloP and Clustal Omega (Pollard et al. 2010; Sievers et al. 2011). Whole exome sequencing data from an anonymous cohort of 5784 proven fathers, representing the general Dutch population visiting the outpatient clinic as a father of a child with severe developmental delay, was used as a control (Wyrwoll et al. 2020).
Mouse models with infertility phenotypes were reviewed in the Mouse Genome Informatics database (http://www.informatics.jax.org/) and through literature searches (https://pubmed.ncbi.nlm.nih.gov/). Testis tissue expression was retrieved from GTex (https://gtexportal.org/home/), Ace-View (https://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/), and BioGPS (http://biogps.org/).
All next-generation sequencing variants were confirmed via the Integrative Genomics Viewer (Thorvaldsdóttir et al. 2013). Top variants were confirmed with Sanger DNA sequencing.
Single-cell RNA-seq data analysis
A processed merged human digital expression matrix data file from Drop-seq experiments on human testicular cells from 4 adult males was obtained from Gene Expression Omnibus (GEO: GSE142585) (Shami et al. 2020). In total, 35,941 transcripts from 13,597 cells were used for global analysis. To identify human testicular cell types, the following known RNA markers were used: spermatogonia (GFRA1, HORMAD1, ID4, ITGA6, LY6K, STRA8, SYCP2, UCHL1, and UTF1), spermatocytes (PIWIL1 and SYCP3), spermatids (ACRV1, PRM1, TNP1, and TSSK6), Leydig cells (IFG1/2 and STAR), endothelial cells (NOSTRIN and VWF), testicular macrophages (CD52, CD163, LYZ, and TYROBP), pericytes (ADIRF, MCAM, PDGFRB, and STEAP4) or myoid cells (ACTA2 and MYH11).
Data were filtered, normalized, and scaled as previously described using Seurat v3.2.2 (Macosko et al. 2015). Principal component analysis (PCA) on the gene expression matrix was used for downstream analysis based on the detection of the “elbow” point on the screen plot and evaluation of jack-straw function heatmaps. Further dimensionality reduction for easy visualization was performed using uniform manifold approximation and projection (UMAP). Louvain–Jaccard clustering was utilized for cluster identification. Expression patterns of conserved marker genes of human testicular cells were analyzed to annotate the biological identity of each cluster. Spermatogonia were further divided into four distinct clusters via unsupervised clustering. The expression of conserved undifferentiated spermatogonia markers (GFRA1, ID4, and UCHL1) and differentiating spermatogonia markers (HORMAD1, KIT, MKI67, STRA8, and TEX11) were used to biologically annotate clusters 1 and 2 (probably representing undifferentiated spermatogonia) and clusters 3 and 4 (representing differentiating spermatogonia). Identified genes were normalized, scaled, and used for PCA and UMAP analysis as above.
Testicular biopsy and histology
Testicular biopsies obtained from SPGF patients were collected under general anesthesia at the Magee Womens Hospital (Pittsburgh, PA), the Centre for Reproductive Medicine and Andrology (Münster, Germany), and Weill Cornell Medical Center (New York, NY). Written informed consent was obtained before surgery. Tissue samples were fixed overnight in Bouin’s solution, washed with 70% ethanol, and embedded in paraffin, using an automatic ethanol and paraffin row (Bavimed Laborgeräte, Birkenau, Germany). For histological analysis, 5-μm sections were stained with periodic acid-Schiff (PAS) reagent and Mayer’s hematoxylin according to the previously published protocol (Brinkworth et al. 1995). Briefly, sections were deparaffinized, rehydrated in a descending ethanol row, and rinsed with water. Cross sections were placed in a freshly prepared 1% periodic acid solution (#1,005,240,100, Carl Roth, Karlsruhe, Germany) for 5–15 min, followed by washing with distilled water. Incubation in Schiff reagent (#1,090,330,500, Merck Millipore, Darmstadt, Germany) was performed for 5–10 min, and sections were washed again. Counterstaining was conducted using Mayer’s hematoxylin (#109,249, Merck Millipore, Darmstadt, Germany) for 30 s and stopped in distilled water. Finally, sections were rinsed with tap water, dehydrated in an increasing ethanol series, and mounted using Merckoglas mounting medium (#103,973, Merck Millipore). Slides were imaged using a PreciPoint M8 Scanning Microscope or Olympus Virtual Slide System Axioskop (Zeiss, Oberkochen, Germany). Histology images are available for patient M13 and M911.
Immunohistochemistry
Bouin’s fixed testis sections were deparaffinized and rehydrated as described earlier (Brieño-Enríquez et al. 2019). Heat-induced epitope retrieval and immunofluorescence were carried out according to Leduc et al. (2008) with the following modifications. An aliquot of freshly prepared 1.5% BSA/0.5% Triton-X100 in PBS was heated to 37 °C prior to blocking at 37 °C for 1 h in a humidification chamber. Tissue sections were incubated with primary antibodies— anti-GCNA 1:25, (HPA023476, Sigma-Aldrich, St. Louis, MO), anti-DDX4 1:100 (ab27591, Cambridge, UK), anti-SOX9 1:100 (AB5535, EMD Millipore, Burlington, MA)—in a humidification chamber overnight at 4 °C. Secondary antibodies included Alexa Fluor 488 1:100 and Alexa Fluor 594 1:100 (115-546-062 and 111-586-144, Jackson ImmunoResearch, West Grove, PA). Nuclei were stained with DAPI and slides were mounted with Vectashield Antifade Mounting Medium (H-1000-10, Vector Laboratories, Burlingame, CA). Images were captured on a Zeiss Axio Imager M2 microscope using Zen software (Zeiss, Oberkochen, Germany).
SprT-like domain 3D modeling and IDR motif search
To identify and model conserved amino acids in the SprT-like domain, human, fruit fly, and roundworm GCNA sequences were obtained from UniProt (accession codes Q96QF7, Q7KW09 and Q23462, respectively). Tertiary structure corresponding to each sequence was predicted using the servers Modeller and HHpred, with crystal structure of the human metalloprotease Spartan (PDBID: 6MDW) used as a template (Sali et al. 1995; Soding et al. 2005; Li et al. 2019). Structural alignment and mapping of relevant residues were performed using PyMOL Molecular Graphics System version 2.0 (Schrodinger 2018).
To identify common motifs within IDRs among GCNA orthologs, IDR sequences were isolated using the MetaDisorder web server (Kozlowski and Bujnicki 2012). Multiple sequence alignments of 24 GCNA IDRs were created and short regions of interest were converted into regular expressions (regex) representing chemical characteristics of the amino acids, such as non-polar or pi-bond-containing residues. Using the CLC-7 suite, matches to regex were identified among the GCNA orthologs. All matches were aligned to determine amino acid ratios, which were converted into predicted motifs for downstream analysis using the MEME suite (Grant et al. 2011) and were represented as logos. Logo visualizations were generated by Seq2Logo (Thomsen and Nielsen 2012). The motif alignment and search tool (MAST) was used to match the regex-based, putative motifs against the GCNA proteins (Bailey and Gribskov 1998). To confirm that the putative motifs are specific to GCNA and not an artifact of intrinsically disordered proteins (IDPs), identified motifs were further tested with MAST against a dataset of experientially determined IDPs retrieved from DisProt (Hatos, Hajdu-Soltész et al. 2020).
Results
Identification of GCNA variants in patients with sporadic SPGF
In an effort to identify genetic causes of SPGF, we first analyzed WES data from sporadic and familial male infertility patients (Pittsburgh and Poznan cohorts, n = 176) for variants in 78 previously reported gene candidates confidently linked to male infertility phenotypes (Oud et al. 2019). Our analysis pipeline did not identify potentially significant variants in these genes. The remaining genes and variants with minor allele frequency < 0.002 (GnomAD v2.1.1) were reviewed for biological role in reproduction and spermatogenesis, function, and testis-specific expression. Of greatest interest were genes that caused an isolated reproductive phenotype in mouse (MGI database). Analysis of knockout models revealed a novel gene candidate involved in genome integrity, X-linked germ-cell nuclear antigen, GCNA. A follow-up study of 2049 international male infertility patients identified several additional GCNA SNVs (Suppl. Table 1). In total, potentially significant GCNA variants were identified in 7 of 2225 patients with SPGF phenotypes ranging from non-obstructive azoospermia to cryptozoospermia (Table 1). All identified variants had extremely low MAF of ≤ 0.0005 in the general male population and 6 of 7 were not found in an anonymous cohort of 5784 Dutch biological fathers (χ2 = 14.2346; p = 0.000161). Variants were visualized in IGV and confirmed with Sanger sequencing (Suppl. Figure 1).
Table 1.
Clinical information for patients with potentially disease-causing GCNA variants
| # | ID | Age | Phenotype | Semen volume | Sperm conc. | FSH | LH | T | PRL | Karyotype | AZF deletions |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GEMINI-166 | 29 | NOA | 3.5 | 0 | 23.9a | 7.4a | 29.5a | 15a | 46,XY | neg |
| 2 | AUS1 | 28 | Crypto | No data | 0** | 7.5a | 1.8a | 8.4a | No data | 46,XY | neg |
| 3 | M1831 | 27 | Crypto | 8.9 | 0** | 14.3 | 5 | 10.5 | 109 | 46,XY | neg |
| 4 | M1410 | 37 | NOA* | 6.8 | 0 | 5.3 | 2.8 | 8.8 | 233 | 46,XY | neg |
| 5 | M13 | 29 | NOA | 3.6 | 0 | 8.9 | 4.2 | 16.1 | 121 | 46,XY | neg |
| 6 | M911 | 32 | NOA | 6.0 | 0 | 14.3 | 9.9 | 12.7 | 358 | 46,XY | neg |
| 7 | GEMINI-433 | 24 | NOA | 4.8 | 0 | 13 | 6.1 | 8.7 | no data | 46,XY | neg |
GCNA variants were identified in 7 males with spermatogenic failure in a study of 2225 participants. Phenotype is based on presence or absence of sperm in the ejaculate. Reproductive hormone levels also vary among patients with FSH ranging from normal to significantly elevated, normal LH, T from low to normal, and no evidence of hyperprolactinemia. No structural or numerical chromosome aberrations were identified. Normal semen volume ≥ 1.5 ml. Normal sperm concentration ≥ 15 × 106/ml. Unless otherwise noted, normal reference hormone values are as follows: Follicle Stimulating Hormone (FSH) 1–7 U/L; Luteinizing Hormone (LH) 2–10 U/L; Testosterone (T) > 12 nmol/l; Prolactin (Prl) < 500 mU/L
Crypto cryptozoospermia, NOA non-obstructive azoospermia
Atypical NOA with normal FSH and LH levels
Rare sperm found in semen pellet
Reference values: FSH < 7.6 IU/L; LH 1.6–8 IU/L; PRL 2–18 IU/L; T > 12 nmol/l
Gene expression and localization of normal GCNA in human testis
To elucidate the germ cell-specific expression profile of GCNA, we first examined a single-cell RNA-seq dataset from cryopreserved normal adult human testis. Results from 35,941 transcripts derived from 13,597 cells were used for global analyses to identify human testicular cell types (spermatogonia, spermatocytes, spermatids, Leydig cells, endothelial cells, testicular macrophages, pericytes, or myoid cells). Analyses of sequencing results indicate that GCNA RNA is expressed predominantly in spermatogonia (Suppl. Fig. 2).
Single-cell transcriptomes from 3297 spermatogonia were partitioned out for downstream analysis. Using unsupervised clustering, we further divided spermatogonia into four distinct clusters. We then utilized the expression of conserved germ cell markers to biologically annotate resultant clusters (Suppl. Fig. 3). Data indicate that clusters 1 and 2 likely represent undifferentiated spermatogonia while clusters 3 and 4 represent differentiating spermatogonia. We observed broad expression in all clusters, with the most abundant in cluster 4, indicating that GCNA RNA is primarily expressed in differentiating spermatogonia in humans (Fig. 1a–d). Immunohistochemical analysis of control testicular tissue showed that GCNA protein is produced throughout spermatogenesis, from spermatogonia to elongated spermatids, in normal adult human testis (Fig. 2).
Fig. 1.
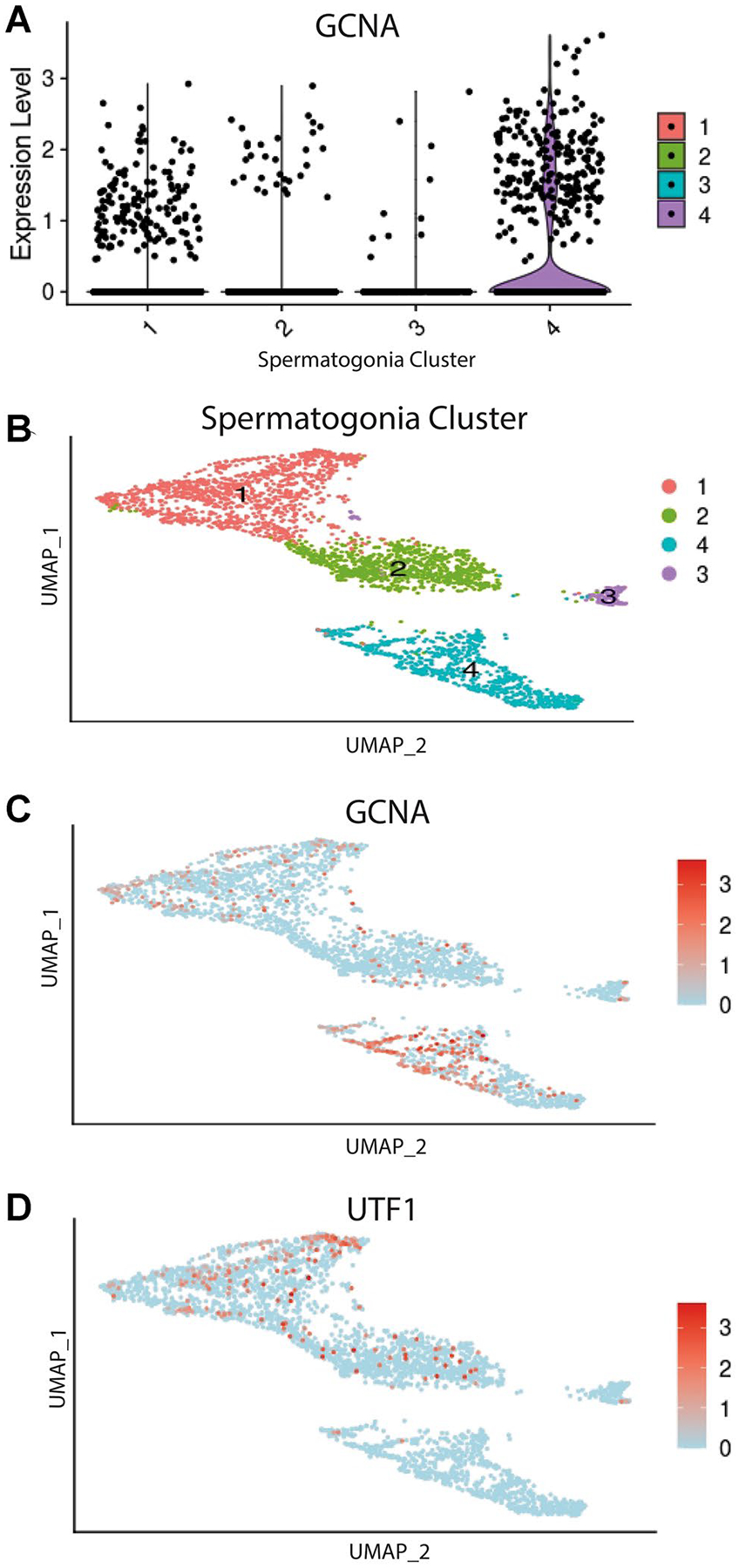
Single-cell transcriptome profiling of adult human spermatogonia. a Violin plot showing expression level of GCNA mRNA across the spermatogonia clusters. b UMAP plot of re-clustered human spermatogonia in distinct clusters representing undifferentiated (clusters 1 and 2) and differentiating spermatogonia (clusters 3 and 4) (see Supplemental Materials for single-cell transcriptome clustering of all testicular cells and expression patterns of common markers utilized to distinguish spermatogonia). Cells demonstrating positive expression shown in red with no or little expression in blue. c UMAP plot showing GCNA RNA expression predominantly in clusters representing differentiating spermatogonia. d UMAP plot showing known RNA marker UTF1 expression primarily in clusters representing undifferentiated spermatogonia
Fig. 2.
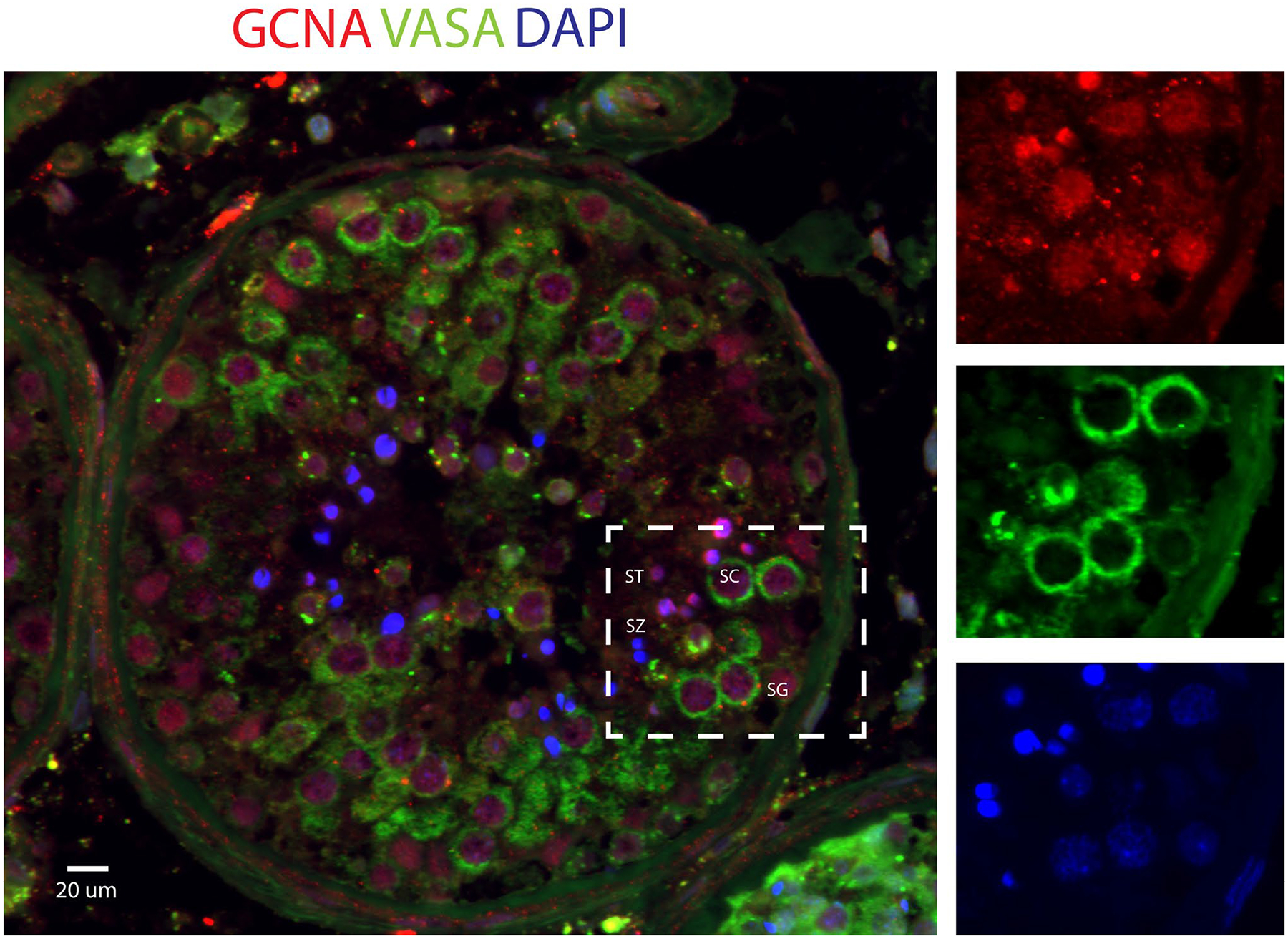
GCNA protein expression in normal adult human seminiferous tubules. Immunofluorescent detection of GCNA and VASA (DDX4) in Bouin-fixed adult human testis confirms GCNA is present in all spermatogenesis cell types, including spermatogonia (SG), spermatocytes (SC), and spermatids (ST), but absent in visible mature spermatozoa (SZ)
Pathogenicity classification of GCNA variants observed in SPGF
To aid in the assessment of identified variant effects on GCNA function, we utilized computational prediction tools and ACMG classification guidelines (Table 2). Accordingly, variant p.Ala115ProfsTer7, identified in an atypical (normal FSH and LH levels) non-obstructive azoospermia (NOA) individual, was determined to be “likely pathogenic” as it creates an early frameshift and premature termination codon in the small ubiquitin-like modifier (SUMO)-interacting motif (SIM) domain, likely resulting in protein truncation (Borgermann et al. 2019) (Fig. 3).
Table 2.
Potentially disease-causing GCNA variants observed in patients with spermatogenic failure
| Sample | ID | Pheno type | Origin | NT change | AA change | GnomAD | Controls | Domain | ACMG |
|---|---|---|---|---|---|---|---|---|---|
| 1 | GEMINI-166 | NOA | USA | c.49G > A | p.Asp17Asn | 0.000 | 0 | N-terminus | VUS |
| 2 | AUS1 | Crypto | Australia | c.226G > A | p.Val76Met | 0.00004045 | 0 | IDR (SIM) | VUS |
| 3 | M1831 | Crypto | Turkey | c.306C > A | p.Ser102Arg | not found | 0 | IDR (SIM) | VUS |
| 4 | M1410 | NOA* | Croatia | c.343del | p.Ala115ProfsTer7 | not found | 0 | IDR (SIM) | Likely pathogenic |
| 5 | M13 | NOA | Germany | c.883 T > C | p.Ser295Pro | 0.00004388 | 1 | IDR | VUS |
| 6 | M911 | NOA | Germany | c.1976C > G | p.Ser659Trp | 0.0001912 | 0 | SprT-like | VUS |
| 7 | GEMINI-433 | NOA | Estonia | c.1990C > T | p.Arg664Cys | 0.0004944 | 0 | SprT-like | VUS |
Seven males with spermatogenic failure from 2225 study participants were identified with potentially disease-causing GCNA variants. Phenotype is based on presence or absence of sperm in the ejaculate. Minor allele frequency values for males obtained from GnomAD v2.1.1 (Broad Institute). “Controls” column refers to number of times variant was observed in 5784 Dutch fathers. Domain reflects variant position within the protein. ACMG refers to variant classification according to American College of Medical Genomics and Genetics guidelines
Crypto cryptozoospermia, NOA non-obstructive azoospermia, NT nucleotides, AA amino acid, IDR Intrinsically disordered region, SIM SUMO interacting motif, SprT-like Spartan like, VUS variant of uncertain significance
Atypical NOA with normal FSH and LH levels (see Table 1)
Fig. 3.
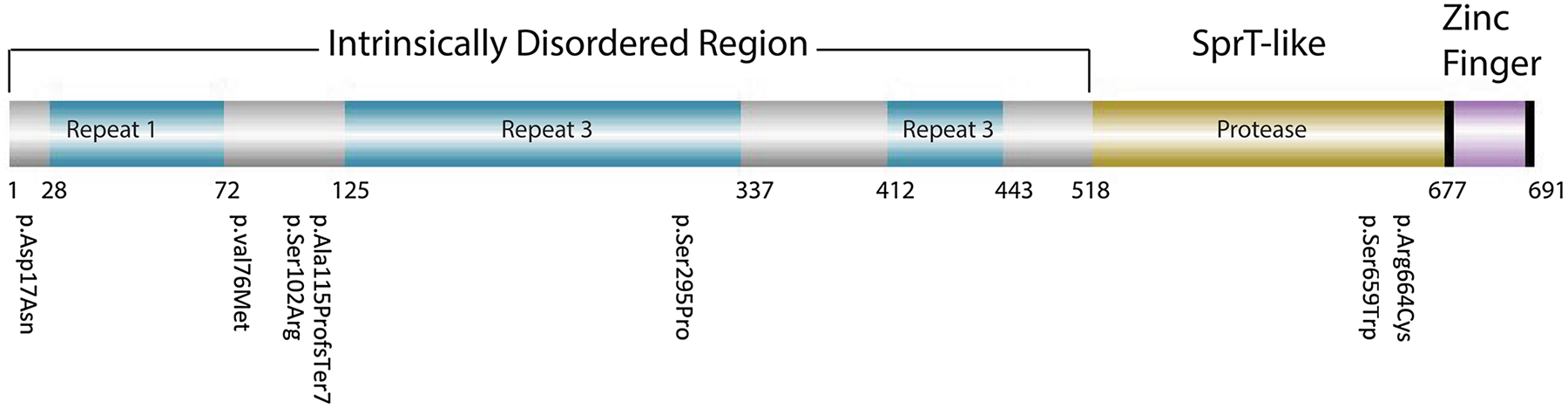
Diagram of human GCNA protein domains and variant locations. Schematic depiction of human GCNA protein with known domains (intrinsically disordered region, Spartan like, and zinc finger) and amino acid coordinates. Potentially disease-causing variants observed in 7 patients with SPGF are labeled and correspond to the IDR and SprT-like domains (see also Table 2). Repeat 1 = 2 exon repeat, Repeat 2 = DD(N/S)DDS(E/D)(A/V)P 21x, Repeat 3 = DD(N/S)DDS(E/D)(A/V)P 21x (Carmell, Dokshin et al. 2016)
Variant p.Ser659Trp, located in the SprT-like domain, was found in an azoospermic man. Ser659 is a highly conserved amino acid of which Trp substitution is expected to be “damaging”, “probably damaging”, or “disease-causing” by SIFT, PolyPhen, and MutationT@ster, respectively (Fig. 4a, c). A moderately conserved p.Arg664Cys variant, also in the SprT-like domain, was observed in an NOA patient and predicted as “possibly damaging” by PolyPhen with a CADD score of 16.11 (Fig. 4b, c). Comparison of primary sequences and secondary structures, as well as 3D modeling, revealed striking similarity in the SprT-like domain among multiple GCNA orthologues, suggesting a conserved function throughout evolution (Fig. 4d).
Fig. 4.
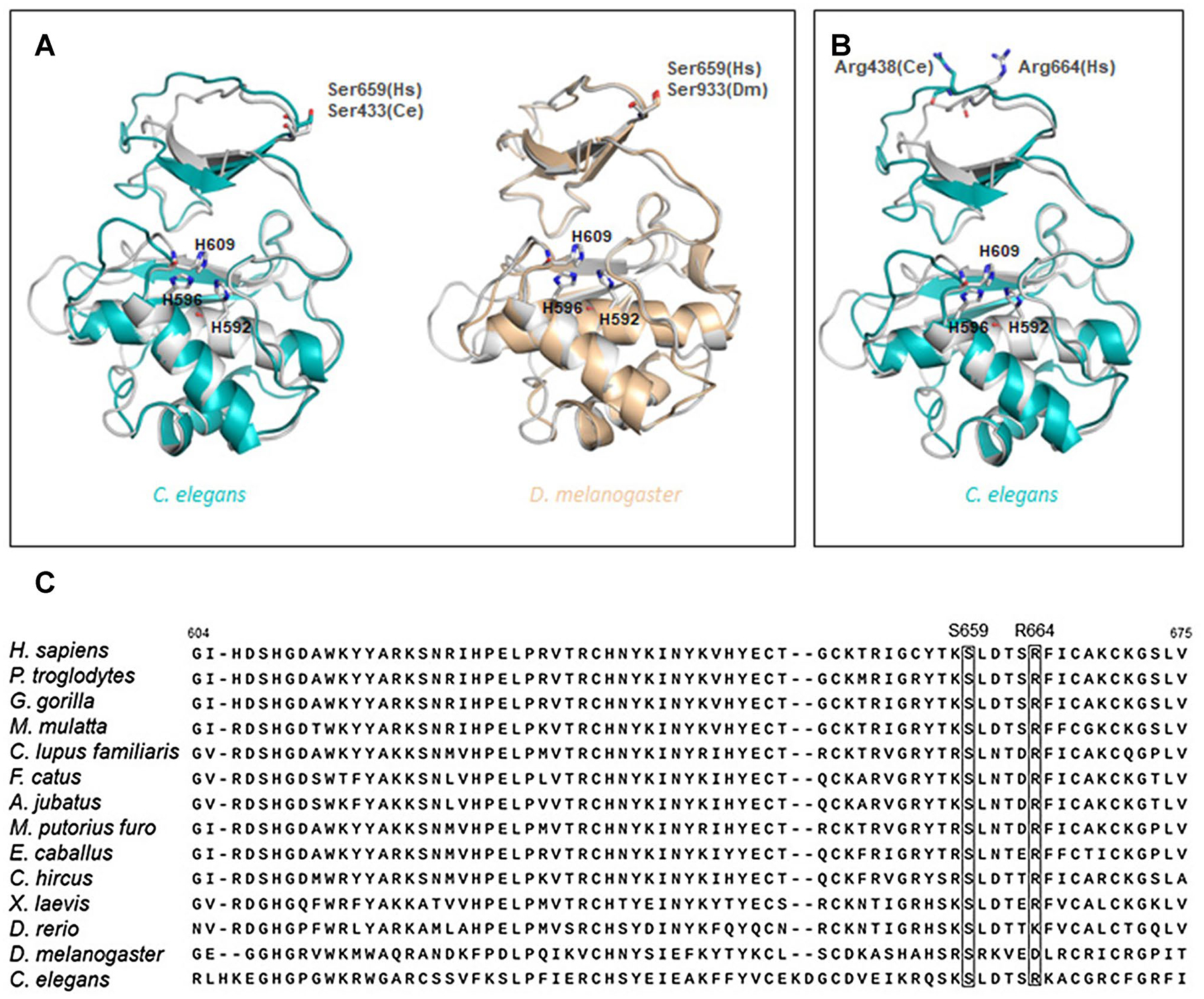
3D Modeling and conservation of GCNA variant positions in SprT-like domain residues. Three dimensional models depict a highly conserved aa residue Ser659 in H. sapiens (Hs) and C. elegans (Ce, left) and H. sapiens and D. melanogaster (Dm, right) which was altered in a Sertoli cell only individual with variant p.Ser659Trp and b moderately conserved aa residue Arg664 in H. sapiens and C. elegans which was altered in an azoospermic individual with variant p.Arg664Cys. Human catalytic histidines (H592, H596 and H609) are represented by sticks. c Partial amino acid sequences for multiple species of the conserved SprT-like protease domain also demonstrate conservation of affected amino acids (see also Table 2)
N-terminus variant p.Asp17Asn, found in NOA patient GEMINI-166, results in a significant change in amino acid (aa) composition, from charged to neutral, which may alter GCNA’s secondary structure with potentially functional consequences. This substitution is predicted to be “intolerant” by MetaDome, “damaging” by SIFT, and “probably damaging” according to PolyPhen and with a CADD score of 16.16.
Conventional prediction tools were less applicable for 2 missense variants in the consensus SUMO interacting motif (aa 72–125) within the IDR of two individuals with cryptozoospermia (Table 2, Fig. 4). Variant p.Val76Met lies in a predicted consensus motif of aliphatic residues (Suppl. Fig. 4A, Suppl. Table 2). Variant p.Ser102Arg may impact GCNA post-translational modifications.
Conventional prediction tools were uninformative for SNVs located in IDR segments without known motifs due to the inherently high variability of the domain between orthologs. To assess the impact of these missense variants, we performed a thorough search of consensus motifs and analyzed key binding sites and post-translational modifications (Suppl. Fig. 4, Suppl. Table 2). Variant p.Ser295Pro is located in a predicted consensus IDR motif that contains both aliphatic residues and sequential pi-bonds (Suppl. Fig. 4B). Of note, this variant was also observed in 1 out of 5784 biological fathers.
Analysis of testicular biopsy histology images revealed Sertoli-cell only phenotypes associated with p.Ser659Trp (Fig. 5) and p.Ser295Pro (data not shown).
Fig. 5.
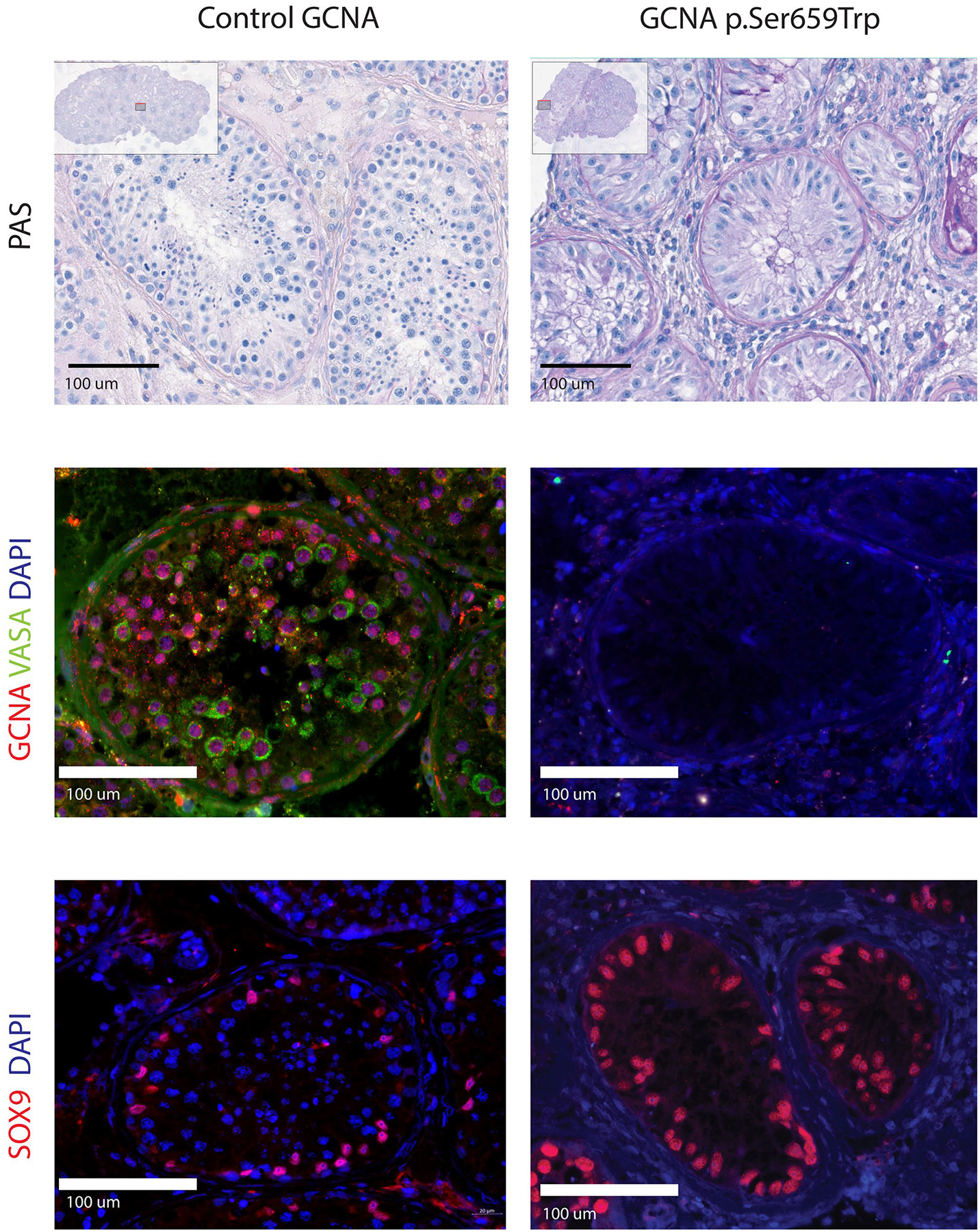
Histological and immunohistochemical imaging of human seminiferous tubules with normal and likely altered GCNA expression. Appearance of human seminiferous tubule in controls (left) and Sertoli cell-only phenotype (right). PAS staining shows presence of all testicular germ cell stages in control and presence of only Sertoli cells in individual harboring GCNA variant p.Ser659Trp in the Spartan-like domain (row 1). GCNA and VASA colocalize in normal spermatogonia, spermatocytes, and spermatids but are not seen in Sertoli cells confirming germ cell specific expression (row 2). Both germ cells stained with DAPI and somatic Sertoli cells labelled with SOX9 are present in individuals with normal GCNA but only Sertoli cells are seen in the seminiferous tubules of the patient with variant p.Ser659Trp in the Spartan-like domain (row 3)
Discussion
The production of genetically high-quality sperm is fundamental for fertility in sexually reproducing species. Dysfunctional spermatogenesis leads to infertility in approximately 8% of men and 15% of couples worldwide (Organization 2000). Yet, despite the high global incidence of male infertility, its basis is frequently undetermined (idiopathic). As such, primary male infertility is often difficult to counsel with limited treatment options. While substantial evidence from over 600 animal models of infertility suggests a strong genetic component in male infertility, translation to human reproductive medicine is lagging (Oud et al. 2019). Encouragingly, multiple limitations related to the high genetic heterogeneity of animal knockout models and low incidence of human mutations can seemingly be overcome by comprehensive genomic studies that include large patient cohorts with broad clinical etiologies, examine numerous gene candidates, and take into account multiple inheritance modes (Alhathal et al. 2020, Wyrwoll et al. 2020, Xavier et al. 2021). Here, we successfully utilized this approach, using genomic data from a massive cohort of over 2200 infertile males. Our analysis identified GCNA as a plausible gene candidate that associates with SPGF and male infertility in seven unrelated individuals.
Recent animal models have implicated GCNA in several critical germ cell roles including resolution of DNA–protein crosslinks (meiosis and mitosis) and regulation of cell division (meiosis and embryonic mitosis) (Dokshin et al. 2020). It is proposed that GCNA is essential for the integrity of the heritable genome and germ line immortality. Loss of Gcna leads to progressive accumulation of genomic aberrations in successive generations, decline in germ cell production, and infertility in mice, flies, worms, and zebrafish (Carmell et al. 2016; Bhargava et al. 2020; Dokshin et al. 2020). Interestingly, while GCNA may have conserved biological roles across species, the protein is composed largely of a uniquely variable N-terminus intrinsically disordered region. Across species, IDRs are variable in length but highly enriched in acidic amino acid content. Interestingly, despite IDR sequence variability among species, recurring motifs have been identified in IDRs in multiple species (Suppl. Fig. 4, Suppl. Table 2). Mouse GCNA is composed almost exclusively of IDRs and binds SUMO (Carmell et al. 2016). Knockout male mice show major disruptions in meiosis, suggesting GCNA IDRs are critical for maintaining germ cell genome integrity. Loss of mouse GCNA is proposed to result both in increased DNA damage, in part due to poor resolution of topoisomerase II DNA–protein crosslinks, and also in a reduced number of crossing-over events, resulting in chromosomal aberrations which ultimately lead to meiotic arrest, reduced cell numbers, and/or abnormal head morphology in surviving sperm (Dokshin et al. 2020). Interestingly, the primary proposed function of mouse IDRs is proteolysis mediated by binding polySUMO and the MRE protein complexes. This theory is in line with critical roles of GCNA in promoting resolution of DNA double-strand breaks and homologous recombination (HR) in meiosis as well as chromatin condensation post-meiotically. Hypothesized roles of GCNA domains together with the variety of mutation sites described herein could explain the range of phenotypic features observed; while some patients (M13 and M911) show complete loss of germ cells (SCO); occasionally, rare sperm with abnormal head morphology was seen in patient (AUS1), resembling sperm features observed in male knockout mice (Dokshin et al. 2020).
The large number of genes reported to cause male infertility in animal models suggests that many genes responsible for human male infertility have yet to be identified. Since it is estimated that any individual gene will likely have a small contribution to overall SPGF load, it is not surprising that the average frequency of GCNA variants identified here is low (< 0.4%) and is in line with such estimation. GCNA SNV c.343del (p.Ala115ProfsTer7 in the SIM) fulfills ACMG criteria for classification as likely pathogenic, while the remainder are currently of variant(s) of uncertain significance. Nonetheless, all variants should be further examined for reliable functional evidence in human cells and/or animal models. Testing of human GCNA variants in mice is complicated because mice do not have the SprT-like or Zinc finger domains and the rest of the gene is the intrinsically disordered region where it is not possible to identify human/mouse orthologous sequences. Moreover, independent studies of human GCNA in patients with male infertility could provide adequate proof of the pathogenicity of these SNVs, as animal models may only partially correspond to human physiology. Also, such functional studies of human GCNA SNVs will assist with subtle clinical features, potentially offering “reverse phenotyping” in addition to indicating the origins of male infertility.
Recent reports indicate that some individuals with mutations in the Spartan protein present clinically with genome instability and progeria-like Ruijs-Aalfs syndrome (Lessel et al. 2014; Maskey et al. 2014). Similarly, evidence of gradual progressive accumulation of genomic errors was shown in SprT-containing protein fly Gcna knockout model and premature aging was seen in late-generation gcna-1 mutant C. elegans (Bhargava et al. 2020). As such, GCNA mutations affecting the SprT-like domain like p.Ser659Trp and p.Arg664Cys observed here may induce genome instability and/or a premature aging phenotype in humans. Additionally, considering GCNA’s role in genome stability in meiosis, we propose a potential effect of mild missense GCNA variants on predisposition to chromosome aberrations (increased chromosome fragility and genome instability) and/or mild SPGF phenotypes like reduced sperm number or morphology defects. We speculate that the identified p.Ser295Pro variant in IDR observed in one SPGF patient and a single control might be a contributing factor to inaccurate resolution of meiotic bivalents in homologous recombination, promoting chromosome aberrations at a low, but progressively accumulating rate of genome damage in each generation, similarly to the phenomenon noted in mouse, fly, and worm GCNA models (Bhargava et al. 2020). It will be of great interest to perform follow-up semen analysis of all GCNA patients initially presenting with reduced germ cells, as well as their offspring, to ascertain potential long-term effects on chromosome aberrations, longevity, and other features of the progeria spectrum. If the proposed linkage is correct, the value of this phenotypic expansion in future studies cannot be overestimated. Indeed, it has also been clinically recognized that men with severe forms of male infertility are at increased risk of all forms of cancer (Schlegel et al. 2021). The mechanism for increased risk of neoplastic disease has not been elucidated but could occur from genomic instability through pathways described above.
In conclusion, here we present the first study of human GCNA, a germ cell-specific genome stability protein, in the context of spermatogenic failure and male fertility. Since identified variants affected functional domains with distinct roles, effects of the respective mutations likely lead to different molecular and phenotypic features of SPGF. Such phenotypic diversity is consistent with reports of Gcna knockouts in multiple animal models and suggest that human GCNA variants lead to diverse but specific phenotypic features, extending the role of GCNA as a key regulator of germ cell genome integrity. We speculate that the function of human GCNA is sensitive to even small missense changes which can lead to increased DNA damage load over generations and ultimately cause heritable germ cell chromosome aberrations and/or infertility, as has been demonstrated in animal models.
Supplementary Material
Acknowledgements
We wish to thank the participants and clinical staff for making this research study possible. We would also like to recognize Dr. Andrea Berman at the University of Pittsburgh, who provided expert assistance with GCNA 3D modeling. Additionally, we would like to acknowledge Magee-Womens Research Institute scientific editor Bruce Campbell for carefully proofreading the article.
Funding
This study was supported by The Eunice Kennedy Shriver NICHD Grant HD080755 (ANY), the Magee-Womens Research Institute University of Pittsburgh Start Up Fund (ANY), PA DoH Grant SAP4100085736 (ANY), NIH P50 Specialized Center Grant HD096723 (KO, ANY, DC, PNS, KH, and MBE), German Research Foundation Clinical Research Unit ‘Male Germ Cells’ grant DFG CRU326 (FT), National Science Centre in Poland, grants no.: 2017/26/D/NZ5/00789 (AM) and 2015/17/B/NZ2/01157; NCN 2020/37/B/NZ5/00549 (MK), Magee-Womens Research Institute University of Pittsburgh, Faculty Fellowship Award and NICHD T32 HD087194 (JH), GM125812 (MB), GM127569 (MB, JLY, and ANY), NIH R00H090289 (MABE), National Health and Medical Research Council Project grant APP1120356 (MKOB, JAV, and DC), UUKi Rutherford Fund Fellowship (BJH), Estonian Research Council, grants IUT34-12 and PRG1021 (ML), and The Netherlands Organization for Scientific Research grant no.: 918-15-667 as well as an Investigator Award in Science from the Wellcome Trust grant no.: 209451 (JAV). Computational analysis was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided.
Appendix
GEMINI Consortium Members
Department of Genetics, Oregon National Primate Research Center, Oregon Health & Science University, Beaverton, OR, USA
Donald F. Conrad, Liina Nagirnaja
Andrology and IVF Laboratory, Department of Surgery (Urology), University of Utah School of Medicine, Salt Lake City, UT, USA
Kenneth I. Aston, Douglas T. Carrell, James M. Hotaling, Timothy G. Jenkins
(1) Hudson Institute of Medical Research and the Department of Obstetrics and Gynaecology, Monash University, Clayton, Victoria, Australia; (2) Monash IVF and the Hudson Institute of Medical Research, Clayton, Victoria, Australia
Rob McLachlan
School of Biological Sciences, Monash University, Clayton, Victoria, Australia
Moira K. O’Bryan
Department of Urology, Weill Cornell Medicine, New York, NY, USA
Peter N. Schlegel
Department of Urology, Stanford University School of Medicine, Stanford, CA 94,305, USA
Michael L. Eisenberg
Department of Urology, Medical College of Wisconsin, Milwaukee, WI, 53226, USA
Jay I. Sandlow
Washington University in St Louis, School of Medicine, St Louis, MO, USA
Emily S. Jungheim, Kenan R. Omurtag
(1) i3S—Instituto de Investigação e Inovação em Saúde, Universidade do University of Porto.
(2) IPATIMUP—Instituto de Patologia e Imunologia Molecular da Universidade do Porto, Porto, Portugal.
(3) Serviço de Genética, Departamento de Patologia, Faculdade de Medicina da Universidade do Porto, Porto, Portugal
Alexandra M. Lopes1,2, Susana Seixas1,2, Filipa Carvalho1,3, Susana Fernandes1,3, Alberto Barros1,3
(1) Departamento de Genética Humana, Instituto Nacional de Saúde Dr Ricardo Jorge, Lisboa, Portugal
(2) ToxOmics, Faculdade de Ciências Médicas, Universidade Nova de Lisboa, Portugal
(3) Centro de Medicina Reprodutiva, Maternidade Dr. Alfredo da Costa, Lisboa, Portugal
João Gonçalves1,2, Iris Caetano1, Graça Pinto3, Sónia Correia3
Institute of Biomedicine and Translational Medicine, University of Tartu, 51010 Tartu, Estonia
Maris Laan
Andrology Center, Tartu University Hospital, 50406 Tartu, Estonia
Margus Punab
Department of Growth and Reproduction, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark
Ewa Rajpert-De Meyts, Niels Jørgensen, Kristian Almstrup
(1) Department of Experimental and Clinical Biomedical Sciences, University of Florence, Florence, Italy; (2) Andrology Department, Fundacio Puigvert, Instituto de Investigaciones Biomédicas Sant Pau (IIB-Sant Pau), Barcelona, Spain
Csilla G. Krausz
Division of Urology, Department of Surgery, Mount Sinai Hospital, University of Toronto, Toronto, ON, Canada
Keith A. Jarvi
Footnotes
Membership of the GEMINI Consortium is provided in the Appendix.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00439-021-02287-y.
Conflict of interest The author(s) report no conflicts of interest associated with the outcome or publication of the research described.
Ethical approval All study participants provided informed consent and were enrolled under respective IRB protocols, PRO10030036 (Pittsburgh), OKB-5-2/15, 772/15, and 1003/18 (Poznan), 2010-578-f-S (Münster), 0102004794 (New York) (GEMINI Consortium), 254/M-17, 21.12.2015 (Tartu) (GEMINI Consortium), or approval by the human ethics committees of Monash Surgical Private Hospital (Clayton), Monash Medical Centre and Monash University (Melbourne).
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A, Baskaran S, Parekh N, Cho CL, Henkel R, Vij S, Arafa M, Panner Selvam MK, Shah R (2021) Male infertility. Lancet 397(10271):319–333 [DOI] [PubMed] [Google Scholar]
- AlAsiri S, Basit S, Wood-Trageser MA, Yatsenko SA, Jeffries EP, Surti U, Ketterer DM, Afzal S, Ramzan K, Faiyaz-Ul Haque M, Jiang H, Trakselis MA, Rajkovic A (2015) Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J Clin Invest 125(1):258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhathal N, Maddirevula S, Coskun S, Alali H, Assoum M, Morris T, Deek HA, Hamed SA, Alsuhaibani S, Mirdawi A, Ewida N, Al-Qahtani M, Ibrahim N, Abdulwahab F, Altaweel W, Dasouki MJ, Assiri A, Qabbaj W, Alkuraya FS (2020) A genomics approach to male infertility. Genet Med 22(12):1967–1975 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Gribskov M (1998) Combining evidence using p-values: application to sequence homology searches. Bioinformatics (Oxford, England) 14(1):48–54 [DOI] [PubMed] [Google Scholar]
- Bhargava V, Goldstein CD, Russell L, Xu L, Ahmed M, Li W, Casey A, Servage K, Kollipara R, Picciarelli Z, Kittler R, Yatsenko A, Carmell M, Orth K, Amatruda JF, Yanowitz JL, Buszczak M (2020) GCNA preserves genome integrity and fertility across species. Dev Cell 52(1):38–52.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgermann N, Ackermann L, Schwertman P, Hendriks IA, Thijssen K, Liu JC, Lans H, Nielsen ML, Mailand N (2019) SUMOylation promotes protective responses to DNA-protein crosslinks. EMBO J. 10.15252/embj.2019101496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieño-Enríquez MA, Moak SL, Abud-Flores A, Cohen PE (2019) Characterization of telomeric repeat-containing RNA (TERRA) localization and protein interactions in primordial germ cells of the mouse†. Biol Reprod 100(4):950–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkworth MH, Weinbauer GF, Schlatt S, Nieschlag E (1995) Identification of male germ cells undergoing apoptosis in adult rats. J Reprod Fertil 105(1):25–33 [DOI] [PubMed] [Google Scholar]
- Carmell MA, Dokshin GA, Skaletsky H, Hu Y-C, van Wolfswinkel JC, Igarashi KJ, Bellott DW, Nefedov M, Reddien PW, Enders GC, Uversky VN, Mello CC, Page DC (2016) A widely employed germ cell marker is an ancient disordered protein with reproductive functions in diverse eukaryotes. Elife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Jeon S, Choi M, Lee MH, Park M, Lee DR, Jun KY, Kwon Y, Lee OH, Song SH, Kim JY, Lee KA, Yoon TK, Rajkovic A, Shim SH (2010) Mutations in SOHLH1 gene associate with nonobstructive azoospermia. Hum Mutat 31(7):788–793 [DOI] [PubMed] [Google Scholar]
- Cooke HJ, Saunders PT (2002) Mouse models of male infertility. Nat Rev Genet 3(10):790–801 [DOI] [PubMed] [Google Scholar]
- Desai S, Wood-Trageser M, Matic J, Chipkin J, Jiang H, Bachelot A, Dulon J, Sala C, Barbieri C, Cocca M, Toniolo D, Touraine P, Witchel S, Rajkovic A (2017) MCM8 and MCM9 nucleotide variants in women with primary ovarian insufficiency. J Clin Endocrinol Metab 102(2):576–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokshin GA, Davis GM, Sawle AD, Eldridge MD, Nicholls PK, Gourley TE, Romer KA, Molesworth LW, Tatnell HR, Ozturk AR, de Rooij DG, Hannon GJ, Page DC, Mello CC, Carmell MA (2020) GCNA interacts with spartan and topoisomerase II to regulate genome stability. Dev Cell 52(1):53–68.e56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27(7):1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatos A, Hajdu-Soltész B, Monzon AM, Palopoli N, Álvarez L, Aykac-Fas B, Bassot C, Benítez GI, Bevilacqua M, Chasapi A (2020) DisProt: intrinsic protein disorder annotation in 2020. Nucleic Acids Res 48(D1):D269–D276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99(4):877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski LP, Bujnicki JM (2012) MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinformatics 13(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz C, Riera-Escamilla A (2018) Genetics of male infertility. Nat Rev Urol 15(6):369–384 [DOI] [PubMed] [Google Scholar]
- Leduc F, Maquennehan V, Nkoma GB, Boissonneault G (2008) DNA damage response during chromatin remodeling in elongating spermatids of mice. Biol Reprod 78(2):324–332 [DOI] [PubMed] [Google Scholar]
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JC, Smith KR, Oehler J, Cabrera E, Freire R, Pope K, Nahid A, Norris F, Leventer RJ, Delatycki MB, Barbi G, von Ameln S, Högel J, Degoricija M, Fertig R, Burkhalter MD, Hofmann K, Thiele H, Altmüller J, Nürnberg G, Nürnberg P, Bahlo M, Martin GM, Aalfs CM, Oshima J, Terzic J, Amor DJ, Dikic I, Ramadan K, Kubisch C (2014) Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet 46(11):1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Raczynska JE, Chen Z, Yu H (2019) Structural insight into DNA-dependent activation of human metalloprotease spartan. Cell Rep 26(12):3336–3346.e3334 [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161(5):1202–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, Machida YJ (2014) Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun 5:5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (2016) The ensembl variant effect predictor. Genome Biol 17(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Hasuike S, Yogev L, Maduro MR, Ishikawa M, Westphal H, Lamb DJ (2003) Azoospermia in patients heterozygous for a mutation in SYCP3. Lancet 362(9397):1714–1719 [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S (2001) Predicting deleterious amino acid substitutions. Genome Res 11(5):863–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization WH (2000) WHO Manual for the Standardized Investigation, Diagnosis, and Management of the Infertile Male, Cambridge University [Google Scholar]
- Oud MS, Volozonoka L, Smits RM, Vissers L, Ramos L, Veltman JA (2019) A systematic review and standardized clinical validity assessment of male infertility genes. Hum Reprod 34(5):932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20(1):110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47(D1):D886–d894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23(3):318–326 [DOI] [PubMed] [Google Scholar]
- Schilit SLP, Menon S, Friedrich C, Kammin T, Wilch E, Hanscom C, Jiang S, Kliesch S, Talkowski ME, Tüttelmann F, MacQueen AJ, Morton CC (2020) SYCP2 translocation-mediated dysregulation and frameshift variants cause human male infertility. Am J Hum Genet 106(1):41–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel PN, Sigman M, Collura B, De Jonge CJ, Eisenberg ML, Lamb DJ, Mulhall JP, Niederberger C, Sandlow JI, Sokol RZ, Spandorfer SD, Tanrikut C, Treadwell JR, Oristaglio JT, Zini A (2021) Diagnosis and treatment of infertility in men: AUA/ASRM guideline part I. Fertil Steril 115(1):54–61 [DOI] [PubMed] [Google Scholar]
- Schrodinger L (2018) The PyMOL Molecular Graphics System. Version 2.0 https://pymol.org/2/
- Schultz N, Hamra FK, Garbers DL (2003) A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci USA 100(21):12201–12206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) Mutation-Taster evaluates disease-causing potential of sequence alterations. Nat Methods 7(8):575–576 [DOI] [PubMed] [Google Scholar]
- Shami AN, Zheng X, Munyoki SK, Ma Q, Manske GL, Green CD, Sukhwani M, Orwig KE, Li JZ, Hammoud SS (2020) Single-cell RNA sequencing of human, macaque, and mouse testes uncovers conserved and divergent features of mammalian spermatogenesis. Dev Cell 54(4):529–547.e512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Schimenti JC (2015) The genetics of human infertility by functional interrogation of SNPs in mice. Proc Natl Acad Sci U S A 112(33):10431–10436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33(Web Server issue):W244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MCF, Nielsen M (2012) Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res 40(W1):W281–W287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2):178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tüttelmann F, Ruckert C, Röpke A (2018) Disorders of spermatogenesis: perspectives for novel genetic diagnostics after 20 years of unchanged routine. Med Genet 30(1):12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Tu C, Nie H, Meng L, Li Y, Yuan S, Zhang Q, Du J, Wang J, Gong F, Fan L, Lu GX, Lin G, Tan YQ (2019) Biallelic mutations in CFAP65 lead to severe asthenoteratospermia due to acrosome hypoplasia and flagellum malformations. J Med Genet 56(11):750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiel L, Baakman C, Gilissen D, Veltman JA, Vriend G, Gilissen C (2019) MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum Mutat 40(8):1030–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood-Trageser MA, Gurbuz F, Yatsenko SA, Jeffries EP, Kotan LD, Surti U, Ketterer DM, Matic J, Chipkin J, Jiang H, Trakselis MA, Topaloglu AK, Rajkovic A (2014) MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am J Hum Genet 95(6):754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrwoll MJ, Temel Ş G, Nagirnaja L, Oud MS, Lopes AM, van der Heijden GW, Heald JS, Rotte N, Wistuba J, Wöste M, Ledig S, Krenz H, Smits RM, Carvalho F, Gonçalves J, Fietz D, Türkgenç B, Ergören MC, Çetinkaya M, Başar M, Kahraman S, McEleny K, Xavier MJ, Turner H, Pilatz A, Röpke A, Dugas M, Kliesch S, Neuhaus N, Aston KI, Conrad DF, Veltman JA, Friedrich C, Tüttelmann F (2020) Bi-allelic mutations in M1AP are a frequent cause of meiotic arrest and severely impaired spermatogenesis leading to male infertility. Am J Hum Genet 107(2):342–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier MJ, Salas-Huetos A, Oud MS, Aston KI, Veltman JA (2021) Disease gene discovery in male infertility: past, present and future. Hum Genet 140(1): 7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Silber S, Leu NA, Oates RD, Marszalek JD, Skaletsky H, Brown LG, Rozen S, Page DC, Wang PJ (2015) TEX11 is mutated in infertile men with azoospermia and regulates genome-wide recombination rates in mouse. EMBO Mol Med 7(9):1198–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsenko AN, Georgiadis AP, Ropke A, Berman AJ, Jaffe T, Olszewska M, Westernstroer B, Sanfilippo J, Kurpisz M, Rajkovic A, Yatsenko SA, Kliesch S, Schlatt S, Tuttelmann F (2015) X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med 372(22):2097–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.