

Abstract
Acute lung injury induced by ischemia–reperfusion (I/R)-associated pulmonary inflammation is associated with high rates of morbidity. Despite advances in the clinical management of lung disease, molecular therapeutic options for I/R-associated lung injury are limited. Zinc finger protein 36 (ZFP36) is an AU-rich element-binding protein that is known to suppress the inflammatory response. A ZFP36 binding site occurs in the 3ʹ UTR of the cAMP‐response element-binding protein (CREB) binding protein (CREBBP) gene, which is known to interact with apoptotic proteins to promote apoptosis. In this study, we investigate the involvement of ZFP36 and CREBBP on I/R-induced lung injury in vivo and in vitro. Intestinal ischemia/reperfusion (I/R) activates inflammatory responses, resulting in injury to different organs including the lung. Lung tissues from ZFP36-knockdown mice and mouse lung epithelial (MLE)-2 cells were subjected to either Intestinal I/R or hypoxia/reperfusion, respectively, and then analyzed by Western blotting, immunohistochemistry, and real-time PCR. Silico analyses, pull down and RIP assays were used to analyze the relationship between ZFP36 and CREBBP. ZFP36 deficiency upregulated CREBBP, enhanced I/R-induced lung injury, apoptosis, and inflammation, and increased I/R-induced lung fibrosis. In silico analyses indicated that ZFP36 was a strong negative regulator of CREBBP mRNA stability. Results of pull down and RIP assays confirmed that ZFP36 direct interacted with CREBBP mRNA. Our results indicated that ZFP36 can mediate the level of inflammation-associated lung damage following I/R via interactions with the CREBBP/p53/p21/Bax pathway. The downregulation of ZFP36 increased the level of fibrosis.
Subject terms: Transcription, Respiratory tract diseases
Introduction
Acute lung injury, resulting from systemic inflammatory responses as a consequence of ischemia–reperfusion (I/R), has limited therapeutic options and is associated with high morbidity [1, 2]. Injury to the lung from I/R can occur through a global response from thoracic surgery to other organs, such as the liver [3], heart [4], kidneys [5], and intestine [6]. Intestinal ischemia–reperfusion (I/R) injury develops when the blood fow to the intestines decreases, followed by the reestablishment of the blood supply to the ischemic tissue. Intestinal I/R injury results in intestinal mucosal barrier dysfunction, which may cause severe local and systemic infammation [6]. The inflammation triggered by I/R involves many proinflammatory factors including neutrophils and cytokines [7]. Neutrophils that are recruited to the lungs following I/R produce an excessive amount of free radicals and reactive oxygen species (ROS) to cause further injury and necrosis [8].
Although the inflammatory process in acute lung injury is well-studied, less is known about the role played by apoptosis and fibrosis. However, drugs that ameliorate the inflammatory processes associated with I/R can reduce levels of fibrosis in the lung and alleviate injury [9]. Elevated levels of transforming growth factor (TGF)-β1 and tumor necrosis factor (TNF)-α in the lung are associated with increased levels of fibrosis [10]. However, the role of TGF-β1 in fibrosis is contradictory. Although TGF-β1 is thought to reduce fibrosis by regulating neutrophil apoptosis through IL-6 [11], it is also believed to promote fibrosis and apoptosis through Egr-1 [12, 13]. Overexpression of TNF-α following induction by inflammatory factors is a well-documented characteristic of fibrosis [14, 15]. TNF-α is mainly derived from macrophages and accumulates rapidly in response to injury [16, 17]. In conjunction with interleukins (ILs), TNF-α is known to activate the NF-κB and JAK/STAT pathway in response to lung injury to induce the expression of several major chemokines including CXCL13, which is involved in pulmonary fibrosis [18].
Zinc finger protein 36 (ZFP36, also known as tristetraprolin) is an AU-rich element-binding protein that suppresses the inflammatory response [19]. ZFP36 promotes mRNA decay by binding to mRNA 3ʹUTR and is known to target TNF-α [20]. Mice that are deficient in ZFP36 have high levels of TNF-α and are known to develop severe inflammatory syndrome as a consequence [21]. Moreover, overexpressing ZFP36 in mouse embryonic fibroblasts inhibits the induction of p65/NF-κB by cAMP‐response element-binding protein (CREB) binding protein (CREBBP) [22]. CREB and CREBBP are associated with the regulation of several proteins involved in apoptosis, such as BCL2 and Bax [23, 24]. The inhibition of CREBBP is known to protect against apoptotic injury during I/R, and it has been proposed that the binding of CREBBP to CREB triggers apoptosis [25].
Thus, there is no research about the relationship between ZFP36 and CREBBP in I/R. In this study, we investigate the involvement of ZFP36 and CREBBP in I/R-induced lung injury and the interaction of ZFP36 with CREBBP mRNA 3ʹUTR in mouse lung epithelial (MLE) cells and a murine model of I/R.
Results
ZFP36 reduces the severity of intestinal I/R-induced acute lung injury
To study the involvement of ZFP36 in I/R-induced acute lung injury we first created an intestine ischemia model with reperfusion in C57BL/6 mice. The injury to lung tissue, represented by arterial blood partial pressure of oxygen (PaO2), lung water content, and bronchoalveolar lavage fluid (BALF) protein content, increased significantly with reperfusion and then stabilized after 60 min (Fig. S1A–C). Therefore, 60 min of intestine ischemia followed by 60 min of reperfusion was selected as the best representation of lung damage in the mouse model. After 60 min of reperfusion, hematoxylin and eosin (H&E)-stained tissue and measurements of IL-1β, TNF-α, and IL-6 levels indicated significant lung damage with alveolar edema and inflammatory cellular sequestration (Fig. S1D–G).
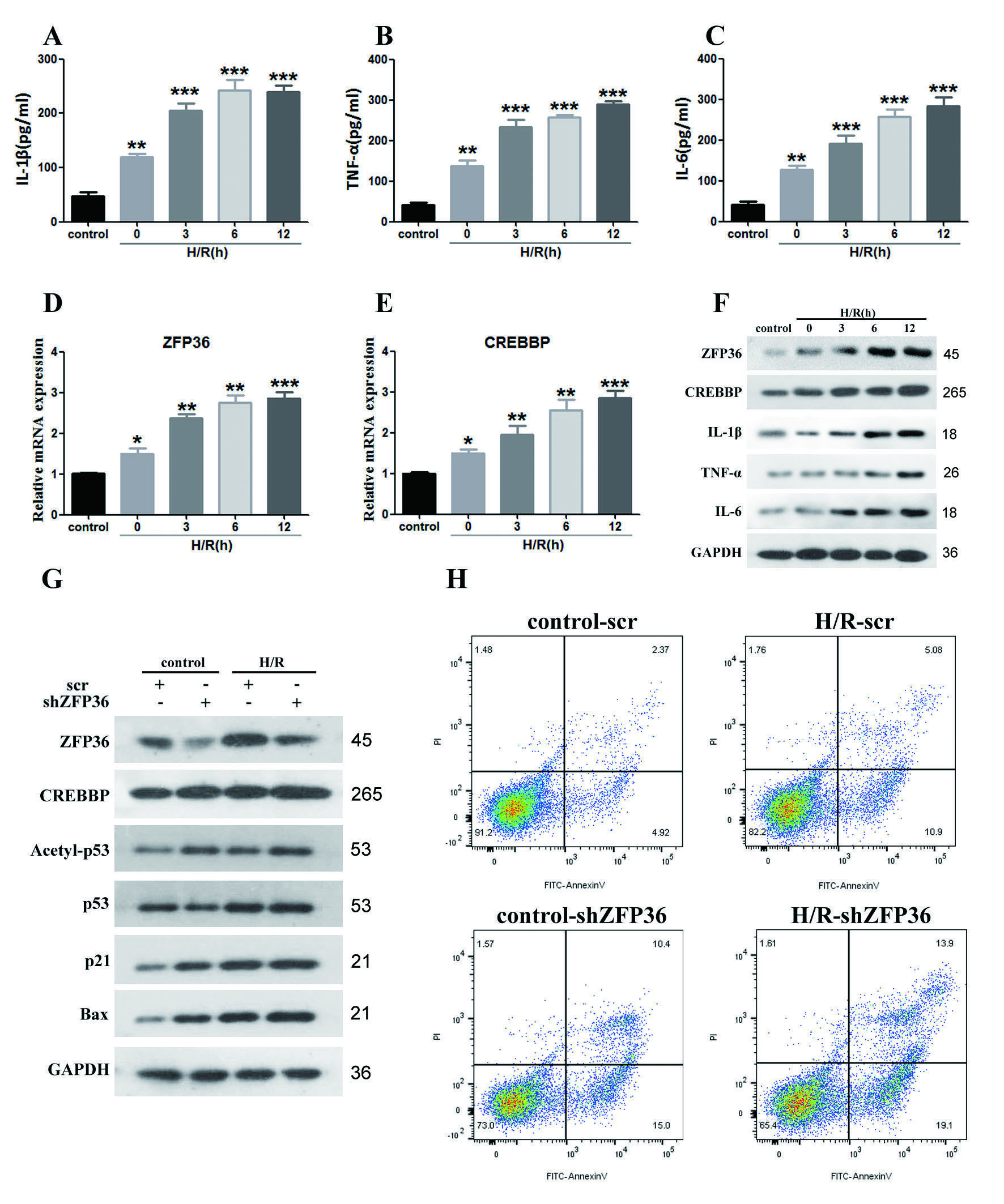
We used the intestinal I/R-induced acute lung injury in mice to assess the levels of ZFP36 in lung tissue. ZFP36 mRNA and protein expression gradually increased compared with the sham group as the length of reperfusion increased (p < 0.05, p < 0.01, p < 0.001) (Fig. 1A). High levels of ZFP36 were detected by immunohistochemical staining in the lung tissues of the I/R-induced mouse model compared with sham-operated mice (Fig. 1B). When the expression of ZFP36 is inhibited by RNA interference, the protein levels of IL-1β, TNF-α, and IL-6 increase significantly, indicating that the inflammatory response is elevated when ZFP36 is suppressed (Fig. 1C–F). This is also demonstrated in H&E-stained lung sections, with increased alveolar edema in tissue when the expression of ZFP36 is suppressed (Fig. 1G). Arterial blood PaO2 was lowered on suppression of ZFP36 (p < 0.05, p < 0.01), whereas lung water content and the protein content of BALF was significantly increased in shZFP36 group (p < 0.05, p < 0.01, p < 0.001) (Fig. 1H–J). This indicates that the severity of lung injury is increased when ZFP36 is suppressed leading to reduced availability of blood oxygen and increased inflammation. Overall, our results suggest that ZFP36 is upregulated during the process of lung injury and suppresses inflammatory responses by mediating the regulation of inflammatory related genes.
Fig. 1. Role of ZFP36 in intestinal ischemia–reperfusion (I/R)-induced acute lung injury.

C57BL/6 mice were subjected to 60 min of intestinal ischemia followed by 0, 30, 60, and 90 min of reperfusion as indicated. Sham mice were included as the control. A ZFP36 mRNA and protein expression in lung tissues were analyzed by RT-qPCR and Western blot (n = 6 per group, **P < 0.01). B Immunohistochemical staining of lung tissues for ZFP36 of sham and I/R 60 min. Scale bars: 50 μm. C–F Levels of IL-1β (C), TNF-α (D), and IL-6 (E) were measured by ELISA and Western blotting (F). G H&E staining of lung tissues. Red arrows outline collapsed alveoli, blue arrows outline multiple inflammatory cells infilitration, black arrows outline bronchial hemorrhage. Scale bars: 50 μm. H–J Arterial blood PaO2 (H), lung water content (I), and BALF protein content (J) were measured.
ZFP36 interacts with CREBBP mRNA to prevent the promotion of intestinal I/R-induced acute lung injury
Next, we assessed the involvement of CREBBP in the intestinal I/R-induced acute lung injury model. CREBBP mRNA expression and protein levels were significantly increased in the lung tissue of the intestinal I/R-induced model (p < 0.05, p < 0.01, p < 0.001) (Fig. 2A) and elevated levels of CREBBP were found in I/R lung tissue sections (Fig. 2B). The inhibition of CREBBP by RNA interference resulted in the increased availability of oxygen in arterial blood (p < 0.05, p < 0.01) and reduced level of lung injury as indicated by lung water content and BALF protein content (p < 0.05, p < 0.01, p < 0.001) (Fig. 2C–E). Furthermore, CREBBP was associated with an increased level of cell death in I/R injured tissue as demonstrated in H&E lung tissue sections and by TUNEL-positive cells (p < 0.05) (Fig. 2F, G). These results indicate that CREBBP could be involved in the apoptosis of cells in the lungs following I/R acute lung injury.
Fig. 2. Role of CREBBP in intestinal ischemia–reperfusion (I/R)-induced acute lung injury.

A CREBBP mRNA and protein expression in lung tissues were analyzed by RT-qPCR and Western blotting (n = 6 per group, **P < 0.01). B Immunohistochemical stain of lung tissues for CREBBP of sham and I/R 60 min group. Scale bars: 50 μm. C–E Arterial blood PaO2 (C), lung water content (D), and BALF protein content (E) were measured. F Representative H&E staining of lung sections. Scale bars: 50 μm. G TUNEL assay was performed on lung sections from each group and TUNEL-positive cells were determined.
We used a catRAPID fragment-based prediction assay to assess whether ZFP36 interacts with the 3ʹUTR of CREBBP. A strong interaction with high confidence levels was identified in the binding assay (interaction propensity, 49; discriminative power, 94%) (Fig. 3A). Western blot analysis of RNA pull down and an immunoprecipitation assay with ZFP36-coated beads confirmed the ZFP36/CREBBP interaction in MLE-2 cells (p < 0.001) (Fig. 3B, C). We also performed a luciferase assay with the 3ʹUTR of CREBBP or ZFP36 either increased or decreased expressed in MLE-2 cells. The underexpression of ZFP36 resulted in greater luciferase activity associated with the mRNA 3ʹUTR of the gene encoding CREBBP whereas overexpression reduced luciferase activity (p < 0.01) (Fig. 3D, F). Moreover, the remaining time of CREBBP mRNA was higher in MLE-2 cells when ZFP36 was underexpressed compared to when ZFP36 was overexpressed (p < 0.05, p < 0.01) (Fig. 3E, G), which indicates that ZFP36 could promote the decay of CREBBP mRNA by binding to it.
Fig. 3. ZFP36 destabilizes CREBBP mRNA by binding to it.

A catRAPID fragment-based prediction assays of interaction between ZFP36 and CREBBP identified a strong interaction with high confidence levels (interaction propensity = 49 and discriminative power = 94%). B Western blotting analysis followed by RNA pull-downs, incubated with lysed lung epithelial-2 cell supernatant. GAPDH was used as a control. C Fold enrichment of CREBBP expression in RIP assays performed with ZFP36-coated beads. D, F Lung epithelial-2 cells underexpressing (D) or overexpressing (F) ZFP36 were stably transfected with luciferase constructs carrying the 3ʹUTR of the gene encoding CREBBP or empty vector (pGL3). Luciferase activities were measured and normalized to the activities obtained in pGL3-transfected cells (n = 3 in every group, **P < 0.01). E, G Remaining CREBBP mRNA levels were measured in lung epithelial-2 cells by real-time PCR (n = 3 in every group).
Effect of ZFP36 silencing on acute lung injury induced inflammation and apoptosis
To confirm our finding that ZFP36 could prevent CREBBP-induced lung injury through mRNA degradation, we measured the effect of altering the expression of ZFP36 on the level of CREBBP mRNA in the intestinal I/R-induced lung injury mouse model. CREBBP mRNA expression and protein levels were significantly increased in the lung tissue of the I/R model when ZFP36 expression was knocked down (p < 0.05) (Fig. 4A). The severity of lung injury (measured by arterial blood PaO2, lung water content, and BALF protein content) (p < 0.05, p < 0.01) (Fig. 4B–D) and inflammatory response (measured by levels of IL-1β, TNF-α, and IL-6 levels) were highest in the lung tissue of the I/R model with ZFP36 knockdown (Fig. 4B–G). In contrast, downregulating the expression of CREBBP by RNA interference significantly reduced the severity of lung injury. H&E-stained lung sections of the mice with I/R confirmed that alveolar edema was the most pronounced in the absence of ZFP36 (Fig. 4H).
Fig. 4. Effect of ZFP36 and CREBBP deficiency on ischemia–reperfusion (I/R)-induced lung damage and inflammation in vivo.

A CREBBP mRNA and protein expression in lung tissues of mice transfected with shZFP36 or empty vector (scr) were analyzed by RT-qPCR (n = 6 per group, **P < 0.01) and Western blot. B–D Arterial blood PaO2 (B), lung water content (C), and the protein content of BALF (D) were measured. E–G Levels of IL-1β (E), TNF-α (F), and IL-6 (G) were measured by ELISA. H H&E staining of lung sections, scale bar: 50 μm.
To reaffirm our findings, we repeated the experiment in MLE-2 cells subjected to hypoxia followed by regeneration (H/R) and further investigated the expression of apoptosis-related proteins and cell viability (Fig. S2). When the expression of ZFP36 is downregulated, the protein levels of CREBBP, Bax, p21, and acetyl-p53 are increased and cell viability is reduced, indicating that ZFP36 inhibits apoptosis via the CREBBP/p53/p21/Bax pathway in vitro. We repeated the experiment in the I/R mouse model with the expression of ZFP36 or CREBBP downregulated (Fig. 5). The expression of apoptosis-related proteins and TUNEL assays performed on lung sections from the mice demonstrated that ZFP36 inhibits I/R-induced apoptosis through the CREBBP/p53/p21/Bax pathway (p < 0.05, p < 0.01, p < 0.001) (Fig. 5B).
Fig. 5. ZFP36 inhibits ischemia–reperfusion (I/R)-induced apoptosis via the CREBBP/p53/p21/Bax pathway.

A Expression levels of apoptosis-related protein were analyzed by Western blotting. B TUNEL assay was performed on lung sections from each group and TUNEL-positive cells were determined.
ZFP36 protects against intestinal I/R-induced lung fibrosis by inhibiting epithelial–mesenchymal transition (EMT)
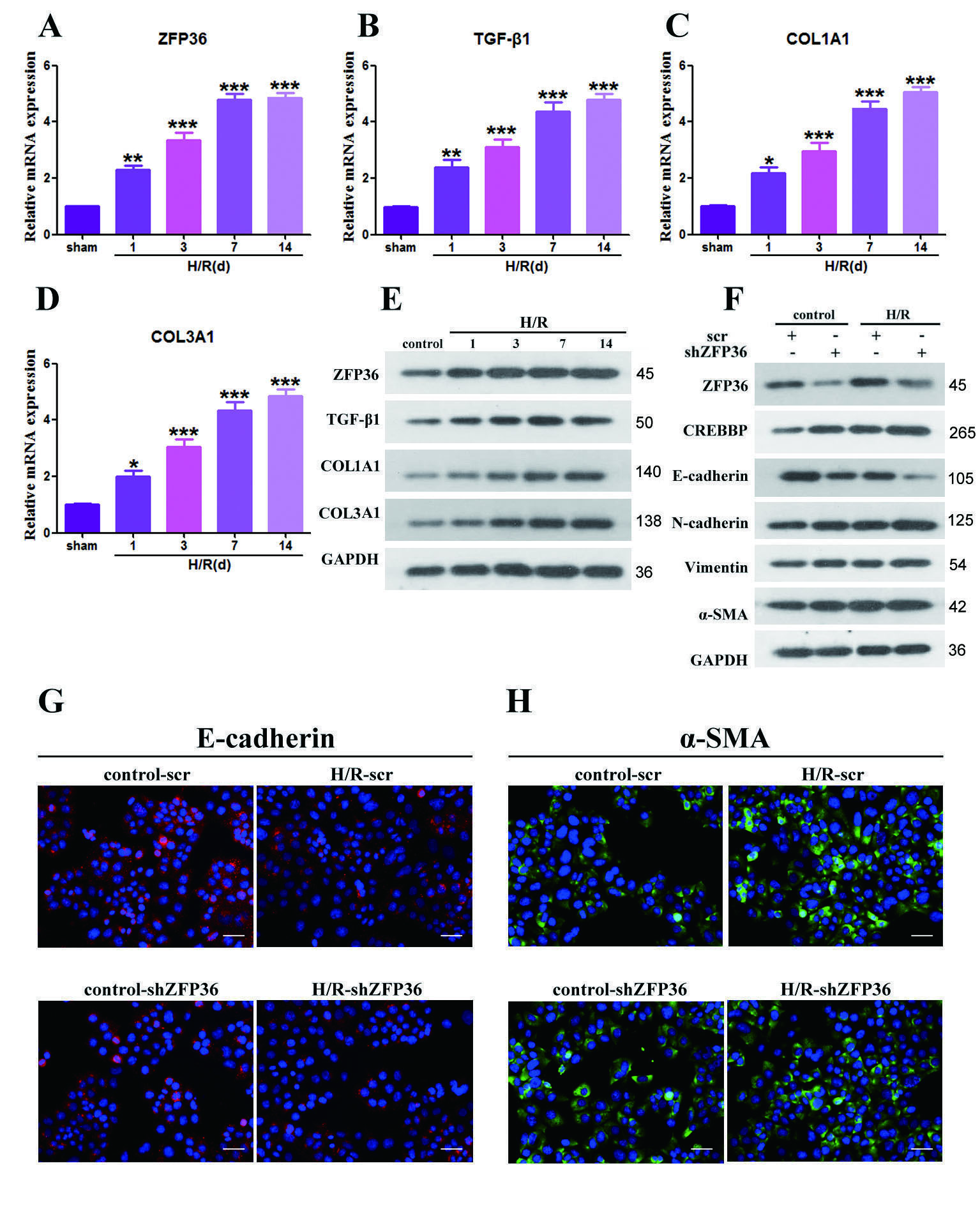
To determine whether ZFP36 could alleviate intestinal I/R-induced lung fibrosis associated lung injury, proteins related to fibrosis (TGF-β1, COL1A1, and COL3A1) were measured in the I/R mouse model 14 days after the I/R procedure (Fig. 6A–E). There was a significant increase in the accumulation of fibrosis-related proteins in the lung tissue of mice 14 days after I/R (p < 0.01, p < 0.001). The detection of ZFP36 by the immunohistochemical staining of lung tissue in mice with ZFP36 knockdown revealed that high levels of ZFP36 were related to reduced levels of alveolar edema and lung injury (Fig. 6F). Intensely stained collagen fibers surrounding the vessels and bronchioles in the lung tissue in Masson trichrome-stained sections indicated a greater presence of fibrosis in the I/R mice with ZFP36 knockdown (Fig. 6G, H). Experiments in vitro confirmed that the downregulation of ZFP36 increased the level of fibrosis (Fig. S3). Proteins involved in fibrosis and EMT all indicated that the silencing of ZFP36 increased the level of fibrotic activity following I/R-induced injury.
Fig. 6. Role of ZFP36 in intestinal ischemia–reperfusion (I/R)-induced lung fibrosis.

C57BL/6 mice were subjected to 60 min intestine ischemia followed by 60 min reperfusion, and then detections were performed at 1, 3, 7, and 14 days. Sham mice were included as a control. A–D ZFP36 (A), TGF-β1 (B), COL1A1 (C), and COL3A1 (D) mRNA expression in lung tissues of each group were analyzed by RT-qPCR (n = 6 per group, **P < 0.01) and Western blot. E ZFP36, TGF-β1, COL1A1, and COL3A1 protein expression in lung tissues of each group by Western blotting. F Immunohistochemical staining of lung tissues of mice with stable knockdown of ZFP36 or scr for sham and I/R after 14 days. Scale bars: 50 μm. G, H H&E stain (G) and Masson trichrome (H)-stained lung sections. Scale bar: 50 μm.
Discussion
Acute lung injury derived from pulmonary inflammation following I/R has a complex etiology [26, 27]. Presently, there are no markers to predict the severity of lung injury following I/R and there are no molecular-based therapies either [28]. However, some proinflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6) are differentially regulated following I/R and several of these are negatively correlated with the expression of ZFP36 [29]. ZFP36 has been found to suppress inflammatory responses by destabilizing the mRNA of genes encoding proteins associated with inflammation [21, 30]. Therefore, we aimed to investigate the molecular role of ZFP36 behind the development of lung injury in an animal model of I/R.
Our findings that ZFP36 is upregulated in lung injury derived from intestinal I/R, and suppression of ZFP36 led to increased levels of proinflammatory proteins (IL-1β, TNF-α, and IL-6) and severity of lung injury is similar to other studies [31]. In this study, we examined the role of ZFP36 in mRNA degradation by assessing its interactions with CREBBP, which promotes apoptosis following I/R and possesses an AU-rich 3ʹUTR ZFP36 binding site. We confirmed that ZFP36 interacts with the 3ʹUTR binding site in CREBBP. We also found that ZFP36 is negatively correlated with CREBBP. Our results suggest that ZFP36 is upregulated during the process of lung injury and suppresses inflammatory responses by regulating the mRNA stability of CREBBP. We found that ZFP36 can mediate the inflammatory response by destabilizing RNA as reported previously in independent studies [31–33]. Marchese et al. proposed that ZFP36 degrades mRNA through the recruitment of the Ccr4/Caf1/Not deadenylase complex and that ZFP36 is recruited by the activation of p38-MK2 signaling [34].
To determine the pathway involved in the suppression of I/R-induced injury promoted by the ZFP36/CREBBP interaction, we assessed the expression and activation of proteins involved in inflammation-mediated apoptosis. We found that downregulating the expression of ZFP36 could increase the protein levels of CREBBP, Bax, p21, and acetyl-p53 and reduce cell viability. The degradation of CREBBP mRNA by ZFP36 suppresses the expression and activation of apoptotic-related proteins. Therefore, ZFP36 could inhibit apoptosis through the CREBBP/p53/p21/Bax pathway. Similarly, in a rat myocardial I/R injury model, Yang et al. [25] found that downregulating of CREBBP resulted in decreased p53 acetylation activity and levels of Bax and p21. However, the downregulation of CREBBP, in this case, did not occur by the degradation of CREBBP mRNA but by the competitive endogenous binding of microRNA.
Finally, we examined whether ZFP36 could alleviate the fibrosis-related to I/R-induced lung injury by measuring markers associated with fibrosis and EMT in mice with ZFP36 knockdown. Pulmonary fbrosis is a disease induced by lung injury, which may develop as the result of repeated stimuli, with early cycles of injury to alveolar epithelial and endothelial cells, followed by inflammation and attempted repair [35]. The levels of fibrosis and EMT were increased when ZFP36 was knockdown (Fig. S3). Montorsi et al. [30] reported similar results; ZFP36 inhibits the expression of transcription factors that are involved in EMT and is negatively correlated with the Wnt/β-catenin pathway in colorectal cancer. The suppression of EMT by ZFP36 is a recurring pattern in other cancers, such as ovarian cancer, hepatocellular carcinoma, and lung cancer [36–38]. In cancer cells, ZFP36 is believed to act as an EMT suppressor by binding to AU-rich elements in the mRNA 3′UTRs of Twist1 and Snail1 [39].
To summarize, ZFP36 suppressed the inflammatory response in I/R-associated pulmonary inflammation through the destabilization of CREBBP mRNA. Analysis of lung tissues in ZFP36-knockdown mice by Western blotting, immunohistochemistry, and real-time PCR showed that ZFP36 deficiency upregulated CREBBP, enhanced I/R-induced lung injury, apoptosis, and inflammation, and increased I/R-induced lung fibrosis. In silico analyses revealed that ZFP36 is a strong negative regulator of CREBBP mRNA stability. Experimental verification of this association revealed a direct interaction between ZFP36 and an AU-rich 3ʹUTR in CREBBP to regulate cell inflammation and apoptosis negatively through the p53/p21/Bax axis in MLE-2 cells.Thus these results of our study indicate that ZFP36 interacts with the mRNA of CREBBP and may be useful as a molecular marker or have a potential role in alleviating inflammation-associated lung damage.
Materials and methods
Animal model of intestinal ischemia/reperfusion-induced acute lung injury
Eight-week-old C57BL/6 J mice (purchased from the Chinese Science Academy) were used in accordance with the guidelines specified by the Animal Care and Usage Committee of Tongji University. The animals were fasted with free access to water for 24 h before the I/R procedure. After animals were sedated with an intraperitoneal injection of sodium pentobarbital (50 mg/kg) the superior mesenteric artery was clamped with an atraumatic microvascular clip. After 60 min the clamps were removed, and intestinal reperfusion was established for the specified times. The procedure was replicated in sham-operated mice without vascular clamping.
Real-time PCR
Total RNA was extracted from tissue using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Real-time PCR was performed using an ABI Prism 7,500 system (PE Applied Biosystems, Waltham, MA, USA) and the following conditions: 95 °C for 5 min; 45 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s; then a final extension of 72 °C for 1 min. U6 was used as an internal control and the 2−ΔΔCt method was used to calculate the relative expression.
Western blot analysis
The protein levels of ZFP36, CREBBP, IL-1β, TNF-α, IL-6, TGF-β1, COL1A1, and COL3A1 were determined by Western blot analysis. Protein was first extracted from cells and lung tissue using RIPA buffer containing protease and phosphatase inhibitors (Beyotime, Wuhan, China) for 30 min. Equivalent samples of protein (40 μg) were separated by 10% SDS-polyacrylamide electrophoresis and then transferred to a PVDF membrane (Millipore, Bedford, MA, USA). Membranes were first blocked in 5% non-fat dry milk for 2 h and then incubated overnight at 4 °C with relevant primary antibodies following manufacturers’ recommendation. Finally, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (C13091, Applygen Technologies Inc., China, 1:1000) at room temperature for 1 h and immunoreactive proteins were visualized by using an enhanced chemiluminescence detection kit (Pierce, Rockford, IL, USA). Images of visualized blot bands were acquired and Gapdh was used as the loading control. The primary antibodies are listed as follows: ZFP36 (#71632, Cell Signaling Technology, USA, 1:1000), CREBBP (ab2832, Abcam, UK, 1:2000), IL-1β (ab205924, Abcam, UK, 1:1000), TNF-α (ab66579, Abcam, UK, 1:1000), IL-6 (MAB406, R&D Systems, Minneapolis, USA, 1:1000), TGF-β1 (ab179695, Abcam, UK, 1:5000), COL1A1 (ab254113, Abcam, UK, 1:1000), and COL3A1 (ab7778, Abcam, UK, 1:1000) and GAPDH (ab8245, Abcam, UK, 1:5000).
RNA immunoprecipitation
We used a Magna RIP RNA-binding protein immunoprecipitation kit (Millipore, Burlington, MA, USA) to assess the interaction of ZFP36 with CREBBP mRNA 3ʹUTR. Co-immunoprecipitation was performed using antibody toward ZFP36 (#71632, Cell Signaling Technology, USA, 1:1000). RNA was quantified by qRT-PCR.
Pull down
RNA was first labeled with biotin using a Biotin-RNA Labeling Mix (Roche, Basel, Switzerland) following the manufacturer’s instructions. Cells were sonicated in lysis buffer (20 mM Tris-HCl, pH7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, and a cocktail of protease, phosphatase, and RNase inhibitors) and then centrifuged for 10 min at 13,000 × g at 4 °C. The supernatant was collected and incubated with 2 μg biotin-labeled RNA for 2 h at 4 °C. Dynabeads (Invitrogen, Carlsbad, CA, USA) were added to the sample and incubated for a further 1 h at 4 °C. The interacting proteins were isolated according to the manufacturer’s instructions and then subjected to SDS-PAGE.
Histopathological analysis
Lung tissue was fixed in 10% buffered formalin, embedded in paraffin, and then sectioned into 5 μm slices. The tissue sections were stained with H&E or Masson’s trichrome stain and then assessed by light microscopy. Lung tissue was scored according to severity (0–3, with 3 as severe). The presence of intra-alveolar hemorrhage and debris, cellular hyperplasia, hyperemia, and congestion were considered in the representation of acute lung injury.
TUNEL assay
Apoptosis was determined by using a TUNEL-based colorimetric apoptosis detection system (Dead End, Promega Corporation, Madison, WI, USA) according to the manufacturer’s instructions. The percentage of TUNEL-positive cells in 20 random fields was used to calculate the rate of apoptosis.
Immunofluorescence and immunohistochemical staining
After deparaffinization, lung tissue sections were permeabilized with 0.1% Triton X-100 and blocked with bovine serum albumin (5%). Tissue samples were then incubated with primary antibodies overnight at 4 °C. After several washes in phosphate-buffered saline, sections were incubated with green or red-fluorescent Alexa Fluor IgG Ab (Invitrogen). Nuclei were stained with DAPI and then images were obtained using a fluorescence microscope (Olympus, Tokyo, Japan).
Cell culture and treatment
MLE-2 cells were cultured in RPMI 1640 supplemented with 5% fetal bovine serum at 37 °C and 5% CO2. ZFP36 and CREBBP shRNA and expression vectors were obtained from Gene Pharma (Shanghai, China). Vectors and shRNA were transfected into cells using Lipofectamine RNAi MAX (Invitrogen) following the manufacturer’s instructions,. MLE-2 cells were subjected to 2 h of hypoxia by incubation in an anaerobic chamber at 85% N2, 5% H2, 10% CO2, and 35 °C. Cells were removed from the anaerobic chamber and regenerated for 0, 3, 6, and 12 h in a normal incubator in fresh media.
Analysis of apoptosis by flow cytometry
To determine apoptosis under each experimental condition, cells were double-stained with Annexin V-FITC and propidium iodide for 15 min, and viability was assessed by flow cytometric analysis (BD Biosciences, San Jose, CA, USA).
Assay of lung tissue cytokines
Frozen lung tissues were homogenized on ice using a homogenizer and centrifuged at 4000 g for 10 min at 4 °C. Levels of tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) in the supernatant were assayed according to the manufacturer’s instructions using ELISA kits (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
All results are expressed as the means ± standard deviation (SD). Statistical analysis was performed using the Student’s t-test, and one-way analysis of variance (ANOVA) was used for multiple comparisons. P-values <0.05 indicate a statistically significant difference.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors are grateful to National Natural Science Foundation of China, the Shanghai Pujiang Talent Plan with Class A, the Construction of Integrated Chinese and Western Medicine in General Hospital in Shanghai, and the Shanghai Science and Technology Commission project.
Author contributions
Conception: Xiaoping Zhang, Yingchuan Li; Article Design: Yingchuan Li, Xuan Zhao; Acquisition of data: Yongmei Cao, Weifeng Huang, Fang Wu; Analysis of data: Yongmei Cao, Weifeng Huang, Jiawei Shang; Interpretation of data: Yongmei Cao, Weifeng Huang, Weifeng Huang, and Feng Ping; Manuscript preparation: Yongmei Cao, Jiawei Shang, Wei Wang; Final approval of manuscript: All authors read and approved the final manuscript.
Funding
The present study was supported by the Shanghai Pujiang Talent Plan with Class A (18PJ1409200), the Construction of Integrated Chinese and Western Medicine in General Hospital in Shanghai (ZHYY-ZXYJHZX-2-2017010), the Shanghai Science and Technology Commission project (18ZR1424800), Clinical Technology Innovation Project of Shanghai Shenkang Hospital Development Center (SHDC12020122), Key Discipline Project of Shanghai Public Health System Construction (GWV-10.1-XK23), the national natural Science Foundation of China (grant No. 8156010205).
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Ethics statement
The research was approved by the Ethics Committee of Tongji University.
Footnotes
Edited by S. Lavandero
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yingchuan Li, Email: yingchuan.li@sjtu.edu.cn.
Xuan Zhao, Email: zhaoxuan1026@hotmail.com.
Xiaoping Zhang, Email: zxpkxy@tongji.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41419-021-03950-y.
References
- 1.Laubach VE, Sharma AK. Mechanisms of lung ischemia-reperfusion injury. Curr Opin Organ Transplant. 2016;21:246–52. doi: 10.1097/MOT.0000000000000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Hengst WA, Gielis JF, Lin JY, Van Schil PE, De Windt LJ, Moens AL. Lung ischemia-reperfusion injury: a molecular and clinical view on a complex pathophysiological process. Am J Physiol Heart Circ Physiol. 2010;299:H1283–1299. doi: 10.1152/ajpheart.00251.2010. [DOI] [PubMed] [Google Scholar]
- 3.Nastos C, Kalimeris K, Papoutsidakis N, Tasoulis MK, Lykoudis PM, Theodoraki K, et al. Global consequences of liver ischemia/reperfusion injury. Oxid Med Cell Longev. 2014;2014:906965. doi: 10.1155/2014/906965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou X, Jiang R, Dong Y, Wang L. Remote ischemic preconditioning attenuates cardiopulmonary bypass-induced lung injury. PloS One. 2017;12:e0189501. doi: 10.1371/journal.pone.0189501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehrotra P, Collett JA, Gunst SJ, Basile DP. Th17 cells contribute to pulmonary fibrosis and inflammation during chronic kidney disease progression after acute ischemia. Am J Physiol Regul Integr Comp Physiol. 2018;314:R265–R273. doi: 10.1152/ajpregu.00147.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JH, Kim J, Chun J, Lee C, Im JP, Kim JS. Role of iRhom2 in intestinal ischemia-reperfusion-mediated acute lung injury. Sci Rep. 2018;8:3797. doi: 10.1038/s41598-018-22218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huffmyer JL, Groves DS. Pulmonary complications of cardiopulmonary bypass. Best Pract Res Clin Anaesthesiol. 2015;29:163–75. doi: 10.1016/j.bpa.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari RS, Andrade CF. Oxidative stress and lung ischemia-reperfusion injury. Oxid Med Cell Longev. 2015;2015:590987. doi: 10.1155/2015/590987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito M, Chen-Yoshikawa TF, Suetsugu K, Okabe R, Takahagi A, Masuda S, et al. Pirfenidone alleviates lung ischemia-reperfusion injury in a rat model. J Thorac Cardiovasc Surg. 2019;158:289–96. doi: 10.1016/j.jtcvs.2018.08.098. [DOI] [PubMed] [Google Scholar]
- 10.Yue X, Shan B, Lasky JA. TGF-beta: titan of lung fibrogenesis. Curr Enzym Inhib 2010; 6:10.2174/10067. [DOI] [PMC free article] [PubMed]
- 11.Ganeshan K, Johnston LK, Bryce PJ. TGF-beta1 limits the onset of innate lung inflammation by promoting mast cell-derived IL-6. J Immunol. 2013;190:5731–8. doi: 10.4049/jimmunol.1203362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim KK, Sheppard D, Chapman HA. TGF-beta1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol 2018; 10:a022293. [DOI] [PMC free article] [PubMed]
- 13.Sullivan BP, Cui W, Copple BL, Luyendyk JP. Early growth response factor-1 limits biliary fibrosis in a model of xenobiotic-induced cholestasis in mice. Toxicol Sci. 2012;126:267–74. doi: 10.1093/toxsci/kfr311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson EM, Williams A, Yauk CL, Vincent R. Overexpression of tumor necrosis factor-alpha in the lungs alters immune response, matrix remodeling, and repair and maintenance pathways. Am J Pathol. 2012;180:1413–30. doi: 10.1016/j.ajpath.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 15.Malaviya R, Laskin JD, Laskin DL. Anti-TNF alpha therapy in inflammatory lung diseases. Pharmacol Ther. 2017;180:90–98. doi: 10.1016/j.pharmthera.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Zhang S, Cao H, Wang H, Sun C, Liu S, et al. Deficiency of p38alpha in macrophage ameliorates d-galactosamine/TNF-alpha-induced acute liver injury in mice. FEBS J. 2017;284:4200–15. doi: 10.1111/febs.14294. [DOI] [PubMed] [Google Scholar]
- 17.Nakawaki M, Uchida K, Miyagi M, Inoue G, Kawakubo A, Kuroda A, et al. Sequential CCL2 expression profile after disc injury in mice. J Orthop Res. 2020;38:895–901. doi: 10.1002/jor.24522. [DOI] [PubMed] [Google Scholar]
- 18.Bellamri N, Viel R, Morzadec C, Lecureur V, Joannes A, de Latour B, et al. TNF-alpha and IL-10 control CXCL13 expression in human macrophages. J Immunol. 2020;204:2492–502. doi: 10.4049/jimmunol.1900790. [DOI] [PubMed] [Google Scholar]
- 19.Clark AR, Dean JL. The control of inflammation via the phosphorylation and dephosphorylation of tristetraprolin: a tale of two phosphatases. Biochem Soc Trans. 2016;44:1321–37. doi: 10.1042/BST20160166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahmoud L, Moghrabi W, Khabar KSA, Hitti EG. Bi-phased regulation of the post-transcriptional inflammatory response by Tristetraprolin levels. RNA Biol. 2019;16:309–19. doi: 10.1080/15476286.2019.1572437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–54. doi: 10.1016/S1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 22.Liang J, Lei T, Song Y, Yanes N, Qi Y, Fu M. RNA-destabilizing factor tristetraprolin negatively regulates NF-kappaB signaling. J Biol Chem. 2009;284:29383–90. doi: 10.1074/jbc.M109.024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao Y, Zhang JJ. Propofol induces oxidative stress and apoptosis in vitro via regulating miR-363-3p/CREB signalling axis. Cell Biochem Funct. 2020;38:1119–28. doi: 10.1002/cbf.3572. [DOI] [PubMed] [Google Scholar]
- 24.Cai X, Fu H, Wang Y, Liu Q, Wang X. Depletion of GPSM1 enhances ovarian granulosa cell apoptosis via cAMP-PKA-CREB pathway in vitro. J Ovarian Res. 2020;13:136. doi: 10.1186/s13048-020-00740-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Chen L, Yang J, Ding J, Li S, Wu H, et al. MicroRNA-22 targeting CBP protects against myocardial ischemia-reperfusion injury through anti-apoptosis in rats. Mol Biol Rep. 2014;41:555–61. doi: 10.1007/s11033-013-2891-x. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Yao W, Yuan D, Huang F, Liu Y, Luo G, et al. Effects of Connexin 32-mediated lung inflammation resolution during liver ischemia reperfusion. Digestive Dis Sci. 2020;65:2914–24. doi: 10.1007/s10620-019-06020-8. [DOI] [PubMed] [Google Scholar]
- 27.Maltesen RG, Buggeskov KB, Andersen CB, Plovsing R, Wimmer R, Ravn HB, et al. Lung protection strategies during cardiopulmonary bypass affect the composition of bronchoalveolar fluid and lung tissue in cardiac surgery patients. Metabolites. 2018;8:54.. doi: 10.3390/metabo8040054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butt Y, Kurdowska A, Allen TC. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med. 2016;140:345–50. doi: 10.5858/arpa.2015-0519-RA. [DOI] [PubMed] [Google Scholar]
- 29.Patial S, Blackshear PJ. Tristetraprolin as a therapeutic target in inflammatory disease. Trends Pharmacol Sci. 2016;37:811–21. doi: 10.1016/j.tips.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montorsi L, Guizzetti F, Alecci C, Caporali A, Martello A, Atene CG, et al. Loss of ZFP36 expression in colorectal cancer correlates to wnt/ ss-catenin activity and enhances epithelial-to-mesenchymal transition through upregulation of ZEB1, SOX9 and MACC1. Oncotarget. 2016;7:59144–57. doi: 10.18632/oncotarget.10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joe Y, Kim SK, Chen Y, Yang JW, Lee JH, Cho GJ, et al. Tristetraprolin mediates anti-inflammatory effects of carbon monoxide on lipopolysaccharide-induced acute lung injury. Am J Pathol. 2015;185:2867–74. doi: 10.1016/j.ajpath.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Tu Y, Wu X, Yu F, Dang J, Wang J, Wei Y, et al. Tristetraprolin specifically regulates the expression and alternative splicing of immune response genes in HeLa cells. BMC Immunol. 2019;20:13. doi: 10.1186/s12865-019-0292-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tollenaere MAX, Tiedje C, Rasmussen S, Nielsen JC, Vind AC, Blasius M, et al. GIGYF1/2-driven cooperation between ZNF598 and TTP in posttranscriptional regulation of inflammatory signaling. Cell Rep. 2019;26:3511–21. doi: 10.1016/j.celrep.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 34.Marchese FP, Aubareda A, Tudor C, Saklatvala J, Clark AR, Dean JL. MAPKAP kinase 2 blocks tristetraprolin-directed mRNA decay by inhibiting CAF1 deadenylase recruitment. J Biol Chem. 2010;285:27590–27600. doi: 10.1074/jbc.M110.136473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panganiban RA, Day RM. Hepatocyte growth factor in lung repair and pulmonary fibrosis. Acta Pharmacol Sin. 2011;32:12–20. doi: 10.1038/aps.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Ding N, Guo J, Xia J, Ruan Y. Dysregulation of TTP and HuR plays an important role in cancers. Tumour Biol. 2016;37:14451–61. doi: 10.1007/s13277-016-5397-z. [DOI] [PubMed] [Google Scholar]
- 37.Sanduja S, Blanco FF, Young LE, Kaza V, Dixon DA. The role of tristetraprolin in cancer and inflammation. Front Biosci. 2012;17:174–88. doi: 10.2741/3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brennan SE, Kuwano Y, Alkharouf N, Blackshear PJ, Gorospe M, Wilson GM. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res. 2009;69:5168–76. doi: 10.1158/0008-5472.CAN-08-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoon NA, Jo HG, Lee UH, Park JH, Yoon JE, Ryu J, et al. Tristetraprolin suppresses the EMT through the down-regulation of Twist1 and Snail1 in cancer cells. Oncotarget. 2016;7:8931–43. doi: 10.18632/oncotarget.7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.