

Abstract
Purpose
To identify a pathogenic gene mutation in a female infertility proband characterized by empty follicle syndrome (EFS) and explore the genetic cause of EFS.
Methods
Whole exome sequencing (WES) was performed to identify the candidate pathogenic mutation. Sanger sequencing was used to validate the mutation in family members. The pathogenicity of the identified variant and its possible effects on the protein were evaluated with in silico tools. Immunofluorescence staining was used to study the possible mechanism of the mutation on affected oocyte.
Results
We identified a family with a novel homozygous nonsense mutation in zona pellucida 1 (ZP1) (c.199G > T [p.Glu67Ter]). Based on bioinformatics analysis, the mutation was predicted to be pathogenic. This variant generates a premature stop codon in exon 2 at the 199th nucleotide, and was inferred to result in a truncated ZP1 protein of 67 amino acids at the ZP-N1 domain. An in vitro study showed that the oocyte of the EFS proband was degenerated and the zona pellucida was absent. Additionally, the mutant ZP1 proteins were localized in the cytoplasm of the degenerated oocyte but not at the surface.
Conclusions
The novel mutation in ZP1 is a genetic cause of female infertility characterized by EFS. Our finding expands the genetic spectrum for EFS and will help justify the EFS diagnosis in patients.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10815-021-02136-x.
Keywords: Zona pellucida 1, Mutation, Empty follicle syndrome, In vitro fertilization, Whole-exome sequencing
Introduction
Empty follicle syndrome (EFS) is a condition where no oocytes are retrieved from mature ovarian follicles even after repeated aspiration and washing, despite adequate response to ovarian stimulation in in vitro fertilization (IVF) [1]. EFS was first reported by Coulam et al. in 1986 [2] and classified by Stevenson and Lashen et al. in 2008 [3] as “genuine” (GEFS) and “false” EFS (FEFS) based on beta-human chorionic gonadotropin (β-HCG) levels on the day of oocyte retrieval. FEFS patients present with a low β-hCG level, indicating iatrogenic or pharmacological problems and can have a successful IVF outcome with adjustments to the ovarian stimulation protocol. A condition where a few mature or immature oocytes are recovered from mature follicles has been described as a borderline form of EFS by some literatures [4–7]. Like patients with FEFS, patients with the borderline form of EFS may achieve pregnancy through the use of rescue protocols. However, while GEFS patients have an optimal β-hCG level on retrieval day, they are unlikely to respond to any rescue protocols. The estimated prevalence of GEFS is 0.016% among patients who underwent IVF [8]. Although GEFS is rare, it is a severe condition for patients who suffer from it. Understanding the etiology of GEFS is imperative for the accurate diagnosis and treatment of these patients.
The cause of GEFS is still not well understood, but is commonly associated with ovarian aging, dysfunctional folliculogenesis, or genetic defects [9, 10]. To date, several studies have reported on the genetic defects responsible for the occurrence of GEFS. GEFS-related genes include the luteinizing hormone/chorionic gonadotropin receptor (LHCGR, MIM: 152790) [9, 11–13], the zona pellucida glycoprotein 3 (ZP3, MIM: 182889) [14], and the zona pellucida glycoprotein 1 (ZP1, MIM: 19500) [15–21]. Mutations in these genes can impair LH signaling or the ZP assembly, leading to GEFS. Some advancements have been made in our understanding of GEFS, but many cases cannot currently be explained by known genetic mutations. More investigations of novel pathogenic variants are necessary to further our knowledge of GEFS.
The zona pellucida (ZP) is an extracellular matrix that surrounds the oocyte [22]. It performs a vital role in oogenesis, fertilization, and the protection of preimplantation embryos [23–25]. Previous studies have presented diverse phenotypes of ZP gene mutations that manifest as an abnormal ZP, immature oocytes, or even EFS. Among the EFS-related genes, ZP1 has the closest relationship with EFS. Since the first report in 2019, 11 ZP1 mutations have been identified in EFS cases [15–21]. However, more reports are needed for broadening our understanding of the genotype and phenotype relationships of EFS patients.
In this study, we report on a patient with a clinical manifestation of EFS and identified a novel homozygous nonsense mutation in the ZP1 gene. This instance of EFS appears to follow the recessive inheritance pattern according to pedigree analysis results. Genetic analysis showed this pathogenic mutation is located in the ZP-N1 domain and therefore may cause the truncated ZP1 protein at the N terminal. In vitro studies indicated that this mutation might result in degenerated oocytes by preventing ZP assembly and secretion. Based on previous literature and our own findings, we herein propose a precise definition for ZP-associated EFS to distinguish this EFS subtype from other EFS types.
Materials and methods
This study was approved by the ethics committee of the First Affiliated Hospital of Nanjing Medical University. All participants signed the informed consent form before enrollment in this study.
Patients
The proband (Fig. 1, (II-3)) was enrolled at the Reproductive Center of the First Affiliated Hospital of Nanjing Medical University. She was 30 years old and is the third sister in a Han Chinese family. Their ancestral history was provided by the proband. Her parents had a non-consanguineous marriage, but their ancestors inhabited a rural village with little immigration for hundreds of years. She was diagnosed with unexplained primary infertility at the age of 29, after 7 years of unprotected sexual intercourse with her husband. She has had regular menstrual cycles since menarche at 15 years of age. Her basal hormone levels were normal, and other infertility-related examinations did not display any abnormalities. Her husband’s semen analyses were normal based on WHO’s standards [26]. The couple had no family history of infertility or aberrant karyotypes.
Fig. 1.

ZP1 mutation in the proband with empty follicle syndrome (EFS). a Pedigree of the patient with EFS. Male family members are denoted by squares, female family members by circles, and the EFS patient by filled circles. The arrow indicates the proband (II-3). Genotypes of the mutation c.199 G > T are noted below the symbols. b Sanger sequencing verification of ZP1 mutation, c.199 G > T. The proband carried the homozygous nonsense mutation, and the arrow shows the mutation site. Her parents harbored the heterozygous allele. c–f Analysis of oocytes morphology. c, d Normal cumulus oocyte complex and oocyte from the control undergoing intracytoplasmic sperm injection (ICSI) due to male infertility. e, f Cumulus oocyte complex and a degenerated oocyte after removing cumulus cells from the proband.
Given the unexplained infertility, she underwent 3 cycles of IVF treatment. The first two attempts were performed in another hospital, while the last attempt was performed in our hospital. In the first two cycles, ovarian stimulation using the long gonadotropin-releasing hormone (GnRH) agonist protocol in the first cycle and progestin-primed ovarian stimulation (PPOS) protocol in the second produced 14 cumulus-oocyte complexes (COCs), 12 of which were empty. The remaining two contained degenerated oocytes. In the third attempt, the PPOS protocol was implemented [27]. Medroxyprogesterone acetate (MPA, 8 mg/d; Xianju Pharmaceutical Co., China) was administered on day 3 of the menstrual cycle. On the same day, human menopausal gonadotropin (hMG, Shanghai Lizhu Pharmacy Co., China) was administered at a dose of 225 IU per day for 6 days, and 300 IU per day for 3 days. After a total gonadotropin dose of 2250 IU was administered, 8 leading follicles (≥ 14 mm) among a total of 13 follicles were identified on the trigger day (Supplemental Fig. 1a and b). Patient monitoring revealed a serum estradiol level of 7208.2 pmol/L. Oocyte retrieval was performed 36 h after a dual trigger with human chorionic gonadotropin (10000 IU; Shanghai Lizhu Pharmacy Co., China) and GnRH agonist (triptorelin, 0.2 mg; Ipsen, France). The serum β-HCG level was 287.8 IU/L on the day of oocyte retrieval. A total of 8 COCs were retrieved after repeated aspiration and flushing, but no recognizable oocytes could be identified. After removal of the cumulus cells, one degenerated oocyte lacking the ZP was found. Three IVF attempts resulting in EFS lead to us considering this case a GEFS case (Supplemental Table 1).
The control COCs and oocytes for the in vitro studies were from a patient undergoing intracytoplasmic sperm injection (ICSI) due to male infertility. The patient was 25 years old and had regular menstrual cycles. She had no family history of aberrant karyotypes and her infertility tests were normal. She was given a depot GnRHa protocol for ovarian stimulation and obtained 20 mature oocytes and several germinal vesicle-stage (GV) oocytes. The morphologies of these oocytes were normal. Since the mature oocytes were too precious, the GV oocytes were used for in vitro study after obtaining informed consent from the couple.
Morphologic studies
The collected COCs were placed in a rinse dish (35 mm culture dish) containing 2 ml Sydney IVF Gamete Buffer (COOK) to wash off erythrocytes, and were then transferred into a 4-well dish [four-well plate with 1.0 ml Sydney IVF Fertilization Medium (COOK)]. The COCs were deposited into droplets containing 80 U/mL hyaluronidase (SAGE, In-vitro Fertilization, Inc.) for 1–2 min to remove the cumulus cells. The degenerated oocytes were examined using an inverted microscope (Eclipse TE2000-S; Nikon, Japan) with a magnification of × 40, × 100, or × 200.
Exome sequencing and analyses
We performed whole-exome sequencing (WES) for the proband and her parents. Genomic DNA was extracted from blood samples using a DNeasy Blood & Tissue Kit (Qiagen). WES samples were prepared using an IDT xGen Exome Research Panel V1.0 (Integrated DNA Technologies). The sequencing library’s quantity was evaluated using a Qubit 2.0 fluorometer (Thermo Fisher Scientific). The size and quality of the libraries were measured using a 2100 Bioanalyzer High Sensitivity DNA Assay (Agilent Technologies). The qualified libraries were loaded onto an Illumina NovaSeq platform (Illumina, San Diego, USA). FASTQ files were aligned to the human genome reference (hg19) using a BWA v0.7.13 [28]. Single nucleotide variants and indels were genotyped from recalibrated BAM files with a GATK 4.0 [29] and were annotated by ANNOVAR [30] over multiple databases containing population frequency, HGVS variant description, phenotype or disease and variant functional prediction. Variants were then categorized as benign, likely benign, unknown significance, likely pathogenic, or pathogenic based on the American College of Medical Genetics (ACMG) guidelines [31]. Copy number variants were labelled using a DNAcopy R package [32], filtered and grouped following ACMG guidelines [33] and manually checked using an Integrative Genomics Viewer [34].
Variant analyses and identification
To validate the mutation revealed by WES, sanger sequencing was performed in the proband and her parents. The primers were as follows: ZP1-E2F:GCCCAAGACCACACAACAAA; and ZP1-E2R: TCTTCGGCTTTGGCTCCTTT. Polymerase chain reaction (PCR) was performed to amplify the mutation region, and the resulting products underwent Sanger sequencing using an ABI 3100 DNA analyzer (Applied Biosystem, Foster City, CA, USA). The allele frequencies of the variant were assessed using a genome aggregation database (gnomAD, https://gnomad.broadinstitute.org/). The pathogenicity of the identified ZP1 variant was analyzed with the SIFT algorithm and the Mutation Taster. NNSplice was used to assess splicing mutations.
In vitro studies of oocytes with immunofluorescence
Immunostaining was used to compare the localization of ZP1 mutant proteins in the degenerated oocytes with that of the control oocytes. The oocytes were fixed in 4% paraformaldehyde, washed in PBS containing BSA, and permeabilized with 0.5% Triton X-100 at room temperature. Oocytes were blocked with 5% BSA and then incubated with mouse anti-ZP1 primary antibodies (sc-365435; Santa Cruz, CA, USA) at 1:100 at 4 °C overnight. ZP1 (sc-365435) is a mouse monoclonal antibody against amino acids 40-271 mapping the N-terminal of human ZP1. On the following day, the cells were washed three times in PBS and were then incubated with Donkey anti-Mouse IgG (H + L) ReadyProbes™ Secondary Antibodies, Alexa Fluor 488 (Cat# R37114, Invitrogen, USA) at 1:200 for 1 h at room temperature. To visualize the DNA, the oocytes were mounted on slides and stained with Hoechst 33342 containing anti-fade (Cat# C1026, Beyotime, China). Imaging was performed on a confocal laser scanning microscope (Zeiss, LSM 700).
Results
Phenotypes in patients
Twenty-two COCs were obtained from the patient, 19 of which were empty and 3 of which contained degenerated abnormal oocytes in the three failed IVF attempts. We examined the COCs retrieved from the last IVF attempt and found the granulosa cells of the COCs were irregular and loose with no recognizable oocytes (Fig. 1e; Supplemental Fig. 1c and d). After digestion with hyaluronidase to remove the granulosa cells immediately following follicular aspiration, one degenerated oocyte was discovered as an abnormally shaped cell mass with no zona pellucida (see Fig. 1f).
Genetic and bioinformatic analyses
To determine the potential genetic cause of EFS, we performed a WES analysis of the proband and identified the mutation responsible for the patient’s phenotype. The variant was a homozygous nonsense mutation c.199G > T (p.Glu67Ter) in exon 2 of the ZP1 gene (NM_207341). Sanger sequencing was performed to validate the mutation (Fig. 1b). The results revealed that the proband carried the ZP1 homozygous mutation (II-3) and her parents harbored heterozygous alleles (I-1 and I-2), indicating an autosomal recessive pattern of inheritance (Fig.1a). Multiple sequence alignment showed a 100% conservation of the mutation site in ZP1 orthologs across different species (Fig. 2b). This novel mutation (c.199G > T) represented a low allele frequency in ExAC and gnomAD database, and the frequency of homozygotes was zero (Table 1). We evaluated the predicted protein function of the novel mutation using in silico tools, including the SIFT algorithm, Mutation Taster, and NNSplice (Table 1). The results confirmed the novel homozygous ZP1 nonsense mutation (c.199G > T) is pathogenic.
Fig. 2.

Location and conservation of the mutation size in ZP1, and immunofluorescence in oocytes. a Schematic diagram of ZP1 with function domain and position of the identified mutation. ZP1 gene contains 12 exons. The position of the nonsense mutation c.199G > T is in exon 2 of the ZP1 gene, resulting in the 67th amino acid Glutamate (Glu) replaced by a premature STOP codon, as indicated by the arrow. The human ZP1 protein (638aa) includes N-terminal signal peptide, ZP-N1 domain, Trefoil domain, ZP-N domain, ZP-C domain, cleavage site, and C-terminal transmembrane domain. The nonsense ZP1 mutation the proband carried led to a truncated ZP1 protein at 67 amino acid, which comprises N-terminal signal domain and a small fragment of N1 domain. b Conservation of the mutation site in ZP1 was studied across different species. c Oocyte images with confocal laser scanning microscope. The Fluorescent signals of ZP1 (green) and nuclei counterstained with DAPI (blue) were imaged separately and merged.
Table 1.
Analysis of the novel ZP1 mutation predicted with in silico tool
| Chromosome11 Co-ordinatesa |
cDNA alteration |
Amino acid alteration | Exon | Mutation | ExACb allele frequency | ExACb homozygotes frequency | GnomADc allele frequency | GnomADc homozygotes frequency | NN splice | SIFT | Mutation taster |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 60636620G > T | c.199G > T | p.Glu67Ter | 2 | Nonsense | 1/121212 | 0 | 1/251402 | 0 | - | - | 1.0 (D)d |
aData are based on hg19. bExAC, Exome Aggregation Consortium; cgnomAD, genome aggregation database.
dThe letter D in the brackets denotes deleterious
Characterization of mutant ZP1 protein
The nonsense mutation c.199G > T resulted in a premature stop codon in exon 2 at the 199th nucleotide, and was inferred to cause a truncated ZP1 protein of 67 amino acids (aa) instead of the full-length 638 aa protein. The human ZP1 protein includes N-terminal signal peptide, ZP-N1 domain, Trefoil domain, ZP-N domain, ZP-C domain, cleavage site, and C-terminal transmembrane domain. The mutant ZP1 protein was truncated at the beginning of ZP-N1 domain, and was expected to contain only the N-terminal signal peptide and a small segment of the ZP-N1 domain. All other domains were absent, resulting in the total loss of ZP1 function (Fig. 2a).
Protein localization
To determine the relationship between ZP1 and EFS, we performed an immunofluorescence analysis of the ZP1 localization in the degenerated oocyte of the proband and compared the results with those of a normal control oocyte. Since mature oocytes were too precious, the control was a GV oocyte of a patient undergoing ICSI due to male infertility. In the proband’s oocyte, mutant ZP1 and the nucleus were diffusely scattered throughout the cytoplasm. In contrast, ZP1 in the control oocyte was concentrated near the periphery of the oocyte and the nucleus was located in the middle of the oocyte (Fig. 2c).
Discussion
In this study, we detected a novel nonsense homozygous mutation c.199G > T (p.Glu67Ter) in the ZP1 gene in a woman with primary infertility characterized by EFS. The inheritance pattern is autosomal recessive. To our knowledge, this is the first study discovering a homozygous ZP1 mutation located in the ZP-N1 domain associated with EFS, advancing the pursuit of the genetic cause for EFS. Based on our study, we speculate the pathogenic mutation of ZP1 may impede the formation of the ZP, ultimately resulting in oocyte degeneration and EFS. In addition, we reviewed the reported genetic defects associated with EFS (Table 2), and proposed a standardized definition for ZP-related EFS based on previous studies and our own findings.
Table 2.
The currently reported genotypes leading to empty follicle syndrome
| Mutated genes | cDNA change | Amino acid change | Mutation type | Genotype | Inheritancea | Reference |
|---|---|---|---|---|---|---|
| LHCGR | c.1199A > G | p.N400S | Missense | Homozygous | AR | Yariz et al. 2011 (9) |
| LHCGR | c.1345G > A | p.Ala449Thr | Missense | Homozygous | AR | Yuan et al. 2017 (11) |
| LHCGR | c.736 C > T | p.Q246* | Nonsense | Homozygous | AR | Chen et al. 2018 (12) |
| LHCGR | c.846dupT | p.R283* | Nonsense | Homozygous | AR | Chen et al. 2018 (12) |
| LHCGR | c.T32C | p.Leu11Pro | Missense | Homozygous | AR | Zhang et al. 2020 (13) |
| LHCGR | c.384-2A > T; c.32_58dupTGAAGCTGCTGCTGCTG CTGCAGCCGC | p.Leu11_Pro19dup |
Splicing; in-frame insertion |
Compound heterozygous | AR | Zhang et al 2020 (13) |
| ZP1 | c.170_174delGCCAG; c.1169_1176delTTTTCCCA |
p.G57Dfs*9; p.I390Tfs*16 |
Frameshift | Compound heterozygous | AR | Sun et al. 2019 (16) |
| ZP1 | c.1228C > T | p.Arg410Trp | Missense | Homozygous | AR | Zhou et al. 2019 (18) |
| ZP1 |
c.181C > T; c.1169-1176del |
p.Arg61Cys; p.Ile390Thrfs*16 | Missense; frameshift | Compound heterozygous | AR | Yuan et al. 2019 (19) |
| ZP1 | c.1129-1130del | p.Val377LeufsTer5 | Frameshift | Homozygous | AR | Dai et al. 2019 (15) |
| ZP1 | c.508del; c.1573-2A > G | p.His170IlefsTer52;NA | Frameshift; splicing | Compound heterozygous | AR | Dai et al. 2019 (15) |
| ZP1 |
c.123C > A; c.1663C > T |
p.Tyr41Ter; p.Arg555Ter |
Nonsense | Compound heterozygous | AR | Dai et al. 2019 (15) |
| ZP1 | c.1510C > T | p.Arg504Ter | Nonsense | Homozygous | AR | Dai et al. 2019 (15) |
| ZP1 | c.1014 + 1G > A | NA | Splicing | Homozygous | AR | Dai et al. 2019 (15) |
| ZP1 |
c.507delC; c.239 G > A; c.241 T > C |
p. His170fs; p. Cys80Tyr; p. Tyr81His |
Frameshift; missense | Compound Heterozygous | AR | Luo et al. 2020 (17) |
| ZP1 | c.769 C > T | p. Q257* | Nonsense | Homozygous | AR | Xu et al. 2020 (21) |
| ZP1 |
c.2 T > A; c.1112 + 1G > T |
p. M1K; p. Val339Aspfs*11 |
Missense; splice site | Compound heterozygous | AR | Liu et al. 2020 (20) |
| ZP1 | c.199G > T | p.Glu67Ter | Nonsense | Homozygous | AR | This study |
| ZP3 | c.400 G > A | p.Ala134Thr | Missense | Heterozygous | AD | Chen et al. 2017 (14) |
aAR autosomal recessive, AD autosomal dominant
The etiology of GEFS has been believed to include ovarian aging, dysfunctional folliculogenesis, or genetic defects [9, 10]. Since many GEFS cases cannot be explained by poor ovarian response, there is a growing interest in genetic evidence and the cause of GEFS. To date, pathogenic variants in three genes have been reported as causative factors for EFS: LHCGR [9, 11–13], ZP1[15–21], and ZP3 [14]. The phenotypes between mutations in the LHCGR and ZP genes present differently. Neither COCs nor oocytes could be identified in LHCGR mutation patients, while COCs could be obtained but only degenerated oocytes or no oocytes could be identified in ZP mutation patients. Our case falls into the latter category. The mechanisms behind these two phenotypes differ depending on the particular mutated gene. LHCGR mutations may impair signaling pathways of LH/hCG receptors, resulting in the tight adherence of COCs to the follicular wall. ZP mutations may inhibit the formation of the ZP, disrupt connections between oocytes and granulosa cells, and result in oocyte degeneration and EFS.
To date, eight studies have reported on 11 mutations in ZP1 and 1 mutation in ZP3 in EFS patients [14–21]. These ZP-related EFS cases shared commonalities with our study, including the majority of the retrieved COCs being empty, and few COCs containing degenerated oocyte cell masses. In addition, none of these cases obtained any intact oocytes, with or without a ZP. Technically, the definition of EFS is “no oocytes” retrieved [1]. However, ZP-related EFS cases can obtain degenerated oocytes [14–21], making the EFS definition obscure. Thus, we propose standardizing the definition of ZP-related EFS as a subtype of EFS characterized by degeneration. It is worth noting that we came upon an early study reporting on ZP mutation cases describing the retrieval of degenerated oocytes and abnormal eggs [35], but as no morphological images were provided in the paper, we could not draw any definitive conclusions from the study about EFS. A precise definition of ZP-related EFS is clearly vitally important for research classification and an accurate diagnosis.
The zona pellucida is the glycoprotein matrix that surrounds the oocyte and is crucial in oogenesis, fertilization and early embryo implantation. The ZP supports oocyte growth and development in mammals [36, 37]. It also has the function of specifically recognizing sperm-egg binding, inducing exocytosis and acrosome reaction of sperm, and preventing multiple sperm fertilization [22, 38, 39]. The glycoprotein ZP1 plays an indispensable role in the formation of the ZP by crosslinking the filament chains containing ZP2-4 [40, 41]. The ZP1 protein contains the N-terminal ZP-N1 domain, but the function of this domain was previously unknown. A recent study revealed that the N-terminal ZP1-N1 domain formed cross-links, which are essential for the assembly and maintenance of the human ZP [42].
In our study, we identified the homozygous nonsense ZP1 mutation (c.199G > T [p.Glu67Ter]) that causes a premature stop codon and leads to a truncated ZP1 of 67 amino acids at the ZP-N1 domain. The truncated ZP1 was predicted to comprise only the N-terminal signal domain and a small fragment of ZP-N1 domain, resulting in the total loss of ZP1 function. We speculate that the defects in the ZP-N1 domain hindered the formation of ZP crosslinks and in turn impaired ZP assembly. Three other ZP1 mutations (c.507delC, c.239G > A, c.241 T > C; c.123C > A, c.1663C > T; c.170_174delGCCAG, c.1169_1176delTTTTCCCA) have also been shown to cause truncated proteins at the N1 domain resulting in EFS. However, these mutation occurred in one allele with the compound heterozygous genotype [15–17]. In contrast, the mutation we examined occurred in two alleles with the homozygous genotype. In addition, in our case, the mutant ZP1 was inferred to be missing the C-terminal domain as well, which would in turn affect cleavage of C-terminal domain, transmembrane localization, and the transportation of proteins inside and outside the ovum [43]. Release of ZP1 proteins to the extracelluar space would be inhibited as well. Thus, we postulate that this homozygous nonsense mutation not only impairs ZP cross-linking but also damages ZP1 secretion, causing the failure of ZP filaments to interconnect communication between the oocyte and cumulus cells, resulting in oocyte degeneration and EFS [44]. In line with the results of our immunofluorescence analysis, the proband’s oocyte presented as an abnormally shaped degenerated cell mass with a disassembled nucleus. Additionally, the mutant ZP1 was spread diffusely throughout the abnormal oocyte. In contrast, ZP1 in the control was concentrated in the ZP surrounding the oocyte. This suggests mutant ZP was trapped inside the cell, impeding the secretion and formation of the ZP. The discovery of this N-terminal homozygous nonsense mutation of ZP1 further confirms the importance of ZP1 function on oocytes growth and development. An in-depth study of the mechanism will require further investigation.
Previous studies showed that ZP gene mutations are associated with oocyte defects and can cause female infertility and EFS [14–21, 35, 43, 45–50]. To date, a total of 29 ZP gene mutations have been identified as a causative factor of oocyte defects, of which ZP1 accounts for 20 (list in Supplemental Table 2). The phenotypes of ZP gene mutations are diverse, ranging from a thin or absent ZP to the most severe manifestations of EFS, like with our case. Understanding the phenotype and genotype relationship of ZP mutations will guide us in developing an optimal treatment plan for patients. According to previous literatures, the phenotypes exhibited dosage-dependent mutation effects, validated both in humans and mice [45, 46]. Specifically, the severity of clinical manifestations depends on what residual function remains in the mutant ZP protein and the complementary effects of one complete allele in heterozygous patients. As reported in literature, 2 cases with missense ZP mutations (ZP1, p.Arg109His; ZP2, p.Cys372Ser) and 1 case with compound heterozygous mutation (ZP1, p.D367G; p.L405fs) could obtain several mature ZP-free oocytes after repeated IVF attempts, resulting in a pregnancy [18, 45, 47]. Therefore, ZP gene sequencing could be a beneficial procedure for patients with abnormal oocytes and EFS, and the treatment plan of the following IVF attempt should take into account the severity of the phenotype and the specific ZP mutation mode. For patients with the most severe phenotypes of EFS caused by ZP gene mutations, including those with EFS carrying the mutation identified in our study, the current optimal treatment choice would be egg donation. Further approach may involve culturing early-stage oocytes in vitro to maturity before they can degenerate. However, this treatment requires further study.
In this study we identified a novel homozygous nonsense ZP1 mutation that causes human EFS. By utilizing pedigree analysis, genetic analysis and an in vitro study of the degenerated oocyte, our study is highly thorough. We also proposed a standardized definition of ZP associated EFS to clearly differentiate the condition from other subtypes of EFS. Our findings will assist physicians in more accurately diagnosing ZP associated EFS patients. Meanwhile, we comprehensively summarized the potential genetic etiology of EFS and the phenotypes and genotypes of the ZP gene mutations. However, this study only reported on one case of ZP associated EFS. More cases may lead to additional discoveries.
Conclusions
We identified a novel homozygous mutation in the ZP1 gene in an EFS patient. Genetic analysis suggests that this ZP1 mutation may cause the truncated ZP1 protein at the N1 domain and prevent ZP assembly and secretion, resulting in degenerated oocytes and the clinical manifestation of EFS. Together with previously reported EFS-related genetic defects, this case expands the genetic spectrum for EFS and highlights the importance of screening for genetic mutations in EFS patients. Furthermore, we propose to unambiguously distinguish ZP-related EFS from other types of EFS and formulate appropriate treatment strategies based on the specific phenotype and genotype of the ZP gene mutation. Further studies are warranted to investigate more genetic factors of EFS and the corresponding molecular mechanism behind the phenotypes.
Supplementary information
{kind=link}
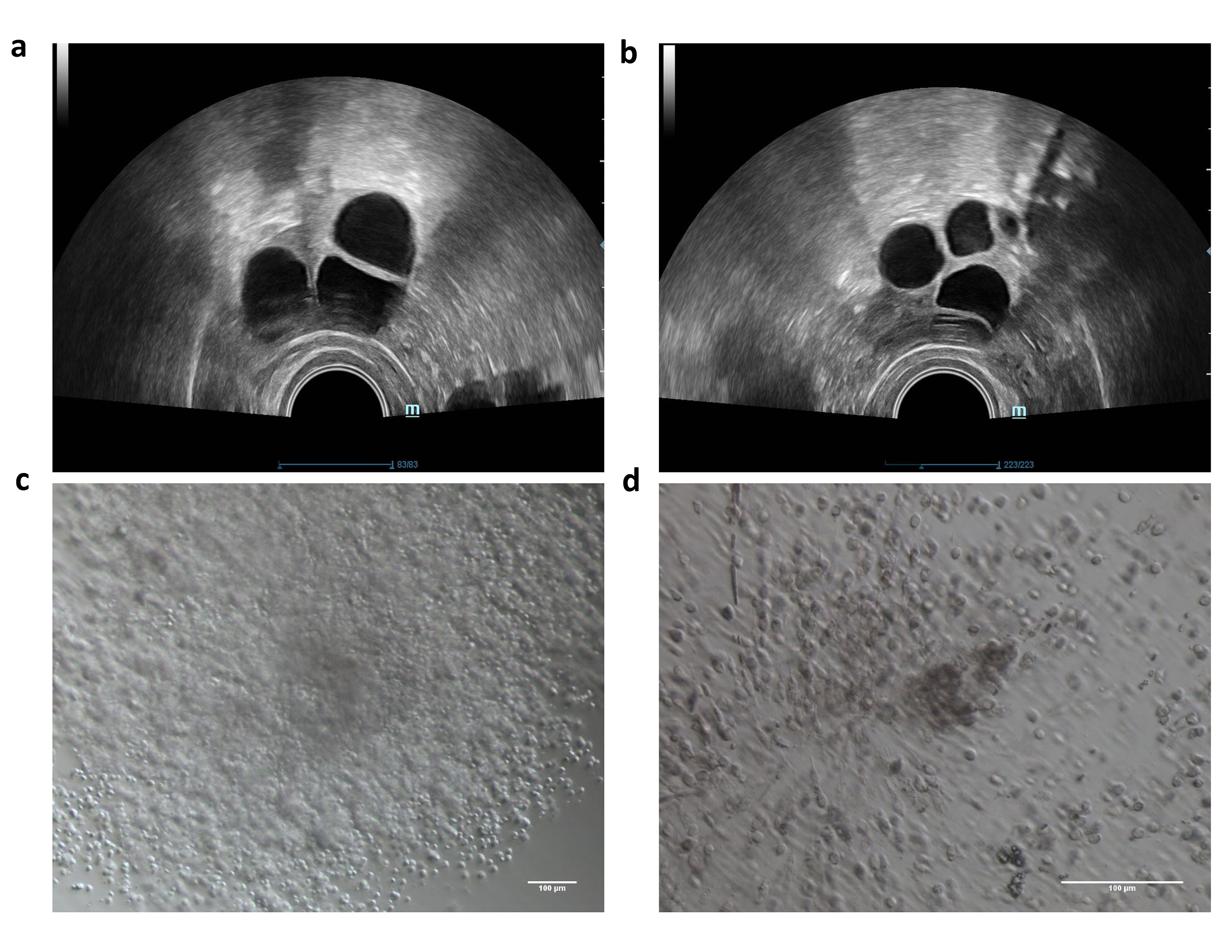
Clinical characterization of the proband. (a,b) Image of bilateral follicles under vaginal ultrasound on Human Chorionic Gonadotropin (HCG) day. (c,d) Empty cumulus oocyte complexes from the proband. (PNG 6295 kb).
(DOCX 27 kb).
Acknowledgements
We acknowledge Anthony Liu for his English edits. The authors thank the patients and their family members for participating in this study.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFC1001004, 2016YFC1000603) and National Nature and Science Foundation of China (81571403, 81401267, 81971374).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declarations
Ethics approval
The ethics committee of the First Affiliated Hospital of Nanjing Medical University approved this study. This study was performed in line with the principles of the Declaration of Helsinki.
Consent to participate
We obtained informed consent from all participants included in the study.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jing Wang and Xiaoyu Yang contributed equally to this work.
References
- 1.Beck-Fruchter R, Weiss A, Lavee M, Geslevich Y, Shalev E. Empty follicle syndrome: successful treatment in a recurrent case and review of the literature. Hum Reprod. 2012;27:1357–1367. doi: 10.1093/humrep/des037. [DOI] [PubMed] [Google Scholar]
- 2.Coulam CB, Bustillo M, Schulman JD. Empty follicle syndrome. Fertil Steril. 1986;46:1153–1155. doi: 10.1016/s0015-0282(16)49898-5. [DOI] [PubMed] [Google Scholar]
- 3.Stevenson TL, Lashen H. Empty follicle syndrome: the reality of a controversial syndrome, a systematic review. Fertil Steril. 2008;90:691–698. doi: 10.1016/j.fertnstert.2007.07.1312. [DOI] [PubMed] [Google Scholar]
- 4.Işik AZ, Vicdan K. Borderline form of empty follicle syndrome: Is it really an entity? Eur J Obstet Gynecol Reprod Biol. 2000;88:213–215. doi: 10.1016/s0301-2115(99)00152-9. [DOI] [PubMed] [Google Scholar]
- 5.Nikolettos N, Asimakopoulos B, Simopoulou M, Al-Hasani S. A borderline form of empty follicle syndrome. Case report. Clin Exp Obstet Gynecol. 2004;31:79–80. [PubMed] [Google Scholar]
- 6.Cao XL, Cao XL, Sun ZG. Borderline form of empty follicle syndrome treated with a novel dual trigger method combined with delayed oocyte retrieval: a case report. World J Clin Cases. 2020;8:825–830. doi: 10.12998/wjcc.v8.i4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song J, Sun Z. A borderline form of empty follicle syndrome treated with a double-trigger of gonadotropinreleasing hormone agonist and human chorionic gonadotropin: a case report. Medicine (Baltimore) 2019;98:213–215. doi: 10.1097/MD.0000000000016213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mesen TB, Yu B, Richter KS, Widra E, DeCherney AH, Segars JH. The prevalence of genuine empty follicle syndrome. Fertil Steril. 2011;96:1375–1377. doi: 10.1016/j.fertnstert.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yariz KO, Walsh T, Uzak A, Spiliopoulos M, Duman D, Onalan G, King MC, Tekin M. Inherited mutation of the luteinizing hormone/choriogonadotropin receptor (LHCGR) in empty follicle syndrome. Fertil Steril. 2011;96:e125–e130. doi: 10.1016/j.fertnstert.2011.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zreik TG, Garcia-Velasco JA, Vergara TM, Arici A, Olive D, Jones EE. Empty follicle syndrome: evidence for recurrence. Hum Reprod. 2000;15:999–1002. doi: 10.1093/humrep/15.5.999. [DOI] [PubMed] [Google Scholar]
- 11.Yuan P, He Z, Zheng L, Wang W, Li Y, Zhao H, Zhang VW, Zhang Q, Yang D. Genetic evidence of genuine’ empty follicle syndrome: a novel effective mutation in the LHCGR gene and review of the literature. Hum Reprod. 2017;32:944–953. doi: 10.1093/humrep/dex015. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Xu X, Kong L, Li P, Zhou F, Zhao S, Xin X, Tan J, Zhang X. Novel homozygous nonsense mutations in LHCGR lead to empty follicle syndrome and 46, XY disorder of sex development. Hum Reprod. 2018;33:1364–1369. doi: 10.1093/humrep/dey215. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Wu L, Diao F, Chen B, Fu J, Mao X, Yan Z, Li B, Mu J, Zhou Z, Wang W, Zhao L, Dong J, Zeng Y, du J, Kuang Y, Sun X, He L, Sang Q, Wang L. Novel mutations in LHCGR (luteinizing hormone/choriogonadotropin receptor): expanding the spectrum of mutations responsible for human empty follicle syndrome. J Assist Reprod Genet. 2020;37:2861–2868. doi: 10.1007/s10815-020-01931-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen T, Bian Y, Liu X, Zhao S, Wu K, Yan L, Li M, Yang Z, Liu H, Zhao H, Chen ZJ. A recurrent missense mutation in ZP3 causes empty follicle syndrome and female infertility. Am J Hum Genet. 2017;101:459–465. doi: 10.1016/j.ajhg.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai C, Chen Y, Hu L, Du J, Gong F, Dai J, et al. ZP1 mutations are associated with empty follicle syndrome: evidence for the existence of an intact oocyte and a zona pellucida in follicles up to the early antral stage. A case report. Hum Reprod. 2019;34:2201–2207. doi: 10.1093/humrep/dez174. [DOI] [PubMed] [Google Scholar]
- 16.Sun L, Fang X, Chen Z, Zhang H, Zhang Z, Zhou P, Xue T, Peng X, Zhu Q, Yin M, Liu C, Deng Y, Hu H, Li N. Compound heterozygous ZP1 mutations cause empty follicle syndrome in infertile sisters. Hum Mutat. 2019;40:2001–2006. doi: 10.1002/humu.23864. [DOI] [PubMed] [Google Scholar]
- 17.Luo G, Zhu L, Liu Z, Yang X, Xi Q, Li Z, Duan J, Jin L, Zhang X. Novel mutations in ZP1 and ZP2 cause primary infertility due to empty follicle syndrome and abnormal zona pellucida. J Assist Reprod Genet. 2020;37:2853–2860. doi: 10.1007/s10815-020-01926-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Z, Ni C, Wu L, Chen B, Xu Y, Zhang Z, Mu J, Li B, Yan Z, Fu J, Wang W, Zhao L, Dong J, Sun X, Kuang Y, Sang Q, Wang L. Novel mutations in ZP1, ZP2, and ZP3 cause female infertility due to abnormal zona pellucida formation. Hum Genet. 2019;138:327–337. doi: 10.1007/s00439-019-01990-1. [DOI] [PubMed] [Google Scholar]
- 19.Yuan P, Li R, Li D, Zheng L, Ou S, Zhao H, Zhang Q, Wang W. Novel mutation in the ZP1 gene and clinical implications. J Assist Reprod Genet. 2019;36:741–747. doi: 10.1007/s10815-019-01404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu M, Shen Y, Zhang X, Wang X, Li D, Wang Y. Novel biallelic loss-of-function variants in ZP1 identified in an infertile female with empty follicle syndrome. J Assist Reprod Genet. 2020;37:2151–2157. doi: 10.1007/s10815-020-01855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Q, Zhu X, Maqsood M, Li W, Tong X, Kong S, et al. A novel homozygous nonsense ZP1 variant causes human female infertility associated with empty follicle syndrome (EFS) Mol Genet Genomic Med. 2020;8:1–8. doi: 10.1002/mgg3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wassarman PM. Zona pellucida glycoproteins. J Biol Chem. 2008;283:24285–24289. doi: 10.1074/jbc.R800027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matzuk MM, Burns KH, Viveiros MM, Eppig JJ. Intercellular communication in the mammalian ovary: oocytes carry the conversation. Science. 2002;296:2178–2180. doi: 10.1126/science.1071965. [DOI] [PubMed] [Google Scholar]
- 24.Wassarman PM, Jovine L, Litscher ES. A profile of fertilization in mammals. Nat Cell Biol. 2001;3:E59–E64. doi: 10.1038/35055178. [DOI] [PubMed] [Google Scholar]
- 25.Conner SJ, Lefièvre L, Hughes DC, Barratt CLR. Cracking the egg: increased complexity in the zona pellucida. Hum Reprod. 2005;20:1148–1152. doi: 10.1093/humrep/deh835. [DOI] [PubMed] [Google Scholar]
- 26.World Health Organization . Laboratory manual for the examination and processing of human semen. Cambridge: Cambridge Univ. Press; 2010. [Google Scholar]
- 27.Lu X, Hong Q, Sun LH, Chen Q, Fu Y, Ai A, Lyu Q, Kuang Y. Dual trigger for final oocyte maturation improves the oocyte retrieval rate of suboptimal responders to gonadotropin-releasing hormone agonist. Fertil Steril. 2016;106:1356–1362. doi: 10.1016/j.fertnstert.2016.07.1068. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:1–7. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 33.Brandt T, Sack LM, Arjona D, Tan D, Mei H, Cui H, Gao H, Bean LJH, Ankala A, del Gaudio D, Knight Johnson A, Vincent LM, Reavey C, Lai A, Richard G, Meck JM. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet Med. 2020;22:336–344. doi: 10.1038/s41436-019-0655-2. [DOI] [PubMed] [Google Scholar]
- 34.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang P, Luan X, Peng Y, Chen T, Su S, Zhang C, Wang Z, Cheng L, Zhang X, Wang Y, Chen ZJ, Zhao H. Novel zona pellucida gene variants identified in patients with oocyte anomalies. Fertil Steril. 2017;107:1364–1369. doi: 10.1016/j.fertnstert.2017.03.029. [DOI] [PubMed] [Google Scholar]
- 36.Familiari G, Relucenti M, Heyn R, Micara G, Correr S. Three-dimensional structure of the zona pellucida at ovulation. Microsc Res Tech. 2006;69:415–426. doi: 10.1002/jemt.20301. [DOI] [PubMed] [Google Scholar]
- 37.Wassarman PM, Litscher ES. Biogenesis of the Mouse Egg’s Extracellular Coat, the Zona Pellucida. Curr Top Dev Biol. 2013;102: 243–66. [DOI] [PubMed]
- 38.Bleil JD, Greve JM, Wassarman PM. Identification of a secondary sperm receptor in the mouse egg zona pellucida: role in maintenance of binding of acrosome-reacted sperm to eggs. Dev Biol. 1988;128:376–385. doi: 10.1016/0012-1606(88)90299-0. [DOI] [PubMed] [Google Scholar]
- 39.Pang PC, Chiu PCN, Lee CL, Chang LY, Panico M, Morris HR, et al. Human sperm binding is mediated by the sialyl-lewisx Oligosaccharide on the zona pellucida. Science. 2011;333:1761–1764. doi: 10.1126/science.1207438. [DOI] [PubMed] [Google Scholar]
- 40.Bokhove M, Jovine L. Structure of zona pellucida module proteins. Curr Top Dev Biol. 2018;130:413–442. doi: 10.1016/bs.ctdb.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Louros NN, Chrysina ED, Baltatzis GE, Patsouris ES, Hamodrakas SJ, Iconomidou VA. A common “aggregation-prone” interface possibly participates in the self-assembly of human zona pellucida proteins. FEBS Lett. 2016;590:619–630. doi: 10.1002/1873-3468.12099. [DOI] [PubMed] [Google Scholar]
- 42.Nishimura K, Dioguardi E, Nishio S, Villa A, Han L, Matsuda T, Jovine L. Molecular basis of egg coat cross-linking sheds light on ZP1-associated female infertility. Nat Commun. 2019;10:3086. doi: 10.1038/s41467-019-10931-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H-L, Lv C, Zhao Y-C, Li W, He X-M, Li P, Sha AG, Tian X, Papasian CJ, Deng HW, Lu GX, Xiao HM. Mutant ZP1 in familial infertility. N Engl J Med. 2014;370:1220–1226. doi: 10.1056/NEJMoa1308851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilula NB, Epstein ML, Beers WH. Cell-to-cell communication and ovulation. A study of the cumulus-oocyte complex. J Cell Biol. 1978;78:58–75. doi: 10.1083/jcb.78.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao Q, Zhao C, Zhang X, Zhang H, Lu Q, Wang C, Hu Y, Ling X, Zhang J, Huo R. Heterozygous mutations in ZP1 and ZP3 cause formation disorder of ZP and female infertility in human. J Cell Mol Med. 2020;24:8557–8566. doi: 10.1111/jcmm.15482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu W, Li K, Bai D, Yin J, Tang Y, Chi F, Zhang L, Wang Y, Pan J, Liang S, Guo Y, Ruan J, Kou X, Zhao Y, Wang H, Chen J, Teng X, Gao S. Dosage effects of ZP2 and ZP3 heterozygous mutations cause human infertility. Hum Genet. 2017;136:975–985. doi: 10.1007/s00439-017-1822-7. [DOI] [PubMed] [Google Scholar]
- 47.Chu K, He Y, Wang L, Ji Y, Hao M, Pang W, Liu Y, Sun N, Yang F, Li W. Novel ZP1 pathogenic variants identified in an infertile patient and a successful live birth following ICSI treatment. Clin Genet. 2020;97:787–788. doi: 10.1111/cge.13693. [DOI] [PubMed] [Google Scholar]
- 48.Dai C, Hu L, Gong F, Tan Y, Cai S, Zhang S, Dai J, Lu C, Chen J, Chen Y, Lu G, du J, Lin G. ZP2 pathogenic variants cause in vitro fertilization failure and female infertility. Genet Med. 2019;21:431–440. doi: 10.1038/s41436-018-0064-y. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Shangguan T, Li YY, He W. Infertility due to lack of zona pellucida caused by a compound heterozygous mutation in ZP1 gene. Reprod Dev Med. 2018;2:183–186. [Google Scholar]
- 50.Okutman Ö, Demirel C, Tülek F, Pfister V, Büyük U, Muller J, et al. Homozygous splice site mutation in ZP1 causes familial oocyte maturation defect. Genes (Basel) 2020;11:1–11. doi: 10.3390/genes11040382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical characterization of the proband. (a,b) Image of bilateral follicles under vaginal ultrasound on Human Chorionic Gonadotropin (HCG) day. (c,d) Empty cumulus oocyte complexes from the proband. (PNG 6295 kb).
(DOCX 27 kb).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.