

Abstract
Background
Occurrence at a young age is known to be associated with unique clinical features in colorectal cancer (CRC). However, the genomic differences between young and old patients with CRC are not well elucidated and, to the best of our knowledge, have never been investigated in a Chinese population.
Methods
Tumor tissue samples from 29 young (age ≤50 years) and 46 old (age >50 years) patients with CRC were collected. Targeted sequencing of 808 cancer‐related genes was conducted to characterize the genomic landscape for Chinese CRC.
Results
Overall, mutational profiles exhibited notable differences between the two groups. In particular, APC and PIK3CA mutations were more frequently observed in old patients (p = 0.009 and p = 0.012, respectively), while SMAD4 mutations tended to occur in young patients (p = 0.054). Mutation loci distributions of KRAS in the young cohort differed from those in the old cohort, and a higher frequency of KRAS codon 12 mutations was potentially associated with a young age (p = 0.076). The frequencies of clinically actionable alterations were analyzed by defined age categories, which unveiled a distinctive targeted genomic profile in the young group. Furthermore, among patients with mismatch repair‐proficient (pMMR) CRC, tumor mutation burden (TMB) was positively correlated with age (Pearson's r = 0.306, p = 0.011), and genomic alterations associated with high TMB in young patients differentiated from those in old patients.
Conclusions
These findings revealed different molecular characterization between young and old Chinese patients with CRC, which may provide novel insights for the personalized treatment of CRC.
Keywords: Chinese, colorectal cancer, genomic landscape, old, young
This is the first NGS study to compare genomic profiles between young and old Chinese cohorts with CRC. The results demonstrated that young patients with CRC presented with some unique molecular features. Moreover, the comprehensive investigation of clinically actionable mutations and TMB will contribute to the improvement of personalized therapy and clinical management in CRC.

1. INTRODUCTION
Colorectal cancer (CRC) is the third most common type of cancer worldwide, with an estimated 1.8 million new cases and 800,000 deaths reported in 2018. 1 CRC is generally considered to be a disease that occurs in patients >50 years, and young patients only account for a small proportion of all the patients with CRC. 2 Although the incidence and mortality rate for patients >50 years of age have declined due to preventive screening, accumulating evidence indicates that CRC incidence is rising in the population of individuals <50 years of age. 3 , 4 The increasing prevalence of CRC among young individuals has led to a heavy burden on patients, their families, and society.
Patients with early‐onset CRC are more likely to exhibit distinctive biological behaviors, such as distal colon cancer and rectal cancer, low‐grade of differentiation, advanced stage, and development of metastatic disease. 5 , 6 , 7 , 8 Multiple studies have reported that patients diagnosed with CRC at <50 years old harbor a poor prognosis compared with old patients. 9 , 10 From the molecular perspective, the pathogenesis of early‐onset CRC is well characterized in patients with inherited CRC syndromes, in which some germline mutations of cancer susceptibility genes are identified. 11 , 12 , 13 , 14 , 15 However, hereditary CRC syndromes only account for a minority of young‐onset CRC cases, and knowledge regarding somatic mutation profiles in early‐onset CRC is limited. Therefore, a comprehensive molecular profile in young CRC patients should be defined, which will help enhance understanding of the unique biology and improve the treatment options for this population.
Recently, next‐generation sequencing (NGS) has led to considerable advances in the analysis of genomic alterations in cancer research and clinical application. 16 , 17 In order to better understand the disease biology and determine potentially distinct molecular features for young patients with CRC, the present study comprehensively investigated the molecular signatures from young and old Chinese patients with CRC by NGS, which may provide crucial knowledge regarding the genomics of this population.
2. MATERIALS AND METHODS
2.1. Patients and sample collection
A total of 75 patients with CRC were enrolled in the present study between September 2017 and May 2019, including 29 young patients (age ≤50 years) and 46 old patients (age >50 years). Tissue samples of these patients were collected for molecular testing. Eligible patients were determined based on the following inclusion criteria: i) Patients were diagnosed with CRC, and ii) ≥150 ng DNA from each tissue sample was successfully extracted.
2.2. DNA extraction and library construction
DNA was extracted from tissue samples using the QIAamp Genomic DNA kit (Qiagen GmbH). The quantification and quality of the DNA were assessed using Qubit 2.0 fluorimeter with ds DNA HS assay kit (Thermo Fisher Scientific, Inc.) and the Agilent 2100 BioAnalyzer (Agilent Technologies, Inc.). Sequencing libraries were prepared based on the Illumina standard library construction instructions (Illumina, Inc.).
2.3. NGS
The library was hybridized with a targeted panel, including 808 cancer‐related genes, which was enriched for the coding regions and selected introns of genes with known relevance to CRC. Subsequently, the target‐enriched library was sequenced on HiSeq2500 NGS platform (Illumina, Inc.). After the removal of the low‐quality sequencing data, reads were aligned to the human genome reference (hg19) using Burrows‐Wheeler Aligner (BWA, version 0.7.12). PCR duplicates were marked using the MarkDuplicates function in Picard tools (version 2.1.0). Base recalibration was conducted using GATK software (version 3.8). MuTect2 (version 1.1.7) was used to identify single nucleotide variant (SNV) and small insertion or deletion (INDEL), and ≥5 variant supporting reads were required for their filtering. All variants were annotated using ANNOVAR. Copy number variant calling was conducted using CONTRA software (version 2.0.8).
2.4. Statistical analysis
The data was analyzed using R (version 3.5.1) and GraphPad Prism (version 5; GraphPad Software, Inc.). Associations between gene mutation status and clinical characteristics were analyzed using Fisher's exact test or χ 2 test. Correlations between variables were assessed using Pearson's correlation coefficient. Differences in continuous variables were evaluated by Student's t‐test. A two‐sided p < 0.05 was considered as statistically significant.
3. RESULTS
3.1. Patient features
In the present study, a total of 29 young patients (age, ≤50 years) and 46 old patients (age, >50 years) diagnosed with CRC were enrolled. In the young cohort, the median age at diagnosis was 40 years (range, 16–50 years). In the old cohort, the median age at diagnosis was 65 years (range, 51–84 years). There was no difference in the clinical stage between the two groups. Further analysis demonstrated CRC was more likely to occur in the old male patients, although no statistically significant differences were observed (p = 0.082). The clinical and pathological characteristics of the patients are listed in Table 1.
TABLE 1.
Clinical characteristics of young and old patients with colorectal cancer
| Characteristics | Total (n = 75) | Young (n = 29) | Old (n = 46) | p value |
|---|---|---|---|---|
| Age, year, median (range) | 56 (16–84) | 40 (16–50) | 65 (51–84) | <0.001 |
| Gender, n (%) | ||||
| Male | 43 (57.3%) | 13 (44.8%) | 30 (65.2%) | 0.082 |
| Female | 32 (42.7%) | 16 (55.2%) | 16 (34.8%) | |
| T stage, n (%) | ||||
| 1 | 3 (4.0%) | 0 (0) | 3 (6.6%) | 0.131 |
| 2 | 2 (2.7%) | 2 (6.9%) | 0 (0) | |
| 3 | 42 (56.0%) | 19 (65.5) | 23 (50.0%) | |
| 4 | 20 (26.6%) | 6 (20.7%) | 14 (30.4%) | |
| Unknown | 8 (10.7%) | 2 (6.9%) | 6 (13.0%) | |
| N stage, n (%) | ||||
| 0 | 21 (28.0%) | 8 (27.6%) | 13 (28.3%) | 0.151 |
| 1 | 28 (37.3%) | 15 (51.7%) | 13 (28.3%) | |
| 2 | 18 (24.0%) | 4 (13.8%) | 14 (30.4%) | |
| Unknown | 8 (10.7%) | 2 (6.9%) | 6 (13.0%) | |
| M stage, n (%) | ||||
| 0 | 39 (52.0%) | 15 (51.7%) | 24 (52.2%) | 0.658 |
| 1 | 28 (37.3%) | 12 (41.4%) | 16 (34.8%) | |
| Unknown | 8 (10.7%) | 2 (6.9%) | 6 (13.0%) | |
| Clinical stage, n (%) | ||||
| I | 2 (2.7%) | 0 (0) | 2 (4.3%) | 0.565 |
| II | 13 (17.3%) | 4 (13.8%) | 9 (19.6%) | |
| III | 24 (32.0%) | 11 (37.9%) | 13 (28.3%) | |
| IV | 28 (37.3%) | 12 (41.4%) | 16 (34.8%) | |
| Unknown | 8 (10.7%) | 2 (6.9%) | 6 (13.0%) | |
| MMR status, n (%) | ||||
| pMMR | 68 (90.7%) | 26 (89.7%) | 42 (91.3%) | 0.811 |
| dMMR | 7 (9.3%) | 3 (10.3%) | 4 (8.7%) | |
3.2. Genomic profile of chinese CRC
Samples from all 75 patients were profiled by targeted sequencing, and all exhibited at least one genetic alteration. The most commonly mutated genes were TP53 (64%), APC (55%), KRAS (49%), PIK3CA (17%), and FBXW7 (13%) (Figure 1). Compared with TCGA data, a significantly higher number of somatic mutations were observed in CDKN1B (p = 0.029), and significantly fewer mutations in APC and AMER1 were identified among the Chinese CRC patient cohort (p = 0.002 and p = 0.030, respectively). Furthermore, potentially statistical differences were identified in the frequencies of PIK3CA and SOX9 mutations between TCGA and Chinese cohort (p = 0.060 and p = 0.087, respectively). Although the KRAS mutation rate was higher in the Chinese cohort compared with TCGA cohort, no significant difference was observed (p = 0.162) (Table S1).
FIGURE 1.
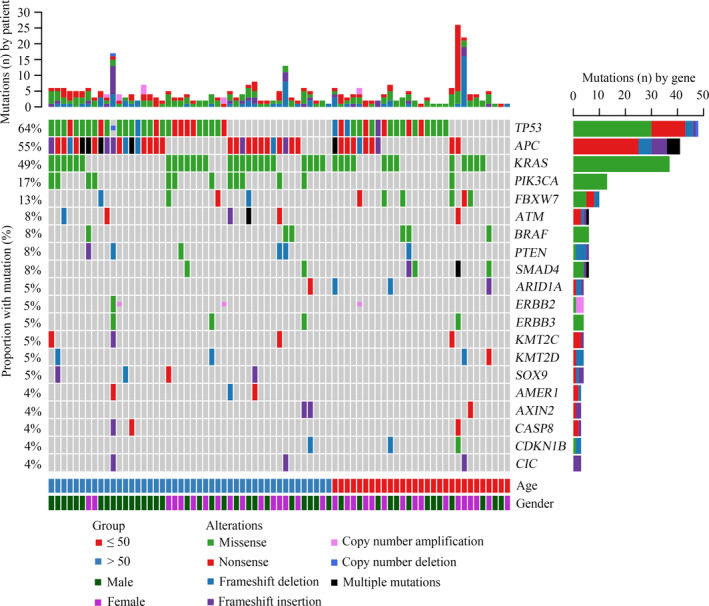
Landscape of genomic alterations among 75 Chinese patients with colorectal cancer (CRC). Genetic mutations were identified by targeted next‐generation sequencing of the tumor tissues of patients with CRC. Abbreviations: Multiple mutations, mutant numbers more than 2
Among all the patients with CRC, TP53, APC, and KRAS co‐mutations were observed in 10 patients (13.3%), and co‐alterations of TP53/APC, TP53/KRAS, and APC/KRAS were present in 27 (36.0%), 20 (26.7%) and 19 patients (25.3%), respectively. Evolutionary studies of CRC have shown that TP53, APC, and KRAS mutations are the most dominant clonal mutations 18 , 19 , 20 ; thus, it was hypothesized that there may be a correlation among the mutant allele frequencies of these genes in individual patients. As expected, a significant linear relationship was observed (Figure 2A). Further analysis demonstrated that the mutant types of TP53, APC, and KRAS demonstrated significant differences (Figure S1A), and TP53 missense mutations or indels tended to coexist with APC mutations (Figure S1B). According to the genomic profile, pairwise associations between somatic events in CRC were further investigated. Mutual exclusivity between mutations in KRAS and BRAF, and co‐mutations of PTEN and BRAF, PTEN and CIC, PIK3R1 and ERBB3, PIK3R1 and CAPS8, PIK3CA and KMT2C, and APC and ATM were observed (Figure 2B).
FIGURE 2.
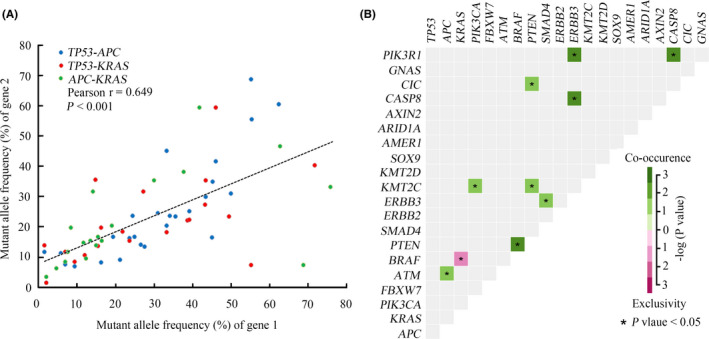
Comprehensive analysis of the associations between genomic mutations of CRC. A, Correlation analysis between mutant allele frequencies in TP53, APC, and KRAS. B, A pairwise association plot for mutant genes in CRC
3.3. Differences in mutation spectrums between young and old CRC groups
The genomic profiles of young and old groups were compared, and notable differences between them were observed. In the young group, the most commonly mutated genes were TP53 (66%), KRAS (41%), APC (34%), FBXW7 (21%), SMAD4 (14%), ARID1A (10%), and BRAF (10%) (Figure 3A). In the old group, the most frequently mutated genes were APC (67%), TP53 (63%), KRAS (54%), PIK3CA (26%), ATM (11%), PTEN (11%), FBXW7 (9%), and SOX9 (9%) (Figure 3B). Although TP53, APC, and KRAS were the most commonly mutated genes in both two groups, the mutation rates and ranking orders of them were different. Statistical analysis revealed that the frequencies of APC and PIK3CA mutations in the old group were significantly higher compared with that in the young group (p = 0.009 and p = 0.012, respectively) (Figure 4A,B). SMAD4 alterations were potentially associated with young age (p = 0.054) (Figure 4C). In addition, FBXW7 mutations were more likely to occur in young patients, although no statistically significant difference was observed (p = 0.137) (Figure S2A). Furthermore, the incidence of KRAS mutations between the two groups exhibited no significant difference (p = 0.274) (Figure S2B). The mutation loci distributions of KRAS were evaluated. In the young group, KRAS mutations were mainly distributed in codon 12 (Figure S3A). However, in the old group, KRAS mutations were distributed in different codons (Figure S3B). Further analysis indicated that the frequencies of the mutation loci distributions of KRAS between the young and old groups exhibited a potential statistical difference (p = 0.076) (Figure 4D).
FIGURE 3.
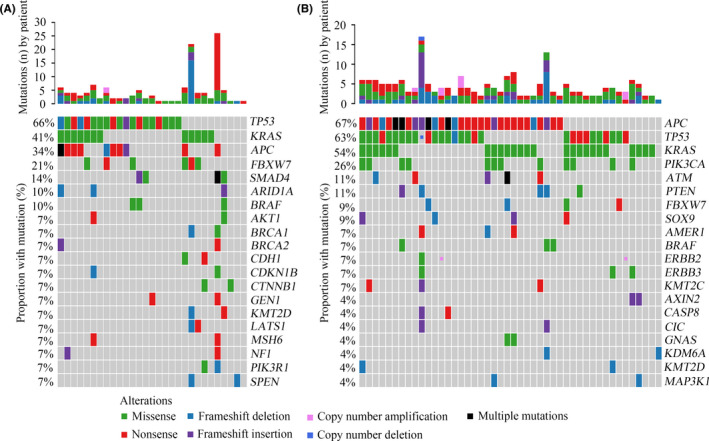
Genomic profiling between young and old patients with CRC. A, Genetic mutations in the young CRC group. B, Genetic mutations in the old CRC group
FIGURE 4.
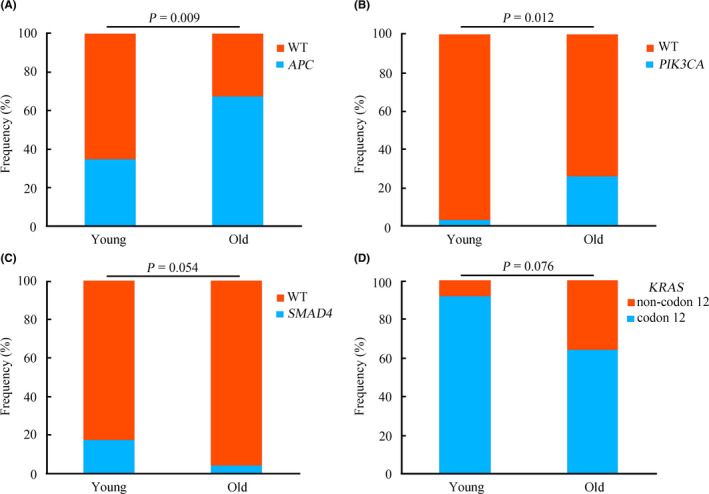
Distributions of representative genomic alterations between young and old groups with CRC. The comparative analysis of frequencies of APC (A), PIK3CA (B), SMAD4 (C), and KRAS (D) mutations between young and old patients. Abbreviations: WT, wild type
3.4. Profiles of clinically actionable mutations
In the present study, actionable alterations, defined as molecular targets for new drugs, as well as existing drugs that could guide treatment decisions for patients were evaluated. 21 , 22 Overall, 148 actionable mutations were detected from 58 patients (77.3%). Alterations in KRAS, PIK3CA, FBXW7, ATM, BRAF, and PTEN were the most common targets observed in the present study (Figure S4). Among all the patients, 69.0% of young cases harbored at least one actionable alteration. The most common actionable mutations in this group occurred in the following: KRAS (41%), FBXW7 (21%), ARID1A (10%), AKT1 (10%), and BRAF (10%) (Figure 5A). Additionally, 82.6% of old patients harbored at least one actionable alteration, and the most common actionable mutations occurred in the following: KRAS (54%), PIK3CA (26%), PTEN (11%), and ATM (11%) (Figure 5B). Based on all the actionable mutations identified, further analysis revealed a significant difference between the young and old groups regarding the targeted genomic profile (p = 0.008) (Figure 5C).
FIGURE 5.
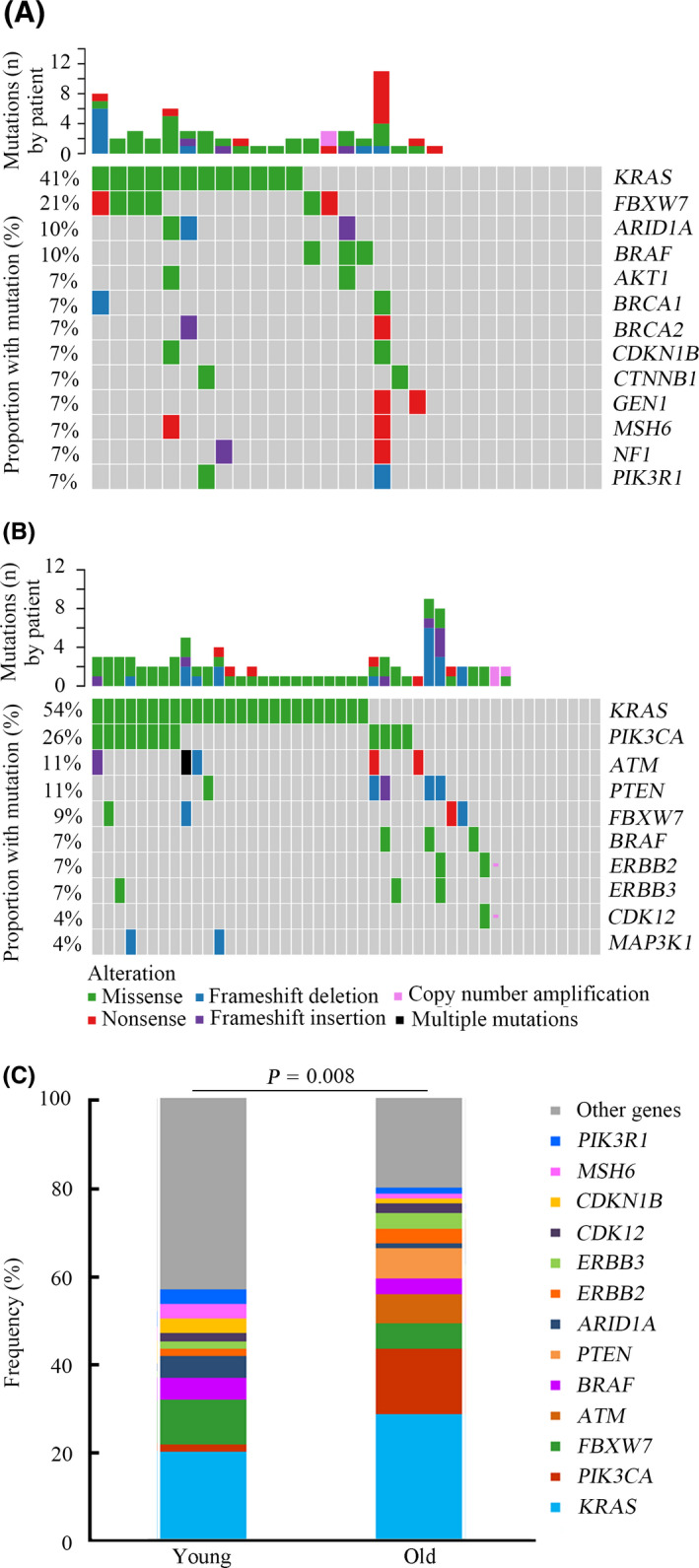
Overview of targeted genomic profiles between young and old groups with CRC. A, The targeted genomic profile in the young patients with CRC. B, The targeted genomic profile in the old patients with CRC. C, The comparative analysis of prevalence of targeted genomic alterations between the young and old patients
Pathway enrichment analysis was next performed according to the actionable mutations, to investigate functionally aberrant pathways. The most enriched pathways associated with actionable mutations in CRC included the MAPK (34.5%), PI3K (25.7%), and DDR (DNA damage repair) (18.9%) signaling pathways. Additionally, the ERBB, cell cycle, chromatin remodeling, and Wnt signaling pathways were also observed to be associated (Figure S5A). The frequencies of different pathways were then compared between the young and old groups, which revealed no significant difference between the two groups (p = 0.527) (Figure S5B). Notably, the proportions of mutated genes in the PI3K signaling pathway were significantly different between the two groups (p = 0.014) (Figure 6A). Previous work has reported that the DDR system comprises different pathways, including mismatch repair (MMR), base excision repair (BER), check point factors (CPF), Fanconi anemia (FA), homologous recombination repair (HRR), nucleotide excision repair (NER), nonhomologous end‐joining (NHEJ), and DNA translesion synthesis (TLS). 23 , 24 The composition of the DDR system between the two groups was then further analyzed, which demonstrated that HRR, MMR, CPF, and FA were the most common DDR pathways in CRC. Among these, HRR and MMR mainly occurred in the young group; however, CPF was more likely to occur in the old group (p = 0.022) (Figure 6B).
FIGURE 6.
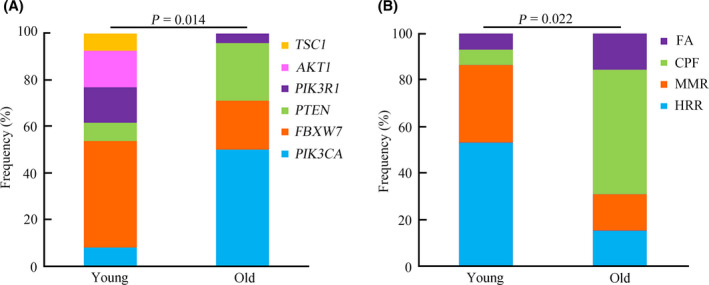
Comprehensive analysis of the composition of PI3K and DDR signaling pathways between young and old groups with CRC. A, Comparative analysis of the composition of the PI3K signaling pathway between young and old patients. B, Comparative analysis of the composition of the DDR signaling pathway between young and old patients
3.5. Tumor mutation burden analysis
Numerous studies have indicated that TMB is a potential biomarker to predict the efficacy of immune‐checkpoint inhibitors in various cancer types. 25 , 26 , 27 In advanced CRC, dMMR/MSI‐H is the standard marker for immunotherapy. Although previous work has reported dMMR/MSI‐H was dramatically correlated with a high TMB, 28 it is not known whether TMB is associated with other factors, except for dMMR/MSI‐H, in CRC. In the present study, the TMB was calculated using with the total number of nonsynonymous alterations (single‐nucleotide variants and small insertions or deletions) for each patient. 29 The TMB in patients with and without dMMR was then compared. As expected, patients with dMMR exhibited a significantly higher TMB compared with patients with pMMR (p = 0.002) (Figure S6). Among patients with pMMR, the association between TMB and age was investigated, and a significantly linear relationship was observed (Pearson r = 0.306, p = 0.011) (Figure 7A). Furthermore, the TMB in the old group was remarkably higher compared with that in the young group (p = 0.042) (Figure 7B). In CRC, the association between genomic mutations and TMB is not clear. Therefore, the present study further analyzed the influence of gene mutations on TMB among young and old patients with pMMR CRC. In this study, young patients with APC, KRAS, FBXW7, and ARID1A mutations were more likely to exhibit a high TMB (p < 0.05) (Figure 8A–D). In contrast, old patients with PIK3CA and ATM mutations harbored strikingly high TMB (p < 0.05) (Figure 8E, F).
FIGURE 7.
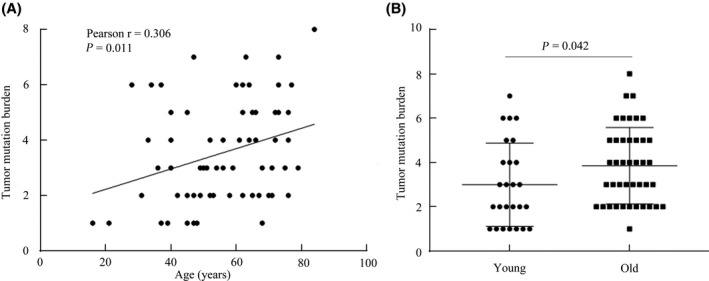
Association between TMB and age in CRC. A, Correlation analysis between TMB and age in pMMR patients with CRC. B, Comparison of TMB between young and old patients with CRC. Abbreviations: TMB, tumor mutation burden; pMMR, DNA mismatch repair‐proficient
FIGURE 8.
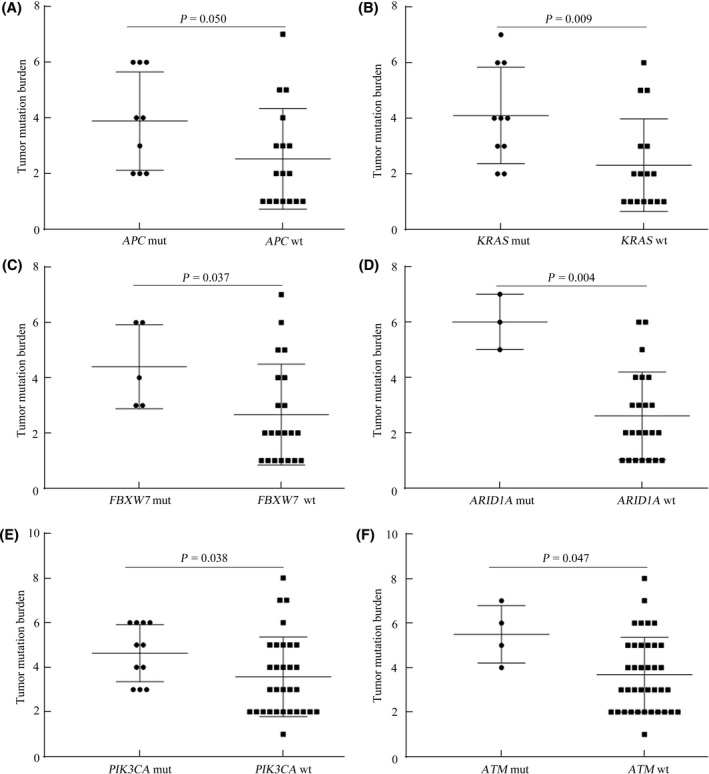
Relationship between TMB and certain specific gene mutations in pMMR patients with CRC. A‐D, Comparison of TMB between patients with and without APC, KRAS, FBXW7, and ARID1A in young patients with CRC. E and F, Comparison of TMB between patients with and without PIK3CA and ATM in old patients with CRC
4. DISCUSSION
An increasing incidence of CRC in young patients has attracted attention from the public, and caused a heavy burden on patients and societies. Although a number of studies have indicated that CRC in the young population harbors unique clinical features, 5 , 6 , 7 , 8 the genomic signatures of this population are not well understood. Based on the comprehensive analysis of young and old Chinese patients with CRC, the present study provides novel insights into the differences of genomic profiles between the two groups.
The present study observed the differences in the prevalence of several driver gene mutations between Chinese and TCGA cohorts, indicating a specific mutational spectrum in Chinese patients with CRC. The majority of CRC patients carried mutations in two genes of TP53/APC/KRAS, mutant allele frequencies of which displayed a significant linear correlation, confirming that mutations in these genes regularly co‑occur as clonal events in the evolution of CRC. The patterns of co‐mutation and mutual exclusivity were further explored, which indicated that the PI3K signaling pathway tends to co‐exist with another signaling pathway in CRC.
The genomic landscape in the young group exhibited notable differences from that of the old group. Significantly, a higher prevalence of APC and PIK3CA mutations were identified in old patients, which was consistent with a previous study. 30 Additionally, SMAD4 mutation was more likely to occur in the young group. Numerous studies reported that SMAD4 mutation tends to be a higher risk feature associated with poor prognosis in CRC. 31 , 32 , 33 According to previous studies, young patients with CRC exhibited a shorter survival time. 9 , 10 The present data provided a novel insight into the differences in prognosis between young and old CRC groups from a molecular perspective. A previous study has reported that KRAS mutations were more commonly observed in old patients with CRC. 30 In the current study, no differences in KRAS mutation frequencies between the two groups were observed, which may be attributed to the difference in race. However, based on the mutation loci distributions of KRAS, it was identified that young patients predominantly carried KRAS codon 12 mutations. By contrast, KRAS mutations in old patients tended to be distributed in different loci. A number of studies have demonstrated that mutations in KRAS codon 12 are associated with poor survival in CRC. 34 , 35 Based on the present results, the distribution of KRAS mutations may further help explain the difference in prognosis in young and old CRC patients.
Targeted therapies and immunotherapies have markedly improved the clinical outcome for certain patients with advanced CRC. However, limited understanding of CRC‐related molecular biomarkers and indicated limitations of certain anticancer drugs hinders the development of such therapies and corresponding clinical trial design. Therefore, comprehensive analysis of clinically actionable mutations that could potentially guide therapeutic decisions is essential to promote the treatment of patients with CRC. The present results revealed 77.3% of patients carried at least one clinically actionable alteration, which indicates the importance of comprehensive molecular testing and the necessity of “basket” clinical trials in which patients can enroll according to their actionable alterations. In addition, although no difference was observed in pathways related to actionable mutations between young and old groups, the molecular compositions of some pathways were different between them, further demonstrating the significance of comprehensive elaborate genetic testing and personalized therapy.
Currently, the clinical association of MSI‐H/dMMR has been highlighted by the fact that it is an effective predictive biomarker for the efficacy of immune checkpoint inhibitors. 36 , 37 In advanced CRC with MSI‐H/dMMR, given the encouraging effect of immunotherapies, PD‐1 inhibitors have been approved as first line therapy and beyond by the current National Comprehensive Cancer Network guidelines. However, MSI‐H only occurs in approximately 15% of CRC cases. 38 Therefore, there is an urgent need for additional biomarkers that can identify patients likely to respond to PD‐1/PD‐L1 inhibition. Multiple studies have revealed that the TMB is a potential biomarker to predict the response to immune‐checkpoint inhibitors. 25 , 26 , 27 Owing to the defect of the MMR system, MSI‐H/dMMR tumors tend to harbor a high TMB, whereas not every CRC with high TMB exhibits MSI‐H/dMMR signature. Therefore, a full understanding of TMB and its correlation with other factors is of great significance to guide personalized therapy and promote the clinical management of patients with CRC. Notably, the overall TMB in the pMMR CRC cohort increased with increasing age, and similar results have been observed in other cancer types. 39 Previous reports investigating the mutation profile within certain cancers have demonstrated a positive relationship between the age of cancer diagnosis and the mutant numbers, indicating that the mutation process may occur at a constant rate over time and eventually lead to an increased mutational burden. 40 , 41 , 42 Furthermore, the present study also comprehensively evaluated the correlation between TMB and specific genetic mutations in young and old patients with pMMR CRC. We found that somatic mutations associated with high TMB between the two groups showed some differences. For instance, APC and KRAS mutations were observed to be associated with high TMB in young patients, whereas PIK3CA mutations were associated with high TMB in old patients. Currently, no significant associations between these frequently mutated genes and high TMB were found in CRC. Additionally, ARID1A and ATM alterations classified as DDR mutations were related to high TMB in young and old patients, respectively. Although numerous studies have revealed the correlation between DDR alterations and high TMB in CRC, 43 , 44 our results further confirmed these findings and revealed different mutations in DDR genes associated with high TMB between young and old tumors. The findings in the present study recognized unique molecular features in future immune therapy trials for pMMR CRC, and highlighted the importance of personalized immunotherapy in this population. Therefore, these data may provide more possibilities for developing novel biomarkers that could predict the efficacy of immune checkpoint inhibitor therapies in CRC and further implied that the biomarkers need to be considered differently between young and old groups.
5. CONCLUSIONS
To the best of our knowledge, this is the first NGS study to compare genomic profiles between young and old Chinese cohorts with CRC. The results demonstrated that young patients with CRC presented with some unique molecular features. In addition, the comprehensive investigation of clinically actionable mutations and TMB will contribute to the improvement of personalized therapy and clinical management in CRC.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All participants provided written informed consent before sample collection. This study was approved by the ethical committee of the Affiliated Sir Run Run Shaw Hospital of Zhejiang University School of Medicine.
CONFLICT OF INTEREST
H.N.W, L.F, and S.B.C are employees of Acornmed Biotechnology Co., Ltd. The other authors have declared that no competing interest exists.
AUTHOR CONTRIBUTIONS
S.D, F.W, H.Q.C, and X.Z designed the study. L.S, B.J.B, and K.K.C enrolled the patients and collected the corresponding clinical information. H.N.W, L.F, and S.B.C conducted the data analysis. F.W, H.Q.C, and X.Z wrote the paper. Other authors discussed and commented on the manuscript. All authors read and approved the final manuscript.
Supporting information
Supplementary Material
Fei Wang, Huanqing Cheng, and Xiao Zhang contributed equally to the study.
Funding information
This study was supported by the National Natural Science Foundation of China (grants no. 81703076 and 82072628).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Schellerer VS, Merkel S, Schumann SC, et al. Despite aggressive histopathology survival is not impaired in young patients with colorectal cancer: CRC in patients under 50 years of age. Int J Colorectal Dis. 2012;27(1):71‐79. [DOI] [PubMed] [Google Scholar]
- 3. Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young‐onset colorectal cancer: a call to action. Mayo Clin Proc. 2014;89(2):216‐224. [DOI] [PubMed] [Google Scholar]
- 4. Murphy CC, Sandler RS, Sanoff HK, Yang YC, Lund JL, Baron JA. Decrease in incidence of colorectal cancer among individuals 50 years or older after recommendations for population‐based screening. Clin Gastroenterol Hepatol. 2017;15(6):903‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Connell JB, Maggard MA, Livingston EH, Yo CK. Colorectal cancer in the young. Am J Surg. 2004;187(3):343‐348. [DOI] [PubMed] [Google Scholar]
- 6. Siegel RL, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States. J Natl Cancer Inst. 2017;109(8):1974‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perea J, Alvaro E, Rodríguez Y, et al. Approach to early‐onset colorectal cancer: clinicopathological, familial, molecular and immunohistochemical characteristics. World J Gastroenterol. 2010;16(29):3697‐3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yantiss RK, Goodarzi M, Zhou XK, et al. Clinical, pathologic, and molecular features of early‐onset colorectal carcinoma. Am J Surg Pathol. 2009;33(4):572‐582. [DOI] [PubMed] [Google Scholar]
- 9. Lieu CH, Renfro LA, de Gramont A, et al. Association of age with survival in patients with metastatic colorectal cancer: analysis from the ARCAD Clinical Trials Program. J Clin Oncol. 2014;32(27):2975‐2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanford SD, Zhao F, Salsman JM, Chang VT, Wagner LI, Fisch MJ. Symptom burden among young adults with breast or colorectal cancer. Cancer. 2014;120(15):2255‐2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Losi L, Di Gregorio C, Pedroni M, et al. Molecular genetic alterations and clinical features in early‐onset colorectal carcinomas and their role for the recognition of hereditary cancer syndromes. Am J Gastroenterol. 2005;100(10):2280‐2287. [DOI] [PubMed] [Google Scholar]
- 12. Schofield L, Watson N, Grieu F, et al. Population‐based detection of Lynch syndrome in young colorectal cancer patients using microsatellite instability as the initial test. Int J Cancer. 2009;124(5):1097‐1102. [DOI] [PubMed] [Google Scholar]
- 13. Balaguer F, Castellví–Bel S, Castells A, et al. Identification of MYH mutation carriers in colorectal cancer: a multicenter, case‐control, population‐based study. Clin Gastroenterol Hepatol. 2007;5(3):379‐387. [DOI] [PubMed] [Google Scholar]
- 14. Pilarski R, Stephens JA, Noss R, Fisher JL, Prior TW. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan‐Riley‐Ruvalcaba syndrome clinical features. J Med Genet. 2011;48(8):505‐512. [DOI] [PubMed] [Google Scholar]
- 15. Farrington SM, Tenesa A, Barnetson R, et al. Germline susceptibility to colorectal cancer due to base‐excision repair gene defects. Am J Hum Genet. 2005;77(1):112‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shyr D, Liu Q. Next generation sequencing in cancer research and clinical application. Biol Proced Online. 2013;15(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gagan J, Van Allen EM. Next‐generation sequencing to guide cancer therapy. Genome Med. 2015;7(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim T‐M, An CH, Rhee J‐K, et al. Clonal origins and parallel evolution of regionally synchronous colorectal adenoma and carcinoma. Oncotarget. 2015;6(29):27725‐27735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braxton DR, Zhang R, Morrissette JD, Loaiza‐Bonilla A, Furth EE. Clinicopathogenomic analysis of mismatch repair proficient colorectal adenocarcinoma uncovers novel prognostic subgroups with differing patterns of genetic evolution. Int J Cancer. 2016;139(7):1546‐1556. [DOI] [PubMed] [Google Scholar]
- 20. Imperial R, Ahmed Z, Toor OM, et al. Comparative proteogenomic analysis of right‐sided colon cancer, left‐sided colon cancer and rectal cancer reveals distinct mutational profiles. Mol Cancer. 2018;17(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carr TH, McEwen R, Dougherty B, et al. Defining actionable mutations for oncology therapeutic development. Nat Rev Cancer. 2016;16(5):319‐329. [DOI] [PubMed] [Google Scholar]
- 22. Shen C, Meric‐Bernstam F, Su X, Mendelsohn J, Giordano S. Prevalence of actionable mutations and copy number alterations and the price of a genomic testing panel. Oncotarget. 2016;7(44):71686‐71695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z, Zhao J, Wang G, et al. Co‐mutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Cancer Res. 2018;78(22):6486‐6496. [DOI] [PubMed] [Google Scholar]
- 25. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hellmann MD, Ciuleanu T‐E, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093‐2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goodman AM, Sokol ES, Frampton GM, Lippman SM, Kurzrock R. Microsatellite‐stable tumors with high mutational burden benefit from immunotherapy. Cancer Immunol Res. 2019;7(10):1570‐1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hellmann MD, Nathanson T, Rizvi H, et al. Genomic features of response to combination immunotherapy in patients with advanced non‐small‐cell lung cancer. Cancer Cell. 2018;33(5):843‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lieu CH, Golemis EA, Serebriiskii IG, et al. Comprehensive genomic landscapes in early and later onset colorectal cancer. Clin Cancer Res. 2019;25(19):5852‐5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oyanagi H, Shimada Y, Nagahashi M, et al. SMAD4 alteration associates with invasive‐front pathological markers and poor prognosis in colorectal cancer. Histopathology. 2019;74(6):873‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wasserman I, Lee LH, Ogino S, et al. SMAD4 loss in colorectal cancer patients correlates with recurrence, loss of immune infiltrate, and chemoresistance. Clin Cancer Res. 2019;25(6):1948‐1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xourafas D, Mizuno T, Cloyd JM. The impact of somatic SMAD4 mutations in colorectal liver metastases. Chin Clin Oncol. 2019;8(5):52. [DOI] [PubMed] [Google Scholar]
- 34. Jones RP, Sutton PA, Evans JP, et al. Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br J Cancer. 2017;116(7):923‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bai B, Shan L, Xie B, et al. Mutations in KRAS codon 12 predict poor survival in Chinese patients with metastatic colorectal cancer. Oncol Lett. 2018;15(3):3161‐3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science. 2017;357(6349):409‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer‐the stable evidence. Nat Rev Clin Oncol. 2010;7(3):153‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pan X, Ji X, Zhang R, et al. Landscape of somatic mutations in gastric cancer assessed using next‐generation sequencing analysis. Oncol Lett. 2018;16(4):4863‐4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alexandrov LB, Jones PH, Wedge DC, et al. Clock‐like mutational processes in human somatic cells. Nat Genet. 2015;47(12):1402‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blokzijl F, de Ligt J, Jager M, et al. Tissue‐specific mutation accumulation in human adult stem cells during life. Nature. 2016;538(7624):260‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parikh AR, He Y, Hong TS, et al. Analysis of DNA damage response gene alterations and tumor mutational burden across 17,486 tubular gastrointestinal carcinomas: implications for therapy. Oncologist. 2019;24(10):1340‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Song Y, Huang J, Liang D, et al. DNA damage repair gene mutations are indicative of a favorable prognosis in colorectal cancer treated with immune checkpoint inhibitors. Front Oncol. 2021;10:549777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.