

SUMMARY
Female human pluripotent stem cells (hPSCs) routinely undergo inactive X (Xi) erosion. This progressive loss of key repressive features follows the loss of XIST expression, the long non-coding RNA driving X inactivation, and causes reactivation of silenced genes across the eroding X (Xe). To date, the sporadic and progressive nature of erosion has obscured its scale, dynamics, and key transition events. To address this problem, we perform an integrated analysis of DNA methylation (DNAme), chromatin accessibility, and gene expression across hundreds of hPSC samples. Differential DNAme orders female hPSCs across a trajectory from initiation to terminal Xi erosion. Our results identify a cis-regulatory element crucial for XIST expression, trace contiguously growing reactivated domains to a few euchromatic origins, and indicate that the late-stage Xe impairs DNAme genome-wide. Surprisingly, from this altered regulatory landscape emerge select features of naive pluripotency, suggesting that its link to X dosage may be partially conserved in human embryonic development.
Graphical abstract

In brief
Reactivation of the silenced X in human female iPSC/ESCs compromises their utility. Bansal et al. perform an integrated genomics analysis to reveal a prevalent X erosion trajectory that they validate in long-term culture. Starting with XIST loss, this trajectory indicates that reactivation may spread contiguously from escapees to silenced genes.
INTRODUCTION
Human pluripotent stem cells (hPSCs) of embryonic (ESC) or induced (iPSC) origin resemble the inner cell mass (ICM) of the early post-implantation embryo and have become a cornerstone for modeling human disease in vitro (Chamberlain, 2016). However, their tremendous potential for future allogenic or autologous cell replacement therapies (Attwood and Edel, 2019) is limited by challenges to their genetic (Laurent et al., 2011; Martins-Taylor and Xu, 2012; Merkle et al., 2017; Popp et al., 2018) and epigenetic stability (Bar and Benvenisty, 2019; Panopoulos et al., 2017; Perrera and Martello, 2019), specifically in regard to DNA methylation (DNAme) at imprinted and X-linked genes. Female-derived hPSCs in particular may be expected to lag behind male hPSCs in some applications due to progressive reactivation of their inactive X (Xi), a process termed Xi erosion (XIE) (Geens and Chuva De Sousa Lopes, 2017; Mekhoubad et al., 2012; Wutz, 2012).
In contrast to female hPSCs that reflect a late epiblast (or “primed”) cell state after completed X chromosome inactivation (XCI), female ESCs and iPSCs of the mouse approximate the ICM of the (“naive”) pre-implantation embryo, and harbor 2 active X (Xa) chromosomes (Khan et al., 2017). To transition from naive to primed pluripotency, mouse ESCs must silence all supernumerary X chromosomes to maintain only a single Xa per diploid genome (van Bemmel et al., 2016; Furlan and Rougeulle, 2016; Schulz et al., 2014). This obligate coupling of pluripotency state to X dosage has propelled most of our current understanding of random XCI (Payer, 2016; Pinter, 2016). However, recent studies of peri-implantation embryos across a range of mammals have shed light on important species-specific differences in XCI establishment (Chen et al., 2016b; Okamoto et al., 2011; Petropoulos et al., 2016; Ramos-Ibeas et al., 2019; Tachibana et al., 2012).
XIE in female hPSCs is an in vitro- and species-specific defect in XCI maintenance that is best understood as two mechanistically distinct observations. First, post-XCI mouse ESC-derived epiblast-like cells faithfully maintain Xist expression and gene silencing of the Xi (Chen et al., 2016b), whereas female primed hPSCs show a strong tendency to lose XIST expression (Shen et al., 2008; Silva et al., 2008). Second, even upon deletion of Xist, the vast majority of genes on the mouse Xi remain stably silenced on account of long-term and self-propagating DNAme of promoters and CpG islands (Csankovszki et al., 1999, 2001; Pasque et al., 2014), with some context-specific exceptions (Adrianse et al., 2018; Yildirim et al., 2013; Zhang et al., 2007). Moreover, even a brief period (<3 days) of mouse Xist expression is sufficient to irreversibly silence genes long-term (Wutz and Jaenisch, 2000). In contrast, female hPSCs that have lost XIST expression have been demonstrated to undergo frequent, progressive, and irreversible X reactivation (Anguera et al., 2012; Lengner et al., 2010; Mekhoubad et al., 2012; Tchieu et al., 2010; Tomoda et al., 2012; Vallot et al., 2015).
Despite the relevance of hPSCs to X-linked disease modeling and future cell replacement therapies, our mechanistic understanding of XIE has remained limited, with most studies confined to a small number of lines. As a result, descriptions of XIE have ranged from minor (Shen et al., 2008) to major (Nazor et al., 2012) to whole Xi reactivation (Mekhoubad et al., 2012), indicating that the progressive nature of this phenomenon may require analysis across a larger number of female hPSC lines.
To address this knowledge gap, we performed an integrated analysis of available DNAme, chromatin accessibility, and gene expression data across 421 hPSC samples. Differential DNAme across the eroding X (Xe) orders female hPSCs across a trajectory of XIE, which we replicate in a long-term continuous passaging experiment. We find that XIE spreads in contiguous fashion from a few euchromatic regions that escape XCI and that terminal XIE results in female-specific DNA hypomethylation that triggers some markers of naive pluripotency. Our analysis is relevant to understanding and staging the epigenetic fidelity of female hPSC disease models and reveals the impact of X chromosome dosage on embryonic stage-specific pluripotency.
RESULTS
Female hPSC samples trace a common path through XIE
To assemble a comprehensive view of X chromosome methylation dynamics in human pluripotency, we collated Illumina BeadChip (450K, MethylEpic) data from suitable hPSC studies in GEO (Banovich et al., 2018; Butcher et al., 2016; Garitaonandia et al., 2015; Nazor et al., 2012; Nishizawa et al., 2016; Salomonis et al., 2016; Takasawa et al., 2018; Zdravkovic et al., 2015). After removal of hPSC lines with incorrectly listed sex chromosomes (see Method details), the resulting superset spanned 471 hPSC lines (Figure S1A). Because X-linked DNAme (β values) in female hPSCs are averaged between Xi and Xa, probes in genes subject to XCI center around ~0.5 (Figure 1A). Male hPSC lines cluster together in principal-component analysis (PCA) (Figure 1B), whereas PC1 reflects the large range of average X chromosome DNAme of female hPSCs (Figures S1C and S1E). There was little to no association with the originating study, hESC/iPSC category, or hPSC identifier on either PC, except for an overrepresented cluster of the predominant H9 hESC model that skewed overall analysis and was removed (see Method details; Figures S1B and S1D). This balanced superset of 178 male and 221 remaining female hPSC lines revealed excess female-specific variance in X-linked probes, without sex differential across autosomal probes (Figures 1C and 1D). This confirms that XIE cannot result from overt sex differences in genome-wide DNAme (Nazor et al., 2012), and enables the identification of XIE-relevant probes on the basis of their significant excess variance across female hPSCs relative to male hPSCs (Figures S1F and S1G). Significantly varying probes (p ≤ 0.01, Brown-Forsythe test) on the X have a bimodal distribution that reflects genes subject to XCI (β value of ~0.5), and likely escapee genes that are hypomethylated on both Xa and Xi, with a wide range of probes between these 2 peaks (Figure S1H). Average X DNAme in female hPSCs correlates almost perfectly with the sex-variant probes, exceeding its correlations with non-variant X-linked probes or genome-wide DNAme levels (Figures S1I and S1J).
Figure 1. XIE reflected in high X-specific variance in female hPSCs.

(A) β values distribution of probes on the X in female (red) and male (blue) samples.
(B) Principal-component analysis (PCA) of female (red) and male (blue) samples using only X-linked probes with Brown-Forsythe p ≤ 0.01.
(C and D) Density plots of probe β variance on X and autosomes, respectively, with red lines for female samples, and blue for male samples (Kolmogorov-Smirnov [KS] distance indicated, with KS p values of 5.6e–15 and 0.78 for C and D, respectively).
(E) PCA of DMPs across female samples colored by their k-means assigned cluster (see Method details). Gray lines link different passages of the same cell line. Shaded areas represent the 95% confidence ellipse for each cluster.
(F and G) Projection of C19 and C23 iPSC validation samples, using PCs from (E), labeled by passage numbers, and colored as in (E).
(H) Fraction of DMPs demethylated to within 10% of expected DNAme loss captured in each passage of C19 and C23 iPSCs.
(I) DNAme heatmap of high variance X probes in female hPSCs. Samples within each k-means cluster (A–F) are ordered by mean X DNAme. Probes (rows) are grouped by transition (lowest q ≤ 0.05), and β value change (↑ up, ↓ down, or ~ fluctuating irrespective of transition). Cluster transitions are numbered (1–5). Cell lines with data from multiple passages are described below the heatmap, with arrows drawn between samples and the passage numbers marked.
(J) DNAme heatmap for C19 (dark gray) and C23 (light gray) iPSC samples, ordered by cluster and passage as annotated below heatmap.
To stage XIE progression, we used probes with high sex variance to perform K-means clustering of female samples, yielding 6 clusters (Figures 1E and S1K) ordered by their average X DNAme. We then identified all differentially methylated probes (DMPs) between neighboring clusters (Table S2). Despite the inclusion of both iPSCs and ESCs from 7 independent studies, each cluster comprised samples from a minimum of 5 studies (Figure S1L), and DNAme is remarkably similar for any given probe within each cluster, demonstrating that most reactivation events are not sporadic, but reproducible. Moreover, of 9 hPSC lines contributing to more than a single cluster, 8 revealed passage number increases consistent with this cluster order, suggesting that they reflect a common XIE timeline (Figures 1E and 1I). To validate this trajectory, we performed a continuous culture experiment over 24 weekly passages starting from 2 isogenic female euploid hPSC lines (C19 and C23), which expressed XIST at early passage (Figure S1M) and matched cluster A samples in X-linked DNAme (Figures 1F–1J). With increasing passages, our validation samples re-trace the described XIE trajectory, transitioning from cluster A via B to C, a span that covers 163/221 (74%) of the published hPSC samples (Figures 1F–1J). Importantly, DNAme loss in transition-1-associated DMPs precedes the demethylation of transition-2 DMPs (Figures 1H–1J), whereas nearly all transition-3, −4, and −5 DMPs maintain DNAme status. This pattern is consistent with our most eroded samples grouping with cluster C, and demonstrates that our described XIE trajectory follows time in culture.
Overall, we see a clear, systematic pattern of DNAme changes that illustrate stepwise XIE progression (Figure 1I), the degree of which is evident in the significant increase in the fraction of the inferred Xe allele (LAF, lesser allele fraction) across heterozygous X-linked variants in hPSC samples with RNA sequencing (RNA-seq) data (Figure S2A). Probes losing DNAme far outnumber probes gaining DNAme, with promoter-associated probes pre-dominating among demethylated DMPs (Figure S2B). Of 450 X-linked genes that are subject to XCI (promoter-associated β between 0.3 and 0.7 in cluster A), only 7 (1.6%) maintain their DNAme in cluster F (Table S2), suggesting that most genes subject to XCI can reactivate in XIE.
DNAme in XIST and the FIRRE tandem repeat rises as chromatin accessibility drops at XIE onset
With this progressive staging of XIE in hand, we parsed stepwise DNAme changes occurring at each transition between clusters A and F. Analysis of transition-1 (cluster A–B) reveals that DMPs reside in specific regions (Figure 2A, blue line) in a pattern that matches neither overall probe density (gray, 9,191 probes), nor the 1,412 probes that fail the DMP threshold (q > 0.05; Figure S2C). For transition-1, 8 DMPs stand out in degree and significance of increasing DNAme (Figures 2B and 2C), with 5 DMPs in XIST and 3 in the tandem repeat of FIRRE. Loss of XIST expression has been suggested as an early or causative event for XIE (Anguera et al., 2012; Mekhoubad et al., 2012; Shen et al., 2008; Silva et al., 2008; Vallot et al., 2015), but its mechanistic basis has remained unclear. Of the 9 probes spanning XIST, 5 change in transition-1, 2 of which are located in its core promoter (Hendrich et al., 1997). The other 3 DMPs are located in a likely enhancer in exon 1 (~1.2 kb from the XIST promoter), 2 of which directly overlap binding sites for the YY1 transcription factor (Figures 2C and 2F). The YY1 cognate motif is conserved across a range of mammalian XIST homologs, and YY1 is necessary for Xist expression in mouse ESCs (Makhlouf et al., 2014). YY1 binding is sensitive to DNAme (Kim et al., 2003), and in differentiated cells, only the silent XIST allele on the Xa features methylated YY1 cognate motifs (Chapman et al., 2014). As these same YY1 sites gain DNAme in transition-1, the major overlapping assay of transposase accessible chromatin (ATAC) peak is lost and XIST expression drops by >2 orders of magnitude (Figures 2F and 2D). Chromatin accessibility and DNAme of this CpGII/P2 element are therefore strongly anti-correlated (r = −0.91, p < 1e–6) and jointly reflective of XIST expression (Figure 2E). A small group of samples with intermediate XIST levels, assigned to cluster B due to early XIE changes (4/14), likely reflects mixed XIST+ and XIST− hPSC cultures (Kim et al., 2014; Vallot et al., 2015). Conversely, 2 outliers assigned to cluster A despite low XIST levels (2/13), only 1 of which maintained intermediate CpGII/P2 DNAme and accessibility (Figure 2E), may have been collected just before the onset of XIE.
Figure 2. Differential DNAme analysis on Xe distinguishes early from late XIE.

(A) Density plot of probes across X coordinates reveal transition 1 probes (1↓↑, blue) to concentrate irrespective of overall probe density (gray-shaded area), in contrast to fluctuating probes (~, black). KS distances quantify similarity to frequency distribution of the null probe density distribution (* KS test p value = 5.09e–11 for blue transition-1 probes).
(B) Volcano plot of transition-1 differentially methylated probes (DMPs), colored by X coordinate (legend above the plot). Top increasing DMPs annotated by gene name.
(C) DNAme of XIST and FIRRE probes across clusters A–F. Lines to (F) and (G) indicate the position of probes in relation to the genes and their annotated regions (* indicates DMPs from B).
(D) XIST and FIRRE expression (VST + batch-corrected/log2-transformed), respectively, for all samples with available RNA-seq data (all but cluster E).
(E) XIST CpGII/P2 ATAC peak accessibility score over DNAme (β) values, with symbol size indicating XIST expression (VST + batch-corrected/log2-transformed) across all A–F samples with all 3 datapoints.
(F and G) ATAC-seq coverage for XIST and FIRRE, respectively. Horizontal lines below coverage indicate called peaks (gray, black), including differential, closing peaks (red). Bottom panels of (F) and (G) relate DMPs and ATAC peaks to cis-regulatory sites for XIST and FIRRE, respectively.
Three other top-ranking DMPs increasing in DNAme map to the FIRRE locus, which is also the only X-linked gene outside XIST to lose ATAC peaks in transition-1 (Figures 2C and 2G). This evolutionarily conserved tandem repeat transcribes a long non-coding RNA (ncRNA) primarily from the Xa, and forms long-range CCCTC-binding factor (CTCF)-mediated chromatin loops with 2 other tandem repeats, DXZ4 and ICCE, on the Xi (Bansal et al., 2020; Fang et al., 2019; Froberg et al., 2018). The 3 FIRRE DMPs gaining DNAme reside inside the tandem repeat, characterized by closing ATAC peaks, while probes near its major promoter decrease in DNAme (Figures 2C and 2G). The tandem repeat DMPs of FIRRE correlate strongly (r = 0.94, p < 1e–6) with those in the CpGII/P2 site of XIST, whereas FIRRE expression increases significantly (Figure 2D). In summary, the tandem repeat DNAme and chromatin accessibility changes of FIRRE mirror the changes of XIST in transition-1 and suggest that this conserved ncRNA gene uniquely reflects early XIE.
Contiguous progression of XIE originates near escapee genes
While XIST-proximal genes appeared to remain refractory to the early loss of DNAme (Figure 2A), the genes near FIRRE (130 Mb, magenta), XACT and DXZ4 (110–115 Mb, blue/lavender) on the long arm of the X (Xq) and on the escapee-rich short arm (Xp), experience the earliest and most significant loss of DNAme in XIE. Tracking transition-specific significant DMPs (Figure 3A, colors) and cumulative DNAme changes (gray), we find that XIE appears to emanate from these multiple chromosomal regions, which also correlate significantly with those in transitions-1 and -2 of our validation set (p < 1.4e–49; Figure S2D). XIE then spreads in contiguous fashion across the intervening regions of the X chromosome during subsequent transitions (Figure 3A). This wave of DNAme loss is mirrored by progressive (~2-fold) gains in chromatin accessibility in the same regions (Figure 3B, inset). Curiously, DMPs and opening ATAC peaks specific to early transitions reside in H3K27me3-rich regions of the Xi, which are likely most sensitive to the loss of XIST RNA, while those of later transitions tend to fall in H3K9me3-rich regions. Such an association with specific chromatin compartments is observed in iPSCs and ESCs (Figure S2E) and best reflected in the progressive shift of DMP and differential ATAC peak density correlating with H3K27me3 levels to H3K9me3 in late XIE (Figures 3C and 3F). In summary, these DNAme and chromatin accessibility patterns strongly suggest that XIE is dictated by linear spreading of reactivation from multiple points along the chromosome.
Figure 3. Contiguous spread of XIE from euchromatic origins on the Xi.
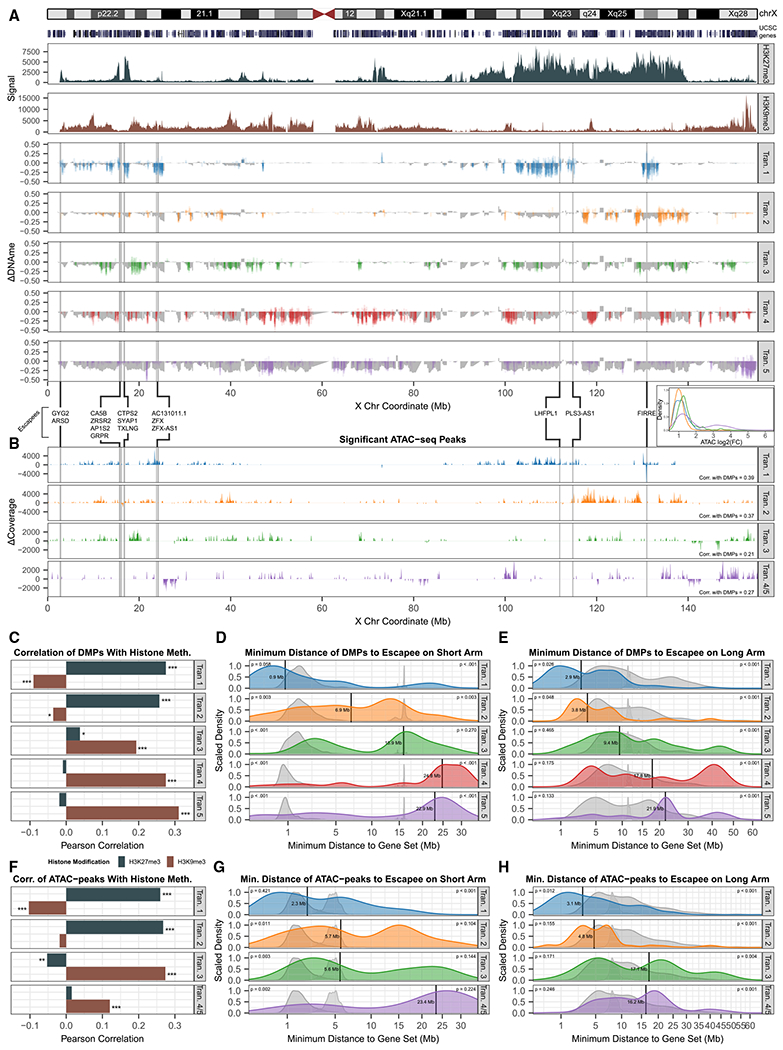
(A) Map of cumulative (gray) and transition-specific DMP (colors) changes in β values across the X plotted underneath Xi-specific H3K27me3 (dark teal) and H3K9me3 (brown) H9 chromatin immunoprecipitation sequencing (ChIP-seq) signal from Vallot et al. (2015). Gray vertical lines indicate escapee locations, determined from DNAme in cluster A, as in Cotton et al. (2015).
(B) Differential ATAC-seq coverage (peak width sum per 100-kb intervals), across increasing and decreasing ATAC peaks, and correlations with DMP map (A) for each transition on bottom right. Transitions 4 and 5 are combined (purple), due to lack of ATAC-seq data for cluster E. Histograms of log2 fold change (log2FC) of differential peaks for each transition are shown on the top right (inset).
(C) Pearson correlations of transition-specific DMP changes (colored in A) to H3K27me3 (dark teal) and H3K9me3 levels (brown).
(D and E) DMP distance distributions relative to escapee locations for each transition on Xp and Xq, respectively, with black vertical lines denoting median distance. Null distance distributions using permuted escapees (broad gray) or permuted DMPs (narrow gray) plotted underneath actual distributions. Calculated p values (two-tailed rank test against the permutated distributions) indicated on the top left (permuted escapees) and top right (permuted DMPs) of each plot.
(F) Pearson correlations of transition-specific ATAC-peaks (B) to H3K27me3 (dark teal) and H3K9me3 levels (brown).
(G and H) ATAC-peak distance distributions relative to escapee locations for each transition on Xp and Xq, respectively, with black vertical lines denoting median distance.
In tracing such contiguous XIE to its origins, we noticed that early DMPs and opening ATAC peaks map near genes known to escape XCI, on both the escapee-rich Xp and the Xq from 2 regions in particular (Figures 3A and 3B): (1) the ~5-Mb focal domain centered on FIRRE, and (2) the ~15-Mb wide domain to the left of the DXZ4 macrosatellite (near PLS3-AS1), previously shown to require DXZ4 for H3K27me3 maintenance (Darrow et al., 2016). To confirm and characterize this pattern quantitatively, we identified bona fide escapee genes in hPSC lines of cluster A using the approach of Cotton et al. (2015). We identified 15 genes that, based on DNAme, are likely to escape XCI in non-eroding, XIST+ hPSCs (Figures S2F–S2H). In stark contrast to genes subject to XCI, these genes feature largely hypomethylated promoters and hypermethylated gene bodies due to their biallelic expression (Figure S2I). We therefore tested whether the minimal linear distance from Xp or Xq escapees segregated DMPs or differential ATAC peaks by their transition (Figures S2J–S2O). These cumulative distributions suggest that XIE traverses between ~3 and 9 Mb for most transitions, with a median escapee distance of ~15 Mb (DNAme) and ~10 Mb (ATAC) at the XIE mid-point in transition-3, and reaching H3K9me3 chromatin last in transitions-4 and -5 (Figures 3A–3C and 3F).
To determine the significance of this observation, we performed 2 permutation tests for each transition. In the first, we randomized the 15 escapee labels across all of the genes represented by probes, while in the second test we randomized the transition labels for each DMP/ATAC peak. The resulting distributions of these permutations are represented as broad (randomized escapees) and sharp (randomized DMPs/ATAC peaks) gray background distributions (Figures 3D, 3E, 3G, and 3H). On both Xq and Xp, DMPs and opening ATAC peaks associated with transition-1 (blue distribution) are significantly closer to the 15 escapees than at least 1 of their randomized permutation sets. At subsequent transitions, the real median DMP/ATAC peak-escapee distance progressively approaches and eventually surpasses both randomized distance sets. In summary, the progressively increasing linear distance of DMPs and ATAC peaks to escapees serves as a strong predictor for which genes are most likely to undergo XIE early versus late, which suggests that XIE is likely to spread in contiguous fashion.
Terminal XIE is associated with global DNA hypomethylation
While changes in the first 3 transitions primarily affect the X, genome-wide DNAme and expression analysis reveals an emergent and dominant trans-effect as XIE nears completion: relative to clusters A–D, hPSC samples of cluster F show moderate but statistically significant autosomal hypomethylation compared to other clusters and male samples (Figure 4A), This global reduction of DNAme is paralleled by the biased upregulation of autosomal genes (Figures 4B and 4C). Such global effects are particularly striking when compared to largely X-specific changes in transitions−1 to −3, with some early loss of autosomal DNAme starting in transition-4, and rapidly amplifying in transition-5 (Figure S3), when a plethora of DMPs across the genome drop to reduce cumulative DNAme.
Figure 4. Global hypomethylation and emergent naive pluripotency markers in terminal XIE.
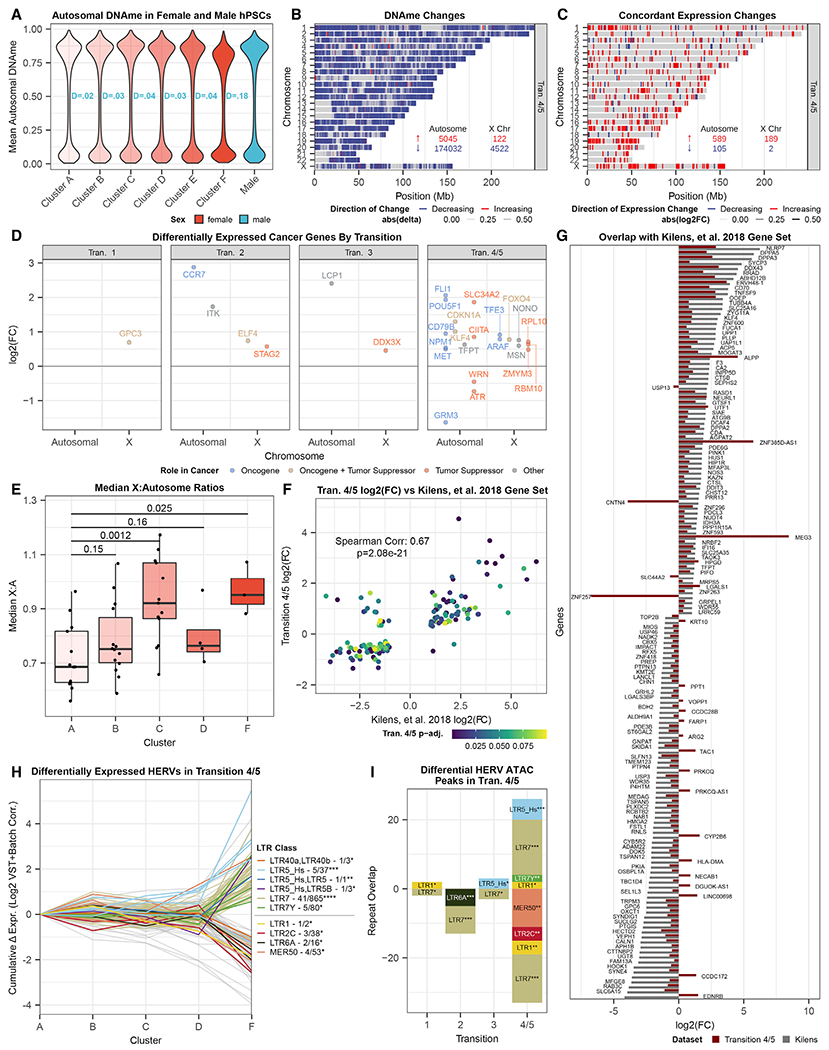
(A) Autosomal methylation distributions across female hPSC clusters (A–F, red shades) and their KS distances relative to male hPSC distribution (teal).
(B) Chromosome-resolved DMPs of the final XIE transitions (4 or 5). Total number of probes changing for autosomes and X listed with colors corresponding to increasing (red) or decreasing (blue) DNAme. The transparency is on a continuous scale based on the β-differential.
(C) As in (B), but plotting differentially expressed genes (DESeq2) with concordant DNAme changes (B) and/or ATAC changes. The transparency is on a continuous scale reflecting the log2FC.
(D) Transition- and autosome/X-resolved differentially expressed genes annotated in the COSMIC cancer gene census (Sondka et al., 2018) (also listed in Table S4).
(E) Median X:autosome ratio for each female sample cluster (Wilcoxon p value relative to cluster A).
(F) Differential expression scatterplot of transition-4/−5-specific genes (y axis) over log2FCs (x axis) of these genes between naive and primed hPSCs from Kilens et al. (2018).
(G) Comparison of expression changes in transition-4/−5-specific differential genes (dark red) relative to corresponding changes (gray) in naive versus primed hPSCs from Kilens et al. (2018).
(H) Cumulative changes (relative to cluster A) in all differentially expressed HERVs identified by Telescope (Bendall et al., 2019) (VST + batch-corrected/log2-transformed). Only significantly overrepresented HERVs (hypergeometric p ≤ 0.1) are labeled by colors (others in gray). Color legend lists total counts of differentially expressed HERV classes (with hypergeometric significance indicated as *p < 0.1, **p < 0.05, ***p < 0.01, ****p < 0.001).
(I) Tally of differentially expressed HERV classes (H) overlapped by significantly differential ATAC peaks (Fisher right-side p value indicated as *p < 0.01, **p < 0.001, ***p < 0.0001). Negative overlaps indicate overlaps by closing ATAC peaks, and positive by opening peaks.
In principle, 2 competing hypotheses for this effect can be considered: (1) cluster F may comprise aberrant hPSC lines that suffer some generalized loss of facultative heterochromatin or (2) terminal XIE may specifically reduce global DNAme, likely via overexpression of X-linked genes that affect DNAme in trans. Arguing against the former, overall autosomal chromatin accessibility drops in transition-4 with far fewer opening than closing peaks (Figure S3), which primarily reside in sparsely accessible non-coding regions and near cell-type-specific clustered paralogous gene families (e.g., olfactory receptors, keratins, cadherins). In contrast, and in support of the latter hypothesis, genome-wide hypomethylation of XaXa mouse ESCs relative to 39,X and 40,XY cells has previously been linked to double-copy dosage of the X-linked Dusp9 gene (Choi et al., 2017). Consistent with this observation, cluster F hPSC samples uniformly show a loss of DNAme at the DUSP9 promoter, paralleled by DUSP9 overexpression in this last transition (Figures S4A–S4C). Autosomal DNAme correlates strongly with DUSP9 methylation in male and female hPSCs, but the slope of this relationship steepens in the context of 2 active DUSP9 copies in cluster F hPSCs (Figure S4A). The expression of mouse Dusp9 from 2 active copies in XaXa ESCs has been shown to affect global DNAme by inhibiting the mitogen-activated protein kinase (MAPK) pathway, resulting in a post-translational decrease in DNMT3A/B and UHRF1 proteins that was not evident in Dnmt3a/b and Uhrf1 transcript levels (Choi et al., 2017). Likewise, neither human ortholog was differentially expressed in hPSC samples of cluster F. Two additional factors signaling via the MAPK pathway (ELK1 [Bruck et al., 2013] and ARAF) reside in another H3K9me3-rich region of the hPSC Xi and increase ~2-fold upon loss of promoter-associated DNAme (Figures S4B and S4C). We also tested whether oncogenes are preferentially activated (Anguera et al., 2012), but found only a handful of bona fide oncogenes specific to the last transition on X (ARAF, TFE3) and autosomes (MET, NPM1, CD79B, POUF5F1 [OCT4], FLI1) alike (Figure 4D). Consistently, there was only a single cancer-associated term enriched among the X-linked genes differentially expressed in transition-5 (Table S4), arguing against a strong oncogene-driven selective benefit for hPSCs undergoing early XIE.
While MAPK inhibition is essential for the in vitro induction of naive pluripotency (Guo et al., 2017; Theunissen et al., 2016) it also abrogates global DNAme and erases imprinting (Pastor et al., 2016). Motivated by the observation that reduced inhibition of MAPK signaling is sufficient for naive pluripotency (Di Stefano et al., 2018), we tested whether the modest attenuation of DNAme levels in cluster F hPSCs is reflected in expression differences of their pluripotency genes. To this end, we compared gene expression profiles of isogenic naive and primed hPSCs derived in parallel (Kilens et al., 2018) to genes differentially expressed in cluster F, when the median X:autosomal gene expression ratio peaks (Figure 4E). The overlap (189/1,686 genes) and correlation (ρ = 0.67, p = 2.08e–21) across differentially expressed genes greatly exceeded null expectation (Figures 4F and 4G), with naive pluripotency factors DPPA2/3/5 and KLF4 upregulated alongside Kelch homology (KH) or KH domain-like genes NRLP7 and OOEP (also DPPA5). Moreover, among upregulated genes we find many specifically expressed in 3 embryonic pre-implantation stages (zygote, morula, and blastocyst), including DDIT3, NANOG, POU5F1 (OCT4), TLE6, and ZSCAN4. Upregulation of these genes is also significantly reflected in reduced DNAme (Figure S4D; sign test p < 2.2e–16). Because the overall degree of these changes lagged behind the dynamic range observed in the bona fide naive hPSCs, we also examined whether there were changes in human endogenous retroviral (HERV) elements, which are expressed in stage-specific fashion during mammalian pre-implantation development and feature pluripotency factor binding sites in their long terminal repeats (LTRs) (Gerdes et al., 2016; Torres-Padilla, 2020). Retrotranscriptome analysis using Telescope (Bendall et al., 2019) reveals significantly enriched upregulation of HERV-H (LTR7Y p = 2.7e–4) and HERV-K (LTR5_Hs p = 0.003) elements (Figure 4H), which are respectively associated with blastocyst and morula-stage embryos (Göke et al., 2015) and likewise upregulated in naive hPSCs in vitro (Di Stefano et al., 2018; Theunissen et al., 2016; Torres-Padilla, 2020). While these HERV classes were also significantly enriched for opening ATAC peaks in cluster F (with 16 LTR7, 2 LTR7Y, and 6 LTR5_Hs elements), no naive pluripotency-specific upregulation or chromatin opening of hominid SVA elements (Pontis et al., 2019; Theunissen et al., 2016) was observed (Figure 4I). In summary, these changes suggest that despite their more limited hypomethylation relative to bona fide naive hPSCs, cluster F hPSCs appear to regain some specific hallmarks of naive pluripotency (Figures 4F–4I).
DISCUSSION
Our integrated DNAme, ATAC, and expression analysis addresses 3 major challenges that have obscured our understanding of XIE in female hPSC to-date: (1) an incomplete view of human XIST regulation, (2) the progressive nature of XIE coupled with a lack of longitudinal studies, and (3) a consequently large range of reported outcomes, from piecemeal to whole Xi reactivation (Anguera et al., 2012; Bar et al., 2019; Kim et al., 2014; Lengner et al., 2010; Mekhoubad et al., 2012; Nazor et al., 2012; Tchieu et al., 2010; Tomoda et al., 2012; Vallot et al., 2015). In ordering 221 female hPSC DNAme samples from early to late XIE, a trajectory validated by 22 new longitudinal female hPSC samples, this integrated analysis leads us to 3 novel insights regarding this elusive epigenetic phenomenon.
First, we pinpoint a dynamic increase of DNAme and decrease in chromatin accessibility in a cis-regulatory CpG island just 1.2 kb downstream of the major promoter of XIST that coincides with the loss of XIST expression and the very earliest Xi reactivation events (Figure 2). The same “P2” element is 1 of only 3 female-specific DNase hypersensitive sites on the human X, and is fully methylated on the single Xa in male cells (Chapman et al., 2014; Chen et al., 2016a). The corresponding DMPs overlap conserved cognate motifs of transcription factor YY1 that binds in a DNAme-sensitive manner to boost both human and mouse XIST/Xist expression (Makhlouf et al., 2014). Of the 2 remaining female-specific YY1 clusters on the human X (Chapman et al., 2014; Chen et al., 2016a), the tandem repeat of FIRRE also reflects this first XIE transition with some of the most significantly increasing DMPs and the only closing ATAC peaks on the X outside XIST (Figures 2 and 3). FIRRE has been shown to form conserved Xi-specific long-range interactions with the third female-specific YY1 cluster, the macrosatellite DXZ4, which functions as a topological boundary on the Xi (reviewed in Bansal et al., 2020). Both of these non-coding tandem repeat loci maintain focal euchromatin on the otherwise escapee-poor long arm and reside at the center (FIRRE) or periphery (DXZ4) of the earliest reactivation clusters (Figure 3). Interestingly, the same DXZ4-adjacent 15-Mb region was shown to lose H3K27me3 when DXZ4 is deleted in differentiated RPE-1 cells (Darrow et al., 2016). This region also spans XACT, another ncRNA gene associated with XIE initiation in H9 hESCs (Vallot et al., 2015). Looking forward, it will be critical to ascertain the mechanistic basis for increasing DNAme in the P2 element of XIST, and determine whether the female-specific YY1 clusters in FIRRE and DXZ4 are sensitive to the same pluripotency circuit or merely respond to the earliest Xi structural changes upon loss of XIST.
Second, in addition to H3K27me3/H3K9me3 association, we find that the linear distance to the closest escapee is a major predictive factor for whether an XCI-subject gene loses DNAme and becomes more accessible during early or late XIE (Figure 3). To this end, we identified 15 escapee candidates in female hPSCs on the basis of their DNAme states, as in Cotton et al. (2015). On Xq, the novel escapee candidate LHFPL1 (~1 Mb from XACT) joined PLS3-AS1 (~0.25 Mb from DXZ4) and FIRRE, while on Xp, outside PAR1, 12 genes qualified as escapees based on their DNAme. On both Xq and Xp, DMPs associated with later transitions increased in distance from these escapee candidates, and this contiguous spread was also evident in progressive gains of chromatin accessibility (Figure 3). Likewise, our longitudinal experiment identifies early demethylating DMPs near escapees, and shows highly significant correlations with transitions-1 and -2 DMPs (Figure S2D). One plausible mechanistic basis for this pattern is that active chromatin spreads from escapee genes to reactivate silenced genes in a contiguous fashion. In support of this notion, the deletion of an escapee boundary does not abrogate its expression on the mouse Xi, but instead drives the reactivation of neighboring genes (Horvath et al., 2013). In another X reactivation context during mouse iPSC reprogramming, the genes closest to escapees also reactivate before distal genes (Bauer et al., 2020; Janiszewski et al., 2019). The high density of escapees and the large active PAR1 may also predispose the short arm of the human Xi to XIE, which may explain why hPSCs, in contrast to mouse epiblast cells, depend on XIST for the maintenance of XCI.
Third, terminal XIE is strikingly associated with female-specific global DNA hypomethylation (Figure 4), as previously demonstrated in XaXa mouse ESCs (Choi et al., 2017). As in the mouse system, we find that the doubling of DUSP9 dosage, which is specific to the final XIE transition, correlates with autosomal hypomethylation (Figures S3 and S4). We find little evidence for the preferential upregulation of oncogenes before terminal XIE, and conclude that oncogenic selection is unlikely to drive early XIE. Surprisingly, in terminal XIE, when the reactivation of DUSP9 impairs the MAPK pathway and DNAme is reduced genome-wide, we find a highly significant shift toward a naive pluripotent gene expression profile, as primed markers are repressed and naive markers are induced (Figures 4 and S4). While the magnitude of naive marker induction lags behind that observed for bona fide naive hPSCs, either due to homogeneous but low induction of naive markers or an increase in the proportion of cells cycling through a naive-like state, reactivated and opening HERVs match the in vivo equivalent preimplantation stage-specific HERV classes, namely HERV-H (blastocyst) and HERV-K (morula) (Göke et al., 2015). This HERV profile is also activated either transiently during iPSC reprogramming (Ohnuki et al., 2014) or stably upon induction of naive pluripotency (Di Stefano et al., 2018; Theunissen et al., 2016). In mouse ESCs, double X chromosome dosage blocks the exit from naive pluripotency (Schulz et al., 2014), likely by stabilizing the expression of specific pluripotency regulators (Song et al., 2019). As a result, pluripotency exit is coupled to XCI. We therefore interpret the basal induction of naive-specific genes and HERVs in terminal XIE (Figure 4) as hPSCs partially recapitulating this process in reverse, suggesting some conservation of this double X dosage impact on the human naive pluripotency circuit. Intriguingly, the enrichment of 35 reactivated X-linked mitochondrial factors and regulators of oxidative phosphorylation in terminal XIE (Table S4) may hint at a possible link to a third hallmark of naive pluripotency, namely increased mitochondrial respiration (Kilens et al., 2018). In light of recent reports that further stratify the spectrum from naive to primed pluripotency (Cornacchia et al., 2019; Lau et al., 2020; Nakanishi et al., 2019), the observation that human X dosage affects this spectrum raises the important question of why this female/Xa-dosage bias toward naive pluripotency may have been conserved in evolution (Schulz et al., 2014; Song et al., 2019).
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be addressed by the Lead Contact, Stefan Pinter (spinter@uchc.edu).
Materials availability
All unique reagents generated in this study can be requested from the Lead Contact with a completed Materials Transfer Agreement, and be made available upon reasonable request with applicable processing and shipping fees through the University of Connecticut Cell and Genome Engineering Core.
Data and code availability
Validation time-course methylEPIC datasets have been deposited under accession number GSE167883 in the Gene Expression Omnibus (GEO). Analysis code is made available through the github repository: https://github.uconn.edu/sfp-lab/contiguous_x_erosion
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The female fibroblast line AG05278 was obtained from the Coriell Institute (Camden, NJ), and reprogrammed by the University of Connecticut Stem Cell Core using the CytoTune hiPSC 2.0 Sendai Reprogramming Kit (Thermo Fisher, Waltham MA). To confirm the presence of two X chromosomes in each individual hiPSC clone, DNA was extracted from each clone using the Hot-Shot method. Briefly, hiPSCs were incubated with alkaline lysis buffer (25mM NaOH, 0.2mM EDTA pH 12) at 95C for 1 hour and then neutralized with 40mM Tris-HClpH5. PCR was performed using the One Taq Hot Start 2x Master Mix (New England Biolabs, Ipswich, MA) across the short tandem repeat HPRT locus (HPRT_F: ATGCCACAGATAATACACATCCCC, HPRT_R: CTCTCCAGAATAGTTAGATGTAGG), and PCR products were analyzed by agarose gel electrophoresis. Euploid (46,XX) clones were identified by the presence of two products of different lengths. The hiPSCs were cultured on mitotically inactive mouse embryonic fibroblasts in standard hiPSC media (80% DMEM/F12, 20% Knockout Serum Replacement, 1% Glutamax, 1% Non-Essential Amino Acids, 0.1% β-Mercaptoethanol, and 8ng/mL bFGF), and passaged weekly by manual disruption of 10-20 representative colonies. Samples collected across this continuous long-term culture are listed in Table S1, and ranged from passages 8 and 12 to 32 and 35, respectively, for the C19 and C23 hiPSC clones.
METHOD DETAILS
For RNA isolation, the PureLink RNA Mini Kit (Invitrogen, Waltham, MA) was used, and cDNA synthesis was performed with the iScript gDNA Clear cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Real-time PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories, Hercules, CA). XIST expression (XIST_F: AGCAGTTTGCCCTACTAGCTC, XIST_R: TAGCTGTTTGCAGTCCTCAGG) was normalized to TATA-box binding protein TBP (TBP_F: TCACTGTTTCTTGGCGTGTG, TBP_R: TGGCAAACCAGAAACCCTTG) using the ΔCt method. Primer specificity was confirmed by melting curve analysis, and relative expression is calculated as %TBP (100/2ΔCt).
For DNA extraction, cells were pelleted and resuspended in cell lysis buffer (10 mM Tris-HCl, pH 8.0; 100 mM NaCl; 10 mM EDTA, pH 8.0; 1% SDS; 200 μg/ml Proteinase K) and incubated at 55°C overnight. DNA was isolated using Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) (Invitrogen, Waltham, MA), precipitated with isopropanol, washed with 70% ethanol, and resuspended in TE buffer. Genomic DNA was bisulfite-converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA), then labeled and hybridized using the Infinium Methylation EPIC BeadChip Kit (Illumina, San Diego, CA) following standard protocol of each manufacturer, and scanned on a NextSeq 550 system.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data collection, processing, and normalization
We searched GEO for Illumina 450K and 850K Methylation Array data for primed human iPSC and ESC samples and selected all studies with at least ten samples, yielding eight studies with a total of 281 female samples, and 190 male samples (Table S1) (Banovich et al., 2018; Butcher et al., 2016; Garitaonandia et al., 2015; Nazor et al., 2012; Nishizawa et al., 2016; Salomonis et al., 2016; Takasawa et al., 2018; Zdravkovic et al., 2015). Most of the studies had pre-processed methylation data available. We processed the remainder using minfi’s IlluminaNormalization method to mimic the pre-processed data (Aryee et al., 2014), and analyzed the probe set common to both 850K and 450K methylation arrays. We also removed all of the cross reactive probes as identified by Chen et al. (2013) in order to exclude ambiguous methylation signals. We further mapped all probes to the hg38 genome assembly using BLAT and removed any that mapped to multiple regions in the genome (Kent, 2002). After this processing, there were 413,491 probes across the genome, including 9,191 probes on the X.
Mapping the probes to the hg38 genome build also provided updated gene annotations. A gene was assigned to a probe if the probe was within 5kb of the gene, and each probe was annotated as being either “OutsideTranscript5” (−5000 to −1500 of TSS), “TSS1500” (−1500 to −200 of TSS), “TSS200” (−200 to 0 of TSS), “5_UTR” (in the 5′ UTR of the gene), “Body” (in the body of the gene including exons and introns), “3_UTR” (in the 3′UTR of the gene), and “OutsideTranscript3” (5kb past the end of the gene). All positions were annotated according to a canonical transcript for each gene, which was determined to be either the highest transcript support level (TSL) or the highest APPRIS level as annotated by Ensembl (Cunningham et al., 2019). TSL marks the amount of literature support behind a transcript, while APPRIS marks the functional importance of the transcript.
We performed sex prediction using minfi and removed any lines where the predicted sex did not match the annotated sex. In cases where no annotated sex was available in the originating methylation study, it was obtained from Cellosaurus and compared to the predicted sex (Bairoch, 2018). We then performed PCA analyses on the X chromosome probes separately for male and female samples to determine that the data were sufficiently normalized to enable quantitative comparisons between studies (Figure S1A). Among the female samples, H9 hESCs were over-represented in the data and tended to separate from most other female hPSCs. To not skew our analysis to differences between H9 and other cell lines, we removed all 60 H9 samples from our analysis (Figures S1B and S1C). For the male samples, there were 12 outliers in the PCA that were removed (Figures S1D and S1E). This left us with 221 female samples, and 178 male samples.
Clustering samples by variant probes
To identify sites with greater variance in female samples than male samples, we performed a Brown Forsythe (BF) test for each methylation array probe to calculate statistical significance for the difference in variance for the probe between female and male samples. The BF test assigned statistical significance to the differences in variation for each probe between female and male samples. The BF alpha was much more significant for X-probes than for autosomal probes (Figure S1G). The significantly more variant X-probes also have lower DNAme than the non-sig sites (Figure S1H). We selected X-linked sites that had a greater variance in the female samples than the male samples (BF p value ≤ 0.01), and performed a weighted k-means clustering analysis using the scikit-learn python library on the female samples (Pedregosa et al., 2011). The weights for the k-means clustering were based on how many of the cell lines were included in the analysis so that small differences in number of samples per cell line would not skew the k-means calculations. We used the elbow method to determine the appropriate number of clusters to be around six. These clusters were then labeled A-F based on their average X chromosome methylation (most to least methylated). Representative images of how the clusters separated the samples in the PCA is shown in Figure S1K for a k = 4-8.
Clustering probes by progression of XIE
We parsed how the X chromosome probes were changing during XIE, using the minfi package’s dmpFinder function to identify differentially methylated probes between each adjacent sample cluster. Then we divided each DMP on the X chromosome into one of 11 groups (Figure 1I, y axis) based on the most statistically significant transition (smallest q-value ≤ 0.001), and the direction of its methylation change. These clusters are labeled in Figure 1I corresponding to their transition (1-5), and either increase (↑) or decrease (↓) in methylation. The last cluster (labeled “~”) contains probes that do not pass the q-value threshold (0.001) for all the DMP tests, and thus are not considered to be changing during XIE. All X probes and cluster assignments are listed in Table S2. For the 22 samples of our validation set, the 850K Methylation Array DNAme data was processed using minfi as described above. Samples were assigned to clusters based on the same k-means clustering model derived above. Samples were also projected onto the principle components from the meta-analysis PCA to facilitate comparison to the established cluster boundaries, outlined by ellipses as calculated by stat_ellipse method from ggplot2 (Figures 1F and 1G). With longitudinal DNAme data from successive passages of these validation samples, the fraction of transition-specific DMPs changing DNAme in the validation set was quantified at each passage. A DMP was considered “demethylated” in the sample if it reached within 10% of the expected change from cluster A based on the larger analysis. The transition-specific DMP analysis limited to the C19 and C23 validation samples was performed as described above, except we used the q-value cutoff of < 0.05.
ATAC-seq analysis
We aligned the ATAC-seq fastq files using esATAC which serves as a wrapper for AdapterRemoval (Schubert et al., 2016) for adaptor trimming and Bowtie2 (Langmead and Salzberg, 2012) for read alignment. MACS2 (Zhang et al., 2008) was used for peak calling, and DiffBind (Stark and Brown, 2011) was used for differential peak analysis. Differential analysis was done using cluster boundaries as for DMP differential methylation analysis.
Cumulative demethylation and calculating correlation with Xi-specific histone marks
To calculate cumulative methylation changes (gray areas in Figure 3A), we window-averaged all probes that passed the p value threshold (≤0.05) in at least one transition and calculated the difference in methylation compared to cluster A moving through the transitions. In contrast, probes that did not pass the threshold had more fluctuant DNAme changes that did not track with successive transitions (Figure S2B).
We calculated the correlation of the XIE DNAme changes with Xi-specific H3K27me3 and H3K9me3 peaks from Vallot et al. (2015). Since these peaks were already window-averaged in GEO-deposited bed files, we averaged our DNAme data over the same windows (100 kb windows every 50 kb). We used the transition-specific changes (colored lines in Figure 3A) to calculate the Pearson correlation for each transition (Figure 3C). Similarly, we windowed the differential ATAC peaks (Figure 3B), and calculated the Pearson correlation for each transition (Figure 3F).
Annotating genes as escapees
We annotated X chromosome genes as escaping XCI using the DNAme-based escapee calling method reported previously (Cotton et al., 2015) (Figures S2E–S2G). We used all male samples, and the female samples from the cluster A to perform this analysis. First, we removed all X chromosome probes with a mean methylation of > = 0.25 in male samples. To calculate mean DNAme for each gene, we used the TSS for the canonical transcript, and averaged all remaining probes between −500 and +1500 of the TSS. We calculated a mean TSS methylation separately for the male and cluster A female samples. Assessing the genes defined as the escapee training set, as per Cotton et al., and using the same verification method, we found that only 4/12 their “training set escapees” truly escaped XCI in our dataset (CA5B, SYAP1, ZFX, and ZRSR2). These had a mean cluster A female methylation of 0.106, and a mean sex difference of 0.02. A gene is classified as an escapee if its female DNAme is less than the mean female DNAme + (3* SD female DNAme), and if its sex difference is less than the mean sex difference + (3*SD sex difference). Altogether, this approach identified 15 escapee genes (shown in Figure 3A): 12 on Xp, and 3 on Xq. We verified the differences in methylation in the promoter and gene body for escapee and subject genes and found that escapee genes tend to have strongly demethylated promoters and methylation gene bodies on both chromosomes, whereas subject genes have a bimodal distribution of DNAme in both the gene body and the promoter (Figure S1H).
Calculating distances to escapee genes
We calculated the distance of eroding DNAme probes from the escapee genes to test whether erosion may be emanating from escapees. For each DMP that was assigned to a transition, we calculated the distance to the nearest escapee gene. We assessed Xp and Xq separately due to the disparity in the number of escapees on each arm. Additionally, we performed two permutation tests to provide randomized distributions against which to compare transition-specific distributions of actual minimal distances. One test randomized the escapee labels across all genes represented by probes, and the other randomized the transition labels for each DMP. Each test was repeated 1000 times for each transition and each arm of the chromosome to produce the final distributions (Figures 3D and 3E). We repeated this analysis with the differential ATAC peaks instead of the DMPs (Figures 3G and 3H).
Expression analysis
Of the 221 female cell lines we analyzed (after H9 removal), 47 had RNA seq data available. The RNA seq data was downloaded using SRA Toolkit. The FASTQ files were aligned to a hg38 genome assembly (with the PAR region masked on the Y chromosome) using STAR, and reads were counted using HTSeq-count (Anders et al., 2015; Dobin et al., 2013).
Differential expression (DE) analysis was performed using DESeq2 (Table S3) Love et al., 2014). We excluded any genes with fewer than 10 counts. DE was performed based on the methylation-derived sample clusters and treated each transition boundary individually, rather than considering the cluster assignments as a continuous variable. For transition 1, we assigned cluster A samples as one group, and clusters B-F as another, then identified differentially expressed genes using DESeq2. For transition 2, we compared clusters A-B to clusters C-F. For transition 3 we compared A-C to D-F. As there were no available expression data for samples in cluster E, we combined transition 4/5 into one and compared clusters A-D to cluster F. By considering all samples, we filtered differentially expressed genes that are not changed in a permanent/consistent manner throughout XIE. We also used DESeq2 to extract VST normalized expression for XIST, FIRRE, DUSP9, ELK1, REX1, and KLF4, and then ran limma batch correction to adjust for differences in RNA-seq data from different groups. The VST transformation applies a pseudo-log2 transformation on the counts. These VST and batch-corrected values are used for all the expression plots. Median X:Autosome ratios were calculated on FPKM normalized counts using the pairwiseCI package in R.
We extracted allelic read counts of these hPSC RNA-seq datasets from the Skymap database (Tsui et al., 2019) to calculate X-linked allele frequencies. The Xe allele was inferred as the allele with the lower read count across all heterozygous positions (lesser allele frequency, LAF), excluding reads with average base quality under 30, and positions with read depth under 2. Altogether, RNA seq data of 34/47 female hPSC RNA-seq datasets were available on Skymap, representing each of the clusters (A-D,F). Differences in LAF medians was assessed by Mann-Whitney U test p values between clusters A and B-F.
Concordance and enrichment analysis
We considered genes as concordant in methylation and expression when at least one of its methylation array probes changed in the same transition and in the opposite direction as its expression (i.e., decreasing methylation for genes increasing in expression, and vice versa). We further considered a gene concordant if a differential ATAC peak fell within ± 3kb of the gene and changed in the same transition as its expression (all concordant genes are shown in Figure S3C). DNAme or ATAC concordant genes are listed in Table S4, and plotted in Figure S3C.
Using these transition-specific concordant gene sets, we calculated ontology term enrichments in each transition for X chromosome genes, and autosomal genes (Table S4). Using the clusterProfiler R package, we queried the following gene sets: Disease Ontology, DisGeNET, GO, KEGG, MeSH gendoo, MSigDB, Network of Cancer Gene, Reactome, and WikiPathways. For the cancer gene counts (Figure 4F), we used the COSMIC Cancer Gene Census to count how many cancer genes of the various types were in each transition (Sondka et al., 2018).
Telescope HERV analysis
We used Telescope (Bendall et al., 2019) to enumerate the HERV expression for each STAR aligned bam files from the expression analysis. Using the same DESeq2 pipeline from the expression analysis (see above), we calculated differential HERV expression between clusters to obtain the log2-transformed VST+batch-corrected expression for each sample.
ATAC HERV overlap analysis
Using the bedtools fisher tool, we compared the overlap for each repeat from RepeatMasker and our differential ATAC-peaks from each transition. We used the right-sided p value to select overlaps that were enriched in our differential ATAC-peaks, and plotted the number of significant overlaps for HERVs that were also differentially expressed in Transition 4/5 (Figure 4I).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Human FGF-basic recombinant protein | Thermo Fisher | PHG0023 |
| Critical commercial assays | ||
| Infinium methylation EPIC BeadChip kit | Illumina | WG-317-1001 |
| EZ DNA methylation kit | Zymo Research | D5001 |
| PureLink RNA mini kit | Invitrogen | 12183025 |
| Deposited data | ||
| GEO methylEPIC, RNA-seq, ATAC data series (GSE) | Banovich et al., 2018 | GSE89895 |
| GEO methy450K data series (GSE) | Butcher et al., 2016 | GSE59091 |
| GEO methyl450K data series (GSE) | Garitaonandia et al., 2015 | GSE34982 |
| GEO methyl450K data series (GSE) | Nazor et al., 2012 | GSE31848 |
| GEO methyl450K data series (GSE) | Nishizawa et al., 2016 | GSE60924 |
| GEO methyl450K data series (GSE) | Salomonis et al., 2016 | GSE85828 |
| GEO methyl450K data series (GSE) | Takasawa et al., 2018 | GSE73938 |
| GEO methyl450K data series (GSE) | Zdravkovic et al., 2015 | GSE72923 |
| GEO methylEPIC data series (GSE) for C19 and C23 passage experiment | This study | GSE167883 |
| Experimental models: cell lines | ||
| skin fibroblasts | NIA Aging Cell Repository, Coriell Institute | AG05278 |
| Euploid female hiPSC clone C19 from AG05278 | This study | C19 |
| Euploid female hiPSC clone C23 from AG05278 | This study | C23 |
| Oligonucleotides | ||
| XIST_F: AGCAGTTTGCCCTACTAGCTC | This study | XIST_Fwd |
| XIST_R: TAGCTGTTTGCAGTCCTCAGG | This study | XIST_Rev |
| TBP_F: TCACTGTTTCTTGGCGTGTG | This study | TBP_Fwd |
| TBP_R: TGGCAAACCAGAAACCCTTG | This study | TBP_Rev |
| HPRT_F: ATGCCACAGATAATACACATCCCC | This study | HPRT_Fwd |
| HPRT_R: CTCTCCAGAATAGTTAGATGTAGG | This study | HPRT_Rev |
| Recombinant DNA | ||
| CytoTune-iPS 2.0 Sendai reprogramming kit | ThermoFisher Scientific | A16517 |
| Software and algorithms | ||
| minfi | Aryee et al., 2014 | https://bioconductor.org/packages/release/bioc/html/minfi.html |
| tidyverse | Wickham et al., 2019 | https://www.tidyverse.org/ |
| ggsci | https://nanx.me/ggsci/articles/ggsci.html | https://github.com/nanxstats/ggsci |
| limma | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| biomaRt | Durinck et al., 2005 | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| clusterProfiler | Yu et al., 2012 | https://guangchuangyu.github.io/software/clusterProfiler/ |
| numpy | Harris et al., 2020 | https://numpy.org/ |
| pandas | McKinney, 2010 | https://pandas.pydata.org/ |
| sklearn | Pedregosa et al., 2011 | https://scikit-learn.org |
Highlights.
Order of inactive X erosion in human iPSCs/ESCs is recapitulated in long-term passage
XIST regulatory elements reflect loss of XIST expression at onset of X erosion
X erosion trajectory is consistent with contiguous spread of reactivation from escapees
Global hypomethylation and partial naive pluripotency features emerge in late X erosion
ACKNOWLEDGMENTS
We thank the University of Connecticut’s Cell and Genome Engineering Core and Center for Genome Innovation for reprogramming and methylEPIC core services, respectively. This work was supported by NIH grant R35GM124926 to S.F.P.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109215.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adrianse RL, Smith K, Gatbonton-Schwager T, Sripathy SP, Lao U, Foss EJ, Boers RG, Boers JB, Gribnau J, and Bedalov A (2018). Perturbed maintenance of transcriptional repression on the inactive X-chromosome in the mouse brain after Xist deletion. Epigenetics Chromatin 11, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguera MC, Sadreyev R, Zhang Z, Szanto A, Payer B, Sheridan SD, Kwok S, Haggarty SJ, Sur M, Alvarez J, et al. (2012). Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell 11, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, and Irizarry RA (2014). Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood SW, and Edel MJ (2019). iPS-Cell Technology and the Problem of Genetic Instability–Can It Ever Be Safe for Clinical Use? J. Clin. Med 8, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A (2018). The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech 29, 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banovich NE, Li YI, Raj A, Ward MC, Greenside P, Calderon D, Tung PY, Burnett JE, Myrthil M,Thomas SM, et al. (2018). Impact of regulatory variation across human iPSCs and differentiated cells. Genome Res. 28, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal P, Kondaveeti Y, and Pinter SF (2020). Forged by DXZ4, FIRRE and ICCE: how tandem repeats shape the active and inactive X chromosome. Front. Cell Dev. Biol 7, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar S, and Benvenisty N (2019). Epigenetic aberrations in human pluripotent stem cells. EMBO J. 38, e101033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar S, Seaton LR, Weissbein U, Eldar-Geva T, and Benvenisty N (2019). Global Characterization of X Chromosome Inactivation in Human Pluripotent Stem Cells. Cell Rep. 27, 20–29.e3. [DOI] [PubMed] [Google Scholar]
- Bauer M, Vidal E, Zorita E, Pinter SF, Filion GJ, and Payer B (2020). Chromosome Compartments on the Inactive X Guide TAD Formation Independently of Transcription during X-Reactivation. BioRxiv. 10.1101/2020.07.02.177790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall ML, de Mulder M, Iñiguez LP, Lecanda-Sánchez A, Pérez-Losada M, Ostrowski MA, Jones RB, Mulder LCF, Reyes-Terán G, Crandall KA, et al. (2019). Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol 15, e1006453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck T,Yanuka O, and Benvenisty N (2013). Human pluripotent stem cells with distinct X inactivation status show molecular and cellular differences controlled by the X-Linked ELK-1 gene. Cell Rep. 4, 262–270. [DOI] [PubMed] [Google Scholar]
- Butcher LM, Ito M, Brimpari M, Morris TJ, Soares FAC, Ährlund-Richter L, Carey N, Vallier L, Ferguson-Smith AC, and Beck S (2016). Non-CG DNA methylation is a biomarker for assessing endodermal differentiation capacity in pluripotent stem cells. Nat. Commun 7, 10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ (2016). Disease modelling using human iPSCs. Hum. Mol. Genet 25 (R2), R173–R181. [DOI] [PubMed] [Google Scholar]
- Chapman AG, Cotton AM, Kelsey AD, and Brown CJ (2014). Differentially methylated CpG island within human XIST mediates alternative P2 transcription and YY1 binding. BMC Genet. 15, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, and Weksberg R (2013). Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 8, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shi W, Balaton BP, Matthews AM, Li Y, Arenillas DJ, Mathelier A, Itoh M, Kawaji H, Lassmann T, et al. (2016a). YY1 binding association with sex-biased transcription revealed through X-linked transcript levels and allelic binding analyses. Sci. Rep 6, 37324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Schell JP, Benitez JA, Petropoulos S, Yilmaz M, Reinius B, Alekseenko Z, Shi L, Hedlund E, Lanner F, et al. (2016b). Single-cell analyses of X Chromosome inactivation dynamics and pluripotency during differentiation. Genome Res. 26, 1342–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Clement K, Huebner AJ, Webster J, Rose CM, Brumbaugh J, Walsh RM, Lee S, Savol A, Etchegaray JP, et al. (2017). DUSP9 Modulates DNA Hypomethylation in Female Mouse Pluripotent Stem Cells. Cell Stem Cell 20, 706–719.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornacchia D, Zhang C, Zimmer B, Chung SY, Fan Y, Soliman MA, Tchieu J, Chambers SM, Shah H, Paull D, et al. (2019). Lipid Deprivation Induces a Stable, Naive-to-Primed Intermediate State of Pluripotency in Human PSCs. Cell Stem Cell 25, 120–136.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton AM, Price EM, Jones MJ, Balaton BP, Kobor MS, and Brown CJ (2015). Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet 24, 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csankovszki G, Panning B, Bates B, Pehrson JR, and Jaenisch R (1999). Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet 22, 323–324. [DOI] [PubMed] [Google Scholar]
- Csankovszki G, Nagy A, and Jaenisch R (2001). Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J. Cell Biol 153, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S, et al. (2019). Ensembl 2019. Nucleic Acids Res. 47 (D1), D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrow EM, Huntley MH, Dudchenko O, Stamenova EK, Durand NC, Sun Z, Huang S-C, Sanborn AL, Machol I, Shamim M, et al. (2016). Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proc. Natl. Acad. Sci. USA 113, E4504–E4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano B, Ueda M, Sabri S, Brumbaugh J, Huebner AJ, Sahakyan A, Clement K, Clowers KJ, Erickson AR, Shioda K, et al. (2018). Reduced MEK inhibition preserves genomic stability in naive human embryonic stem cells. Nat. Methods 15, 732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, and Huber W (2005). BioMart and Bioconductor: a powerful link between biologyical databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Fang H, Disteche CM, and Berletch JB (2019). X Inactivation and Escape: Epigenetic and Structural Features. Front. Cell Dev. Biol 7, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froberg JE, Pinter SF, Kriz AJ, Jégu T, and Lee JT (2018). Megadomains and superloops form dynamically but are dispensable for X-chromosome inactivation and gene escape. Nat. Commun 9, 5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan G, and Rougeulle C (2016). Function and evolution of the long non-coding RNA circuitry orchestrating X-chromosome inactivation in mammals. Wiley Interdiscip. Rev. RNA 7, 702–722. [DOI] [PubMed] [Google Scholar]
- Garitaonandia I, Amir H, Boscolo FS, Wambua GK, Schultheisz HL, Sabatini K, Morey R, Waltz S, Wang Y-C, Tran H, et al. (2015). Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS ONE 10, e0118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geens M, and Chuva De Sousa Lopes SM (2017). X chromosome inactivetion in human pluripotent stem cells as a model for human development: back to the drawing board? Hum. Reprod. Update 23, 520–532. [DOI] [PubMed] [Google Scholar]
- Gerdes P, Richardson SR, Mager DL, and Faulkner GJ (2016). Transposable elements in the mammalian embryo: pioneers surviving through stealth and service. Genome Biol. 17, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göke J, Lu X, Chan Y-S, Ng H-H, Ly L-H, Sachs F, and Szczerbinska I (2015). Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell Stem Cell 16, 135–141. [DOI] [PubMed] [Google Scholar]
- Guo G, von Meyenn F, Rostovskaya M, Clarke J, Dietmann S, Baker D, Sahakyan A, Myers S, Bertone P, Reik W, et al. (2017). Epigenetic resetting of human pluripotency. Development 144, 2748–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, et al. (2020). Array programming with NumPy. Nature 585, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich BD, Plenge RM, and Willard HF (1997). Identification and characterization of the human XIST gene promoter: implications for models of X chromosome inactivation. Nucleic Acids Res. 25, 2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath LM, Li N, and Carrel L (2013). Deletion of an X-inactivation boundary disrupts adjacent gene silencing. PLoS Genet. 9, e1003952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewski A, Talon I, Chappell J, Collombet S, Song J, De Geest N, To SK, Bervoets G, Marin-Bejar O, Provenzano C, et al. (2019). Dynamic reversal of random X-chromosome inactivation during iPSC reprogramming. Genome Res. 29, 1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ (2002). BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Audergon PNCB, and Payer B (2017). X-chromosome activity in naive human pluripotent stem cells-are we there yet? Stem Cell Investig. 4, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilens S, Meistermann D, Moreno D, Chariau C, Gaignerie A, Reignier A, Lelièvre Y, Casanova M, Vallot C, Nedellec S, et al. ; Milieu Intérieur Consortium (2018). Parallel derivation of isogenic human primed and naive induced pluripotent stem cells. Nat. Commun 9, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kollhoff A, Bergmann A, and Stubbs L (2003). Methylation-sensitive binding of transcription factor YY1 to an insulator sequence within the paternally expressed imprinted gene, Peg3. Hum. Mol. Genet 12, 233–245. [DOI] [PubMed] [Google Scholar]
- Kim K-YY, Hysolli E, Tanaka Y, Wang B, Jung Y-WW, Pan X, Weissman SM, and Park I-HH (2014). X Chromosome of female cells shows dynamic changes in status during human somatic cell reprogramming. Stem Cell Reports 2, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KX, Mason EA, Kie J, De Souza DP, Kloehn J, Tull D, McConville MJ, Keniry A, Beck T, Blewitt ME, et al. (2020). Unique properties of a subset of human pluripotent stem cells with high capacity for self-renewal. Nat. Commun 11, 2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, Lynch C, Harness JV, Lee S, Barrero MJ, et al. (2011). Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 8, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner CJ, Gimelbrant AA, Erwin JA, Cheng AW, Guenther MG, Welstead GG, Alagappan R, Frampton GM, Xu P, Muffat J, et al. (2010). Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell 141, 872–883. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhlouf M, Ouimette JF, Oldfield A, Navarro P, Neuillet D, and Rougeulle C (2014). A prominent and conserved role for YY1 in Xist transcriptional activation. Nat. Commun 5, 4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Taylor K, and Xu R-H (2012). Concise review: genomic stability of human induced pluripotent stem cells. Stem Cells 30, 22–27. [DOI] [PubMed] [Google Scholar]
- McKinney W (2010). Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference (SciPy), pp. 56–61. [Google Scholar]
- Mekhoubad S, Bock C, de Boer AS, Kiskinis E, Meissner A, and Eggan K (2012). Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 10, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M, Mitchell RR, Benoit YD, Orlando L, Reid JC, Shimada K, Davidson KC, Shapovalova Z, Collins TJ, Nagy A, and Bhatia M (2019). Human Pluripotency Is Initiated and Preserved by a Unique Subset of Founder Cells. Cell 177, 910–924.e22. [DOI] [PubMed] [Google Scholar]
- Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Müller F-J, Wang Y-C, Boscolo FS, et al. (2012). Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 10, 620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa M, Chonabayashi K, Nomura M, Tanaka A, Nakamura M, Inagaki A, Nishikawa M, Takei I, Oishi A, Tanabe K, et al. (2016). Epigenetic Variation between Human Induced Pluripotent Stem Cell Lines Is an Indicator of Differentiation Capacity. Cell Stem Cell 19, 341–354. [DOI] [PubMed] [Google Scholar]
- Ohnuki M, Tanabe K, Sutou K, Teramoto I, Sawamura Y, Narita M, Nakamura M, Tokunaga Y, Nakamura M, Watanabe A, et al. (2014). Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl. Acad. Sci. USA 111, 12426–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Patrat C, Thépot D, Peynot N, Fauque P, Daniel N, Diabangouaya P, Wolf J-P, Renard J-P, Duranthon V, and Heard E (2011). Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472, 370–374. [DOI] [PubMed] [Google Scholar]
- Panopoulos AD, Smith EN, Arias AD, Shepard PJ, Hishida Y, Modesto V, Diffenderfer KE, Conner C, Biggs W, Sandoval E, et al. (2017). Aberrant DNA Methylation in Human iPSCs Associates with MYC-Binding Motifs in a Clone-Specific Manner Independent of Genetics. Cell Stem Cell 20, 505–517.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V, Tchieu J, Karnik R, Uyeda M, Sadhu Dimashkie A, Case D, Papp B, Bonora G, Patel S, Ho R, et al. (2014). X chromosome reactivation dynamics reveal stages of reprogramming to pluripotency. Cell 159,1681–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Chen D, Liu W, Kim R, Sahakyan A, Lukianchikov A, Plath K, Jacobsen SE, and Clark AT (2016). Naive Human Pluripotent Cells Feature a Methylation Landscape Devoid of Blastocyst or Germline Memory. Cell Stem Cell 18, 323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payer B (2016). Developmental regulation of X-chromosome inactivation. Semin. Cell Dev. Biol 56, 88–99. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in {P}ython. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- Perrera V, and Martello G (2019). How Does Reprogramming to Pluripotency Affect Genomic Imprinting? Front. Cell Dev. Biol 7, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos S, Edsgärd D, Reinius B, Deng Q, Panula SP, Codeluppi S, Plaza Reyes A, Linnarsson S, Sandberg R, and Lanner F (2016). Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 165, 1012–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter SF (2016). A Tale of Two Cities: How Xist and its partners localize to and silence the bicompartmental X. Semin. Cell Dev. Biol 56, 19–34. [DOI] [PubMed] [Google Scholar]
- Pontis J, Planet E, Offner S, Turelli P, Duc J, Coudray A, Theunissen TW, Jaenisch R, and Trono D (2019). Hominoid-Specific Transposable Elements and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive Human ESCs. Cell Stem Cell 24, 724–735.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp B, Krumbiegel M, Grosch J, Sommer A, Uebe S, Kohl Z, Plötz S, Farrell M,Trautmann U, Kraus C, et al. (2018). Need for high-resolution Genetic Analysis in iPSC: Results and Lessons from the ForIPS Consortium. Sci. Rep 8, 17201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Ibeas P, Sang F, Zhu Q, Tang WWC, Withey S, Klisch D, Wood L, Loose M, Surani MA, and Alberio R (2019). Pluripotency and X chromosome dynamics revealed in pig pre-gastrulating embryos by single cell analysis. Nat. Commun 10, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonis N, Dexheimer PJ, Omberg L, Schroll R, Bush S, Huo J, Schriml L, Ho Sui S, Keddache M, Mayhew C, et al. (2016). Integrated GenomicAnalysis of Diverse Induced Pluripotent Stem Cells from the Progenitor Cell Biology Consortium. Stem Cell Reports 7, 110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Lindgreen S, and Orlando L (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz EG, Meisig J, Nakamura T, Okamoto I, Sieber A, Picard C, Borensztein M, Saitou M, Blüthgen N, and Heard E (2014). The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell 14, 203–216. [DOI] [PubMed] [Google Scholar]
- Shen Y, Matsuno Y, Fouse SD, Rao N, Root S, Xu R, Pellegrini M, Riggs AD, and Fan G (2008). X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc. Natl. Acad. Sci. USA 105, 4709–4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva SS, Rowntree RK, Mekhoubad S, and Lee JT (2008). X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc. Natl. Acad. Sci. USA 105, 4820–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Janiszewski A, De Geest N, Vanheer L, Talon I, El Bakkali M, Oh T, and Pasque V (2019). X-Chromosome Dosage Modulates Multiple Molecular and Cellular Properties of Mouse Pluripotent Stem Cells Independently of Global DNA Methylation Levels. Stem Cell Reports 12, 333–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, and Brown G (2011). DiffBind: differential binding analysis of ChIP-Seq peak data. https://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf. [Google Scholar]
- Tachibana M, Ma H, Sparman ML, Lee H-S, Ramsey CM, Woodward JS, Sritanaudomchai H, Masterson KR, Wolff EE, Jia Y, and Mitalipov SM (2012). X-chromosome inactivation in monkey embryos and pluripotent stem cells. Dev. Biol 377, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasawa K, Arai Y, Yamazaki-Inoue M, Toyoda M, Akutsu H, Umezawa A, and Nishino K (2018). DNA hypermethylation enhanced telomerase reverse transcriptase expression in human-induced pluripotent stem cells. Hum. Cell 31,78–86. [DOI] [PubMed] [Google Scholar]
- Tchieu J, Kuoy E, Chin MH, Trinh H, Patterson M, Sherman SP, Aimiuwu O, Lindgren A, Hakimian S, Zack JA, et al. (2010). Female human iPSCs retain an inactive X chromosome. Cell Stem Cell 7, 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen TW, Friedli M, He Y, Planet E, O’Neil RC, Markoulaki S, Pontis J, Wang H, Iouranova A, Imbeault M, et al. (2016). Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 79, 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda K, Takahashi K, Leung K, Okada A, Narita M, Yamada NAA, Eilertson KEE, Tsang P, Baba S, White MPP, et al. (2012). Derivation conditions impact X-inactivation status in female human induced pluripotent stem cells. Cell Stem Cell 11, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Padilla ME (2020). On transposons and totipotency. Philos. Trans. R. Soc. Lond. B Biol. Sci 375, 20190339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui B, Dow M, Skola D, and Carter H (2019). Extracting allelic read counts from 250,000 human sequencing runs in Sequence Read Archive. Pac. Symp. Biocomput 24, 196–207. [PMC free article] [PubMed] [Google Scholar]
- Vallot C, Ouimette J-F, Makhlouf M, Féraud O, Pontis J, Côme J, Martinat C, Bennaceur-Griscelli A, Lalande M, and Rougeulle C (2015). Erosion of X Chromosome Inactivation in Human Pluripotent Cells Initiates with XACT Coating and Depends on a Specific Heterochromatin Landscape. Cell Stem Cell 16, 533–546. [DOI] [PubMed] [Google Scholar]
- van Bemmel JG, Mira-Bontenbal H, and Gribnau J (2016). Cis- and trans-regulation in X inactivation. Chromosoma 125, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, et al. (2019). Welcome to the Tidyverse. J. Open Source Softw 4, 1686. [Google Scholar]
- Wutz A (2012). Epigenetic alterations in human pluripotent stem cells: a tale of two cultures. Cell Stem Cell 11, 9–15. [DOI] [PubMed] [Google Scholar]
- Wutz A, and Jaenisch R (2000). A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol. Cell 5, 695–705. [DOI] [PubMed] [Google Scholar]
- Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, and Lee JT (2013). Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 152, 727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdravkovic T, Nazor KL, Larocque N, Gormley M, Donne M, Hunkapillar N, Giritharan G, Bernstein HS, Wei G, Hebrok M, et al. (2015). Human stem cells from single blastomeres reveal pathways of embryonic or trophoblast fate specification. Development 142, 4010–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L-FF, Huynh KD, and Lee JT (2007). Perinucleolar targeting of the inactive X during S phase: evidence for a role in the maintenance of silencing. Cell 129, 693–706. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Validation time-course methylEPIC datasets have been deposited under accession number GSE167883 in the Gene Expression Omnibus (GEO). Analysis code is made available through the github repository: https://github.uconn.edu/sfp-lab/contiguous_x_erosion