

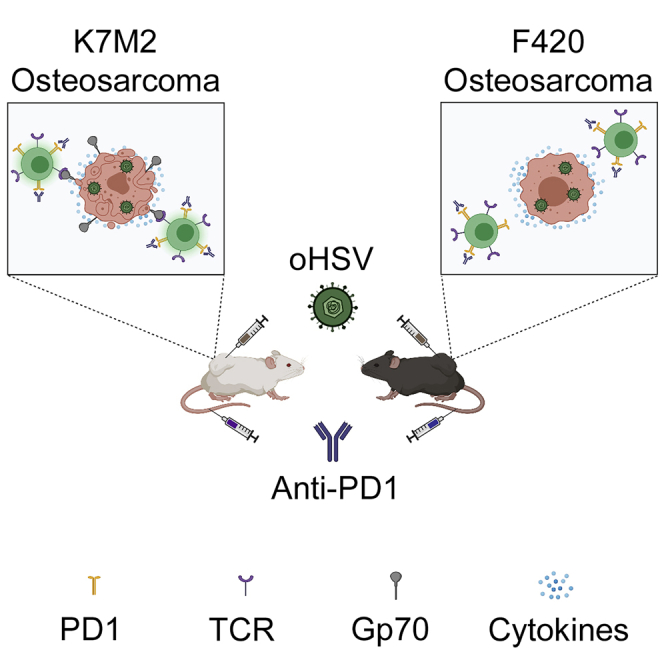
Summary
Osteosarcoma remains one of the deadliest cancers in pediatrics and young adults. We administered two types of immunotherapies, oncolytic virotherapy and immune checkpoint inhibition, to two murine osteosarcoma models and observed divergent results. Mice bearing F420 showed no response, whereas those with K7M2 showed prolonged survival in response to combination therapy. K7M2 had higher expression of immune-related genes and higher baseline immune cell infiltrates, but there were no significant differences in tumor mutational burden or predicted MHC class I binding of nonsynonymous mutations. Instead, we found several mouse endogenous retrovirus sequences highly expressed in K7M2 compared with F420. T cell tetramer staining for one of them, gp70, was detected in mice with K7M2 but not F420, suggesting that endogenous retrovirus proteins are targets for the anti-tumor immune reaction. Given prior observations of endogenous retrovirus expression in human osteosarcomas, our findings may be translatable to human disease.
Subject areas: cancer, immunology
Graphical abstract
Highlights
-
•
K7M2 but not F420 responds to viroimmunotherapy
-
•
K7M2 and F420 exhibit similar tumor mutational burdens
-
•
K7M2 shows much higher expression of endogenous retrovirus genes including gp70
-
•
Tetramer staining suggest gp70 is the immune target in K7M2
Cancer; Immunology
Introduction
Osteosarcoma is the most common bone cancer in children, adolescents, and young adults. Utilizing current standard chemotherapy and surgery, greater than 70% of patients with newly diagnosed, localized disease can achieve long-term remission (Martin and Leavey, 2015). Unfortunately, the prognosis for patients with metastatic or relapsed disease is less favorable with overall survival rates <20% despite multimodal therapy (Kager et al., 2003).
Cancer immunotherapeutics have shown promise in many cancer types including pediatrics (Wedekind et al., 2018a), and a variety of trials are underway for patients with osteosarcoma (Wedekind et al., 2018b). To date, however, robust clinical responses in these patients have been relatively elusive. A major mechanism by which many cancers evade immunity is by expression of immune checkpoints, such as programmed cell death ligand 1 (PD-L1) and its receptor PD-1 (Wolchok et al., 2010, 2013; Reck et al., 2016; Tomita et al., 2017). For osteosarcoma, PD-L1 or PD-1 expression is heterogeneous (Thanindratarn et al., 2019), with patients showing higher PD-L1 or PD-1 expression having a worse prognosis (Koirala et al., 2016; Shen et al., 2014; Lussier et al., 2015b). While these data suggest that osteosarcoma cells may be immunogenic, the exact immunologic targets are unclear, especially given the low tumor mutational burden (<10 mutations/megabase) in osteosarcoma (Chalmers et al., 2017). In a preclinical model of micrometastatic osteosarcoma, mice treated with PD-L1 inhibition alone showed prolonged survival (Lussier et al., 2015b) with even more impressive results when combined with anti-CTLA4 (Lussier et al., 2015a). Unfortunately, in phase 1 clinical trials, no clinical responses were observed in osteosarcoma patients with monotherapy checkpoint inhibition (Merchant et al., 2016; Tawbi et al., 2017), raising doubts regarding the clinical predictive utility of murine models.
An increasingly popular combination immunotherapy being tested in preclinical and clinical settings is oncolytic virotherapy with immune checkpoint inhibition, which results in higher numbers of active cytotoxic T cells in the tumor microenvironment (Ribas et al., 2018; Chesney et al., 2018; Chen et al., 2018). Virotherapy combined with checkpoint inhibition has been shown to broaden the antitumor T cell repertoire (Woller et al., 2015), and new virus constructs are being tested that express transgenes encoding checkpoint inhibitor antibodies (Haines et al., 2020). Several clinical trials of oncolytic viruses are ongoing enrolling patients with osteosarcoma (Wedekind and Cripe, 2020). We previously showed efficacy of combining oncolytic herpes simplex virus (oHSV) with anti-PD1 antibody in preclinical models of pediatric rhabdomyosarcoma (Chen et al., 2017a), although the immunologic targets of such therapies are unclear. For pediatric cancers, most of which exhibit low tumor mutational burdens (Vogelstein et al., 2013), primary target candidates are thought to be normal but aberrantly expressed fetal/developmental genes, oncogenes or cancer testis antigens (Orentas et al., 2012). More recently, expression of repetitive elements and human endogenous retrovirus (HERVs) genes have also been recognized in some adult cancers as potential immunotherapeutic targets, due to their selective epigenetic upregulation in cancer (Curty et al., 2020), including in osteosarcoma (Ho et al., 2017).
Here we report a differential response to immunotherapy in two murine osteosarcoma models. Flow cytometry analysis showed similar T cell infiltrates in response to virotherapy, and genomic analysis showed similar mutational burdens. We found higher NK cells and tumor associated macrophages in the more responsive model, K7M2, as well as higher baseline expression of immune-related genes, suggesting the presence of an immunostimulatory epitope(s). We determined that three specific murine endogenous retrovirus (MERV) genes including murine leukemia virus envelope protein gp70 (Scrimieri et al., 2013) were highly overexpressed in K7M2 but not F420. Immunotherapy stimulated expansion of anti-gp70 T cells only in K7M2, suggesting that murine endogenous retroviral sequences may be tumor-associated therapeutic targets in this model.
Results
Combination immunotherapy slows tumor growth via T cells in K7M2 but not F420 despite equal susceptibility to oncolytic virus infection
We first assessed the antitumor effects of combining intratumoral delivery of the oncolytic virus HSV1716 with an anti-PD-1 antibody. HSV1716 was derived from herpes simplex type 1 strain 17 and attenuated by deletions in both copies of the so-called “neurovirulence” gene (RL1, encoding the γ134.5 protein) (Maclean et al., 1991). We initiated treatment in syngeneic tumor-bearing animals once tumors had grown to a volume of 200-400 mm3 (Figure 1A). In the F420 model, we did not observe any effects on tumor growth or survival in any of the treatment groups compared with controls (Figure 1B; 1C). In contrast, although neither HSV1716 nor anti-PD-1 antibody alone led to tumor growth delays in the K7M2 model, we found that combination therapy was more efficacious, resulting in statistically significant improvement in overall survival compared to other groups (Figure 1D; 1E). Of note, PD-1 inhibition using the same single agent regimen showed mild efficacy in K7M2 in a micrometastatic model following intravenous infusion of cells (not shown) as previously reported by others (Lussier et al., 2015b).
Figure 1.

Combination of oHSV with anti-PD-1 antibody significantly prolongs survival in K7M2 tumor model but does not prolong survival in F420 tumor model
(A) Schematic illustrates the dosing regimens.
(B) F420 tumor volumes of mice treated with PBS (black line; n = 5), oHSV (red line; n = 5), anti-PD-1 (blue line; n = 5), and combination therapy (green line; n = 5). Mice were measured twice per week.
(C) F420 tumor model Kaplan-Meier survival curves for each treatment group. Survival data were evaluated for statistical significance with Log rank Mantel-Cox test and for tumor size in panel f with two-way ANOVA (n = 4 mice per group; ∗p ≤ 0.05).
(D) K7M2 tumor volumes of mice treated with PBS (black line; n = 5), oHSV (red line; n = 4), anti-PD-1 (blue line; n = 5), and combination therapy (green line; n = 6). Mice were measured twice per week.
(C) F420 tumor model Kaplan-Meier survival curves for each treatment group.
(E) K7M2 tumor model Kaplan-Meier survival curves for each treatment group demonstrating efficacy of combination therapy. Survival data were evaluated for statistical significance with Log rank Mantel-Cox test and for tumor size in panel f with two-way ANOVA (n = 4 mice per group; ∗p ≤ 0.05).
(F) Average tumor growth curves of mice treated as in Figure 1A but with IP administration of CD4 or CD8 T cell depleting or control antibodies every four days starting at day −1. Error bars represent standard deviation of the mean.
(G) Cell viability assay at varying MOI of HSV1716 on K7M2 (open triangle) versus F420 (closed square) at Day 3. Error bars represent standard deviation of the mean.
(H) Viral replication assay of F420 (black) and K7M2 (white) with HSV1716 MOI 0.1 at hr 2, 24, 48, and 72. Samples were run in 3 samples per time period with each sample run in triplicate for 9 samples per time period. Error bars represent standard deviation of the mean. Survival data were evaluated for statistical significance with Log rank Mantel-Cox test and for tumor size in panel f with two-way ANOVA (n = 4 mice per group; ∗p ≤ 0.05). ITu: intratumoral; PBS: phosphate-buffered solution; IP: intraperitoneal; anti-PD-1: anti-programmed death 1; ab: antibody; MOI: multiplicity of infection (pfu/cell); pfu: plaque-forming units.
To determine which T cell population(s) contributed to the efficacy of combination therapy in the K7M2 osteosarcoma model, we used antibodies to specifically deplete CD4+ and/or CD8+ T cells in K7M2 tumor-bearing mice treated with either combination therapy or PBS control (Figure 1F). Consistent with our prior results, we observed a clear distinction between combination therapy and isotype control antibody, which was lost in the mice with CD4+ and/or CD8+ depletion. These observations suggest that both populations of CD4+ and CD8+ T cells are necessary for the enhanced efficacy of combination therapy. Presumably, CD4+ cells are required to provide T helper cytokines for the CD8+ effector cells. To rule out a trivial explanation for the differences in efficacy, we found both tumor types express MHC Class I and PD-L1 by flow cytometry (not shown).
We next sought to determine the sensitivity and permissivity of both cell lines to HSV1716 infection to determine if the virolytic effect might be different between the two cell lines. At 72 hr post-infection, we noted that the HSV1716 had little cytotoxic effect on both murine osteosarcoma cell lines until the concentration of HSV1716 reached a multiplicity of infection (MOI) = 10 (Figure 1G). F420 was slightly more sensitive to HSV1716 compared to K7M2. Next, we determined the permissivity to infection from HSV1716 by measuring infectious titers recovered from cell lysates over time. Both cell lines displayed equivalent viral replication kinetics with an approximately 10-fold increase by 24 hr (Figure 1H). These findings are consistent with murine cancer cell lines being less susceptible to human HSV-1 infection compared to human cancer cell lines (neuroblastoma, malignant peripheral nerve sheath tumor) where we routinely observe 1000-fold lower HSV1716 concentrations induce cell killing and 100-1000-fold higher recovery of infectious virus (Currier et al., 2017; Wang et al., 2016). Our prior work with a different oncolytic HSV-1 also showed a similar differential between murine and human cancer cells (rhabdomyosarcoma) (Leddon et al., 2015) and that a human osteosarcoma line was highly susceptible to oncolytic HSV-1 (Currier et al., 2013).
HSV1716 induces similar immune cell infiltrates in both models
Finding no differences in cancer cell autonomous susceptibility to virus that would explain better results in K7M2, we measured immune cell recruitment in each model at baseline and following immunotherapy (Figure 2). We harvested tumors from mice 72 hr after the third and final dose of HSV1716 and the first dose of anti-PD-1 antibody and compared those to control tumors injected with phosphate buffered saline. Overall, K7M2 control tumors showed higher levels of immune cell infiltrates with more CD45 + cells, as well as NK cell and macrophages. However anti-PD1 had very little effects on any cell population in either model, HSV1716 alone or in combination showed higher CD8+ T, CD8+CD44 + T, and exhausted CD8+ T cells (PD-1+, lymphocyte activation gene (LAG)-3+, T cell immunoglobulin and mucin-domain containing (TIM)-3+) cells in both models without a corresponding increase in T-regulatory (T-reg) cells (Foxp3+CD25 + CD4+). NK cells were higher in K7M2 but did not change significantly with therapy, although they were higher in F420 after combination therapy. Tumor-associated macrophages (TAMs) were higher in K7M2 than F420 in the control group and did not change following therapy in F420, whereas they were increased with virotherapy in K7M2 but not in combination therapy.
Figure 2.

oHSV single and oHSV + anti-PD1 combination therapy results in increased CD8+ T cells without an increase in regulatory T cells, similar in both models.
Mice treated as in Figure 1A and sacrificed 3 days after the final dose of oHSV or PBS with a single dose of anti-PD-1 antibody or control antibody given. Single-cell suspensions were obtained from isolated tumors, stained, and then analyzed via flow cytometry. Data show mean and standard error of the mean (SEM, n = 4 per treatment group). Statistical analysis was performed by one-way ANOVA with Tukey-adjusted post hoc tests (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). NK: Natural killer cells; Treg: T-regulatory cells; TAM: Tumor-associated macrophage; PD-1: Programmed cell death 1; CTLA-4: Cytotoxic T-lymphocyte associated protein-4; LAG-3: lymphocyte activation gene-3; TIM-3: T cell immunoglobulin and mucin-domain containing-3.
Despite similar neoantigen characteristics, K7M2 is more immunoreactive at baseline than F420
We used RNA-seq data from untreated tumors and matched germline blood cells to identify mutations that might be neoantigens present in the tumor only and to characterize gene expression profiles in the two models. The average sequencing coverage depth for tumor samples was 80X (K7M2) and 99X (F420) and for blood samples was 89X (K7M2) and 107X (F420). After filtering (see STAR methods), the number of coding somatic variants was 327 in F420 and 380 in K7M2, representing 6.6 and 7.7 mutations per megabase, respectively, similar to numbers found in human osteosarcomas (Chalmers et al., 2017). We detected no gene fusions in F420 but found 9 in K7M2 (Figure 3A), the most notable being between YAP1 and MAML2, which has been shown to activate the hippo signaling pathway and has been described in other cancer cell lines (32). We used pVAC-Seq to identify any somatic mutations for which the corresponding expressed epitope would exhibit strong binding to MHC class I. After filtering to keep variants only with a tumor variant allele frequency ≥20%, a total of 55 unique neoantigens was predicted in F420 tumor and 45 predicted in K7M2 tumor. We considered the best neoepitope candidates as those which had strong binding affinity (≤500 nM) for MHC class I and stronger binding affinity (fold-change > 1) than the counterpart wild-type epitope. Thirty of the 45 (66.7%) in K7M2 and 38 of the 55 (69.1%) in F420 met these criteria, with a median binding affinity of 170.47 nM (range: 8.75-497.98) in F420 and 205.07 nM (range: 11.04-466.68 nM) in K7M2. None of the junctional peptides in the K7M2 fusion genes showed predicted strong binding. Thus, the two models were similar in their mutational burden and predicted neoantigen binding affinities.
Figure 3.

Comparison of F420 and K7M2 genomic and gene expression landscapes
(A) Circos plots show K7M2 contains 9 gene fusions, absent in F420. Mutations are also represented, including single nucleotide variations and insertions/deletions (indels). K7M2 has 380 mutations, while F420 has 327 mutations. Missense variants are shown in black, inframe indels are shown in blue, splice site alterations are shown in green, out of frame indels are shown in red, and nonsense mutations are shown in purple. Mutations considered to be “high coding impact” have been written along the outside of the plot, across from their associated variation; the K7M2 induced tumor contains 25 high coding impact mutations, while the F420 induced tumor has 12 high coding impact mutations.
(B) RNA-seq derived expression values of select inhibitory immune checkpoints prior to oHSV + anti-PD-1 treatment. PD-1: programmed cell death-1; PD-L1: programmed cell death ligand-1; BTLA: B and T lymphocyte associated gene; TIGIT: T cell immunoreceptor with Ig and ITIM domains HAVCR2: hepatitis A virus cellular receptor 2; LAG-3: lymphocyte activation gene 3 protein; ILDR2: immunoglobulin-like domain containing receptor 2.
(C) Circular visualization of gene ontology enrichment terms and gene expression data. Genes with fold-change > 5 in K7M2 vs. F420 were used as input for DAVID (N = 2,185). The top 10 enrichment gene ontology results for “biological processes,” based on DAVID FDR p values, are displayed above each segment of the circle. Dots represent normalized expression values for genes associated with the respective gene ontology ID term. The Z score color for each term represents overexpression (red) and underexpression (blue) of the biological process relative to K7M2.
(D) Heatmap of top 50 most differentially expressed genes between K7M2 and F420 tumor tissues. K7M2 demonstrates higher granzyme and perforin expression compared to F420. R-package heatmap was used for figure generation.
(E) Absolute immune cell proportions in K7M2 and F420 tumors. CIBERSORT values were predicted using normalized RNA-seq expression values as input and the mouse ImmuCC expression matrix as the signature gene file. Treg: T-regulatory cell; NK: Natural killer cell; DC: Dendritic cell
We also mined the RNAseq data to compare characteristics of the immune microenvironment in both tumors. In total, 13,229 protein-coding genes had transcripts assigned. We found that K7M2 had higher expression of the most common inhibitory immune checkpoints, including PD-L1, TIM3, and CD86 (B7) compared to expression in F420, whereas CD276 (B7-H3) was high in F420 (Figure 3B). We used a stringent cutoff for fold-change (absolute value of fold-change > 5) to identify any trends in differential gene expression in one mouse tumor versus the other. A total of 2,185 genes exhibited fold-change > 5 in K7M2 vs. F420. Gene ontology analysis revealed these genes were enriched for biological processes associated with immune system and inflammatory response in K7M2, with the top 10 gene ontology enrichment terms with significant FDR p values from DAVID output shown in Figure 3C. We then analyzed expression data in K7M2 to identify specific immune-related pathways or processes predicted to be activated. We used the normalized expression values and corresponding fold-changes of K7M2 vs. F420 to identify the top 50 most differentially expressed genes (i.e., largest fold-change differences between the two tumors). Using these 50 genes as input for DAVID gene ontology enrichment analysis, the only significant result returned was “cytolysis.” We plotted a heatmap of these 50 genes and observed that genes most differentially overexpressed in K7M2 were associated with cytolysis/apoptosis, specifically the granzyme/perforin pathway (Figure 3D).
We also leveraged the RNAseq data using the CIBERSORT algorithm to deconvolute immune cell populations. Similar to our flow cytometry data (Figure 2), K7M2 had a higher immune cell infiltration in the tumor microenvironment, which was primarily composed of activated NK cells and M0 and M1 macrophages (Figure 3E), consistent with our prior findings using flow cytometry. Taken together, these data support the notion that K7M2 exhibits more immune activation at baseline than F420, suggesting K7M2 harbors one or more immunogenic epitopes not accounted for by our mutational analysis. Consistent with this hypothesis is our observation that tumor establishment and growth rates are less in syngeneic than in athymic nude mice hosts for K7M2 (p = 0.0014) but equal for F420 (data not shown).
Endogenous murine retrovirus envelope gp70 is an immunotherapeutic target in gp70 but not F420
Without an identifiable mutation(s) serving as a neoantigen(s) in K7M2 that might be lacking in F420, we considered endogenous retrovirus sequences as possible candidates. We mined the RNAseq data for murine ERVs and found three genes highly over-expressed in K7M2 compared with F420 (Figure 4A). No information has been reported about two of them regarding reactivation in cancer, but the murine ERV envelope gene, gp70, is well known to be upregulated in murine cancers. We confirmed expression of gp70 mRNA in K7M2, but we were unable to detect it in F420 (Figure 4B). Using gp70 tetramer staining, we were unable to detect anti-gp70 T cells in F420 tumors, but they were readily detectable in all mice in all treatment groups in K7M2 tumors. Combination therapy with oHSV showed higher numbers than in the anti-PD1 alone treatment group. These data show that gp70 is a tumor-associated immunotherapeutic target in K7M2 but not F420, and likely explains the differential antitumor efficacy of immunotherapy between the two models.
Figure 4.

Identification of mouse endogenous retrovirus envelop gp70 as an immunotherapeutic target in K7M2 but not F420
(A) From the RNAseq data set, mouse retroviral transcripts were filtered to remove non-expressed transcripts in either sample. Remaining transcripts were log2-transformed and plotted. K7M2 showed 3 transcripts expressed above any in F420, and those top three highest expressed retroviral transcripts in K7M2 are labeled in both samples. Boxes represent mean and standard deviation of expression of all expressed retroviral transcripts.
(B) RNA was quantified for murine retrovirus envelope gp70 in both cell lines using real-time quantitative PCR. Error bar is standard error of the mean.
(C) Tetramer staining of intratumoral T cells following immunotherapy. Little or no staining was seen in F420, whereas all samples from K7M2 had detectable anti-gp70 T cells in K7M2. Lines are mean with standard error of the mean. The combination group was statistically higher than the anti-PD1 group (p = 0.028).
RNA: ribonucleic acid; gp70: glycoprotein 70.
Discussion
The treatment of osteosarcoma has remained unchanged for the last 20 years despite great efforts with current standard of care including surgery and chemotherapy. With the emerging era of immunotherapeutics, there are increasing numbers of therapies being tested for patients with osteosarcoma. Osteosarcomas variably express immune cell checkpoints (Mochizuki et al., 2019; Lussier et al., 2015b; Koirala et al., 2016), similar to what we found in K7M2 and F420. Unfortunately, PD-L1 or PD-1 expression does not convey response to checkpoint inhibition therapy alone in clinical trials (Wedekind et al., 2018b). Thus, we combined checkpoint inhibition with oncolytic virotherapy in two mouse models of osteosarcoma to elicit a more potent immune response. Indeed, we found antitumor efficacy using the combination in K7M2 but not in F420. In both models, we found higher CD8+ infiltrates following virotherapy alone and in combination with checkpoint inhibition. After analyzing nonsynonymous mutations and gene expression patterns, we ascribed the differential response between the models to the selectively aberrant over-expression in K7M2 of at least one MERV gene, confirmed by tetramer staining of anti-MERV gp70 T cells in K7M2 which were undetectable in F420.
Endogenous retrovirus sequences have long been known to be expressed in murine cancers (Stocking and Kozak, 2008) and are being increasingly recognized as drivers of human cancer (Downey et al., 2015; Curty et al., 2020; Zhang et al., 2019). An analysis of a small number of human osteosarcomas found four HERVs and two repetitive satellite genes over-expressed compared with normal bone cells (Ho et al., 2017). Whether or not the expression of these HERV proteins is immunologically analogous to gp70 and other MERV genes expressed in murine models such as K7M2 is unknown. Nevertheless, gp70 appears to be a murine example of a normal, albeit aberrantly expressed, tumor-associated antigen in a low mutational burden cancer. Because a variety of MERVs appear to be expressed at lower levels, even in F420, other MERVs may be useful in future studies to mimic similar targets at various levels of expression.
Some have suggested the use of viruses as immunoadjuvants to exogenously deliver retrovirus genes to dendritic cells in order to break tolerance to their endogenous cognate proteins (Bermejo et al., 2020). While that may be necessary for less immunogenic proteins, our study showed that the simple use of a virus infection in conjunction with checkpoint inhibition was sufficient in K7M2 to break tolerance in gp70-reactive T cells. Our negative results in F420 also suggest utility in evaluating immunotherapeutic approaches that target MERVs. With the absence of such highly expressed MERVs but many less-expressed MERVs, F420 might be ideal for testing the effectiveness of strategies designed to work with less immunogenic proteins (or at least lower expressed immunogenic proteins, e.g., gp70 in F420).
How well either of these models represents biology associated with immunotherapies in human disease remains an open question. The fact that we utilized flank-based PDX models rather than orthotopic bone or metastatic lung models (sites where disease is commonly found in humans) adds a further element of uncertainty. That said, neither responded well to checkpoint inhibitors alone in this bulky disease setting, consistent with the human experience. Also, the models differed markedly with respect to baseline immunoreactivity, reminiscent of the range of human osteosarcoma in which some lesions are devoid of T cells while others show evidence of an immune response. In fact, there is now substantial evidence that human osteosarcomas vary widely with respect to immunologic characteristics as some but not all cases harbor significant proportions of immune cells (Wu et al., 2020; Majzner et al., 2017; Koirala et al., 2016), increased T cell exhaustion markers correlate with the presence of macrophages (Han et al., 2016), the expression of immune-related genes predicts metastasis and survival (Scott et al., 2018), and low PD-L1 (Koirala et al., 2016) and high CD8/Treg (Fritzsching et al., 2015) are prognostic.
One limitation of the present study is the fact that, unlike human tumor cells, mouse tumor cells are much less susceptible to HSV-1 infection (Leddon et al., 2015). Thus, our results may not fully demonstrate the activity of oHSV via direct oncolysis or tumor immune infiltration that could occur in humans, which may affect treatment with anti-PD-1 antibody. Also, our best clinical responses were short-term stable disease, and all mice eventually showed tumor progression. While we observed an increase in PD1+ CD8+ T cells after virotherapy, we also found higher levels of TIM3+ CD8+ and LAG-3+ CD8+ cells, as well as high expression of TIM-3 mRNA in addition to PD-L1 mRNA in K7M2, so it may be necessary to inhibit those immune checkpoints as well to achieve better results. Also, B7-H3 mRNA was higher in F420 than in K7M2, suggesting it may also be a checkpoint ligand in that model. In K7M2, we also found fewer macrophages with the combination than with virotherapy alone. Because tumor-associated macrophages are known to inhibit T cell function, it is possible that the lower macrophages we detected in combination therapy in K7M2 compared with virotherapy alone contributed to the antitumor efficacy. To fully realize the potential of such immunotherapy, it may also be necessary to counteract other cellular or cytokine-mediated immunosuppression in the tumor microenvironment, as suggested by the finding of multiple different immunosuppressive mechanisms at play in different samples (Wu et al., 2020). In this regard, we previously showed virotherapy with HSV1716 is more effective in murine models of rhabdomyosarcoma when combined with a TGFβ inhibitor (Hutzen et al., 2017).
oHSVs including HSV1716 (Seprehvir) and anti-PD-1 antibody have been separately shown to be safe in the pediatric population (Streby et al., 2017; Tawbi et al., 2017). Combination therapies involving checkpoint inhibition are being conducted in the pediatric population with a few trials in different pediatric cancers (see www.clinicaltrials.gov: NCT03605589, NCT03445858, NCT03837899, NCT03130959, NCT03825367, NCT03907488). Our data suggest the combination of oHSV with checkpoint inhibition might have some activity in patients with osteosarcoma, but, analogous to K7M2, likely only in those cases with evidence of baseline immunoreactivity that implies at least a modicum of pre-existing antitumor immunity. In addition, our findings suggest ERV expression should be explored as a predictive biomarker of immunotherapy response.
Limitations of the study
The study is limited by the use of only two tumor models. In addition, both models were implanted into the flanks of animals and not orthotopically into the bones, so the microenvironments may not be authentic to native osteosarcoma, which may affect the response to immunotherapies. Finally, the models were derived from different mouse strains (C57BL/6 and BALB/c), which may also underlie a differential response to immunotherapies due to differential skewing of the immune system toward Th1 versus Th2 responses.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse PD1 (anti-CD279) | Bio-X-Cell | Clone (RMP1-14); RRID: AB_10949053 |
| Rat IgG2a isotype control antibody | Bio-X-Cell | 2A3; RRID: AB_1107769 |
| Anti-mCD4 for depletions | Bio-X-Cell | Clone GK1.5; RRID: AB_1107636 |
| FITC-anti-mCD4 for surface staining | BioLegend | Clone GK1.5; RRID: AB_312691 |
| APC-anti-CD4 for intracellular staining | BioLegend | Clone GK1.5; RRID: AB_312697 |
| Anti-mCD8 for depletions | Bio-X-Cell | Clone YTS169.4; RRID: AB_10950145 |
| PE-CY7-anti-mCD8a for staining | BioLegend | Clone 53.6-7; RRID: AB_312761 |
| Anti-Phytopthora Ig AFRC MAC 51 control antibody for depletions | In-house (L.Boon) | From hybridoma: https://www.sigmaaldrich.com/US/en/product/sigma/cb_85060405 |
| PE-anti-CD49b | BioLegend | Clone DX5; RRID: AB_313414 |
| Violet 421-anti-CD3e | BioLegend | Clone 145-2C11; RRID: AB_11203705 |
| PerCP/Cy5.5-anti-B220 | BioLegend | Clone RA3-6B2; RRID: AB_893356 |
| APC-anti-CD44 | BioLegend | Clone IM7; RRID: AB_312963 |
| PE-anti-LAG3 | BioLegend | Clone C9B7W; RRID: AB_2133343 |
| APC-anti-Tim3 | BioLegend | Clone RMT3-23; RRID: AB_2561656 |
| PE-anti-CTLA4 | BioLegend | Clone UC10-4B9; RRID: AB_313255 |
| APC-anti-PD1 | BioLegend | Clone 29F.1A12; RRID: AB_2159183 |
| PE-Cy7-anti-F4/80 | BioLegend | Clone BM8; RRID: AB_893478 |
| Violet 421-anti-CD11b | BioLegend | Clone M1/70; RRID: AB_2562904 |
| PerCP-anti-CD11b | BioLegend | Clone M1/70; RRID: AB_2129375 |
| FITC-anti-Foxp3 | eBiosciences | Clone FJK-16s; RRID: AB_465243 |
| PE-anti-CD25 | BD Sciences | Clone 7D4; RRID: AB_1645250 |
| PE-anti-PDL1 | BioLegend | Clone 10F.9G2; RRID: AB_2073556 |
| APC-anti-MHCII | BioLegend | Clone M5/114.15.2; RRID: AB_313329 |
| PE-anti-H2-Kb | eBioscience | Clone AF6-88.5.5.3; RRID: AB_10598797 |
| PE-anti-H2-Kd | eBioscience | Clone SF1-1.1.1; RRID: AB_2043875 |
| APC-anti-H2-Db | BioLegend | Clone KH95; RRID: AB_2565862 |
| FITC-anti-H2-Ld | Invitrogen | Clone 30-5-7S; RRID: AB_2539390 |
| Bacterial and virus strains | ||
| Oncolytic herpes simplex virus | Sorrento Therapeutics (San Diego, CA) | HSV1716 |
| Chemicals, peptides, and Recombinant proteins | ||
| RPMI 1640 | Thermo Fisher Scientific | A1049101 |
| DMEM | Thermo Fisher Scientific | 12430112 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | 26,140 |
| ACK red cell lysis buffer | Lonza | 10-548e |
| Mouse Fc blocking reagent | BD Biosciences | Clone 2.4G2; RRID: AB_394656 |
| Streptavidin-APC | Molecular Probes | SA1005 |
| Biotinylated H2-Ld mouse MuLV gp70 SPSYVYHQF | NIH Tetramer Core Facility (NTCF) | AH1 GP70 peptide |
| Biotinylated H2-Kb mouse MuLV gp70 KSPWFTTL | NIH Tetramer Core Facility (NTCF) | p15E GP70 peptide |
| Critical Commercial assays | ||
| SureSelectXT mouse all exon library kit | Agilent Technologies | G7550 |
| TruSeq Stranded total RNA library kit | Illumina, Inc. | 20020596 |
| MycoAlert mycoplasma detection kit | Lonza | LT07-318 |
| Cell titer 96 Aqueous non-Radioactive cell Proliferation assay | Promega | G5421 |
| RNeasy plus Mini kit | Qiagen | 74,134 |
| SuperScript II reverse Transcriptase | Life Technologies | 18064022 |
| iTaq universal SYBR green supermix kit | Bio-Rad | 1725120 |
| Cell fixation and permeabilization kit | Invitrogen | GAS001S100 and GAS002S100 |
| Experimental models: Cell lines | ||
| F420 osteosarcoma cell line | Kind gift from Jason Yustein | Cells were derived from genetically engineered mouse model reported in Zhao et al. (2015) |
| K7M2 osteosarcoma cell line | ATCC | ATCC Cat# CRL-2836, RRID:CVCL_V455 |
| Experimental models: Organisms | ||
| C57BL/6 female mice | Envigo | C57BL/6NHsd |
| BALB/c female mice | Envigo | Balb/cAnNHsd |
| Deposited data | ||
| Raw and analyzed RNA-seq data | This paper | Gene expression omnibus: GSE166282 |
| Raw tumor exome sequencing data | This paper | Sequence read archive: PRJNA698961 |
| RNA-seq data | This paper | Mendeley: https://doi.org/10.17632/cgswh7x24z.1 |
| Software and algorithms | ||
| bcl2fastq conversion software (version 2.20.0.422) | Illumina, Inc. | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software/downloads.html |
| HaplotypeCaller | Genome Analysis Toolkit (GATK) | https://gatk.broadinstitute.org/hc/en-us/articles/360037225632-HaplotypeCaller |
| Mutect2 | GATK | https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 |
| CHURCHILL | Kelly et al., (2015) | Email lead contact for more information |
| pVACseq | Hundal et al., (2016) | https://github.com/griffithlab/pVACtools |
| Salmon (version 0.9.1) | (Patro et al., 2017) | https://github.com/COMBINE-lab/salmon |
| Mus musculus reference genome | National Center for Biotechnology Information RefSeq | GRCm38.p4_rna.fna |
| Mus musculus endogenous retroviral reference sequences | GEVE | Mmus38.geve.nt_v1.fa |
| gp70 sequence | GenBank | DQ359272.1 |
| DAVID (version 6.8) | (Dennis et al., 2003) | https://david.ncifcrf.gov |
| CIBERSORT | (Newman et al., 2015) | https://cibersort.stanford.edu |
| DESeq2 (version 1.26.0) | (Love et al., 2014) | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GraphPad Prism version 7.0a | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo version 10.0.3 | Tree Star | https://www.flowjo.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Timothy P. Cripe (timothy.cripe@nationwidechildrens.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Section 1: Data. The RNA-sequencing data presented in this publication has been deposited in NCBI's Gene Expression Omnibus (GEO) and is available through accession number GSE166282. The tumor exome sequencing data presented in this publication has been deposited in NCBI's Sequence Read Archive (SRA) and is available through accession number PRJNA698961. RNA-seq expression values for endogenous retroviruses and mouse genes are available through Mendeley at https://doi.org/10.17632/cgswh7x24z.1
Section 2: Code. This paper does not report original code.
Section 3. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental models and details
Cell lines and viruses
We utilized murine osteosarcoma cell lines F420 and K7M2 (Ek et al., 2006; Zhao et al., 2015). F420 was originally derived from a female C57BL/6 mouse transgenic for a mutant p53 under control of a bone-selective promoter (Zhao et al., 2015). K7M2 was derived from a spontaneous metastatic lesion in a female BALB/c background (Ek et al., 2006) (ATCC Cat# CRL-2836, RRID:CVCL_V455). All murine osteosarcoma cell lines were maintained in Dulbecco's Modified Eagle Medium (DMEM, ThermoFisher Scientific) supplemented with 15% heat-inactivated fetal bovine serum (FBS), 1% L-glutamine, 1% nonessential amino acids, 50 μM 2-mercaptoethanol, 100 IU/mL penicillin, and 100 μg/mL streptomycin. Vero cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin. All cell lines used in this study were verified to be free of mycoplasma contamination by the MycoAlert Mycoplasma Detection Kit (LT07-318; Lonza, Allendale, NJ, USA). The HSV1716 oncolytic herpes virus was provided by Virttu Biologics (Glasgow, UK; later acquired by Sorrento Therapeutics, San Diego, CA) and maintained/propagated as previously described (Leddon et al., 2015). HSV1716 is an oncolytic herpes virus derived from HSV-1 strain 17 and attenuated by mutation in the RL1 genes encoding ICP34.5, which confers neurovirulence (11).
Animal studies
All animal studies were approved by Nationwide Children's Hospital Institutional Animal Care and Use Committee (protocol AR12-00074).
Survival studies
F420 osteosarcoma tumors were established by subcutaneously injecting 5 x 106 osteosarcoma cells into the flanks of 6-week-old C57BL/6 mice (Envigo, Frederick, MD). K7M2 tumors were established by transplanting small pieces of sectioned tumor (∼2mm3) into the flanks of 6-week-old BALB/c mice (Envigo). Both sets of mice were all female because both tumors were derived from females. The numbers of animals used in each experiment are shown in the figure legends. Mice were randomized into the different treatment groups, but groups were not blinded. Tumor sizes were measured every other day, and volumes were calculated using length x width2 x π/6, as described previously (Leddon et al., 2015). When tumors reached ∼200-400 mm3 in size, mice were pooled and divided into treatment groups to ensure comparable average tumor burdens. Animals with tumor burdens that exceeded 400 mm3 in size, or who did not reach this threshold within a week of their cohorts, were excluded from further study.
Mice were given fractionated doses of HSV1716 (1 × 108 PFU in 100 μL) or PBS intratumorally every other day for a total of 3 injections. Subsets of mice were given 250 μg of anti-PD-1 (clone RMP1-14) or control antibody (2A3) intraperitoneally twice a week for four weeks, beginning with the last dose of HSV1716 injection (Figure 1A). Tumor sizes were measured twice weekly, unblended, until they reached a volume of 2000 mm3 or a diameter of 2 cm upon which the animal was euthanized.
Immunogenicity studies
Tumors were established by subcutaneously implanting 5 x 106 F420 osteosarcoma cells or K7M2 osteosarcoma cells into the flanks of 6-week-old C57BL/6 or BALB/c mice, respectively. To compare tumor growth without treatment, tumors as above were established in the flanks of 6-week-old nude mice (Envigo).
Method details
PD-1 blockade in vivo
Anti-PD-1 antibody, RMP1-14, and isotype control antibody 2A3, purchased from Bio-X-Cell (West Lebanon, NH), were used in tumor-bearing mice for in vivo PD-1 blockade.
Cell viability (MTS) assay
F420 and K7M2 osteosarcoma cells were plated in 96-well dishes at a density of 3,000 cells/well and incubated at 37°C overnight prior to HSV1716 infection. Viability assays were conducted 96 hr post-treatment using the Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation Assay (G5421; Promega, Madison, WI, USA). Samples were run in triplicate and the data shown are representative of three independent experiments. Results are presented as percent cell survival relative to uninfected control. Error bars represent standard deviation.
In vitro virus replication assays
F420 and K7M2 osteosarcoma cells were plated in 6-well dishes at 2 x 106 cells per well and infected the following day with HSV1716 at a multiplicity of infection of 0.01 infectious virus particles per cell. The infected cells were then collected at 2, 24, 48, and 72 hr postinfection. Samples were freeze-thawed three times and serial dilutions were titrated on Vero cells by standard plaque assay (Leddon et al., 2015). Each sample was run in triplicate and the data shown are representative of three independent experiments. Results are presented as plaque-forming units (pfu) per mL.
Quantitative reverse transcription PCR
Total RNA was isolated from cultured F420 and K7M2 osteosarcoma cells using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA) according to manufacturer instructions. RNA quantity and purity were determined using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Charlotte, NC). 5 μg of total RNA was used to synthesize cDNA using SuperScript II Reverse Transcriptase (Life Technologies, Carlsbad, CA, USA) according to manufacturer instructions. Quantitative real-time PCR was performed using iTaq universal SYBR green supermix kit (Bio-Rad, Hercules, CA) with the Applied Biosystems 7900HT Fast Real-Time PCR System (Beverly Hills, CA). Cycling conditions included the initial step of 2 min of 50°C and 5 min denaturation at 95°C, followed by 40 thermal cycles of denaturation at 95°C for 15 s, annealing at 58°C for 30 s, and elongation at 72°C for 30 s. The comparative quantitation method was used, and the results are represented as fold gene expression relative to gapdh: 2-(Cttarget gene – CtGapdh). Primer sequence for gp70: 5′-AAAGTGACACATGCCCACAA-3′ (forward) and 5′-CCCCAAGAGGCACAATAGAA-3′ (reverse).
Exome sequencing and neoantigen prediction
Exome sequencing
DNA from tumor and blood (normal comparator) were prepared for exome sequencing using Agilent SureSelectXT Mouse All Exon library kit (Santa Clara, CA) and sequenced on an Illumina HiSeq 4000 Sequencing System (San Diego, CA). Reads were aligned to Mus musculus genome reference sequence, build GRCm38 (mm10), and evaluated using a custom in-house pipeline (Kelly et al., 2015). Germline variants were called using the Genome Analysis Toolkit's Haplotypecaller, and somatic variants were called using MuTect2 (Cibulskis et al., 2013). All somatic variants were further filtered for the following: a) variant absent in blood comparator, b) minimum coverage depth in normal and tumor ≥10X, c) number of supporting alternate reads in tumor ≥4, and d) MuTect2 filter = PASS. Somatic variants passing all filters were then annotated using Variant Effect Predictor tool and further filtered to keep only those within exons or within 3 base pairs of a canonical splice site. CIRCOS software package was used to generate circular genome mapping figures (Krzywinski et al., 2009).
pVAC-seq
Personalized variant antigens by cancer sequencing (pVAC-Seq) was used to predict somatic neoantigens (Hundal et al., 2016). Mouse-specific MHC genotypes and a list of non-synonymous somatic mutations annotated with amino acid and transcript sequences for each tumor sample was used as input for the pVAC-Seq pipeline. Binding affinities of all predicted normal and tumor epitopes were predicted with NetMHC and PickPocket (Lundegaard et al., 2008; Zhang et al., 2009). The strongest predicted binding affinity (lowest nM value) from both tools was used to calculate corresponding fold-change compared to predicted binding affinity of the normal, wildtype epitope. All predicted tumor neoantigens were filtered to keep only variants with: tumor variant DNA allele frequency ≥20%.
RNA sequencing and gene expression analysis of tumor tissue
RNA-seq
Total RNA was extracted from tumor tissue without treatment and treated with DNAse and RiboZero capture beads (Illumina, San Diego, CA) to deplete DNA and ribosomal contaminants, respectively. RNA was then used as input for Illumina TruSeq Stranded Total RNA library preparation kit and sequenced on HiSeq 4000. Sequencing reads were processed using STAR-Fusion (https://github.com/STAR-Fusion) to detect putatively expressed gene fusions. Normalization and transcript abundance determination was performed using DESeq packages and a custom in-house pipeline (Kelly et al., 2015).
Gene ontology analysis
Gene Ontology enrichment analysis was performed using DAVID online functional annotation tool (https://david.ncifcrf.gov/), and R-package GOplot was used for data visualization of the results (Walter et al., 2015).
CIBERSORT
The CIBERSORT online tool (https://cibersort.stanford.edu) was used to predict abundance of immune cell types using our RNA-seq gene expression normalized values as input mixture file (Newman et al., 2015). For the signature gene file, we used ImmuCC, a published matrix of microarray expression values for 25 immune cell types from mouse tissue (Chen et al., 2017b).
Cell depletion studies
T cell depletion studies were conducted as previously described (Hutzen et al., 2017). In short, BALB/c mice bearing biflank K7M2 tumors were given intraperitoneal injection of 500 μg of anti-CD4 (GK1.5) and/or anti-CD8 (YTS169.4) antibody every 96 hr. Isotype control mice were similarly injected with 500 μg of anti-Phytopthora Ig AFRC MAC 51 antibody. Specific depletion of the respective T cell subtypes was confirmed by flow cytometry analysis.
Flow cytometry analysis
Single tumor cell preparation and flow cytometric analyses were conducted as described previously (Chen et al., 2017a). In brief, single-cell suspensions from tumors were prepared and lysed with ACK red cell lysis buffer (Lonza) and blocked with 5% mouse Fc blocking reagent (2.4G2; BD Biosciences, San Jose, CA) in FACS buffer (1% FBS and 1 mM EDTA in PBS). Cells were labeled on ice for 30 min with one of the following antibody staining panels. For T and NK cell analysis: CD4-fluorescein isothiocyanate (FITC) (GK1.5), CD49b-phycoerythrin (PE) (DX5), CD8a-PE-Cy7 (53-6.7), CD3e-Violet 421 (145-2C11), B220-PerCP/Cy5.5 (RA3-6B2) and CD44-allophycocyanin (APC) (IM7). For T cell exhaustion markers: CD4-FITC, CD8a-PE-Cy7, CD3e-Violet 421 and B220-PerCP/Cy5.5 with LAG3-PE (C9B7W) and Tim3-APC (RMT3-23) or CTLA4-PE (UC10-4B9) and PD-1-APC (29F.1A12). For tumor associated macrophages: F4/80-PE-Cy7 (BM8) and CD11b-Violet 421 (M1/70). These cells were washed one time with FACS buffer after labeling and fixed in 1% paraformaldehyde. For Foxp3 intracellular staining, mononuclear cells were enriched by Percoll (GE Healthcare Bio-Sciences, Pittsburgh, PA) density gradient centrifugation and stained with cell surface markers including CD4-APC, CD8-PE-Cy7, CD25-PE (7D4), CD11b-PerCP and CD3e-Violet 421 followed by Foxp3-FITC (FJK-16 s) intracellular staining using a cell fixation and permeabilization kit (Invitrogen GAS001S100 and GAS002S100; Thermo Fischer Scientific). BALB/c MuLV GP70 biotinylated monomer (H2-Ld-SPSYVYHQF) and C57B6 MuLV GP70 biotinylated monomer (H2-Kb-KSPWFTTL) were obtained from the NIH tetramer core facility (http://tetramer.yerkes.emory.edu/, human B2M). To produce APC-conjugated GP70 tetramer, each monomer was tetramerized using streptavidin-APC (SA1005, Molecular Probes, Eugene, OR) at a ratio of 1:3 monomer to streptavidin-APC. 10% of the total volume of streptavidin-APC was added to the monomer every 15 min up to 2 hr on ice.
Stained cells were washed one time with FACS buffer and fixed in 0.5% paraformaldehyde. A minimum of 100,000 events were collected and analyzed on a BD FACS LSR II (BD Biosciences). Analysis was carried out using FlowJo software, version 10.0.3 (Tree Star, Ashland, OR).
For surface staining on two murine osteosarcoma tumor cells, adherent tumor cells were trypsinized and washed one time with FACS buffer. Tumor cells were stained with PD-L1-PE (10F.9G2), MHC II-APC (M5/114.15.2), H2-Kb-PE (AF6-88.5.5.3), H2-Kd-PE (SF1-1.1.1), H2-Db-APC (KH95) or H2-Ld-FITC (30-5-7S) individually on ice for 30 min. Stained cells were wash one time with FACS buffer and fixed with 1% paraformaldehyde but otherwise collected and analyzed as described above. All the staining antibodies were purchased from BioLegend (San Diego, CA, USA) except for anti-Foxp3 (eBioscience, San Diego, CA, USA), anti-H2-Ld (Invitrogen), anti-H2-Kb (eBioscience), anti-H2-Kd (eBioscience) and anti-CD25 (BD Sciences).
Retroviral transcript quantification
We generated normalized expression transcript per million (TPM) values as follows: RNA-seq FASTQ files from K7M2 and F420 tumors were used as input for Salmon (version 0.9.1) in mapping-based mode (Patro et al., 2017). For the salmon reference transcriptome, we downloaded the mouse retroviral FASTA Mmus38.geve.nt_v1.fa from the genome-based endogenous viral element database (gEVE) and then manually appended the gp70 (GenBank: DQ359272.1) sequence to the retroviral FASTA before indexing for Salmon use (Nakagawa and Takahashi, 2016). We used standard Salmon parameters with bootstrapping set to 100. TPM values were transformed using log2(TPM +1) calculation.
Quantification and statistical analysis
All statistical analysis was performed using the GraphPad Prism software 7.0a for Mac OS x (GraphPad Software, La Jolla, CA, USA). Kaplan-Meier curves and corresponding log rank Mantel-Cox tests were used to evaluate the statistical differences between groups in survival studies. One-way ANOVA was utilized to assess the statistical difference between groups in the flow cytometry analysis. The T cell depletion studies were analyzed for statistical significance utilizing two-way ANOVA.
Acknowledgments
We thank Jason Yustein (Texas Children's Hospital, Baylor College of Medicine, Houston, Texas) for F420 cells, Julia Love for help with viral preparation and the veterinary staff for animal support. This work was supported by the National Cancer Institute Cancer Moonshot Award U54-CA232561-01A1, St. Baldrick's Foundation, Hyundai Hope On Wheels, Cancer Free Kids Pediatric Cancer Research Alliance and by funds from the Abigail Wexner Research Institute at Nationwide Children's Hospital, the Ohio State University Comprehensive Cancer Center, and the National Institutes of Health under grant number P30 CA016058. We thank the Genomics Shared Resource at The Ohio State University Comprehensive Cancer Center, Columbus, OH.
Author contributions
Conceptualization, M.F.W. and T.P.C.; methodology, M.F.W., K.E.M., C.Y.C., P.Y.W., B.J.H., M.A.C., and T.P.C.; validation, M.F.W., C.Y.C., and M.A.C.; formal analysis, M.F.W., K.E.M., C.Y.C., R.D.R., S.L., B.J.K., D.G., P.W., E.R.M., and T.P.C.; investigation, M.F.W., K.E.M., C.Y.C., P.Y.W., B.J.H., M.A.C., B.N., S.L., B.J.K., D.G., P.W., and E.R.M.; resources, R.D.R., L.B., and J.C.; data curation, M.F.W., K.E.M., C.Y.C., S.L., B.J.K., D.G., and P.W.; writing – original draft, M.F.W., K.E.M., B.J.H., and T.P.C.; writing – review & editing, M.F.W., K.E.M., B.J.H., R.D.R., L.B., J.C., E.R.M., and T.P.C.; visualization, M.F.W., K.E.M., C.Y.C., B.J.H., M.A.C., and T.P.C.; supervision, T.P.C. and E.R.M.; project administration, M.F.W., K.E.M., M.A.C., E.R.M., and T.P.C.; funding acquisition, E.R.M. and T.P.C.
Declarations of interests
J.C. was an employee of Virttu Biologics Ltd (later acquired by Sorrento) during this study. The rest of the authors declare that they have no potential conflicts of interest.
Published: July 23, 2021
References
- Bermejo A., Ragonnaud E., Daradoumis J., Holst P. Cancer associated endogenous retroviruses: ideal immune targets for adenovirus-based immunotherapy. Int. J. Mol. Sci. 2020;21:1–21. doi: 10.3390/ijms21144843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers Z.R., Connelly C.F., Fabrizio D., Gay L., Ali S.M., Ennis R., Schrock A., Campbell B., Shlien A., Chmielecki J. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Hutzen B., Wedekind M.F., Cripe T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother. 2018;7:65–77. doi: 10.2147/OV.S145532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Wang P.Y., Hutzen B., Sprague L., Swain H.M., Love J.K., Stanek J.R., Boon L., Conner J., Cripe T.P. Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci. Rep. 2017;7:2396. doi: 10.1038/s41598-017-02503-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Huang A., Sun J., Jiang T., Qin F.X., Wu A. Inference of immune cell composition on the expression profiles of mouse tissue. Sci. Rep. 2017;7:40508. doi: 10.1038/srep40508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesney J., Puzanov I., Collichio F., Singh P., Milhem M.M., Glaspy J., Hamid O., Ross M., Friedlander P., Garbe C. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J. Clin. Oncol. 2018;36:1658–1667. doi: 10.1200/JCO.2017.73.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currier M.A., Eshun F.K., Sholl A., Chernoguz A., Crawford K., Divanovic S., Boon L., Goins W.F., Frischer J.S., Collins M.H. VEGF blockade enables oncolytic cancer virotherapy in part by modulating intratumoral myeloid cells. Mol. Ther. 2013;21:1014–1023. doi: 10.1038/mt.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currier M.A., Sprague L., Rizvi T.A., Nartker B., Chen C.Y., Wang P.Y., Hutzen B.J., Franczek M.R., Patel A.V., Chaney K.E. Aurora A kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget. 2017;8:17412–17427. doi: 10.18632/oncotarget.14885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curty G., Marston J.L., De Mulder Rougvie M., Leal F.E., Nixon D.F., Soares M.A. Human endogenous retrovirus K in cancer: a potential biomarker and immunotherapeutic target. Viruses. 2020;12:726. doi: 10.3390/v12070726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey R.F., Sullivan F.J., Wang-Johanning F., Ambs S., Giles F.J., Glynn S.A. Human endogenous retrovirus K and cancer: innocent bystander or tumorigenic accomplice? Int. J. Cancer. 2015;137:1249–1257. doi: 10.1002/ijc.29003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek E.T., Dass C.R., Choong P.F. Commonly used mouse models of osteosarcoma. Crit. Rev. Oncol. Hematol. 2006;60:1–8. doi: 10.1016/j.critrevonc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Fritzsching B., Fellenberg J., Moskovszky L., Sapi Z., Krenacs T., Machado I., Poeschl J., Lehner B., Szendroi M., Bosch A.L. CD8(+)/FOXP3(+)-ratio in osteosarcoma microenvironment separates survivors from non-survivors: a multicenter validated retrospective study. Oncoimmunology. 2015;4:e990800. doi: 10.4161/2162402X.2014.990800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines B.B., Denslow A., Grzesik P., Lee J.S., Farkaly T., Hewett J., Wambua D., Kong L., Behera P., Jacques J. ONCR-177, an oncolytic HSV-1 designed to potently activate systemic antitumor immunity. Cancer Immunol. Res. 2020;9:291–308. doi: 10.1158/2326-6066.CIR-20-0609. [DOI] [PubMed] [Google Scholar]
- Han Q., Shi H., Liu F. CD163(+) M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int. Immunopharmacol. 2016;34:101–106. doi: 10.1016/j.intimp.2016.01.023. [DOI] [PubMed] [Google Scholar]
- Ho X.D., Nguyen H.G., Trinh L.H., Reimann E., Prans E., Koks G., Maasalu K., Le V.Q., Nguyen V.H., Le N.T.N. Analysis of the expression of repetitive DNA elements in osteosarcoma. Front. Genet. 2017;8:193. doi: 10.3389/fgene.2017.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundal J., Carreno B.M., Petti A.A., Linette G.P., Griffith O.L., Mardis E.R., Griffith M. pVAC-Seq: a genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016;8:11. doi: 10.1186/s13073-016-0264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutzen B., Chen C.Y., Wang P.Y., Sprague L., Swain H.M., Love J., Conner J., Boon L., Cripe T.P. TGF-beta inhibition improves oncolytic herpes viroimmunotherapy in murine models of rhabdomyosarcoma. Mol. Ther. Oncolytics. 2017;7:17–26. doi: 10.1016/j.omto.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kager L., Zoubek A., Potschger U., Kastner U., Flege S., Kempf-Bielack B., Branscheid D., Kotz R., Salzer-Kuntschik M., Winkelmann W. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J. Clin. Oncol. 2003;21:2011–2018. doi: 10.1200/JCO.2003.08.132. [DOI] [PubMed] [Google Scholar]
- Kelly B.J., Fitch J.R., Hu Y., Corsmeier D.J., Zhong H., Wetzel A.N., Nordquist R.D., Newsom D.L., White P. Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics. Genome Biol. 2015;16:6. doi: 10.1186/s13059-014-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala P., Roth M.E., Gill J., Piperdi S., Chinai J.M., Geller D.S., Hoang B.H., Park A., Fremed M.A., Zang X., Gorlick R. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016;6:30093. doi: 10.1038/srep30093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leddon J.L., Chen C.Y., Currier M.A., Wang P.Y., Jung F.A., Denton N.L., Cripe K.M., Haworth K.B., Arnold M.A., Gross A.C. Oncolytic HSV virotherapy in murine sarcomas differentially triggers an antitumor T-cell response in the absence of virus permissivity. Mol. Ther. Oncolytics. 2015;1:14010. doi: 10.1038/mto.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundegaard C., Lamberth K., Harndahl M., Buus S., Lund O., Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008;36:W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier D.M., Johnson J.L., Hingorani P., Blattman J.N. Combination immunotherapy with alpha-CTLA-4 and alpha-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J. Immunother. Cancer. 2015;3:21. doi: 10.1186/s40425-015-0067-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier D.M., O'neill L., Nieves L.M., Mcafee M.S., Holechek S.A., Collins A.W., Dickman P., Jacobsen J., Hingorani P., Blattman J.N. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J. Immunother. 2015;38:96–106. doi: 10.1097/CJI.0000000000000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean A.R., Ul-Fareed M., Robertson L., Harland J., Brown S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the 'a' sequence. J. Gen. Virol. 1991;72:631–639. doi: 10.1099/0022-1317-72-3-631. [DOI] [PubMed] [Google Scholar]
- Majzner R.G., Simon J.S., Grosso J.F., Martinez D., Pawel B.R., Santi M., Merchant M.S., Geoerger B., Hezam I., Marty V. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer. 2017;123:3807–3815. doi: 10.1002/cncr.30724. [DOI] [PubMed] [Google Scholar]
- Martin A.A., Leavey P.J. Signs and symptoms. In: CRIPE T.P., Y.N., editors. Malignant Pediatric Bone Tumors - Treatment & Malignant. Springer International Publishing; 2015. pp. 1–7. [Google Scholar]
- Merchant M.S., Wright M., Baird K., Wexler L.H., Rodriguez-Galindo C., Bernstein D., Delbrook C., Lodish M., Bishop R., Wolchok J.D. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin. Cancer Res. 2016;22:1364–1370. doi: 10.1158/1078-0432.CCR-15-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K., Kawana S., Yamada S., Muramatsu M., Sano H., Kobayashi S., Ohara Y., Takahashi N., Hakozaki M., Yamada H. Various checkpoint molecules, and tumor-infiltrating lymphocytes in common pediatric solid tumors: possibilities for novel immunotherapy. Pediatr. Hematol. Oncol. 2019;36:17–27. doi: 10.1080/08880018.2019.1578843. [DOI] [PubMed] [Google Scholar]
- Nakagawa S., Takahashi M.U. gEVE: a genome-based endogenous viral element database provides comprehensive viral protein-coding sequences in mammalian genomes. Database (Oxford) 2016;2016:baw087. doi: 10.1093/database/baw087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orentas R.J., Lee D.W., Mackall C. Immunotherapy targets in pediatric cancer. Front. Oncol. 2012;2:1–16. doi: 10.3389/fonc.2012.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck M., Rodriguez-Abreu D., Robinson A.G., Hui R., Csoszi T., Fulop A., Gottfried M., Peled N., Tafreshi A., Cuffe S. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- Ribas A., Dummer R., Puzanov I., Vanderwalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031–1032. doi: 10.1016/j.cell.2018.07.035. [DOI] [PubMed] [Google Scholar]
- Scott M.C., Temiz N.A., Sarver A.E., Larue R.S., Rathe S.K., Varshney J., Wolf N.K., Moriarity B.S., O'brien T.D., Spector L.G. Comparative transcriptome analysis quantifies immune cell transcript levels, metastatic progression, and survival in osteosarcoma. Cancer Res. 2018;78:326–337. doi: 10.1158/0008-5472.CAN-17-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrimieri F., Askew D., Corn D.J., Eid S., Bobanga I.D., Bjelac J.A., Tsao M.L., Allen F., Othman Y.S., Wang S.C., Huang A.Y. Murine leukemia virus envelope gp70 is a shared biomarker for the high-sensitivity quantification of murine tumor burden. Oncoimmunology. 2013;2:e26889. doi: 10.4161/onci.26889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J.K., Cote G.M., Choy E., Yang P., Harmon D., Schwab J., Nielsen G.P., Chebib I., Ferrone S., Wang X. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol. Res. 2014;2:690–698. doi: 10.1158/2326-6066.CIR-13-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocking C., Kozak C.A. Murine endogenous retroviruses. Cell. Mol. Life Sci. 2008;65:3383–3398. doi: 10.1007/s00018-008-8497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streby K.A., Geller J.I., Currier M.A., Warren P.S., Racadio J.M., Towbin A.J., Vaughan M.R., Triplet M., Ott-Napier K., Dishman D.J. Intratumoral injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin. Cancer Res. 2017;23:3566–3574. doi: 10.1158/1078-0432.CCR-16-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawbi H.A., Burgess M., Bolejack V., Van Tine B.A., Schuetze S.M., Hu J., D'angelo S., Attia S., Riedel R.F., Priebat D.A. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18:1493–1501. doi: 10.1016/S1470-2045(17)30624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanindratarn P., Dean D.C., Nelson S.D., Hornicek F.J., Duan Z. Advances in immune checkpoint inhibitors for bone sarcoma therapy. J. Bone Oncol. 2019;15:100221. doi: 10.1016/j.jbo.2019.100221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita Y., Fukasawa S., Shinohara N., Kitamura H., Oya M., Eto M., Tanabe K., Kimura G., Yonese J., Yao M. Nivolumab versus everolimus in advanced renal cell carcinoma: Japanese subgroup analysis from the CheckMate 025 study. Jpn. J. Clin. Oncol. 2017;47:639–646. doi: 10.1093/jjco/hyx049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., Papadoopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter W., Sanchez-Cabo F., Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–2914. doi: 10.1093/bioinformatics/btv300. [DOI] [PubMed] [Google Scholar]
- Wang P.Y., Swain H.M., Kunkler A.L., Chen C.Y., Hutzen B.J., Arnold M.A., Streby K.A., Collins M.H., Dipasquale B., Stanek J.R. Neuroblastomas vary widely in their sensitivities to herpes simplex virotherapy unrelated to virus receptors and susceptibility. Gene Ther. 2016;23:135–143. doi: 10.1038/gt.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind M.F., Cripe T.P. Oncolytic viruses and their potential as a therapeutic opportunity in osteosarcoma. Adv. Exp. Med. Biol. 2020;1258:77–89. doi: 10.1007/978-3-030-43085-6_5. [DOI] [PubMed] [Google Scholar]
- Wedekind M.F., Denton N.L., Chen C.Y., Cripe T.P. Pediatric cancer immunotherapy: opportunities and challenges. Paediatr. Drugs. 2018;20:395–408. doi: 10.1007/s40272-018-0297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind M.F., Wagner L.M., Cripe T.P. Immunotherapy for osteosarcoma: where do we go from here? Pediatr. Blood Cancer. 2018;65:e27227. doi: 10.1002/pbc.27227. [DOI] [PubMed] [Google Scholar]
- Wolchok J.D., Kluger H., Callahan M.K., Postow M.A., Rizvi N.A., Lesokhin A.M., Segal N.H., Ariyan C.E., Gordon R.A., Reed K. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok J.D., Neyns B., Linette G., Negrier S., Lutzky J., Thomas L., Waterfield W., Schadendorf D., Smylie M., Guthrie T., Jr. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- Woller N., Gurlevik E., Fleischmann-Mundt B., Schumacher A., Knocke S., Kloos A.M., Saborowski M., Geffers R., Manns M.P., Wirth T.C. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol. Ther. 2015;23:1630–1640. doi: 10.1038/mt.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.C., Beird H.C., Andrew Livingston J., Advani S., Mitra A., Cao S., Reuben A., Ingram D., Wang W.L., Ju Z. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020;11:1008. doi: 10.1038/s41467-020-14646-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Lund O., Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics. 2009;25:1293–1299. doi: 10.1093/bioinformatics/btp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Liang J.Q., Zheng S. Expressional activation and functional roles of human endogenous retroviruses in cancers. Rev. Med. Virol. 2019;29:e2025. doi: 10.1002/rmv.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Kurenbekova L., Gao Y., Roos A., Creighton C.J., Rao P., Hicks J., Man T.K., Lau C., Brown A.M. NKD2, a negative regulator of Wnt signaling, suppresses tumor growth and metastasis in osteosarcoma. Oncogene. 2015;34:5069–5079. doi: 10.1038/onc.2014.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Section 1: Data. The RNA-sequencing data presented in this publication has been deposited in NCBI's Gene Expression Omnibus (GEO) and is available through accession number GSE166282. The tumor exome sequencing data presented in this publication has been deposited in NCBI's Sequence Read Archive (SRA) and is available through accession number PRJNA698961. RNA-seq expression values for endogenous retroviruses and mouse genes are available through Mendeley at https://doi.org/10.17632/cgswh7x24z.1
Section 2: Code. This paper does not report original code.
Section 3. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.