

Abstract
Down syndrome (DS) is a genetic disorder caused by the triplication of human chromosome 21, which results in neurological and physiological pathologies. These deficits increase during aging and are exacerbated by cognitive decline and increase of Alzheimer’s disease (AD) neuropathology. A nontoxic, noninvasive treatment, maternal choline supplementation (MCS) attenuates cognitive decline in mouse models of DS and AD. To evaluate potential underlying mechanisms, laser capture microdissection of individual neuronal populations of MCS offspring was performed, followed by RNA sequencing and bioinformatic inquiry. Results at ~6 months of age (MO) revealed DS mice (the well-established Ts65Dn model) have significant dysregulation of select genes within the Type 2 Diabetes Mellitus (T2DM) signaling pathway relative to normal disomic (2N) littermates. Accordingly, we interrogated key T2DM protein hormones by ELISA assay in addition to gene and encoded protein levels in the brain. We found dysregulation of adiponectin (APN) protein levels in the frontal cortex of ~6 MO trisomic mice, which was attenuated by MCS. APN receptors also displayed expression level changes in response to MCS. APN is a potential biomarker for AD pathology and may be relevant in DS. We posit that changes in APN signaling may be an early marker of cognitive decline and neurodegeneration.
Keywords: adiponectin, type II diabetes mellitus, Down syndrome, Alzheimer’s disease, trisomy, selective vulnerability
1. Introduction
Triplication of human chromosome 21 (HSA21) in utero causes Down Syndrome (DS), the primary genetic cause of intellectual disability (ID). DS occurs in an estimated 1 in 700 live births, and individuals with DS have multiple systemic deficits, including heart conditions, increased likelihood of leukemia and epilepsy, premature aging, and neurological deficits [1,2,3,4,5]. In recent decades, the lifespan of these individuals has significantly increased, although their healthspan still lags behind [1,4,6,7]. ID in individuals with DS is thought to be due to neurodevelopmental alterations in utero, including gray matter volume reductions, decreased neuronal cell densities, disorganization of laminar structures, astrocyte proliferation and hypertrophy [8]. DS pathology results in dysfunction in motor skills, hippocampal-dependent learning and memory, attentional function, language and communication [9], and cumulates in early onset of Alzheimer’s disease (AD) neuropathology by the mid-third decade of life [3,10,11,12,13,14,15,16]. The majority of adults with DS exhibit AD-like pathology, including neurofibrillary tangles, senile plaques, synaptic dysfunction and basal forebrain cholinergic neuron (BFCN) degeneration [3,10,11,12,13,14,15,16]. To facilitate mechanistic understanding and treatment development, several mouse models of DS and AD have been generated, the most well studied of which is the Ts65Dn (Ts) mouse model [17]. The Ts model recapitulates many of the neurological deficits seen in DS and AD, including hippocampal-dependent learning and memory deficits, BFCN degeneration and septo-hippocampal circuit dysfunction, notably CA1 pyramidal neuron and choline acetyltransferase (ChAT) activity deficits [18,19,20,21,22,23].
In addition to neurological deficits in aging individuals with DS, a higher incidence of metabolic syndrome/insulin resistance (MetS/IR) [24,25] and diabetes mellitus (DM) is observed in both children and adults [26]. A significant proportion of individuals with DS are classified as having adult onset or Type II Diabetes Mellitus (T2DM) [27,28]. MetS/IR as well as T2DM are recognized as risk factors for dementia associated with AD [29]. The protein hormones insulin, leptin and adiponectin (APN) are linked to MetS/IR, T2DM and the associated increased risk for AD pathology [30,31,32,33]. Studies in persons with DS have also shown dysregulation of these protein hormones [24,34]. Ts65Dn mice display MetS/IR pathology in peripheral studies, including increased glucose and decreased insulin levels [35,36]. However, similar studies focusing on the brain have not been performed to date. In brain, insulin is involved in neuronal growth, repair and signaling, and AD subjects display MetS/IR, often in the absence of frank T2DM [37]. In brain, leptin acts on neurons in the hypothalamus, hippocampus and brainstem to equilibrate glucose and lipid metabolism [38]. Rodent studies have shown in the aging brain reduction in the leptin response [39]. Both in vivo and in vitro application of leptin has beneficial effects in neurodegeneration models [40,41,42,43]. APN conveys neuroprotective effects in the brain, including protecting against ischemic brain injury and excitotoxicity [44,45,46], regulation of neurogenesis and proliferation in the hippocampus [47]. APN deficiency using APN−/+ and APN−/− mice leads to reduced dendritic growth and spine density of granule cells in the dentate gyrus of the hippocampus [48]. Knockdown of APN in the brains of aged mice resulted in AD-like pathology and memory impairments [49], indicating that APN may play a critical role in neurodegeneration associated with AD. Studies in the periphery report conflicting findings on APN levels in children [24,50], and adults with DS [51,52,53,54,55,56]. Waragai et al. 2016, linked lower cerebrospinal fluid APN levels in AD with poor cognitive performance [52]. No studies to date have examined APN levels in the brain from individuals with DS or within mouse models of DS.
Treating individuals with DS, as well as ameliorating cognitive decline associated with AD, are major public health issues. The most widely used FDA approved therapeutics for DS and AD are acetylcholinesterase inhibitors, which modulate cholinergic neurotransmission [57]. However, these drugs are ineffective at preventing disease progression and only treat symptoms. Thus, other therapeutic approaches are indicated and there is a current unmet need.
Choline is an essential nutrient which is critical for biosynthesis of the neurotransmitter acetylcholine, is a key substrate of the phosphatidylethanolamine N-methyltransferase (PEMT) pathway, and is the primary dietary source of methyl donors [9,58]. Choline has been shown in rodent models to be essential for fetal brain development, with high levels of choline required for proper neural tube closure and brain development, often depleting maternal stores [43]. A potential treatment modality, maternal choline supplementation (MCS), in which increased levels of choline is delivered to developing pups during gestation and lactation, improves cognitive behaviors in several rodent models [9,58,59]. MCS in the Ts mouse model of DS and AD provides benefits including improved spatial cognition and attentional function, protects BFCNs, and normalizes hippocampal neurogenesis in adult offspring [9,23,60,61,62,63,64,65]. MCS increases ChAT-immunoreactive fiber staining level intensity in the hippocampus prior to BFCN degeneration in trisomic mice [61]. MCS also protects the cholinergic system in an amyloid-beta precursor protein (APP) overexpression model of AD [66]. Several studies using rodent models of neurodevelopmental disorders have demonstrated behavioral and morphologic benefits of MCS [67,68,69,70,71,72], indicating that MCS is neuroprotective and improves behavioral outcomes in multiple disease models. Further, from a translational perspective, MCS administered during the 3rd trimester in normal human pregnancies positively impacts several behaviors in normal infants, including increased performance in attaining developmental milestones [73]. In human plasma samples, decreased choline and choline metabolites were seen in T2DM/obese IR patients compared to obese insulin sensitive individuals, indicating that higher choline and metabolites may confer decreased risk of MetS/IR [74].
We previously identified the T2DM pathway as dysregulated within BFCNs in the Ts mouse at ~6 months of age (MO) [75]. To investigate underlying mechanisms driving T2DM dysregulation in DS, we examined protein hormones and receptors within the T2DM pathway in Ts brain compared to normal disomic (2N) littermates. We postulate dysregulation in the T2DM pathway may be attenuated by MCS. Accordingly, we tested levels of APN, insulin and leptin in the frontal cortex (Fr Ctx) of Ts and 2N littermates. We also examined the effects of MCS on these key protein hormones and APN receptors.
2. Materials and Methods
2.1. Mice and Maternal Diet Protocol
Animal protocols were approved by the Nathan Kline Institute/NYU Grossman School of Medicine Institutional Animal Care and Use Committee (IACUC) in accordance with NIH guidelines. Breeder pairs (female Ts65Dn and male C57Bl/6J Eicher x C3H/HeSnJ F1 mice) were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and mated at the Nathan Kline Institute. Breeder pairs were assigned to receive one of two choline-controlled experimental diets: (i) control rodent diet containing 1.1 g/kg choline chloride (AIN-76A; Dyets Inc., Bethlehem, PA, USA), or (ii) choline-supplemented diet containing 5.0 g/kg choline chloride (AIN-76A; Dyets Inc.), as described previously [23,62,63]. The choline-supplemented diet provides approximately 4.5 times the concentration of choline consumed by the controls and is within the normal physiological range [76]. The control diet supplies an adequate level of choline, so the offspring are not choline deficient. Mice were given ad libitum food and water access [77,78]. Mice were maintained on a 12-h light-dark cycle under temperature- and humidity-controlled conditions. Pups born to choline supplemented (Ts+ and 2N+) or unsupplemented maternal choline (Ts and 2N) dams were weaned on postnatal day 21 and provided ad libitum access to water and the control diet. MCS and unsupplemented pups were housed upon weaning in standard cages (n = 2–4 mice per cage) containing paper bedding and several objects for enrichment (e.g., plastic igloo, t-tube and cotton square). Tail clips were taken and genotyped [79] at weaning and mice were aged to ~6 MO.
2.2. Tissue Preparation
At ~6 MO, mice were sacrificed for brain tissue accession. Mice were given an overdose of ketamine and xylazine and perfused transcardially with ice-cold 0.15 M phosphate buffer to clear blood and other potentially confounding peripheral markers out of the brain [77,78,80,81]. Brain tissues were accessed from unsupplemented normal choline Ts65Dn offspring (Ts; n = 10), MCS Ts65Dn offspring (Ts+; n = 10), unsupplemented normal choline disomic offspring (2N; n = 10) and MCS disomic offspring (2N+; n = 10) male mice with littermates between 2N and Ts mice used when possible (age range: 5.8–6.4 MO, mean age 6.0 MO). In order to obtain enough tissue sample to perform assays, left Fr Ctx tissue from each mouse brain was dissected using standard coordinates from the mouse brain atlas [82]. Dissected brain tissues were either flash frozen or kept on wet ice for homogenization immediately following brain accrual. Tissue was homogenized using ice-cold Tris homogenization buffer (20 mM Tris-Cl (pH 7.4), 1 mM EGTA, 1 mM EDTA and 0.25 M sucrose) with a protease inhibitor cocktail (1:1000, I3786, Sigma-Aldrich, St. Louis, MO, USA and 1 mM PMSF, ThermoFisher, Waltham, MA, USA) using 1.5 mm zirconium beads on Beadbug homogenizer (Benchmark Scientific, Sayreville, NJ, USA) for 30 s at 4000 rpm. Post homogenization, samples were kept on ice and cell debris was spun down at 2500× g for 5 min at 4 °C. Supernatant was aliquoted to fresh tubes for isolation of mRNA, which was performed immediately following homogenization, or for protein assays. Each protein assay was performed using tissue homogenate aliquots stored at −80 °C. RNase-free precautions were employed, and solutions were made with 18.2 mega Ohm RNase-free water (Nanopure Diamond, Barnstead, Dubuque, IA, USA). All consumables were certified RNA/DNA, nuclease and endotoxin free.
2.3. RNA Purification
RNA from Fr Ctx was purified using the miRNeasy Mini kit (Qiagen, Hilden, Germany) according to manufacturers’ specifications. A DNase digestion to remove genomic DNA was performed twice sequentially before the final washes and RNA purification. RNA quality control was performed at a 1:5 dilution to preserve RNA for downstream applications (RNA 6000 Pico kit, Agilent, Santa Clara, CA, USA).
2.4. RT-qPCR
cDNA was synthesized from equal amounts of RNA in a 50 µL sample reaction using random hexamers, as described previously [77,78,80,81,83,84] (Ts, Ts+, 2N, 2N+; n = 10/condition). RT-qPCR was performed using 1 µL of cDNA and Taqman qPCR primers for APN receptor 1 (Adipor1, Mm01291334_mH) and APN receptor 2 (Adipor2, Mm01184032_m1; Life Technologies, Grand Island, NY, USA) in triplicate using a real-time PCR cycler (PikoReal, ThermoFisher), as previously described [77,78,80,81,83,85,86]. The ddCT method was employed to determine relative gene level differences between groups [86,87,88]. Glucuronidase Beta (GusB, Mm01197678_m1) and 45S pre-ribosomal RNA (Rn45s; Rn03928990_g1) PCR products were utilized as controls. Rn45s was selected as the control housekeeping gene for ddCT quantification. Negative controls consisted of the reaction mixture without input RNA. The four study groups (Ts, Ts+, 2N, and 2N+) were compared to identify significant genes along with significant pairs with respect to PCR product synthesis for Adipor1 and Adipor2. For each gene, the PCR product synthesis was modeled as a function of mouse study group (2N, 2N+, Ts, and Ts+), using mixed effects models with random effects to account for the correlation between repeated assays on the same mouse [77,78,80,81,89]. Contrasts of pairwise comparison of interest (e.g., 2N-Ts, 2N-2N+, Ts-Ts+, 2N+-Ts. 2N-Ts+) were subsequently constructed based on the fitted mixed effects model and tested by t-test from the model. p-Values were controlled by the false discovery rate (FDR) [90] due to the multiple comparisons employed. Significance was judged at the level α = 0.05, two-sided.
2.5. Protein Assays
Protein expression analysis was performed using the WES system (Protein Simple, Santa Clara, CA, USA) [91]. Protein samples were diluted in THB buffer 1:100 (w/v), with 1x final concentration of fluorescent molecular weight marker (provided by the manufacturer) and heated to 95 °C for 5 min, then cooled to 4 °C before loading onto the WES system plate with a molecular weight ladder. All blocking reagents, chemiluminescent substrate, separation and stacking matrices (Protein Simple) were dispensed to designated wells. Primary antibodies for ADIPOR1 (MA5-32249; 1:20; ThermoFisher), ADIPOR2 (NBP1-28641; 1:20; Novus Biologicals, Centennial, CO, USA or 14361-1-AP; 1:20; ProteinTech, Rosemont, IL, USA), β-Tubulin III (β-TUB III; MAB1195; 1:50; R&D Systems, Minneapolis, MN, USA) and HRP conjugated secondary antibodies (donkey anti-rabbit (A16029) and donkey anti-goat (A16005); ThermoFisher, 1:50 and rabbit anti-mouse (DM-002); Protein Simple, ready to use) were dispensed to designated wells. Plates were spun for 5 min at 1000× g and loaded onto a WES unit, where separation electrophoresis and immunodetection steps are fully automated within the capillary system. Instrument default settings were used with the exception of the protein loading run time, which was increased from 25 to 35 min. Digital images were analyzed with Compass software (Protein Simple), utilizing dropped lines for peak analysis area calculation. Detected proteins were compared to control β-TUBIII levels and reported as normalized percentage of control. Each protein was performed in triplicate on separate plate runs. Statistical analysis was conducted on each protein normalized to β-TUBIII and modeled as a function of the mouse study group (2N, 2N+, Ts and Ts+; n = 10/genotype/diet), using mixed effects models with random effect to account for the correlation between repeated assays on the same mouse [77,78,80,81,89]. Similar to the RT-qPCR analysis, contrasts of pairwise comparison of interest (e.g., 2N-Ts, 2N-2N+, Ts-Ts+ and 2N+-Ts. 2N-Ts+) were generated using the fitted mixed effects model and tested by t-test from the model with p-values controlled by FDR [90]. Significance was judged at the level α = 0.05, two-sided.
ELISA assays were performed on Fr Ctx tissue homogenates (1:10 w/v in THB buffer) in duplicate for each sample according to the manufacturer’s specifications (Invitrogen) with the exceptions as follows. For the insulin ELISA (EMINS, ThermoFisher), assay standards were added at 3.125 µIU/mL and 1.5625 µIU/mL and samples were run at 1:2 dilution of brain homogenates (w/v). For the APN ELISA (KMP0041, ThermoFisher), an assay standard was added at 0.0625 ng/mL and samples were run at 1:4 dilution of brain homogenates (w/v), as recommended by manufacturer. The Leptin ELISA (KMC2281, ThermoFisher) assay was performed as recommended with 1:4 dilution of brain homogenates (w/v). Statistical analysis was conducted using mixed effects models, as described above [77,78,80,81,89,90]. Significance was judged at the level α = 0.05, two-sided.
3. Results
3.1. Protein Hormone Levels in Trisomic Mice and the Impact of MCS
We previously showed dysregulation within the T2DM pathway in male Ts mice compared to 2N littermates at ~6 MO [75]. Accordingly, we used Fr Ctx tissue, which receives a prominent cholinergic input from BFCNs to interrogate three protein hormones dysregulated in human MetS/IR and T2DM: APN, insulin and leptin [30,31,32,33] in trisomic mice, with the hypothesis that MCS would have a beneficial effect. APN levels were significantly downregulated by approximately two-fold in Ts Fr Ctx (mean 9.53 ng/mL; p < 0.001; Figure 1A) compared to 2N (mean 18.89 ng/mL). MCS had a beneficial effect on trisomic APN levels, as a significant increase was observed in MCS (Ts+) offspring compared to normal choline (Ts) offspring (p < 0.01, Figure 1A). Ts+ APN levels were significantly lower than 2N levels (independent of MCS treatment), indicating that MCS partially rescues APN deficits in this trisomic mouse model (Ts+ mean 14.11 ng/mL; 2N compared to Ts+ p < 0.05; 2N+ compared to Ts+ p < 0.01). MCS had no significant effect on 2N Fr Ctx APN levels (2N mean 18.89 ng/mL versus 2N+ mean 17.59 ng/mL). ELISA assays did not reveal significant differences by genotype or diet for insulin (Figure 1B) or leptin (Figure 1C) in Fr Ctx.
Figure 1.
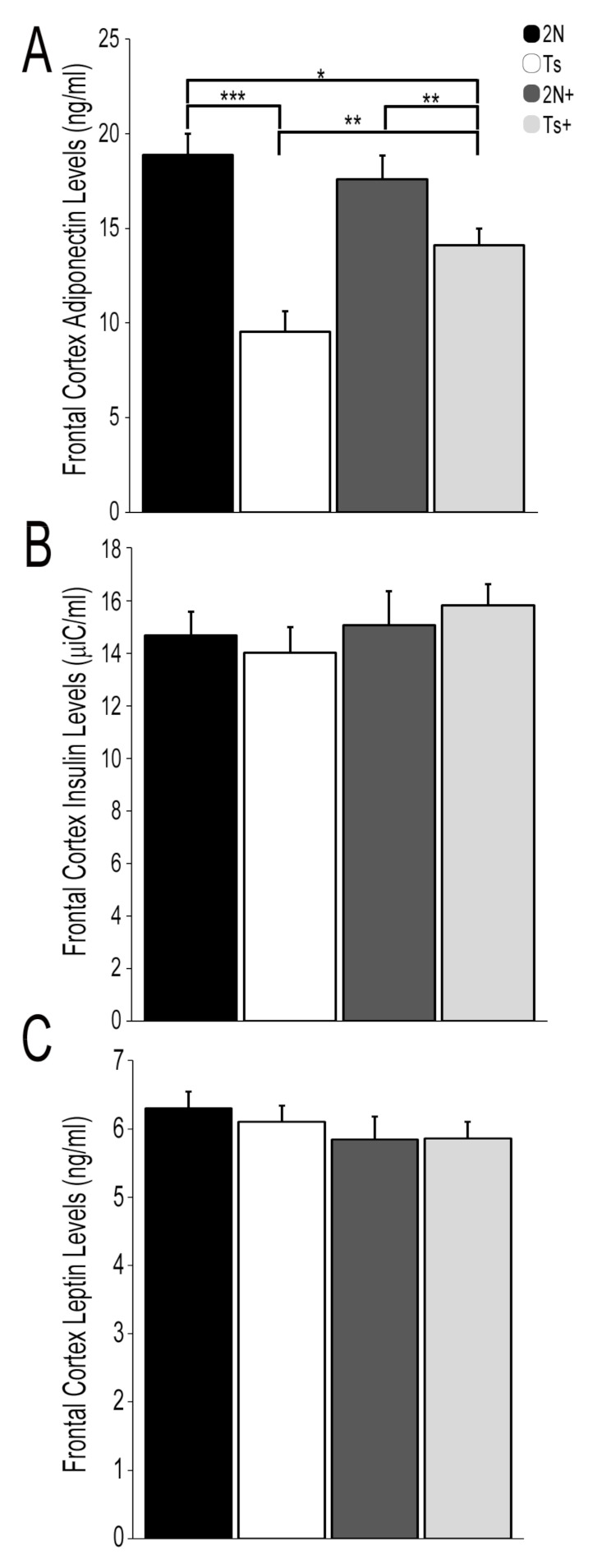
Protein hormone levels for select T2DM pathway members in the Fr Ctx of Ts65Dn mice and 2N littermates in relation to MCS. (A) APN levels were significantly downregulated in Ts mice compared to 2N littermates. Partial rescue was found with MCS in 2N+ and Ts+ mice, as APN levels were upregulated relative to offspring from a choline normal diet. (B) Insulin levels were not differentially regulated by genotype or diet in Fr Ctx. (C) Leptin levels were not differentially regulated by genotype or diet in Fr Ctx (Error bars are standard error of mean (SEM); * p < 0.05, ** p < 0.01, *** p < 0.001).
3.2. APN Receptor mRNA and Protein Levels in Trisomic Mice and the Impact of MCS
APN signals through its receptors Adipor1 and Adipor2 which are expressed within multiple regions of the brain, including Fr Ctx [92,93,94,95]. RT-qPCR analysis revealed no significant differences in Adipor1 or Adipor2 levels by genotype in Fr Ctx (Figure 2A). However, a diet effect was observed as a significant increase in Adipor1 (31.8%; p < 0.001) and Adipor2 (28.2%; p < 0.05) was found in 2N mice due to MCS (e.g., 2N+ versus 2N). PCR product levels were also significantly higher in 2N+ offspring compared to Ts+ offspring (Adipor1, 2N+-Ts+, 16.1%; p < 0.05; Adipor2, 2N+-Ts+, 37.7%; p < 0.05; Figure 2A). A significant increase was seen in Adipor1 PCR product levels by diet in trisomic mice (e.g., Ts+ versus Ts; 23.3%; p < 0.05; Figure 2A), but not for Adipor2 PCR product levels. In terms of Fr Ctx protein levels, no significant differences were observed in ADIPOR1 protein expression by genotype or diet (2N versus Ts; Figure 2B). A significant decrease in ADIPOR1 was found in trisomic choline supplemented offspring compared to normal choline 2N offspring (Ts+ versus 2N; 57.6%; p < 0.05; Figure 2B). ADIPOR2 protein expression levels showed no differences due to genotype (2N versus Ts) or by 2N diet (2N versus 2N+; Figure 2B). A significant decrease in ADIPOR2 expression was observed by diet in trisomic choline supplemented offspring compared to normal choline trisomic offspring (Ts+ versus Ts; 61.1%; p < 0.05; Figure 2B).
Figure 2.
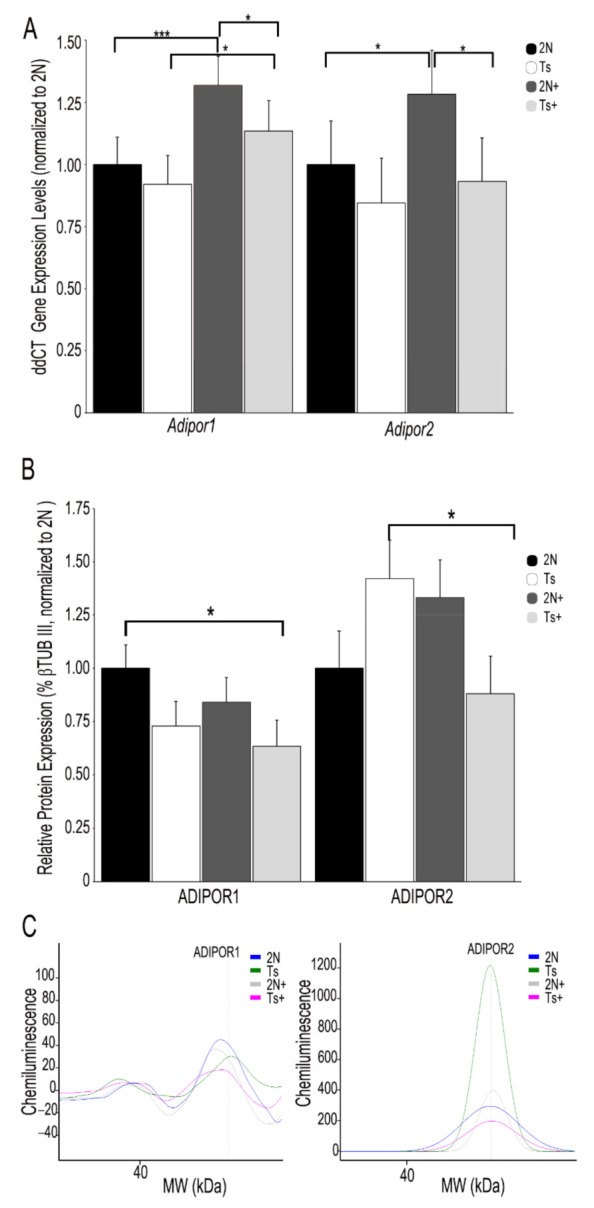
Gene and encoded protein levels of APN receptors ADIPOR1 and ADIPOR2 in the Fr Ctx of Ts65Dn mice and 2N littermates in relation to MCS. (A) Adipor1 and Adipor2 relative PCR product expression (normalized to 2N; ddCT with Rn45s as housekeeping gene) revealed no significant differences by genotype. A significant increase in Adipor1 and Adipor2 was found in 2N mice due to MCS. A significant increase in Adipor1 was also found in Ts mice due to MCS. PCR product levels were significantly higher in 2N+ offspring compared to Ts+ offspring. (B) A significant decrease in ADIPOR1 was found in Ts+ offspring compared to 2N untreated animals. A significant decrease in ADIPOR2 was found in Ts mice due to MCS. (C) Representative digital signatures of each assayed protein raw expression levels (y-axis) and molecular weight (x-axis) from 2N, Ts, 2N+ and Ts+ Fr Ctx tissue homogenates. Key: Error bars = SEM; * p < 0.05, *** p < 0.001.
4. Discussion
We utilized the well-established Ts65Dn mouse model of DS and AD to interrogate potential changes in several key protein hormones in the T2DM pathway within Fr Ctx brain tissue in the context of MCS. Through bioinformatic inquiry, our previous expression profiling study revealed significant dysregulation throughout the T2DM pathway within BFCNs in ~6 MO Ts mice [75], which we postulated was due to changes in protein hormone levels. Prior studies have shown alterations in leptin, insulin and APN can have severe deleterious effects on synaptic plasticity, learning, and memory [37,96]. We demonstrated significant downregulation in Fr Ctx APN levels in trisomic mice which was partially rescued by MCS (Figure 1A). No parallel alterations in insulin (Figure 1B) or leptin (Figure 1C) were observed in Fr Ctx brain tissue either by genotype or diet, indicating that APN is selectively vulnerable in this system. Further, we showed MCS modulates Adipor1 and Adipor2 expression (Figure 2), suggesting that MCS may be a viable treatment for APN-related defects associated with DS and AD.
Downregulation of APN levels in trisomic mice is commensurate with findings in several human studies in DS [34,50], AD [52,56] and mild cognitive impairment with T2DM [97], where decreased APN concentrations are found. Studies in brain tissue from animal models of neurodegeneration show that APN and APN receptors impact signaling and neuronal function in the brain, including neuronal insulin resistance, synaptic plasticity, excitotoxicity, neurodegeneration and cognitive decline [48,49,96,98,99,100,101]. While leptin levels are increased in peripheral studies in persons with DS [24], no studies have reported changes in CNS. Further, insulin levels are decreased in peripheral studies [34], but when examined in human DS brain, there were no significant alterations [102], commensurate with our current findings. Importantly, the present study reveals deficits in APN levels are positively impacted by MCS. MCS is a well-tolerated, low cost easily administered nutrient therapeutic that can be delivered throughout the perinatal period [9,103,104], which has previously shown to have long-term beneficial effects on basocortical and septohippocampal dependent memory tasks [60,63,64,65]. The current results link increased APN levels with MCS, suggesting that early choline supplementation could potentially provide lifelong benefits for individuals with DS and T2DM through an increase in brain APN concentrations, potentially normalizing dysregulation seen in the T2DM pathway. Prior to this study, a question whether peripheral administration of therapeutics could positively impact brain level APN deficiencies was posed [96]. The present translational study in a well-established mouse model of DS and AD indicates that MCS can result in positive functional changes in brain APN levels, suggesting that this early dietary intervention is efficacious in terms of altering APN signaling in the brain.
While APN levels benefited from MCS administration in this model system, we also measured mRNA and protein levels of the APN receptors ADIPOR1 and ADIPOR2. In contrast to APN, no changes in ADIPOR1 or ADIPOR2 mRNA or protein levels were seen due to the genotype in Fr Ctx at ~6 MO (Figure 2), which may reflect compensatory mechanisms seen in these mice in early adulthood, or regional/cellular specificity in gene expression changes. Previous work in an AD mouse model found no changes in Adipor1, but decreased Adipor2 levels in Fr Ctx [100,101]. No significant changes in protein levels for either receptor was reported [100], consistent with our findings in the trisomic mouse model. Knockdown of Adipor1 expression resulted in neurodegenerative phenotypes, spatial learning and memory deficits, and insulin signaling dysfunction in the hippocampus [99]. Further, delivery of an APN nonapeptide mimetic which activates APN receptors restores synaptic function in Adipor1 knockdown mice [101]. In human brain tissue, ADIPOR1 displays high expression in the nucleus basalis of Meynert (NBM) of the basal forebrain [93]. This finding is quite relevant, as cholinergic basal forebrain neurons of the NBM supply cholinergic innervation to the cortical mantle and are selectively vulnerable in DS and AD [3,16,105,106,107]. Therefore, loss of APN expression in trisomic mice may be linked to BFCN degeneration and could be a selective alteration found in basal forebrain [98] or in the septohippocampal and/or basocortical circuits, as evidenced by our current findings in Fr Ctx. The studies conducted by Várhelyi et al., 2017 [100] and Ali et al. 2021 [101] on Adipor1 and Adipor2 suggest that RNA and protein changes are not be expected by genotype and may be driven later in the DS mice by BFCN degeneration or loss of APN expression in the brain.
Increased choline through MCS delivery upregulated Adipor1 levels, but not Adipor2 levels, in trisomic mice (Figure 2A), which may be due to the differential expression levels and interactions of these specific APN receptors within the basocortical circuit. We posit these two APN receptors may have unique expression and signaling properties, as increases in Adipor2 levels, but not Adipor1 levels, were seen in a stress paradigm [100]. Since we found that Adipor1 levels are MCS responsive, this may indicate that BFCN degeneration seen in trisomic mice by ~6 MO may pace or precede changes in Fr Ctx dysfunction including APN signaling.
A caveat of examining APN concentrations in brain is that there are major changes in the functional form and relative amounts dependent on the area tested [96,108], and functional forms were not tested in this paradigm. Measurements of APN and APN receptors in the basal forebrain and other relevant regions in trisomic mice across the lifespan are planned to examine the potential for age-related regional specificity and alterations in a functional form. APN levels here were only assessed in male mice in the present study. Since a study found increased APN plasma concentrations in healthy adults acted as a risk factor for dementia only in females [53], sex differences in APN expression levels must be considered in future studies, especially in the context of BFCNs in the Ts65Dn mouse [22]. Accordingly, an age matched female cohort is in development to evaluate APN and its cognate receptors. Further, studies involving the basocortical and septohippocampal circuit that evaluate potential benefits on behavioral outcomes are also planned. While MCS has shown significant efficacy in multiple mouse models of neurodegenerative disorders [9,69,109,110], additional studies are required in humans as both positive and negative outcomes of clinical trials have been reported [73,111,112,113,114]. Moreover, preclinical and clinical trials are required to determine if MCS will be beneficial in the context of MetS/IR and/or T2DM, including in the study of leptin-based and insulin-derived models of diabetes [115,116]. Additional treatments affecting APN and downstream activators include Os-pep, a novel nonapeptide APN mimetic, which binds to APN receptors [101] and activin, which may be a potential therapeutic target for reduction of amyloidogenic proteins during AD progression [117].
In conclusion, alterations in APN and APN receptors, which may be subtype specific to vulnerable brain regions and functional circuits involved in memory and executive function, namely the septohippocampal and basocortical systems, respectively, could represent novel targets for therapeutic intervention in DS and AD in the context of MetS/IR and T2DM. This initial study provides proof-of-concept for this contention based upon findings in a well-established trisomic model and the significant impact of MCS on APN and Adipor1 levels. Further studies at the basic and translational levels are needed to examine the clinical applications of APN treatment in age-related neurodevelopmental and neurodegenerative disorders.
Acknowledgments
We thank Arthur Saltzman, M.S. for providing expert technical assistance.
Author Contributions
Conceptualization, M.J.A. and S.D.G.; Data curation, M.J.A.; Formal analysis, M.J.A., S.H.L. and S.D.G.; Funding acquisition, S.D.G.; Investigation, M.J.A.; Methodology, M.J.A. and S.H.L.; Project administration, M.J.A. and S.D.G.; Supervision, S.D.G.; Validation, M.J.A.; Visualization, M.J.A.; Writing—original draft, M.J.A.; Writing—review & editing, M.J.A., S.H.L. and S.D.G.; All authors have read and agreed to the published version of the manuscript.
Funding
Funding was provided by support from grants AG014449, AG043375, AG055328, and AG017617 from the National Institutes of Health.
Institutional Review Board Statement
Animal protocols were approved by the Nathan Kline Institute/NYU Grossman School of Medicine IACUC 651 in accordance with NIH guidelines.
Data Availability Statement
Data analyzed within this study are included in this body of the manuscript. Data are also available from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bittles A.H., Bower C., Hussain R., Glasson E.J. The four ages of Down syndrome. Eur. J. Public Health. 2006;17:221–225. doi: 10.1093/eurpub/ckl103. [DOI] [PubMed] [Google Scholar]
- 2.So S.A., Urbano R.C., Hodapp R.M. Hospitalizations of infants and young children with Down syndrome: Evidence from inpatient person-records from a statewide administrative database. J. Intellect. Disabil. Res. 2007;51:1030–1038. doi: 10.1111/j.1365-2788.2007.01013.x. [DOI] [PubMed] [Google Scholar]
- 3.Lott I.T. Neurological phenotypes for Down syndrome across the life span. Chang. Brains Appl. Brain Plast. Adv. Recover. Hum. Abil. 2012;197:101–121. doi: 10.1016/b978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Presson A.P., Partyka G., Jensen K.M., Devine O.J., Rasmussen S.A., McCabe L.L., McCabe E.R. Current estimate of Down syndrome population prevalence in the United States. J. Pediatr. 2013;163:1163–1168. doi: 10.1016/j.jpeds.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mai C.T., Isenburg J.L., Canfield M.A., Meyer R.E., Correa A., Alverson C.J., Lupo P.J., Riehle-Colarusso T., Cho S.J., Aggarwal D., et al. National population-based estimates for major birth defects, 2010–2014. Birth Defects Res. 2019;111:1420–1435. doi: 10.1002/bdr2.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hill D.A., Gridley G., Cnattingius S., Mellemkjaer L., Linet M., Adami H.-O., Olsen J.H., Nyren O., Fraumeni J.F. Mortality and Cancer Incidence Among Individuals with Down Syndrome. Arch. Intern. Med. 2003;163:705–711. doi: 10.1001/archinte.163.6.705. [DOI] [PubMed] [Google Scholar]
- 7.Dick M.B., Doran E., Phelan M., Lott I.T. Cognitive Profiles on the Severe Impairment Battery Are Similar in Alzheimer Disease and Down Syndrome with Dementia. Alzheimer Dis. Assoc. Disord. 2016;30:251–257. doi: 10.1097/WAD.0000000000000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartesaghi R., Guidi S., Ciani E. Is it possible to improve neurodevelopmental abnormalities in Down syndrome? Rev. Neurosci. 2011;22:419–455. doi: 10.1515/rns.2011.037. [DOI] [PubMed] [Google Scholar]
- 9.Strupp B.J., Powers B.E., Velazquez R., Ash J.A., Kelley C.M., Alldred M.J., Strawderman M., Caudill M.A., Mufson E.J., Ginsberg S.D. Maternal choline supplementation: A potential prenatal treatment for Down syndrome and Alzheimer’s disease. Curr. Alzheimer Res. 2016;13:97–106. doi: 10.2174/1567205012666150921100311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Annus T., Wilson L.R., Hong Y.T., Acosta-Cabronero J., Fryer T.D., Cardenas-Blanco A., Smith R., Boros I., Coles J.P., Aigbirhio F.I., et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimer’s Dement. 2016;12:538–545. doi: 10.1016/j.jalz.2015.07.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beacher F., Daly E., Simmons A., Prasher V., Morris R., Robinson C., Lovestone S., Murphy K., Murphy D.G.M. Alzheimer’s disease and Down’s syndrome: An in vivo MRI study. Psychol. Med. 2009;39:675–684. doi: 10.1017/S0033291708004054. [DOI] [PubMed] [Google Scholar]
- 12.Costa A.C. Alzheimer disease: Treatment of Alzheimer disease in Down syndrome. Nat. Rev. Neurol. 2012;8:182–184. doi: 10.1038/nrneurol.2012.40. [DOI] [PubMed] [Google Scholar]
- 13.Coyle J.T., Oster-Granite M.L., Reeves R.H., Gearhart J.D. Down syndrome, Alzheimer’s disease and the trisomy 16 mouse. Trends Neurosci. 1988;11:390–394. doi: 10.1016/0166-2236(88)90075-6. [DOI] [PubMed] [Google Scholar]
- 14.Hartley D., Blumenthal T., Carrillo M., DiPaolo G., Esralew L., Gardiner K., Granholm A.-C., Iqbal K., Krams M., Wisniewski T. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimer’s Dement. 2015;11:700–709. doi: 10.1016/j.jalz.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lott I.T., Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010;9:623–633. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- 16.Mann DM A., Yates P.O., Marcyniuk B. Alzheimer’s presenile dementia, senile dementia of Alzheimer type and Down’s syndrome in middle age form an age related continuum of pathological changes. Neuropathol. Appl. Neurobiol. 1984;10:185–207. doi: 10.1111/j.1365-2990.1984.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 17.Rueda N., Flórez J., Martínez-Cué C. Mouse Models of Down Syndrome as a Tool to Unravel the Causes of Mental Disabilities. Neural Plast. 2012;2012:1–26. doi: 10.1155/2012/584071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cataldo A.M., Peterhoff C.M., Troncoso J.C., Gomez-Isla T., Hyman B.T., Nixon R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000;157:277–286. doi: 10.1016/S0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belichenko P.V., Kleschevnikov A., Masliah E., Wu C., Takimoto-Kimura R., Salehi A., Mobley W.C. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2009;512:453–466. doi: 10.1002/cne.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belichenko P.V., Masliah E., Kleschevnikov A., Villar A.J., Epstein C.J., Salehi A., Mobley W.C. Synaptic structural abnormalities in the Ts65Dn mouse model of down syndrome. J. Comp. Neurol. 2004;480:281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- 21.Granholm AC E., Sanders L.A., Crnic L.S. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Exp. Neurol. 2000;161:647–663. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- 22.Kelley C.M., Powers B.E., Velázquez R., Ash J.A., Ginsberg S.D., Strupp B.J., Mufson E.J. Sex Differences in the Cholinergic Basal Forebrain in the Ts65Dn Mouse Model of Down Syndrome and Alzheimer’s Disease. Brain Pathol. 2013;24:33–44. doi: 10.1111/bpa.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley C.M., Powers B.E., Velazquez R., Ash J.A., Ginsberg S., Strupp B.J., Mufson E.J. Maternal choline supplementation differentially alters the basal forebrain cholinergic system of young-adult Ts65Dn and disomic mice. J. Comp. Neurol. 2014;522:1390–1410. doi: 10.1002/cne.23492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tenneti N., Dayal D., Sharda S., Panigrahi I., Didi M., Attri S.V., Sachdeva N., Bhalla A.K. Concentrations of leptin, adiponectin and other metabolic parameters in non-obese children with Down syndrome. J. Pediatr. Endocrinol. Metab. 2017;30:831–837. doi: 10.1515/jpem-2016-0422. [DOI] [PubMed] [Google Scholar]
- 25.Dierssen M., Fructuoso M., De Lagrán M.M., Perluigi M., Barone E. Down Syndrome Is a Metabolic Disease: Altered Insulin Signaling Mediates Peripheral and Brain Dysfunctions. Front. Neurosci. 2020;14:670. doi: 10.3389/fnins.2020.00670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milunsky A., Neurath P.W. Diabetes mellitus in Down’s syndrome. Arch. Environ. Health Int. J. 1968;17:372–376. doi: 10.1080/00039896.1968.10665244. [DOI] [PubMed] [Google Scholar]
- 27.Taggart L., Coates V., Truesdale-Kennedy M. Management and quality indicators of diabetes mellitus in people with intellectual disabilities. J. Intellect. Disabil. Res. 2012;57:1152–1163. doi: 10.1111/j.1365-2788.2012.01633.x. [DOI] [PubMed] [Google Scholar]
- 28.Capone G., Stephens M., Santoro S., Chicoine B., Bulova P., Peterson M., Jasien J., Smith A.J. Down Syndrome Medical Interest Group (DSMIG-USA) Adult Health Workgroup Co-occurring medical conditions in adults with Down syndrome: A systematic review toward the development of health care guidelines. Part II. Am. J. Med. Genet. Part A. 2020;182:1832–1845. doi: 10.1002/ajmg.a.61604. [DOI] [PubMed] [Google Scholar]
- 29.Leibson C.L., Rocca W.A., Hanson V.A., Cha R., Kokmen E., O’Brien P.C., Palumbo P.J. Risk of Dementia among Persons with Diabetes Mellitus: A Population-based Cohort Study. Am. J. Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- 30.Reguero M., de Cedrón M.G., Wagner S., Reglero G., Quintela J., de Molina A.R. Precision Nutrition to Activate Thermogenesis as a Complementary Approach to Target Obesity and Associated-Metabolic-Disorders. Cancers. 2021;13:866. doi: 10.3390/cancers13040866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C., Cai W., Zhao H., Zhu M., Cui J., Sun Z. Effect of gastric bypass on BMI and lipid metabolism in type 2 diabetes mellitus. Artif. Cells Nanomed. Biotechnol. 2020;48:903–911. doi: 10.1080/21691401.2020.1770263. [DOI] [PubMed] [Google Scholar]
- 32.Trombetta B., Carlyle B.C., Koenig A.M., Shaw L.M., Trojanowski J.Q., Wolk D.A., Locascio J.J., Arnold S.E. The technical reliability and biotemporal stability of cerebrospinal fluid biomarkers for profiling multiple pathophysiologies in Alzheimer’s disease. PLoS ONE. 2018;13:e0193707. doi: 10.1371/journal.pone.0193707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giordano V., Peluso G., Iannuccelli M., Benatti P., Nicolai R., Calvani M. Systemic and Brain Metabolic Dysfunction as a New Paradigm for Approaching Alzheimer’s Dementia. Neurochem. Res. 2006;32:555–567. doi: 10.1007/s11064-006-9125-8. [DOI] [PubMed] [Google Scholar]
- 34.Gutierrez-Hervas A., Gómez-Martínez S., Izquierdo-Gomez R., Veiga O.L., Perez-Bey A., Castro-Piñero J., Marcos A. Inflammation and fatness in adolescents with and without Down syndrome: UP & DOWN study. J. Intellect. Disabil. Res. 2019;64:170–179. doi: 10.1111/jir.12697. [DOI] [PubMed] [Google Scholar]
- 35.Fructuoso M., Rachdi L., Philippe E., Denis R., Magnan C., Le Stunff H., Janel N., Dierssen M. Increased levels of inflammatory plasma markers and obesity risk in a mouse model of Down syndrome. Free Radic. Biol. Med. 2018;114:122–130. doi: 10.1016/j.freeradbiomed.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 36.Peiris H., Duffield M.D., Fadista J., Jessup C.F., Kashmir V., Genders A.J., McGee S.L., Martin A.M., Saiedi M., Morton N., et al. A Syntenic Cross Species Aneuploidy Genetic Screen Links RCAN1 Expression to β-Cell Mitochondrial Dysfunction in Type 2 Diabetes. PLoS Genet. 2016;12:e1006033. doi: 10.1371/journal.pgen.1006033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hölscher C. Insulin Signaling Impairment in the Brain as a Risk Factor in Alzheimer’s Disease. Front. Aging Neurosci. 2019;11:88. doi: 10.3389/fnagi.2019.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J., Yang X., Yu S., Zheng R. The Leptin Signaling. Adv. Exp. Med. Biol. 2018;1090:123–144. doi: 10.1007/978-981-13-1286-1_7. [DOI] [PubMed] [Google Scholar]
- 39.Fernández-Galaz C., Fernández-Agulló T., Campoy F., Arribas C., Gallardo N., Andrés A., Ros M., Carrascosa J.M. Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. J. Endocrinol. 2001;171:23–32. doi: 10.1677/joe.0.1710023. [DOI] [PubMed] [Google Scholar]
- 40.Doherty G., Oldreive C., Harvey J. Neuroprotective actions of leptin on central and peripheral neurons in vitro. Neuroscience. 2008;154:1297–1307. doi: 10.1016/j.neuroscience.2008.04.052. [DOI] [PubMed] [Google Scholar]
- 41.Weng Z., Signore A.P., Gao Y., Wang S., Zhang F., Hastings T., Yin X.-M., Chen J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J. Biol. Chem. 2007;282:34479–34491. doi: 10.1074/jbc.M705426200. [DOI] [PubMed] [Google Scholar]
- 42.Guo Z., Jiang H., Xu X., Duan W., Mattson M.P. Leptin-mediated Cell Survival Signaling in Hippocampal Neurons Mediated by JAK STAT3 and Mitochondrial Stabilization. J. Biol. Chem. 2008;283:1754–1763. doi: 10.1074/jbc.M703753200. [DOI] [PubMed] [Google Scholar]
- 43.Perez-Gonzalez R., Antequera D., Vargas T., Spuch C., Bolós M., Carro E. Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2011;24:17–25. doi: 10.3233/JAD-2011-102070. [DOI] [PubMed] [Google Scholar]
- 44.Wang S., Yao Q., Wan Y., Wang J., Huang C., Li D., Yang B. Adiponectin reduces brain injury after intracerebral hemorrhage by reducing NLRP3 inflammasome expression. Int. J. Neurosci. 2019;130:301–308. doi: 10.1080/00207454.2019.1679810. [DOI] [PubMed] [Google Scholar]
- 45.Jeon B.T., Shin H.J., Kim J.B., Kim Y.K., Lee D.H., Kim K.H., Kang S.S., Cho G.J., Choi W.S., Roh G.S. Adiponectin protects hippocampal neurons against kainic acid-induced excitotoxicity. Brain Res. Rev. 2009;61:81–88. doi: 10.1016/j.brainresrev.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 46.Shen D., Xing S., Chen C. Adiponectin gene polymorphisms contributes to ischemic stroke risk: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2015;16:178–184. doi: 10.1177/1470320314552311. [DOI] [PubMed] [Google Scholar]
- 47.Zhang D., Guo M., Zhang W., Lu X.Y. Adiponectin stimulates proliferation of adult hippocampal neural stem/progenitor cells through activation of p38 mitogen-activated protein kinase (p38MAPK)/glycogen synthase kinase 3β (GSK-3β)/β-catenin signaling cascade. J. Biol. Chem. 2011;286:44913–44920. doi: 10.1074/jbc.M111.310052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang D., Wang X., Lu X.-Y. Adiponectin Exerts Neurotrophic Effects on Dendritic Arborization, Spinogenesis, and Neurogenesis of the Dentate Gyrus of Male Mice. Endocrinology. 2016;157:2853–2869. doi: 10.1210/en.2015-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ng R.C.-L., Cheng O.-Y., Jian M., Kwan J.S.-C., Ho P.W.-L., Cheng K.K.-Y., Yeung P.K.K., Zhou L.L., Hoo R.L.-C., Chung S.K., et al. Chronic adiponectin deficiency leads to Alzheimer’s disease-like cognitive impairments and pathologies through AMPK inactivation and cerebral insulin resistance in aged mice. Mol. Neurodegener. 2016;11:1–16. doi: 10.1186/s13024-016-0136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valentini D., Alisi A., di Camillo C., Sartorelli M., Crudele A., Bartuli A., Nobili V., Villani A. Nonalcoholic Fatty Liver Disease in Italian Children with Down Syndrome: Prevalence and Correlation with Obesity-Related Features. J. Pediatr. 2017;189:92–97.e1. doi: 10.1016/j.jpeds.2017.05.077. [DOI] [PubMed] [Google Scholar]
- 51.Corsi M.M., Dogliotti G., Pedroni F., Galliera E., Malavazos A.E., Villa R., Chiappelli M., Licastro F. Adipocytokines in Down’s syndrome, an atheroma-free model: Role of adiponectin. Arch. Gerontol. Geriatr. 2009;48:106–109. doi: 10.1016/j.archger.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 52.Waragai M., Adame A., Trinh I., Sekiyama K., Takamatsu Y., Une K., Masliah E., Hashimoto M. Possible Involvement of Adiponectin, the Anti-Diabetes Molecule, in the Pathogenesis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2016;52:1453–1459. doi: 10.3233/JAD-151116. [DOI] [PubMed] [Google Scholar]
- 53.Van Himbergen T.M., Beiser A.S., Ai M., Seshadri S., Otokozawa S., Au R., Thongtang N., Wolf P.A., Schaefer E.J., Schaefer E.J. Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and Alzheimer disease: Results from the Framingham Heart Study. Arch. Neurol. 2012;69:594–600. doi: 10.1001/archneurol.2011.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teixeira A.L., Diniz B.S., Campos A.C., Miranda A.S., Rocha N.P., Talib L.L., Gattaz W.F., Forlenza O.V. Decreased Levels of Circulating Adiponectin in Mild Cognitive Impairment and Alzheimer’s Disease. NeuroMolecular Med. 2012;15:115–121. doi: 10.1007/s12017-012-8201-2. [DOI] [PubMed] [Google Scholar]
- 55.Une K., Takei Y.A., Tomita N., Asamura T., Ohrui T., Furukawa K., Arai H. Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer’s disease. Eur. J. Neurol. 2010;18:1006–1009. doi: 10.1111/j.1468-1331.2010.03194.x. [DOI] [PubMed] [Google Scholar]
- 56.Gilbert T., Roche S., Blond E., Bar J.Y., Drai J., Cuerq C., Haution-Bitker M., Ecochard R., Bonnefoy M. Association between peripheral leptin and adiponectin levels and cognitive decline in patients with neurocognitive disorders ≥ 65 years. J. Alzheimer’s Dis. 2018;66:1255–1264. doi: 10.3233/JAD-180533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo L., Tian J., Du H. Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017;57:1071–1086. doi: 10.3233/JAD-160702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeisel S.H., Niculescu M.D. Perinatal choline influences brain structure and function. Nutr. Rev. 2006;64:197–203. doi: 10.1111/j.1753-4887.2006.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meck W.H., Williams C.L. Metabolic imprinting of choline by its availability during gestation: Implications for memory and attentional processing across the lifespan. Neurosci. Biobehav. Rev. 2003;27:385–399. doi: 10.1016/S0149-7634(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 60.Ash J.A., Velazquez R., Kelley C.M., Powers B.E., Ginsberg S., Mufson E.J., Strupp B.J. Maternal choline supplementation improves spatial mapping and increases basal forebrain cholinergic neuron number and size in aged Ts65Dn mice. Neurobiol. Dis. 2014;70:32–42. doi: 10.1016/j.nbd.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelley C.M., Ash J.A., Powers B.E., Velázquez R., Alldred M.J., Ikonomovic M.D., Ginsberg S.D., Strupp B.J., Mufson E.J. Effects of Maternal Choline Supplementation on the Septohippocampal Cholinergic System in the Ts65Dn Mouse Model of Down Syndrome. Curr. Alzheimer Res. 2015;13:84–96. doi: 10.2174/1567205012666150921100515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Powers B.E., Kelley C.M., Velazquez R., Ash J.A., Strawderman M.S., Alldred M.J., Ginsberg S.D., Mufson E.J., Strupp B.J. Maternal choline supplementation in a mouse model of Down syndrome: Effects on attention and nucleus basalis/substantia innominata neuron morphology in adult offspring. Neuroscience. 2017;340:501–514. doi: 10.1016/j.neuroscience.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Powers B.E., Velázquez R., Kelley C.M., Ash J.A., Strawderman M.S., Alldred M.J., Ginsberg S.D., Mufson E.J., Strupp B.J. Attentional function and basal forebrain cholinergic neuron morphology during aging in the Ts65Dn mouse model of Down syndrome. Brain Struct. Funct. 2015;221:4337–4352. doi: 10.1007/s00429-015-1164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Velazquez R., Ash J.A., Powers B.E., Kelley C.M., Strawderman M., Luscher Z.I., Ginsberg S., Mufson E.J., Strupp B.J. Maternal choline supplementation improves spatial learning and adult hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2013;58:92–101. doi: 10.1016/j.nbd.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moon J., Chen M., Gandhy S.U., Strawderman M., Levitsky D.A., MacLean K.N., Strupp B.J. Perinatal choline supplementation improves cognitive functioning and emotion regulation in the Ts65Dn mouse model of Down syndrome. Behav. Neurosci. 2010;124:346–361. doi: 10.1037/a0019590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mellott T.J., Huleatt O.M., Shade B.N., Pender S.M., Liu Y.B., Slack B.E., Blusztajn J.K. Perinatal choline supplementation reduces amyloidosis and increases choline acetyltransferase expression in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE. 2017;12:e0170450. doi: 10.1371/journal.pone.0170450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bearer C., Wellmann K., Tang N., He M., Mooney S.M. Choline Ameliorates Deficits in Balance Caused by Acute Neonatal Ethanol Exposure. Cerebellum. 2015;14:413–420. doi: 10.1007/s12311-015-0691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ross R.G., Hunter S., Hoffman M.C., McCarthy L., Chambers B.M., Law A., Leonard S., Zerbe G.O., Freedman R. Perinatal Phosphatidylcholine Supplementation and Early Childhood Behavior Problems: Evidence forCHRNA7Moderation. Am. J. Psychiatry. 2016;173:509–516. doi: 10.1176/appi.ajp.2015.15091188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scremin O., Roch M., Norman K., Djazayeri S., Liu Y.-Y. Brain acetylcholine and choline concentrations and dynamics in a murine model of the Fragile X syndrome: Age, sex and region-specific changes. Neuroscience. 2015;301:520–528. doi: 10.1016/j.neuroscience.2015.06.036. [DOI] [PubMed] [Google Scholar]
- 70.Stevens K.E., Adams C.E., Yonchek J., Hickel C., Danielson J., Kisley M.A. Permanent improvement in deficient sensory inhibition in DBA/2 mice with increased perinatal choline. Psychopharmacology. 2008;198:413–420. doi: 10.1007/s00213-008-1170-3. [DOI] [PubMed] [Google Scholar]
- 71.Ward B., Agarwal S., Wang K., Berger-Sweeney J., Kolodny N. Longitudinal brain MRI study in a mouse model of Rett Syndrome and the effects of choline. Neurobiol. Dis. 2008;31:110–119. doi: 10.1016/j.nbd.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 72.Ward B.C., Kolodny N.H., Nag N., Berger-Sweeney J.E. Neurochemical changes in a mouse model of Rett syndrome: Changes over time and in response to perinatal choline nutritional supplementation. J. Neurochem. 2009;108:361–371. doi: 10.1111/j.1471-4159.2008.05768.x. [DOI] [PubMed] [Google Scholar]
- 73.Caudill M.A., Strupp B.J., Muscalu L., Nevins J.E.H., Canfield R.L. Maternal choline supplementation during the third trimester of pregnancy improves infant information processing speed: A randomized, double-blind, controlled feeding study. FASEB J. 2018;32:2172–2180. doi: 10.1096/fj.201700692RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al-Sulaiti H., Diboun I., Agha M.V., Mohamed F.F.S., Atkin S., Dömling A.S., Elrayess M.A., Mazloum N.A. Metabolic signature of obesity-associated insulin resistance and type 2 diabetes. J. Transl. Med. 2019;17:1–11. doi: 10.1186/s12967-019-2096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alldred M.J., Penikalapati S.C., Lee S.H., Heguy A., Roussos P., Ginsberg S. Profiling Basal Forebrain Cholinergic Neurons Reveals a Molecular Basis for Vulnerability within the Ts65Dn Model of Down Syndrome and Alzheimer’s Disease. [(accessed on 3 July 2021)];2020 doi: 10.1007/s12035-021-02453-3. in press. Available online: https://assets.researchsquare.com/files/rs-88218/v1/c0c81dd3-d34b-4ed1-8876-c5b1e8b50f8d.pdf?c=1604102440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Detopoulou P., Panagiotakos D.B., Antonopoulou S., Pitsavos C., Stefanadis C. Dietary choline and betaine intakes in relation to concentrations of inflammatory markers in healthy adults: The ATTICA study. Am. J. Clin. Nutr. 2008;87:424–430. doi: 10.1093/ajcn/87.2.424. [DOI] [PubMed] [Google Scholar]
- 77.Alldred M.J., Lee S.H., Petkova E., Ginsberg S.D. Expression profile analysis of vulnerable CA1 pyramidal neurons in young-Middle-Aged Ts65Dn mice. J. Comp. Neurol. 2015;523:61–74. doi: 10.1002/cne.23663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alldred M.J., Lee S.H., Petkova E., Ginsberg S.D. Expression profile analysis of hippocampal CA1 pyramidal neurons in aged Ts65Dn mice, a model of Down syndrome (DS) and Alzheimer’s disease (AD) Brain Struct. Funct. 2015;220:2983–2996. doi: 10.1007/s00429-014-0839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duchon A., Raveau M., Chevalier C., Nalesso V., Sharp A.J., Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: Relevance for modeling down syndrome. Mamm. Genome. 2011;22:674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alldred M.J., Chao H.M., Lee S.H., Beilin J., Powers B.E., Petkova E., Ginsberg S.D. CA1 pyramidal neuron gene expression mosaics in the Ts65Dn murine model of Down syndrome and Alzheimer’s disease following maternal choline supplementation. Hippocampus. 2018;28:251–268. doi: 10.1002/hipo.22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alldred M.J., Chao H.M., Lee S.H., Beilin J., Powers B.E., Petkova E., Ginsberg S.D. Long-term effects of maternal choline supplementation on CA1 pyramidal neuron gene expression in the Ts65Dn mouse model of Down syndrome and Alzheimer’s disease. FASEB J. 2019;33:9871–9884. doi: 10.1096/fj.201802669RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paxinos G., Franklin K.B.J. The Mouse Brain in Stereotaxic Coordinates. Academic Press; Cambridge, MA, USA: 2001. [Google Scholar]
- 83.Alldred M.J., Duff K.E., Ginsberg S.D. Microarray analysis of CA1 pyramidal neurons in a mouse model of tauopathy reveals progressive synaptic dysfunction. Neurobiol. Dis. 2012;45:751–762. doi: 10.1016/j.nbd.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bordi M., Berg M.J., Mohan P.S., Peterhoff C.M., Alldred M.J., Che S., Nixon R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12:2467–2483. doi: 10.1080/15548627.2016.1239003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alldred M.J., Che S., Ginsberg S.D. Terminal Continuation (TC) RNA Amplification Enables Expression Profiling Using Minute RNA Input Obtained from Mouse Brain. Int. J. Mol. Sci. 2008;9:2091–2104. doi: 10.3390/ijms9112091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiang Y., Mullaney K.A., Peterhoff C.M., Che S., Schmidt S.D., Boyer-Boiteau A., Nixon R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA. 2010;107:1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ginsberg S.D., Alldred M.J., Counts S.E., Cataldo A.M., Neve R.L., Jiang Y., Che S. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry. 2010;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.ABI Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR. [(accessed on 3 July 2021)];Appl. Biosyst. Prod. Guide. 2004 :1–60. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms_042380.pdf. [Google Scholar]
- 89.McCulloch C.E., Searle S.R., Neuhaus J.M. Generalized, Linear, and Mixed Models. 2nd ed. John Wiley & Sons; New York, NY, USA: 2011. [Google Scholar]
- 90.Benjamini Y., Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001;29:1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
- 91.Nguyen U., Squaglia N., Boge A., Fung P. The Simple Western™: A gel-free, blot-free, hands-free Western blotting reinvention. Nat. Methods. 2011;8 doi: 10.1038/nmeth.f.353. [DOI] [Google Scholar]
- 92.Rastegar S., Parimisetty A., Cassam-Sulliman N., Narra S.S., Weber S., Rastegar M., Viranaicken W., Couret D., Planesse C., Strähle U., et al. Expression of adiponectin receptors in the brain of adult zebrafish and mouse: Links with neurogenic niches and brain repair. J. Comp. Neurol. 2019;527:2317–2333. doi: 10.1002/cne.24669. [DOI] [PubMed] [Google Scholar]
- 93.Psilopanagioti A., Papadaki H., Kranioti E.F., Alexandrides T., Varakis J.N. Expression of Adiponectin and Adiponectin Receptors in Human Pituitary Gland and Brain. Neuroendocrinology. 2008;89:38–47. doi: 10.1159/000151396. [DOI] [PubMed] [Google Scholar]
- 94. [(accessed on 3 July 2021)]; Available online: https://www.gtexportal.org/home/gene/ADIPOR1.
- 95. [(accessed on 3 July 2021)]; Available online: https://www.gtexportal.org/home/gene/ADIPOR2.
- 96.Forny-Germano L., De Felice F.G., Vieira M.N.D.N. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and Alzheimer’s Disease. Front. Neurosci. 2019;12:1027. doi: 10.3389/fnins.2018.01027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gorska-Ciebiada M., Saryusz-Wolska M., Borkowska A., Ciebiada M., Loba J. Adiponectin, leptin and IL-1 β in elderly diabetic patients with mild cognitive impairment. Metab. Brain Dis. 2016;31:257–266. doi: 10.1007/s11011-015-9739-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Uddin S., Rahman M., Abu Sufian M., Jeandet P., Ashraf G.M., Bin-Jumah M.N., Mousa S.A., Abdel-Daim M.M., Akhtar M.F., Saleem A., et al. Exploring the New Horizon of AdipoQ in Obesity-Related Alzheimer’s Dementia. Front. Physiol. 2021;11 doi: 10.3389/fphys.2020.567678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim M.W., Bin Abid N., Jo M.H., Jo M.G., Yoon G.H., Kim M.O. Suppression of adiponectin receptor 1 promotes memory dysfunction and Alzheimer’s disease-like pathologies. Sci. Rep. 2017;7:1–10. doi: 10.1038/s41598-017-12632-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Várhelyi Z.P., Kálmán J., Oláh Z., Ivitz E.V., Fodor E.K., Sántha M., Datki Z.L., Pákáski M. Adiponectin receptors are less sensitive to stress in a transgenic mouse model of Alzheimer’s disease. Front. Neurosci. 2017;11:199. doi: 10.3389/fnins.2017.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ali T., Rehman S.U., Khan A., Badshah H., Bin Abid N., Kim M.W., Jo M.H., Chung S.S., Lee H.-G., Rutten B.P.F., et al. Adiponectin-mimetic novel nonapeptide rescues aberrant neuronal metabolic-associated memory deficits in Alzheimer’s disease. Mol. Neurodegener. 2021;16:1–22. doi: 10.1186/s13024-021-00445-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tramutola A., Lanzillotta C., Di Domenico F., Head E., Butterfield D.A., Perluigi M., Barone E. Brain insulin resistance triggers early onset Alzheimer disease in Down syndrome. Neurobiol. Dis. 2020;137:104772. doi: 10.1016/j.nbd.2020.104772. [DOI] [PubMed] [Google Scholar]
- 103.Jiang X., West A.A., Caudill M.A. Maternal choline supplementation: A nutritional approach for improving offspring health? Trends Endocrinol. Metab. 2014;25:263–273. doi: 10.1016/j.tem.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 104.Korsmo H.W., Jiang X., Caudill M.A. Choline: Exploring the Growing Science on Its Benefits for Moms and Babies. Nutrients. 2019;11:1823. doi: 10.3390/nu11081823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mufson E.J., Bothwell M., Kordower J.H. Loss of nerve growth factor receptor-containing neurons in Alzheimer’s disease: A quantitative analysis across subregions of the basal forebrain. Exp. Neurol. 1989;105:221–232. doi: 10.1016/0014-4886(89)90124-6. [DOI] [PubMed] [Google Scholar]
- 106.Mufson E.J., Ginsberg S.D., Ikonomovic M.D., DeKosky S.T. Human cholinergic basal forebrain: Chemoanatomy and neurologic dysfunction. J. Chem. Neuroanat. 2003;26:233–242. doi: 10.1016/S0891-0618(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 107.Mufson E.J., Ma S.Y., Dills J., Cochran E.J., Leurgans S., Wuu J., Kordower J.H. Loss of basal forebrain P75NTR immunoreactivity in subjects with mild cognitive impairment and Alzheimer’s disease. J. Comp. Neurol. 2002;443:136–153. doi: 10.1002/cne.10122. [DOI] [PubMed] [Google Scholar]
- 108.Kusminski C.M., McTernan P.G., Schraw T., Kos K., O’Hare J.P., Ahima R., Kumar S., Scherer P.E. Adiponectin complexes in human cerebrospinal fluid: Distinct complex distribution from serum. Diabetologia. 2007;50:634–642. doi: 10.1007/s00125-006-0577-9. [DOI] [PubMed] [Google Scholar]
- 109.Blusztajn J.K., Slack B.E., Mellott T.J. Neuroprotective Actions of Dietary Choline. Nutrients. 2017;9:815. doi: 10.3390/nu9080815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nag N., Berger-Sweeney J.E. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol. Dis. 2007;26:473–480. doi: 10.1016/j.nbd.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 111.Zeisel S.H. Choline: Clinical Nutrigenetic/Nutrigenomic Approaches for Identification of Functions and Dietary Requirements. J. Nutr. Nutr. 2010;3:209–219. doi: 10.1159/000324357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jacobson S.W., Molteno C.D., Meintjes E.M., Senekal M.S., Lindinger N.M., Dodge N.C., Zeisel S.H., Duggan C.P., Jacobson J.L., Carter R. Feasibility and Acceptability of Maternal Choline Supplementation in Heavy Drinking Pregnant Women: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Alcohol. Clin. Exp. Res. 2018;42:1315–1326. doi: 10.1111/acer.13768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jacobson S.W., Carter R.C., Molteno C.D., Stanton M.E., Herbert J.S., Lindinger N.M., Jacobson J.L. Efficacy of maternal choline supplementation during pregnancy in mitigating adverse effects of prenatal alcohol exposure on growth and cognitive function: A randomized, double-blind, placebo-controlled clinical trial. Alcohol. Clin. Exp. Res. 2018;42:1327–1341. doi: 10.1111/acer.13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cheatham C.L., Goldman B.D., Fischer L.M., Da Costa K.-A., Reznick J.S., Zeisel S.H. Phosphatidylcholine supplementation in pregnant women consuming moderate-choline diets does not enhance infant cognitive function: A randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2012;96:1465–1472. doi: 10.3945/ajcn.112.037184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen H., Charlat O., Tartaglia L.A., Woolf E.A., Weng X., Ellis S.J., Lakey N.D., Culpepper J., More K.J., Breitbart R.E., et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in db/db Mice. Cell. 1996;84:491–495. doi: 10.1016/S0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 116.Brüning J.C., Gautam D., Burks D.J., Gillette J., Schubert M., Orban P.C., Kahn C.R. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 117.Hashimoto M., Ho G., Sugama S., Takenouchi T., Waragai M., Sugino H., Masliah E. Possible Role of Activin in the Adiponectin Par-adox-Induced Progress of Alzheimer’s Disease. J. Alzheimers Dis. 2021;81:451–458. doi: 10.3233/JAD-210206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data analyzed within this study are included in this body of the manuscript. Data are also available from the corresponding author upon request.