

This phase 1b/2a nonrandomized clinical trial assesses combination of a type II KIT inhibitor with a conformation-complementary, active-state (type I) KIT inhibitor and broad mutation coverage and global disease control.
Key Points
Question
Is cotargeting the active and inactive conformations of KIT associated with clinical benefit in refractory gastrointestinal stromal tumor (GIST)?
Findings
In this phase 1b/2a nonrandomized clinical trial of 39 patients with GIST, combining PLX9486, a selective type I KIT inhibitor targeting activation-loop mutations, with sunitinib, a type II inhibitor targeting adenosine triphosphate–binding pocket mutations, showed activity with a progression-free survival of 12.1 months in patients with refractory GIST.
Meaning
The data suggest that combining type I and type II KIT inhibitors was associated with clinical benefit and support the further evaluation of the combination in refractory GIST.
Abstract
Importance
Many cancer subtypes, including KIT-mutant gastrointestinal stromal tumors (GISTs), are driven by activating mutations in tyrosine kinases and may initially respond to kinase inhibitors but frequently relapse owing to outgrowth of heterogeneous subclones with resistance mutations. KIT inhibitors commonly used to treat GIST (eg, imatinib and sunitinib) are inactive-state (type II) inhibitors.
Objective
To assess whether combining a type II KIT inhibitor with a conformation-complementary, active-state (type I) KIT inhibitor is associated with broad mutation coverage and global disease control.
Design, Setting, and Participants
A highly selective type I inhibitor of KIT, PLX9486, was tested in a 2-part phase 1b/2a trial. Part 1 (dose escalation) evaluated PLX9486 monotherapy in patients with solid tumors. Part 2e (extension) evaluated PLX9486-sunitinib combination in patients with GIST. Patients were enrolled from March 2015 through February 2019; data analysis was performed from May 2020 through July 2020.
Interventions
Participants received 250, 350, 500, and 1000 mg of PLX9486 alone (part 1) or 500 and 1000 mg of PLX9486 together with 25 or 37.5 mg of sunitinib (part 2e) continuously in 28-day dosing cycles until disease progression, treatment discontinuation, or withdrawal.
Main Outcomes and Measures
Pharmacokinetics, safety, and tumor responses were assessed. Clinical efficacy end points (progression-free survival and clinical benefit rate) were supplemented with longitudinal monitoring of KIT mutations in circulating tumor DNA.
Results
A total of 39 PLX9486-naive patients (median age, 57 years [range, 39-79 years]; 22 men [56.4%]; 35 [89.7%] with refractory GIST) were enrolled in the dose escalation and extension parts. The recommended phase 2 dose of PLX9486 was 1000 mg daily. At this dose, PLX9486 could be safely combined with 25 or 37.5 mg daily of sunitinib continuously. Patients with GIST who received PLX9486 at a dose of 500 mg or less, at the recommended phase 2 dose, and with sunitinib had median (95% CI) progression-free survivals of 1.74 (1.54-1.84), 5.75 (0.99-11.0), and 12.1 (1.34-NA) months and clinical benefit rates (95% CI) of 14% (0%-58%), 50% (21%-79%), and 80% (52%-96%), respectively.
Conclusions and Relevance
In this phase 1b/2a nonrandomized clinical trial, type I and type II KIT inhibitors PLX9486 and sunitinib were safely coadministered at the recommended dose of both single agents in patients with refractory GIST. Results suggest that cotargeting 2 complementary conformational states of the same kinase was associated with clinical benefit with an acceptable safety profile.
Trial Registration
ClinicalTrials.gov Identifier: NCT02401815
Introduction
The identification of activating genetic alterations in protein kinases and the development of corresponding therapeutic inhibitors have revolutionized the treatment of many malignant neoplasms.1 However, treatment resistance commonly develops owing to the selective expansion of tumor cells with resistance mutations. Such resistance has emerged during the treatment of gastrointestinal stromal tumor (GIST), a malignant mesenchymal neoplasm that arises in the walls of the gastrointestinal tract from precursors of the interstitial cells of Cajal.2 The most common primary mutations in GIST involve exons 9 or 11 of the KIT gene,3 which encode the extracellular and juxtamembrane domains of the stem cell factor receptor KIT.4 Imatinib, an inhibitor of exon 9–mutated and exon 11–mutated KIT,5,6,7 is the first-line systemic therapy for patients with metastatic GIST.8 However, the majority of imatinib-treated patients with advanced disease ultimately relapse due to outgrowth of clones with imatinib-resistant KIT mutations.9,10,11 These secondary mutations are typically found in the adenosine triphosphate (ATP)–binding pocket, encoded by exons 13 and 14, or the activation loop (A-loop), encoded by exons 17 and 18.4 Second-line sunitinib is active against ATP-binding pocket mutations but not A-loop mutations.12,13 Third-line regorafenib14 was shown to have activity against a subset of exon 17 mutations in a small phase 2 study,15 but the drug may also act through other signaling pathways (eg, angiogenesis).16
The inability of these tyrosine kinase inhibitors (TKIs) to inhibit both A-loop and ATP-binding pocket mutations lies in the structural mechanism of inhibition. Imatinib, sunitinib, and regorafenib are all type II inhibitors that bind to the inactive conformation of the kinase in which the N-terminal portion of the A-loop, termed the aspartate-phenylalanine-glycine (DFG) motif, is displaced (DFG-out) to expose an allosteric back pocket.5,13 Our x-ray crystallographic analysis showed that D816V, a representative A-loop mutation, stabilized the catalytically active, DFG-in conformation of KIT kinase that precludes the binding of type II inhibitors to the allosteric pocket. This structural insight was used to design PLX9486, a selective type I KIT inhibitor with activity against DFG-in A-loop mutations. Herein, we describe the identification, characterization, and early clinical development of PLX9486 in patients with advanced, refractory GIST. Single-agent dose escalation demonstrated a favorable safety profile and preliminary antitumor activity consistent with the inhibition profile of PLX9486. The main goal of this study was to determine whether PLX9486 could be combined with a type II inhibitor, such as sunitinib, to target a broad spectrum of oncogenic KIT mutations.
Methods
Structure-Based Discovery and Preclinical Characterization
X-ray crystallography study (eTable 1 in Supplement 1) of KITD816V provided detailed molecular understanding of acquired resistance to standard-of-care TKIs and enabled the discovery of PLX9486, a next-generation KIT inhibitor highly specific to the DFG-in conformation promoted by the A-loop mutations. The in vivo efficacy of PLX9486 alone and in combination with sunitinib was evaluated in patient-derived xenograft models representing clonal heterogeneity of GIST. See eMethods in Supplement 1 for additional details.
Study Design, Participants, Procedures, and Conduct
The type I KIT inhibitor PLX9486 was evaluated in a first-in-human, multicenter, 2-part, open-label phase 1b/2a study. Part 1 evaluated the safety and pharmacokinetics (PK) of PLX9486 and established the maximum tolerated dose/recommended phase 2 dose in patients with advanced solid tumors. Extension part 2e assessed the safety and preliminary activity of PLX9486 in combination with sunitinib in patients with refractory GIST.
Eligible patients (parts 1 and 2e) were aged 18 years or older; had advanced solid tumors that progressed following standard therapy, treatment-refractory disease, or no effective standard therapeutic option; had an Eastern Cooperative Oncology Group performance status of 0 to 2; and had adequate hematologic, hepatic, and renal function, and, additionally, for part 2e, a left ventricular ejection fraction greater than 50%. Eligible patients with GIST (parts 1 and 2e) had histologically confirmed, locally advanced, metastatic, and/or unresectable disease and prior failure of or intolerability to imatinib with an archival pathology report containing KIT mutational status.
Both part 1 (PLX9486 monotherapy) and part 2e (PLX9486-sunitinib combination) included a dose-escalation stage following the standard “3+3” design. PLX9486 formulated as 50 mg tablets was administered continuously in 28-day dosing cycles. The starting dose level of PLX9486 in part 1 was 250 mg/d once daily. Subsequent cohorts were at 350, 500, and 1000 mg/d once daily, and 500 mg twice daily. In part 2e, PLX9486 dose escalation commenced at 50% of the monotherapy recommended phase 2 dose in combination with sunitinib 25 mg/d. PLX9486 was then increased to 100% of the recommended phase 2 dose, and in the last cohort, sunitinib was increased to 37.5 mg/d. Treatment continued until disease progression, unacceptable toxic effects, or withdrawal of consent.
The study was performed in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Committee on Harmonization of Technical Requirements for Pharmaceuticals for Human Use. The study protocol (Supplement 2) was approved by the review boards of all participating institutions and the US Food and Drug Administration. All patients provided written informed consent before any study procedures were conducted. Race/ethnicity (self-identified) was assessed for demographic data collection purposes only. A list of the participating sites is provided in eAppendix in Supplement 1.
Outcomes, End Points, Assessments, and Safety
The primary objective for parts 1 and 2e was to assess the PK, safety, tolerability, and maximum tolerated dose/recommended phase 2 dose of PLX9486 monotherapy and combination of PLX9486 with sunitinib in patients with advanced solid tumors including GIST. Safety assessments included the recording of vital signs, laboratory testing, urinalysis, physical examination, and electrocardiography. Treatment-emergent adverse events (TEAEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03. Dose-limiting toxic effect (DLT) was defined as any nonhematologic adverse event (AE) or laboratory toxic effect of grade 3 or greater despite adequate supportive care, grade 3 or greater neutropenia or thrombocytopenia, or grade 4 anemia during cycle 1. The recommended phase 2 dose was defined as the highest dose level at which fewer than 33% of participants experienced a DLT, or in the absence of DLT, it was established based on toxic effects, PK, and convenience of dosing. The plasma concentrations of PLX9486 (parts 1 and 2e) and sunitinib (part 2e) were analyzed using the noncompartmental method. Participants in selected part 1 cohorts participated in a PK substudy to obtain complete information on the PK profile of PLX9486.
Secondary objectives included the following outcome assessments: overall response rate (overall response rate = complete response [CR] + partial response [PR]) using Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1, duration of response, clinical benefit rate (CBR = CR + PR + stable disease that lasted for at least 16 weeks), progression-free survival (PFS), and overall survival (OS). Tumor measurements (both target lesions and nontarget lesions) were performed by a radiologist using RECIST 1.1 guidelines.
Exploratory objectives included biomarker analysis in peripheral blood using circulating tumor DNA (ctDNA) analysis. At protocol-specified time points, frozen K2EDTA plasma samples were collected. Cell-free DNA was isolated, prepared into libraries, and analyzed with the PlasmaSELECT 64 assay (Personal Genome Diagnostics).
Statistical Analysis and Considerations
The sample sizes of the different parts of the study were based on clinical rather than statistical considerations. Clinical end points and descriptive statistics were summarized according to the statistical analysis plan. Exact binomial 95% CIs (2-sided) were provided for category responses (eg, CBR). The PFS and OS were analyzed by Kaplan-Meier methods with median values and corresponding 95% CIs calculated. The Cox proportional hazards regression was used to investigate the contributions of various factors to PFS. Statistical analyses were performed with SAS, version 9.4 (SAS Institute) and R, version 4.0.0 (R Core Team). The log-rank test (2-sided) was used to compare the survival times between 2 treatment groups, and the level of significance was .05.
Results
Type I Inhibitor PLX9486 Complements Type II KIT Inhibitors In Vitro and In Vivo
Exploiting the structural plasticity of kinases to target the oncogene-specific conformations has become an important strategy for designing safe and effective cancer drugs. We used x-ray crystallography to understand the association of a representative A-loop mutation, D816V, with the structure of KIT. In the crystal structure, KITD816V was found in the activated, DFG-in state (Figure 1) incompatible with the binding of type II inhibitors, such as pexidartinib (eFigure 1A in Supplement 1).17,18 The 7-azaindole scaffold of pexidartinib was redesigned by installing a phenyl group at the 2-position to flip the binding orientation by 180° (eFigure 1A in Supplement 1). Further optimization yielded PLX9486 (Figure 1; eFigure 1A and 1B in Supplement 1), a potent KITD816V inhibitor (half-maximal inhibitory concentration = 1.1 nM) with desirable kinome selectivity (eTable 2 in Supplement 1) and a mutant inhibition profile complementary to type II inhibitors imatinib and sunitinib (eFigure 1C and eTable 3 in Supplement 1). In vivo, PLX9486 demonstrated single-agent antitumor effect in a GIST patient-derived xenograft model that harbored KIT mutations in both exons 11 and 17 (eFigure 1D in Supplement 1) and strong synergy with sunitinib in a GIST patient-derived xenograft model harboring concurrent KIT exon 13 and exon 17 mutations (eFigure 1E in Supplement 1).
Figure 1. Type I Inhibitor PLX9486 Binds to Mutant KITD816V With High Atomic Efficiency.

The aspartate to valine substitution in KITD816V promotes hydrophobic packing between V816 and Y646, locking the activation look (A-loop) in an extended, aspartate-phenylalanine-glycine–-in conformation conducive to adenosine triphosphate and substrate binding.
Patient Characteristics and Disposition
Between March 2015 and February 2019, a total of 39 patients with solid tumors (median age, 57 years [range, 39-79 years]; 22 [56.4] men), 35 (89.7%) with GIST, were enrolled into parts 1 (n = 24) and 2e (n = 15) of this study (Figure 2). Three patients enrolled in part 1 were subsequently reenrolled in part 2e, expanding the safety population of part 2e to 18. Demographic and baseline characteristics of all patients enrolled in the study are in eTable 4 in Supplement 1. All 35 PLX9486-naive patients with GIST had received imatinib; 30 (86%) had also received sunitinib, and 27 (77%) had been treated with all 3 standard lines of therapies (eFigure 2 in Supplement 1). On enrollment, 31 patients with GIST were reported to have tumors with at least 1 KIT mutation. When the ctDNA from baseline plasma samples of the 35 patients was analyzed, 20 had tumors with detectable KIT exon 9 and 11 mutations that confirmed the screening reports (eFigure 2 in Supplement 1). Baseline ctDNA analysis confirmed 10 of the KIT exon 13, 14, 17, and 18 mutations that had been reported during screening but also identified previously unknown mutations. In this cohort of heavily pretreated patients, tumor mutations in the A-loop were detected more frequently than in the ATP-binding pocket at baseline (eFigure 2 in Supplement 1).
Figure 2. Trial Profile.
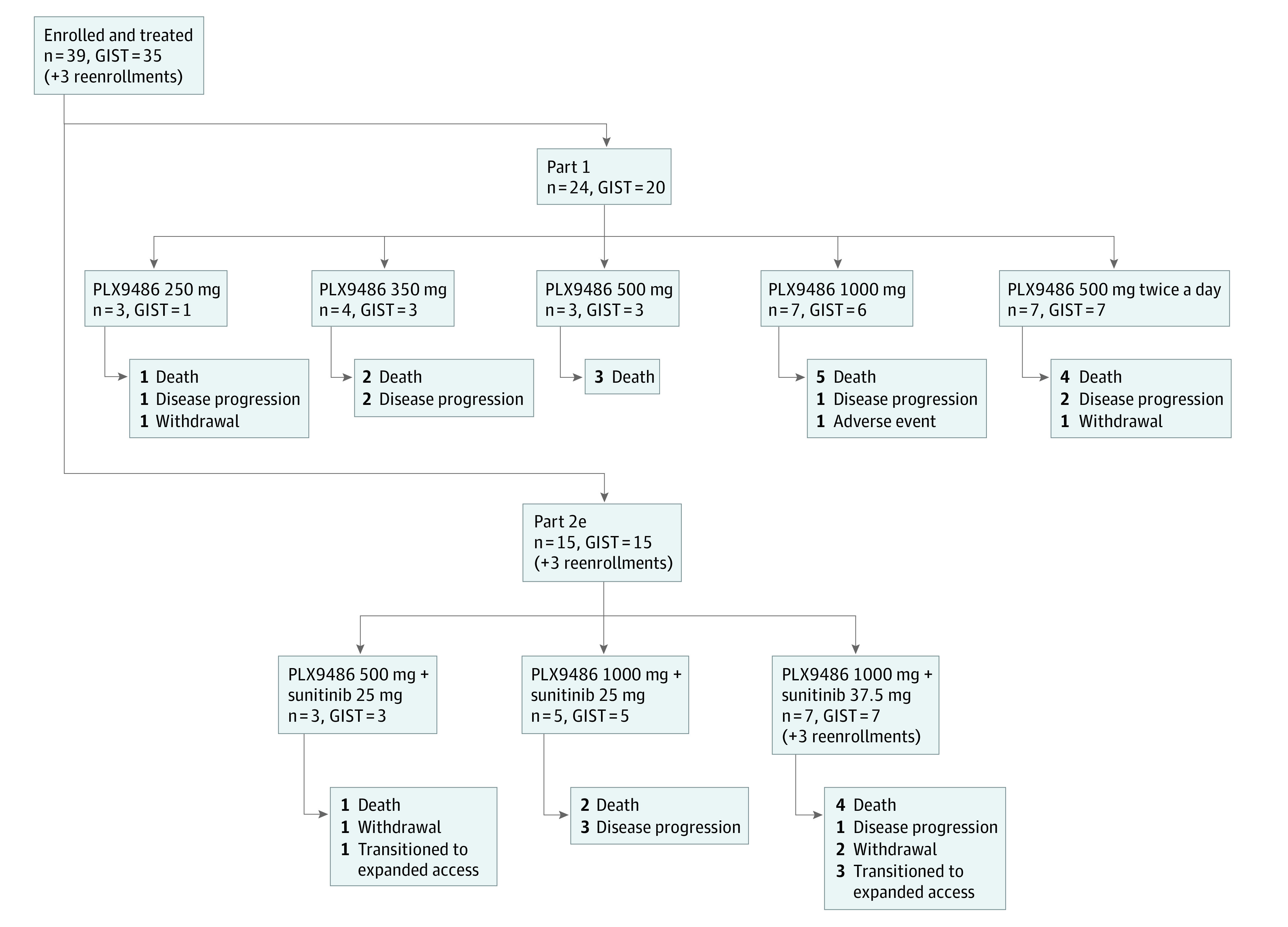
The study enrolled 39 PLX9486-naive patients and reenrolled 3 patients from previous cohorts in part 2e. Reasons for study discontinuation are provided for each cohort. Disease progression includes both progressive disease per Response Evaluation Criteria in Solid Tumors, version 1.1, and clinical progression.
Drug Exposure and Safety
The 39 patients originally enrolled in the study and 3 reenrolled patients were included in safety evaluation (eFigure 3 and eTable 5 in Supplement 1). In part 1 (n = 24), the mean (range) duration of PLX9486 treatment was 148 (14-670) days. Monotherapy with PLX9486 was well tolerated with mostly grade 1 to 2 AEs (eTable 6 in Supplement 1). Common TEAEs were fatigue, aspartate aminotransferase increase, diarrhea, and nausea. The most common TEAEs that were grade 3 or higher included anemia (4 patients, 16.5%) and hyperuricemia (3 patients, 12.5%). In part 2e (n = 18), the mean (range) duration of PLX9486 and sunitinib treatment was 337 (27-825) days and 307 (28-825) days, respectively. Combining PLX9486 with sunitinib was associated with the AE profile expected for each of the single agents.19 The top doses of both agents (1000 mg/d PLX9486 and 37.5 mg/d sunitinib, continuously) were well tolerated. Common TEAEs in the combination of PLX9486 with sunitinib were diarrhea, aspartate aminotransferase/alanine aminotransferase increase, nausea, and vomiting (eTable 7 in Supplement 1). The most common TEAEs that were grade 3 or higher included anemia (5 patients, 27.8%), hypophosphatemia (3 patients, 16.7%), diarrhea, fatigue, hypertension, and lymphopenia (each 2 patients, 11.1%).
Of all patients enrolled in the study, only 1 patient (1000 mg once daily in part 1) had an AE (grade 3 anemia) that met the criteria for DLTs. The AE was resolved in 5 days with no change in PLX9486 dosing.
Pharmacokinetics
All patients enrolled in the study were evaluated for PK. In part 1, PLX9486 showed a modest increase in exposure at steady state between 250 and 1000 mg/d doses (eFigure 4A in Supplement 1). As further increase in dose was not expected to significantly increase the exposure, 1000 mg once daily was selected as the recommended phase 2 dose for single-agent PLX9486. In part 2e, sunitinib coadministration was associated with improved oral uptake of PLX9486 (eFigure 4A in Supplement 1). Sunitinib showed a dose-proportional increase in steady-state exposure from 25 to 37.5 mg (eTable 8 in Supplement 1). However, this study was not powered to assess the statistical significance of the interactions between the 2 agents. Six patients participated in a single-dose PK substudy. Plasma concentrations of PLX9486 reached maximum approximately 24 hours after the dose, followed by a monophasic decline with a mean terminal half-life of 71 hours (eFigure 4B and eTable 9 in Supplement 1).
Efficacy
Of the 35 PLX9486-naive patients with GIST, 34 (19 in part 1 and 15 in part 2e) were evaluable for efficacy (1 patient from part 1 withdrew from the study without a postbaseline tumor assessment). Monotherapy with PLX9486 at 500 mg or lower daily doses (n = 7) had no objective responses and a CBR of 14% (95% CI, 0%-58%) (Figure 3A). However, at 1000-mg daily dose (n = 12), 1 patient achieved a confirmed PR (7.4-month duration), and the CBR reached 50% (95% CI, 21%-79%). A higher response rate was observed in the 15 patients treated with the combination of PLX9486 and sunitinib with 1 confirmed CR (eFigure 5 in Supplement 1), 2 confirmed PRs, and a CBR of 80% (95% CI, 52%-96%) (Figure 3A). All 3 responses in part 2e exceeded 1 year (18.4, 18.3, and 12.3 months) in duration (Figure 3B). Median PFS was 1.74 (95% CI, 1.55-1.84) months for patients with GIST treated with 500 mg or less PLX9486 monotherapy, and 5.75 (95% CI, 0.99-11.0) months for those receiving daily doses of 1000 mg (Figure 3C). The hazard ratio for progression was 0.35 (95% CI, 0.12-1.0; log-rank test P = .051) in favor of the 1000-mg daily dose. Addition of sunitinib (25 or 37.5 mg) to PLX9486 (500 or 1000 mg) observed a median PFS of 12.1 (95% CI, 1.35-not available) months (Figure 3C). The HRs were 0.14 (95% CI, 0.04-0.44; P < .001) and 0.40 (95% CI, 0.16-0.99; P = .048) when compared with 500-mg or less and 1000-mg PLX9486, respectively. Overall survivals for the 3 groups followed the same trend; the median OS was 2.96, 11.34, and 18.11 months, respectively (eFigure 6 in Supplement 1). However, differences in median OS were not statistically significant between the groups.
Figure 3. Clinical Responses and Duration.
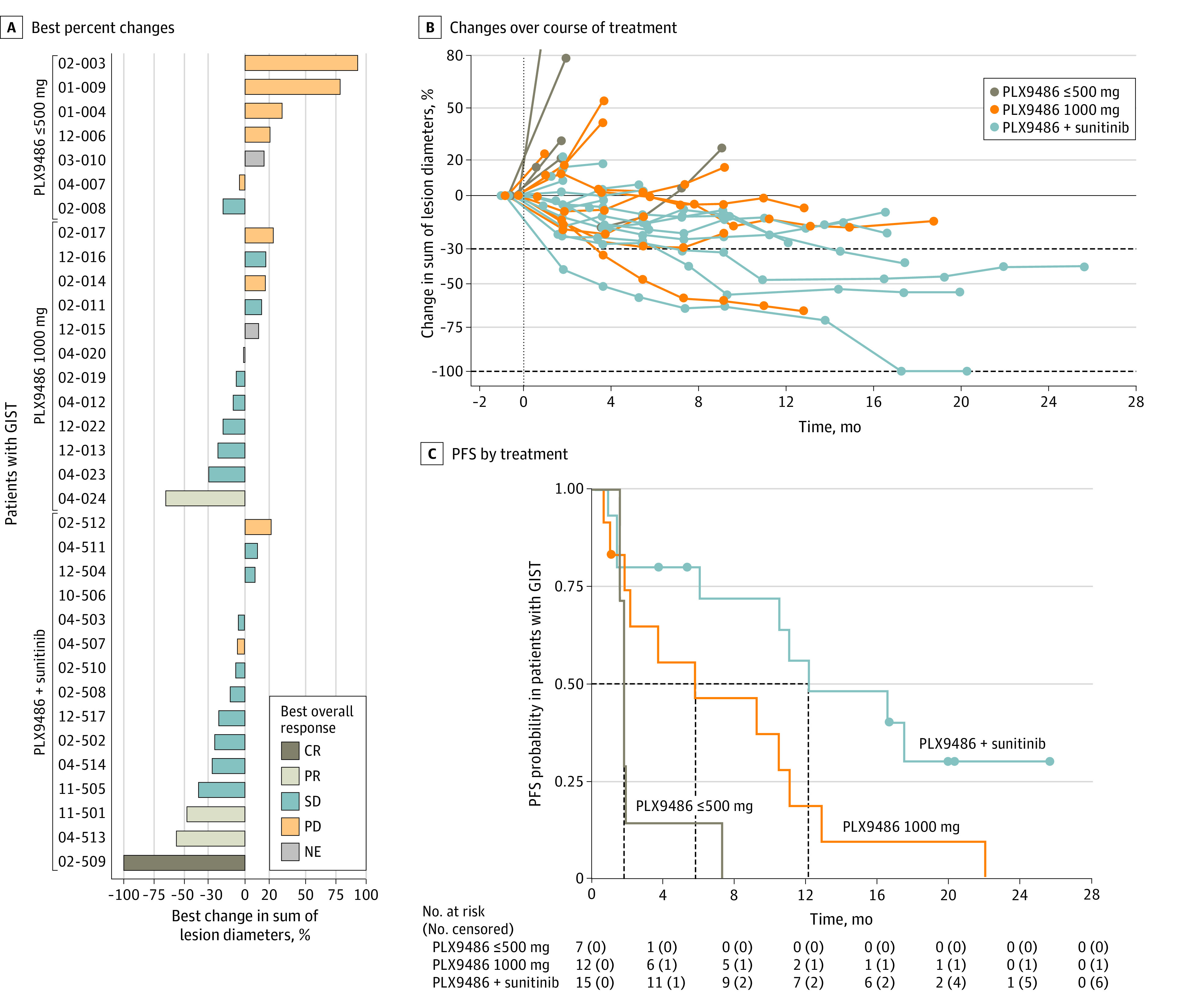
A, Waterfall plot of best percent changes for sums of target lesion diameters for 34 evaluable PLX9486-naive patients with gastrointestinal stromal tumor (GIST) grouped by treatment (PLX9486 ≤ 500 mg, PLX9486 1000 mg, or PLX9486 plus sunitinib). B, Changes in sums of target lesion diameters over the course of treatment. C, Progression-free survival (PFS) by treatment. CR indicates complete response; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease.
Circulating Tumor DNA Analysis
In the 20 patients with tumors with detectable KIT mutations in ctDNA at baseline (eFigure 2 in Supplement 1), an association emerged between the best overall response in the sum of target lesions and the initial change in percentage of mutant reads (eFigure 7A and 7B in Supplement 1). Analysis of the association of the initial change in ctDNA as a continuous variable adjusted for baseline ctDNA with PFS confirmed that decreases in ctDNA are associated with a lower risk of disease progression (eFigure 7C in Supplement 1).
Temporally, levels of circulating exon 17 and 18 mutations decreased early on in patients who experienced clinical benefit on receiving PLX9486 treatment (eFigure 8 in Supplement 1, patients 12-022, 12-013, 11-501, and 02-502), indicating that PLX9486 can suppress tumor subclones that are dependent on A-loop mutations. Levels of exon 13 and 14 mutations were not changed by PLX9486 monotherapy (eg, eFigure 8A in Supplement 1, patient 02-017). By contrast, when PLX9486 was combined with sunitinib, initial decreases in exon 13–mutant and exon 14–mutant alleles were observed (eFigure 8B in Supplement 1, patient 04-507) and coincided with clinical benefit (stable disease). Across all the ctDNA-positive patients, initial changes in ctDNA levels suggest that PLX9486 inhibited clones that harbor A-loop mutations and the addition of sunitinib broadened the spectrum of targetable resistant KIT mutations in the ATP-binding pocket to include exons 13 and 14.
Discussion
Refractory GIST provides an ideal model for exploring the combination of complementary kinase inhibitors targeting different conformations of a driver kinase as a novel approach to addressing clonal heterogeneity in resistance. Tyrosine kinase inhibitor–resistant GISTs harbor mutations in both the ATP-binding pocket and the A-loop and present multiple resistance subclones that limit the clinical activity of monotherapy that inhibits only a subset of the mutations.20,21,22 PLX9486 is a highly selective type I TKI with activity against primary KIT mutations (exons 9 and 11) and A-loop mutations (exons 17 and 18). The safety profile of PLX9486 makes it a desirable partner in combination with TKIs with complementary KIT mutant inhibition profiles, especially type II inhibitors, such as sunitinib, that have activities against ATP-binding pocket mutations (exons 13 and 14). Our preliminary data show that PLX9486 and sunitinib can be safely combined to achieve an improved clinical outcome in patients with GIST with heavily pretreated disease. The median PFS was 12.1 months for all patients (n = 15) and 11.6 months for patients who previously progressed on sunitinib (n = 10; eFigure 9 in the Supplement 1). The median PFS in a subset of patients with 3 or more prior therapies (n = 9) remains 11 months (eFigure 10 in Supplement 1), comparing favorably with the historical data with ripretinib monotherapy (6.3-month median PFS) in a similar patient population.23 Ripretinib has recently been approved as fourth-line treatment for GIST. Though a type II inhibitor, ripretinib was shown to inhibit a broader set of KIT mutations (including A-loop mutations).24
Advances in cell-free DNA analysis have provided a useful tool for detecting and quantifying tumor-specific mutations in patients with cancer. This analysis enables longitudinal monitoring of multiple metastatic lesions with less-invasive serial blood collections as well as identification of emerging resistance mutations in tumors in patients with GIST.25,26 We showed that KIT mutations can be detected in the ctDNA of patients with GIST, and the level of change in mutant reads may indicate disease activity. In conjunction with radiographic measurement, ctDNA analysis supports that the PLX9486-sunitinib combination may provide broad inhibition of KIT mutant receptors in patients with refractory GIST.
Limitations
However, the small sample size and the heterogeneous nature of this disease limits our interpretation of the outcomes of the combination with particular drug-resistant subclones. Further studies of PLX9486 and sunitinib in randomized clinical trials will help evaluate whether proactive complementary inhibition of multiple mechanisms of TKI resistance leads to superior clinical outcome as compared with single-agent treatment.
Conclusions
In this study, a combination of type I, PLX9486, and type II, sunitinib, KIT inhibitors was well tolerated in patients with advanced GIST. Moreover, a broad spectrum of conformationally distinct oncogenic resistance mutations may be targeted by this combination.
eAppendix. Institutions and Collaborators
eMethods.
eResults.
eFigure 1. Structure-based discovery and preclinical characterization of PLX9486
eFigure 2. Patient baseline characteristics
eFigure 3. Duration of treatments
eFigure 4. Pharmacokinetics of PLX9486
eFigure 5. Complete response seen in subject 02-509 with 3 prior therapies treated at RP2D of PLX9486 plus 37.5 mg sunitinib
eFigure 6. Overall survival for 34 evaluable GIST patients by study part
eFigure 7. Correlation between ctDNA analysis and clinical response
eFigure 8. Selected ctDNA and tumor lesion profiles over time for patients in Part 1 and Part 2e
eFigure 9. Progression-free survival for 15 GIST patients in Part 2e by prior sunitinib treatment status
eFigure 10. Progression-free survival for 26 GIST patients who received ≥ 3 lines of prior therapy (imatinib, sunitinib, and regorafenib)
eTable 1. Crystallographic data and refinement statistics
eTable 2. IC50 values in KIT biochemical assays (nM)
eTable 3. IC50 values in KIT autophosphorylation assays in cells (μM)
eTable 4. Summary of demographics and baseline characteristics
eTable 5. Summary of TEAEs reported by >10% of subjects overall by SOC and preferred term
eTable 6. Summary of treatment emergent adverse events that occurred in ≥ 10% of subjects in Part 1
eTable 7. Summary of treatment emergent adverse events that occurred in ≥ 15% of subjects in Part 2e
eTable 8. Sunitinib PK parameters in Part 2e
eTable 9. Pharmacokinetic parameters of PLX9486 after receiving a single 350 mg dose of PLX9486
eReferences.
Trial Protocol
Data Sharing Statement
References
- 1.Gross S, Rahal R, Stransky N, Lengauer C, Hoeflich KP. Targeting cancer with kinase inhibitors. J Clin Invest. 2015;125(5):1780-1789. doi: 10.1172/JCI76094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878. doi: 10.1038/nrc3143 [DOI] [PubMed] [Google Scholar]
- 3.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580. doi: 10.1126/science.279.5350.577 [DOI] [PubMed] [Google Scholar]
- 4.Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007;130(2):323-334. doi: 10.1016/j.cell.2007.05.055 [DOI] [PubMed] [Google Scholar]
- 5.Mol CD, Dougan DR, Schneider TR, et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279(30):31655-31663. doi: 10.1074/jbc.M403319200 [DOI] [PubMed] [Google Scholar]
- 6.McAuliffe JC, Hunt KK, Lazar AJ, et al. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol. 2009;16(4):910-919. doi: 10.1245/s10434-008-0177-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta A, Roy S, Lazar AJ, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci U S A. 2010;107(32):14333-14338. doi: 10.1073/pnas.1000248107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472-480. doi: 10.1056/NEJMoa020461 [DOI] [PubMed] [Google Scholar]
- 9.Chen LL, Trent JC, Wu EF, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004;64(17):5913-5919. doi: 10.1158/0008-5472.CAN-04-0085 [DOI] [PubMed] [Google Scholar]
- 10.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764-4774. doi: 10.1200/JCO.2006.06.2265 [DOI] [PubMed] [Google Scholar]
- 11.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12(6):1743-1749. doi: 10.1158/1078-0432.CCR-05-1211 [DOI] [PubMed] [Google Scholar]
- 12.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352-5359. doi: 10.1200/JCO.2007.15.7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gajiwala KS, Wu JC, Christensen J, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A. 2009;106(5):1542-1547. doi: 10.1073/pnas.0812413106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demetri GD, Reichardt P, Kang YK, et al. ; GRID study investigators . Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302. doi: 10.1016/S0140-6736(12)61857-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeh CN, Chen MH, Chen YY, et al. A phase II trial of regorafenib in patients with metastatic and/or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget. 2017;8(27):44121-44130. doi: 10.18632/oncotarget.17310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Jiang T, Wang W, Piao D. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumor after failure with imatinib and sunitinib treatment: a meta-analysis. Medicine (Baltimore). 2017;96(48):e8698. doi: 10.1097/MD.0000000000008698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tap WD, Wainberg ZA, Anthony SP, et al. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N Engl J Med. 2015;373(5):428-437. doi: 10.1056/NEJMoa1411366 [DOI] [PubMed] [Google Scholar]
- 18.Kim TS, Cavnar MJ, Cohen NA, et al. Increased KIT inhibition enhances therapeutic efficacy in gastrointestinal stromal tumor. Clin Cancer Res. 2014;20(9):2350-2362. doi: 10.1158/1078-0432.CCR-13-3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338. doi: 10.1016/S0140-6736(06)69446-4 [DOI] [PubMed] [Google Scholar]
- 20.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216(1):64-74. doi: 10.1002/path.2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serrano C, Mariño-Enríquez A, Tao DL, et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer. 2019;120(6):612-620. doi: 10.1038/s41416-019-0389-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serrano C, Leal A, Kuang Y, et al. Phase I study of rapid alternation of sunitinib and regorafenib for the treatment of tyrosine kinase inhibitor refractory gastrointestinal stromal tumors. Clin Cancer Res. 2019;25(24):7287-7293. doi: 10.1158/1078-0432.CCR-19-2150 [DOI] [PubMed] [Google Scholar]
- 23.Blay JY, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923-934. doi: 10.1016/S1470-2045(20)30168-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith BD, Kaufman MD, Lu WP, et al. Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell. 2019;35(5):738-751.e9. doi: 10.1016/j.ccell.2019.04.006 [DOI] [PubMed] [Google Scholar]
- 25.Serrano C, Vivancos A, López-Pousa A, et al. Clinical value of next generation sequencing of plasma cell-free DNA in gastrointestinal stromal tumors. BMC Cancer. 2020;20(1):99. doi: 10.1186/s12885-020-6597-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arshad J, Roberts A, Ahmed J, et al. Utility of circulating tumor DNA in the management of patients with GI stromal tumor: analysis of 243 patients. JCO Precis Oncol. 2020;2020(4):66-73. doi: 10.1200/PO.19.00253 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Institutions and Collaborators
eMethods.
eResults.
eFigure 1. Structure-based discovery and preclinical characterization of PLX9486
eFigure 2. Patient baseline characteristics
eFigure 3. Duration of treatments
eFigure 4. Pharmacokinetics of PLX9486
eFigure 5. Complete response seen in subject 02-509 with 3 prior therapies treated at RP2D of PLX9486 plus 37.5 mg sunitinib
eFigure 6. Overall survival for 34 evaluable GIST patients by study part
eFigure 7. Correlation between ctDNA analysis and clinical response
eFigure 8. Selected ctDNA and tumor lesion profiles over time for patients in Part 1 and Part 2e
eFigure 9. Progression-free survival for 15 GIST patients in Part 2e by prior sunitinib treatment status
eFigure 10. Progression-free survival for 26 GIST patients who received ≥ 3 lines of prior therapy (imatinib, sunitinib, and regorafenib)
eTable 1. Crystallographic data and refinement statistics
eTable 2. IC50 values in KIT biochemical assays (nM)
eTable 3. IC50 values in KIT autophosphorylation assays in cells (μM)
eTable 4. Summary of demographics and baseline characteristics
eTable 5. Summary of TEAEs reported by >10% of subjects overall by SOC and preferred term
eTable 6. Summary of treatment emergent adverse events that occurred in ≥ 10% of subjects in Part 1
eTable 7. Summary of treatment emergent adverse events that occurred in ≥ 15% of subjects in Part 2e
eTable 8. Sunitinib PK parameters in Part 2e
eTable 9. Pharmacokinetic parameters of PLX9486 after receiving a single 350 mg dose of PLX9486
eReferences.
Trial Protocol
Data Sharing Statement