

Abstract
Background
Pulmonary alveolar proteinosis is a rare interstitial lung disease characterized by accumulating surfactant materials in the alveoli. The autoimmune form is by far the most common in adults, while in the pediatric age group, the vast majority of cases are congenital. We report a case of an adolescent patient diagnosed with autoimmune pulmonary alveolar proteinosis, which is unusual in this age group.
Case presentation
A-15 year-old Saudi male presented to the emergency department with a history of shortness of breath and low oxygen saturation. High-resolution computed tomography of his chest showed a global crazy-paving pattern. Autoantibodies against granulocyte-macrophage colony-stimulating factor were detected in his serum. A diagnosis of the autoimmune form of pulmonary alveolar proteinosis was confirmed after excluding other possible causes. The patient improved after he underwent whole lung lavage under general anesthesia, and he was independent of oxygen therapy after 6 months of follow-up.
Conclusion
The autoimmune form of pulmonary alveolar proteinosis is rare in the pediatric age group and should be considered when no apparent cause of this disease was found. Whole lung lavage should be the first treatment modality offered in this setting with close follow-up and monitoring.
Keywords: Autoimmune pulmonary alveolar proteinosis, Whole lung lavage, Bronchoalveolar lavage, Crazy-paving pattern, GM-CSF, Case report
Background
Pulmonary alveolar proteinosis (PAP) is a rare disease caused by abnormal accumulation of surfactant material in alveoli [1]. Several forms of PAP exist: primary PAP (either autoimmune or hereditary), secondary PAP due to exposure to a high level of dust (such as silica) or underlying infections or malignancy, and congenital PAP due to defect in the production of surfactant [2, 3].
Primary PAP can be autoimmune (most common) or hereditary [defect in the receptor of colony-stimulating factor 2 receptor alpha and beta (CSF2RA and CSF2RB)] [4, 5]. Autoimmune PAP (aPAP) develops because of circulating autoantibodies against granulocyte-macrophage colony-stimulating factor (GM-CSF), and most affected individuals present to clinical attention in their fourth or fifth decade of life with progressive shortness of breath (SOB) and productive cough [6, 7].
In this report, we describe a case of a 15-year-old patient diagnosed as a case of aPAP, which is a rare cause of PAP in this age group [8].
Case presentation
A 15-year-old boy not known to have prior medical illness presented to our hospital emergency department (ED) with a history of shortness of breath upon climbing stairs and blue discoloration of his lips and extremities. There was no history of cough, chest pain, palpitation, fever, or constitutional symptoms. Cyanosis was first noted by his parents on his hands 3 months before the recent presentation. There was no significant exposure to household or environmental fumes, dust, or mineral oils. The father of the patient has rheumatoid arthritis, and his mother has multiple sclerosis. Other aspects of history were unremarkable, including drug history; specifically, the patient was asked about using any immunosuppressant medication.
On physical examination (P/E), the patient was conscious, oriented, and alert but appeared ill and had peripheral and central cyanosis as well as finger clubbing. He was afebrile but tachypneic (respiratory rate 35 breaths per minute), with room air oxygen saturation of 68%, which improved to 92% with 10 L of oxygen therapy delivered through a nonbreathable mask. Chest auscultation revealed diffuse bilateral crackles with a decrease in air entry bilaterally. The rest of the P/E was unremarkable.
Laboratory investigations in the ED were normal (Table 1) apart from polycythemia and elevated lactate dehydrogenase (LDH). On chest X-ray, extensive bilateral alveolar scattered and coalescent alveolar infiltrates involving both lung fields were evident (Fig. 1). The patient was admitted to the general ward for further evaluation and management. After admission to the general ward, further investigations were conducted, including inflammatory marker and tuberculosis tests, which all returned normal (Table 1).
Table 1.
Laboratory findings
| Inspection item | Reference range | ||
|---|---|---|---|
| Hematology | |||
| Hematocrit | 62.4 | % | 40–50 |
| Hemoglobin | 21.6 | g/dL | 13–17.5 |
| RBCs | 7.53 | 106/μL | 4.5–6.2 |
| WBCs | 6.04 | 103/μL | 4–10 |
| Differential counts | |||
| Eosinophils | 0.1 | 103/μL | 0.02–0.5 |
| Lymphocytes | 3.42 | 103/μL | 1–3 |
| Monocytes | 0.64 | 103/μL | 0.2–1 |
| Neutrophils | 1.84 | 103/μL | 2–7 |
| Platelets | 217 | 103/μL | 150–400 |
| ESR | 0 | per hour | 0–15 |
| PT | 11.80 | sec | 11–16 |
| aPTT | 32.16 | sec | 26–40 |
| INR | 1.09 | – | 0.8–1.3 |
| Biochemistry | |||
| Na | 144 | mmol/L | 135–135 |
| K | 4.37 | mmol/L | 3.5–5.3 |
| Ca | 9.01 | mg/dL | 8.8–10.2 |
| Cr | 0.59 | mg/dL | 0.5–1.3 |
| Urea | 15.6 | mg/dL | 10–50 |
| RBG | 89 | mg/dL | 70–110 |
| ALT | 10.5 | U/L | 00–41 |
| AST | 31.4 | U/L | 00–37 |
| GGT | 20 | U/L | 10–60 |
| ALP | 113 | IU/L | 40–113 |
| Albumin | 3.95 | g/dL | 3.4–4.8 |
| Total protein | 6.2 | g/dL | 6.4–8.3 |
| Total bilirubin | 1.12 | mg/dL | 0–1.10 |
| Direct bilirubin | 0.24 | mg/dL | 0–0.2 |
| LDH | 587 | U/L | 135–225 |
| Serology | |||
| CRP | Negative | mg/dL | 0–0.8 |
| RF | Negative | IU/mL | 0–15 |
| Serum anti-GM-CSF | Positive | – | Negative |
| c-ANCA | Negative | – | Negative |
| p-ANCA | Negative | – | Negative |
| ACPA | Negative | – | Negative |
| ANA | Negative | – | Negative |
| HIVa | Negative | – | Negative |
| HBsAg | Negative | – | Negative |
| HBsAb | 25.1 | mIU/mL | 10–300 |
| HCVb | Negative | – | Negative |
| Other | |||
| PPD | 0 | mm | 0 |
| BALF culture | Negative | – | Negative |
| BALF for TBc | Negative | – | Negative |
| PAS stain | Positive | – | Negative |
aUsing fourth-generation antigen and antibodies combination HIV-1/2 immunoassay
bAnti-HCV antibodies immunoassay
cBy acid fast bacilli smear, culture, and polymerase chain reaction
ACPA anti-citrullinated peptide antibodies, ALP alkaline phosphatase, ALT alanine transaminase, ANA antinuclear antibodies, Anti-GM-CSF anti-granulocyte-macrophage colony-stimulating factor, aPTT activated partial thromboplastin time, AST aspartate transaminase, c-ANCA cytoplasmic antineutrophil cytoplasmic autoantibodies, Cr creatinine, ESR erythrocyte sedimentation rate, GGT gamma-glutamyl transaminase, HBsAb hepatitis B surface antibody, HBsAg hepatitis B surface antigen, HCV hepatitis C virus, HIV human immunodeficiency virus; INR international normalization rate, LDH lactate dehydrogenase, p-ANCA perinuclear antineutrophil cytoplasmic antibodies, PAS stain periodic acid–Schiff, PPD purified protein derivative, PT prothrombin time, RBC red blood cells, RBG random blood glucose, RF rheumatoid factor, WBC white blood cells
Fig. 1.
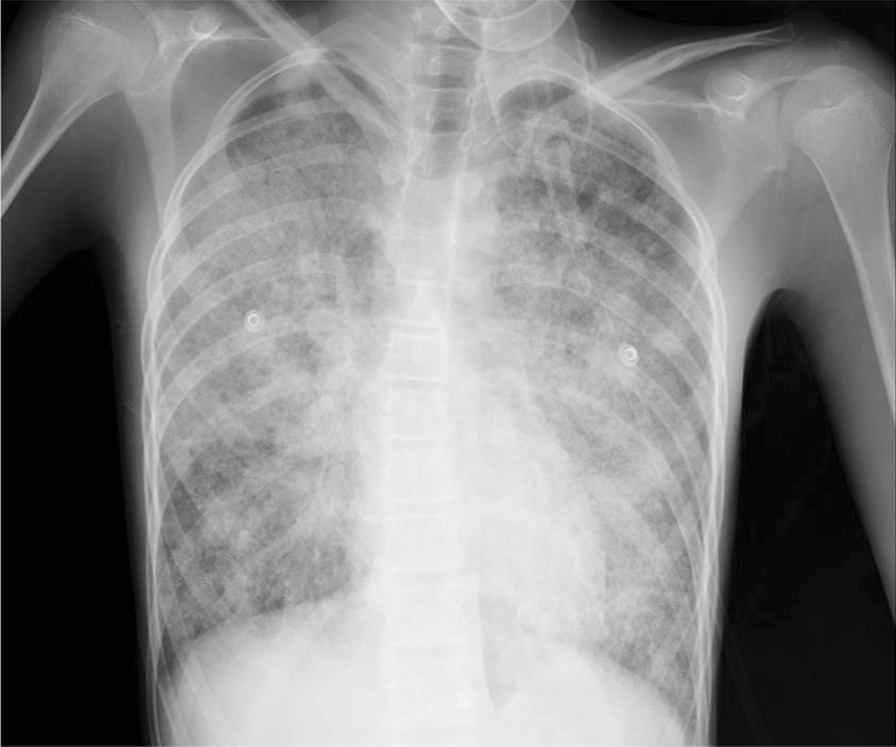
Chest X-ray showing extensive bilateral alveolar scattered and coalescent alveolar infiltrate involving both lung fields
High-resolution computed tomography (HRCT) of the chest showed extensive diffuse bilateral thickening of the lung interstitium with superimposed interlobular septal thickening and a typical “crazy-paving” appearance (Fig. 2). A pulmonary function test was not done. Collectively, these findings indicate a possible diagnosis of PAP.
Fig. 2.
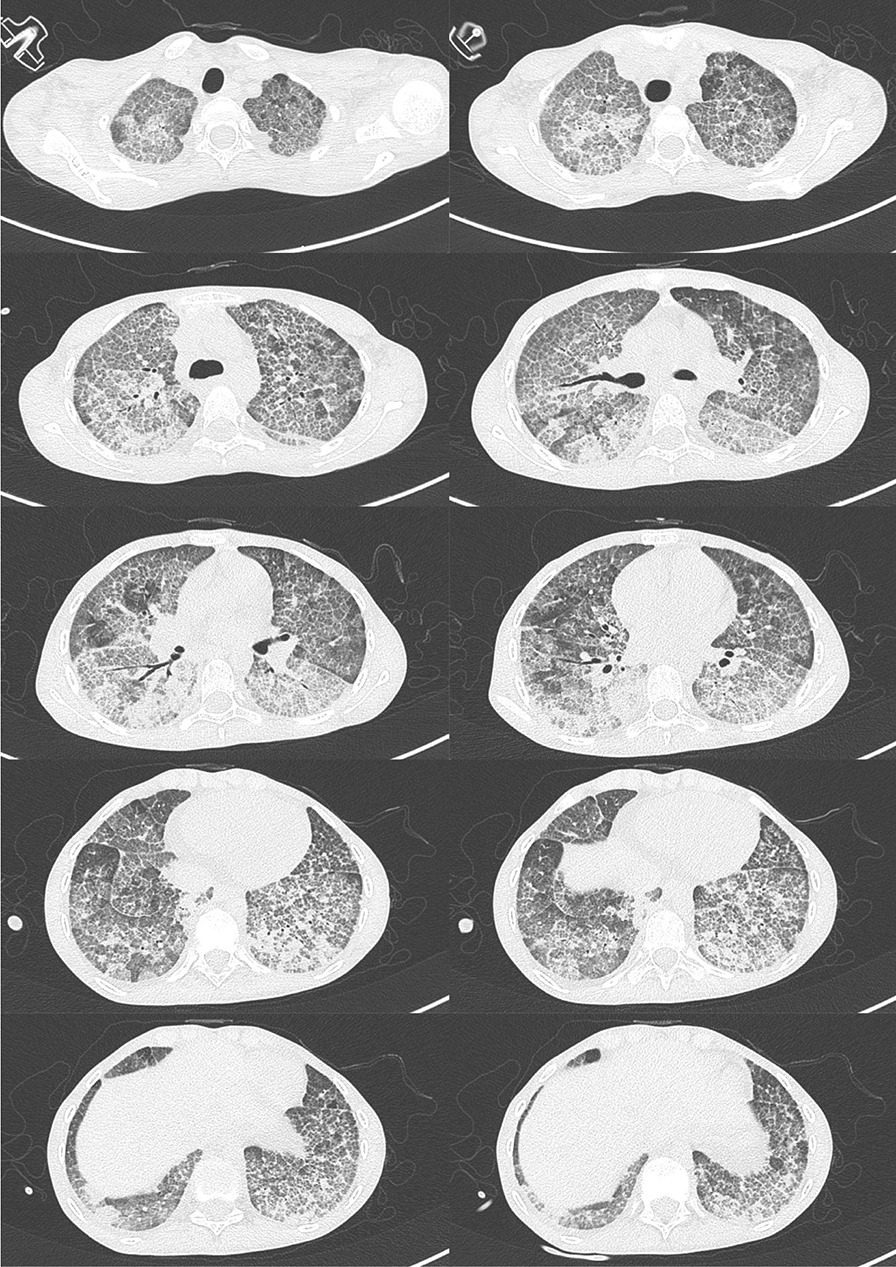
High-resolution tomography of chest showing extensive diffuse bilateral thickening of the lung interstitium with superimposed interlobular septal thickening consistent with “crazy paving” pattern
The patient then underwent flexible bronchoscopy under general anesthesia, and an apparent milky fluid was aspirated and sent for laboratory testing (Fig. 3). Given the lungs’ abundant secretions, a therapeutic whole lung lavage (WLL) of both lungs was planned for the next day. WLL was done for two separate sessions, starting with the left lung, then followed by the right lung after 3 days, and we did the same procedure steps for both. When the patient arrived at the operating room at the first session, electrocardiographic and invasive arterial pressure monitoring was established. He wore a nonbreathable mask, and oxygen saturation was maintained at >90%. Anesthesia was induced with propofol 25 mg, sevoflurane 2–3%, and fentanyl 100 μg and maintained with sevoflurane 1% infusion. He was intubated with a double-lumen endotracheal tube size 32 Fr, and its correct position was confirmed by fiberoptic bronchoscopy. Oxygen saturation was 100% on intermittent positive pressure ventilation mood with a volume control of 350 mL, positive end-expiratory pressure of 5 cm H2O, and airway pressure of 40 cm H2O, which were continuously monitored, and regular arterial blood gas analysis. WLL was performed with the patient in the right lateral position on the operating table, and we did sequential lavage with warm saline solution at body temperature followed by passive drainage under gravity. An experienced physiotherapist performed manual chest vibration and percussion. Cycles were repeated until 10 L of total lavage volume was used and clear fluid effluent was obtained (Fig. 4). The procedure lasted approximately 4 hours. The patient was transferred to the intensive care unit for ventilatory support, where he was extubated within 12 hours. Manual chest physiotherapy techniques and positioning maneuvers were continued postoperatively. Finally, a right-sided WLL was planned for within the next 72 hours with the same procedure. After completing the WLL, the patient reported a dramatic improvement in the SOB but still required oxygen therapy.
Fig. 3.
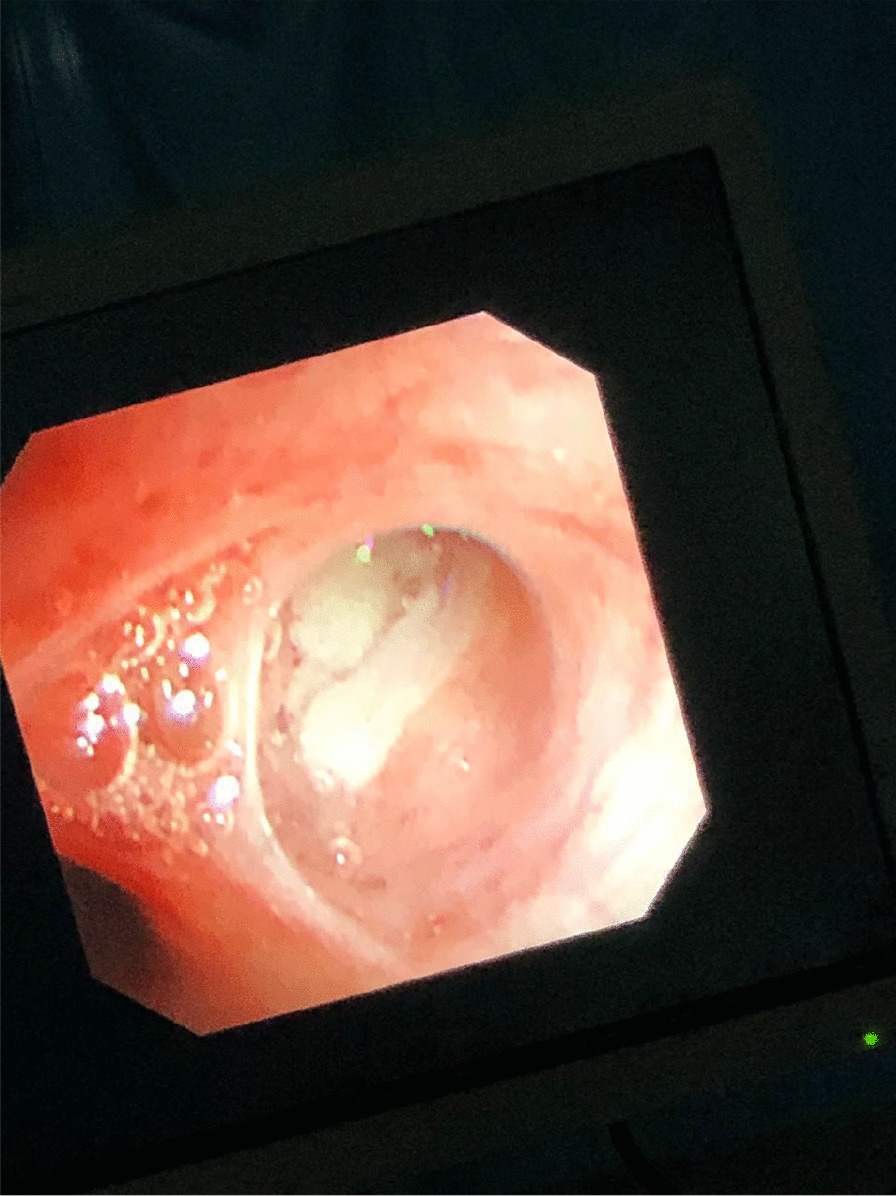
A milky appearance of the secretions throughout all the airway
Fig. 4.
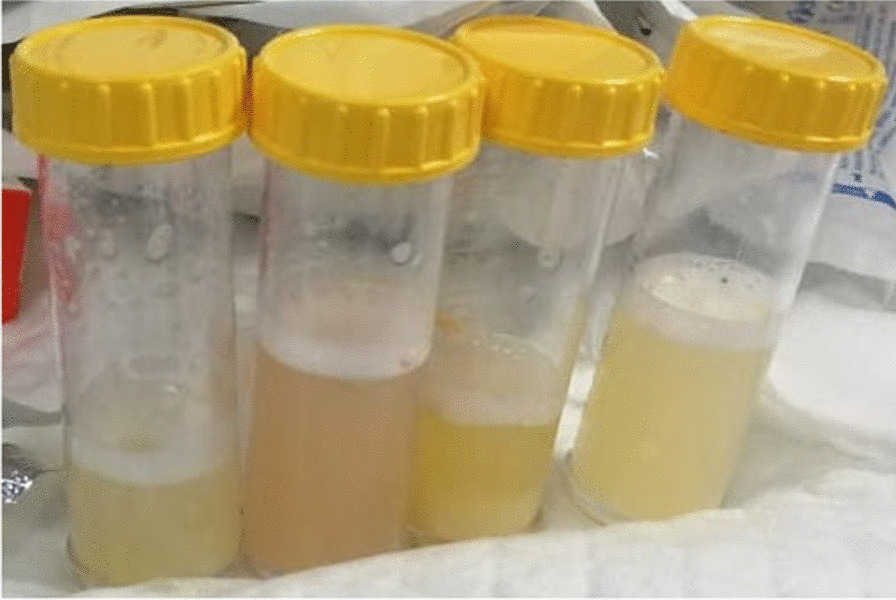
Milky appearance of the bronchoalveolar lavage fluid
Cytological examination of the bronchoalveolar lavage fluid (BALF) revealed multiple eosinophilic globules on the background of granular material and inflammatory cells, including macrophages and lymphocytes. They are periodic acid–Schiff (PAS) stain positive (Fig. 5). PAP diagnosis was confirmed, and antibodies against GM-CSF and necessary immunological and infectious workup were requested (Table 1). Antibodies against GM-CSF returned positive in the patient serum. Microbiological investigations of BALF returned negative, including culture studies. Given the absence of clinical and laboratory evidence of hematological, rheumatological, and infectious diseases, and the presence of anti-GM-CSF antibodies in the patient serum and BALF, the diagnosis of aPAP was established. The patient was discharged from the hospital on oxygen therapy, and he maintained oxygen saturation of >93% on 5 L of oxygen for 1 month, then gradually tapered off until he was completely weaned off the oxygen therapy. After 6 months of follow-up, the patient’s oxygen saturation was > 93% on room air, and the symptoms completely disappeared.
Fig. 5.
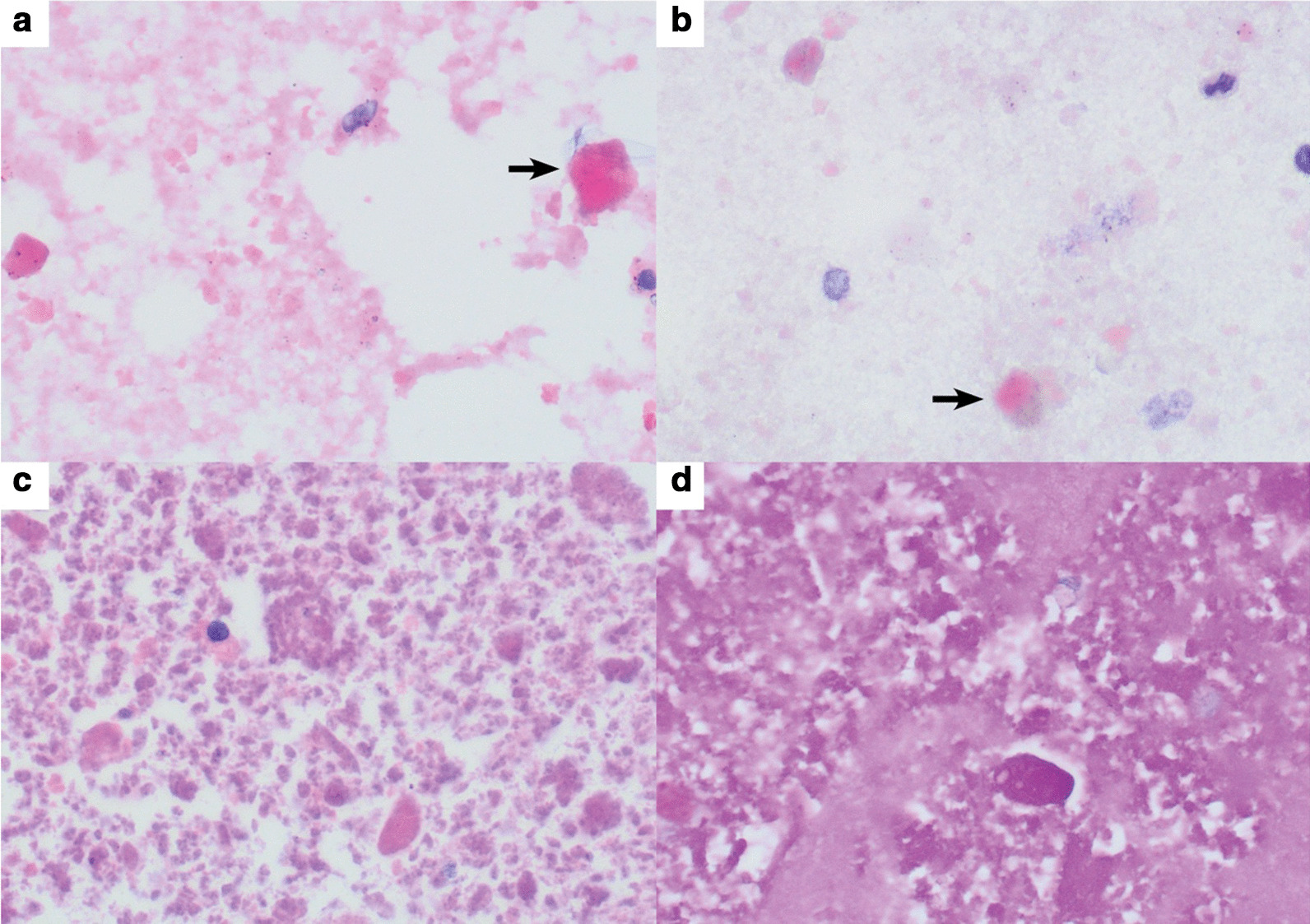
Bronchoalveolar lavage smears showing many dense globules (arrows) in background of finely granular eosinophilic material using (a) hematoxylin and eosin stain (H&E) ×400 and (b) Papanicolaou stain ×400. Cell block of bronchoalveolar lavage showing granular and globular eosinophilic material that was Periodic acid–Schiff (PAS) positive using (c) H&E stain ×400 and (d) PAS stain ×400
Discussion
Pulmonary alveolar proteinosis is a rare disease first described in 1958 [9]. Since that time, our understanding has improved given advances achieved in the molecular and pathological tools available to clinicians. In contrast to adult PAP, which is usually caused by an autoimmune process [10], most PAP cases in children and adolescents are caused by genetic defects leading to abnormal synthesis of alveolar surfactant [11].
PAP classification is not consistent, and multiple nomenclatures are present for different disease forms [3]. PAP can be categorized into two major categories (Fig. 6). The first category is characterized by impaired surfactant clearance, while the second category’s leading cause is defective surfactant metabolism/production.
Fig. 6.

Classification of pulmonary alveolar proteinosis based on underlying cause. GM-CSF granulocyte-macrophage colony-stimulating factor
The disorders caused by impaired surfactant clearance account for most PAP cases in adults [12]. Impaired surfactant clearance manifests because of either diminished GM-CSF function, also known as primary PAP, or decreased number/impaired function of alveolar macrophage, also known as secondary PAP [13]. Primary PAP is further classified into autoimmune and hereditary. Autoimmune PAP is the most common form of PAP in adults and is characterized by circulating autoantibodies against GM-CSF in the patient’s serum and/or BALF [4, 6]. Hereditary PAP is caused by defects in genes encoding GM-CSF receptors [13]. Infection, malignancy, immunodeficiencies, rheumatological diseases, or inhalation of chemicals or minerals can lead to secondary PAP [6, 14].
GATA binding protein 2 is a transcription factor that is encoded by the GATA2 gene. It is essential for normal hematopoiesis [15]. Haploinsufficiency GATA2 gene causes deficiency in GATA binding protein 2, which has heterogeneous presentation including hematologic disorders (such as familial myelodysplastic syndrome/acute myeloid leukemia), immunodeficiency, secondary PAP, and dermatologic disorders (such as panniculitis and erythema nodosum) [16–19]. We exclude the possibility of GATA2 mutation in our patient as a cause of PAP for two reasons. First, the clinical history (including family history) and laboratory findings were not consistent with the clinical presentation of GATA2 gene mutation. However, we cannot exclude GATA2 mutation as some patients may be initially asymptomatic and have completely normal laboratory studies [20, 21]. Second, the patient has autoantibodies in his serum against GM-CSF, which has an accuracy of almost 100% [22, 23]. Taken collectively, the patient’s clinical presentation and laboratory investigations support the diagnosis of primary PAP (with aPAP subtype), not secondary PAP.
Autoimmune PAP is rarely reported in children and adolescents. It may be underdiagnosed given the rarity of the disease and because the test of anti-GM-CSF is expensive and not widely available [10]. We conducted a PubMed search using the following terms: “pulmonary alveolar proteinosis” in combination with “children,” “adolescent,” and “pediatric.” We found 13 cases of pediatric aPAP reported in the literature (Table 2). We cannot exclude the possibility of publication bias. At the onset of the symptoms, the mean age of patients was 12 (±3.04) years; the majority of them were female (64.28%) [24–36].
Table 2.
Summary of autoimmune pulmonary alveolar proteinosis cases reported in children and adolescents
| Case | Age (years)a | Gender | Presenting symptoms | Chronic diseases | Treatment | Outcome | Country |
|---|---|---|---|---|---|---|---|
| Latzin et al. [36] | 11b | M | Exertional SOB | NR | WLL | Partially improved | France |
| Price et al. [35] | 13 | M |
Mild exertional SOB Productive cough FTT |
No |
WLL Inhaled rhGM-CSF |
Improved | Canada |
| Yamamoto et al. [34] | 9 | F | Dry cough | No |
WLL Inhaled rhGM-CSF |
Improved | Japan |
| Robinson et al. [33] | 16 | F | Mild SOB | No |
WLL Inhaled rhGM-CSF |
Improved | USA |
| DiBlasi et al. [32] | 13 | M |
SOB Cough Fatigue Weight loss |
NR | WLL | Improved | USA |
| Strickler et al. [31] | 13 | F |
SOB Dry cough Fever Exercise intolerance Fatigue |
No | WLL | Improved | Chile |
| Sideris et al. [30] | 6 | F | NR | Niemann–Pick disease |
WLL Inhaled rhGM-CSF |
Improved | USA |
| Trukalj et al. [29] | 10 | M |
Frequent respiratory infections Cough |
No |
WLL Inhaled rhGM-CSF Bilateral lung transplantation |
Improved | Croatia |
| Gajewska et al. [28] | 14 | F |
Exertional SOB Cough |
No |
WLL Inhaled rhGM-CSF |
Improved | Denmark |
| Sirin et al. [27] | 15 | F | Exertional SOB | No |
WLL Inhaled rhGM-CSF |
Improved | Turkey |
| Feld et al. [26] | 16 | F |
Exertional SOB Cough Weight loss |
Raynaud syndrome |
WLL Inhaled rhGM-CSF |
Improved | USA |
| Meka et al. [25] | 17 | F |
Exertional SOB Cough |
No | WLL | NR | USA |
| Shivji et al. [24] | 13 | F | Exertional SOB | No | WLL | Improved | Canada |
| Current report | 15 | M |
Exertional SOB Dry cough |
No | WLL | Improved | Saudi Arabia |
aAge of patient at onset of symptoms
bDisease onset was at age of 11 years. However, the bronchoalveolar lavage fluid was obtained and analyzed at age of 24 years with evidence of elevated level of anti-GM-CSF
F female, FFT failure to thrive, M male, NR not reported, rhGM-CSF recombinant human granulocyte-macrophage colony-stimulating factor, SOB shortness of breath, WLL whole lung lavage
The clinical presentation of aPAP typically follows an insidious and progressive course [37]. Among the reported cases of pediatric aPAP, the following symptoms were reported (Table 2): exertional shortness of breath (78%), cough (dry or productive) (64%), fatigue (14%), fever, weight loss, failure to thrive, and recurrent respiratory infections (7% each) [24–36]. Lung crackles, finger clubbing, cyanosis, and respiratory distress may be noted upon clinical examination [7]. There are no characteristics laboratory findings for the diagnosis of aPAP except an elevated level of anti-GM-CSF [2]. The reported sensitivity and specificity of anti-GM-CSF in diagnosing aPAP is 100% [22, 23]. However, it is expensive and not widely available. Our patient had polycythemia secondary to chronic hypoxemia and mildly elevated LDH, which has been reported for some patients [38, 39]. Radiological findings are not specific for aPAP but can help narrow the possible diagnosis [40, 41]. On chest X-ray (CXR), there are bilateral patchy infiltrates with or without bronchogram, and on HRCT, there is a crazy-paving pattern (ground-glass opacities superimposed on septal thickening), which is reported to be more common in aPAP than in other forms of PAP [42–44]. Diagnosis of aPAP is suspected when typical radiological findings are present in HRCT along with supportive clinical presentation and exclusion of possible secondary causes that can cause PAP [2, 37]. A definitive diagnosis of aPAP required the presence of typical histopathological findings from a lung biopsy or BALF and detection of anti-GM-CSF antibodies in patients’ serum and/or BLAF [2, 10]. A stepwise approach for diagnosing aPAP has been suggested by some experts [10].
Management of aPAP depends on the clinical status of the patient. Asymptomatic patients may be closely observed and monitored without specific treatment [37]. For symptomatic patients, WLL is the most effective form of treatment and, for a long time, considered the “standard of care” [45, 46]. Data suggest that the introduction of the WLL to the treatment plan of PAP significantly improved 5-year survival [45]. After WLL, patients usually show a remarkable improvement in their clinical and functional status [47, 48]. No drug has been approved for the treatment of aPAP in any country [49]. Given the GM-CSF dysfunction due to neutralizing autoantibodies, efforts were made to address this pathological process by administering recombinant GM-CSF (rhGM-CSF) [50]. Nevertheless, this approach is off-label, expensive, not widely available, and less effective than WLL when used as monotherapy [49, 51, 52]. On account of these limitations, rhGM-CSF therapy is mostly used as augmentation therapy for patients with unsatisfactory responses to first-line treatment [2, 53]. The efficacy of rhGM-CSF depends on the route of administration, dose, and duration of therapy. A 2018 meta-analysis showed that nebulized rhGM-CSF is more effective than subcutaneous rhGM-CSF [54]. A subsequent randomized controlled trial (RCT) has confirmed this conclusion and further supports the role of inhaled rhGM-CSF as adjustment treatment to WLL for aPAP [49, 51]. The response rate appeared to increase with a continuous and prolonged course of treatment proportionally [49, 51, 55]. It is important to mention that we are not aware of randomized trials of rhGM-CSF therapy in children or adolescents. Trials of rhGM-CSF excluded this age group; hence, there are limited data on this therapy’s safety and efficacy in the pediatric age group and adolescents [26–30, 33–35].
The optimum salvage treatment for refractory cases in the pediatric age group and adolescents is unknown. Targeting autoantibodies with rituximab and plasmapheresis have been used with variable success in adult patients [56–59]. Lung transplantation is a more aggressive and invasive intervention and considered the last-resort option. There is one case report of successful treatment of aPAP in a 4-year-old child who underwent bilateral lung transplantation after failure of inhaled rhGM-CSF (Table 2) [29]. However, the patient developed monomorphic post-transplant lymphoproliferative disease (PTLD) associated with Epstein–Barr virus infection and posterior reversible encephalopathy syndrome (PRES). More studies are needed to elucidate the exact role of these invasive and aggressive interventions in treating children and adolescents with aPAP. Corticosteroids have a long history of safety and efficacy in treating multiple autoimmune diseases, and it may seem conceivable that they are useful in aPAP treatment. However, limited data refuted this proposal and showed the contrary by increasing morbidity and mortality [60]. Hence, they have no role in the treatment of aPAP.
Conclusion
PAP is a rare interstitial lung disease with multiple types and clinical presentations. aPAP is not the usual form in children and adolescents. However, it should be considered in the differential diagnosis after excluding more common causes such as congenital and secondary forms. WLL should be the first-line treatment with or without inhaled rhGM-CSF.
Acknowledgements
We thank the patient and his family for allowing us to share his clinical data, Dr. Abdullah Algarni, MBBS, for medical writing support of an earlier version of the manuscript, and Mr. Abdullah Alsimyr, CEO of RBC laboratory, and Mr. Shan Navas from Bioscientia GmbH for their help with the anti-GM-CSF test.
Abbreviations
- ACPA
Anti-citrullinated peptide antibodies
- ALP
Alkaline phosphatase
- ALT
Alanine transaminase
- ANA
Antinuclear antibodies
- Anti-GM-CSF
Anti-granulocyte-macrophage colony-stimulating factor
- aPAP
Autoimmune pulmonary alveolar proteinosis
- aPTT
Activated partial thromboplastin time
- AST
Aspartate transaminase
- BALF
Bronchoalveolar lavage fluid
- c-ANCA
Cytoplasmic antineutrophil cytoplasmic autoantibodies
- Cr
Creatinine
- ED
Emergency department
- ESR
Erythrocyte sedimentation rate
- FFT
Failure to thrive
- GGT
Gamma-glutamyl transaminase
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- H&E
Hematoxylin and eosin stain
- HBsAb
Hepatitis B surface antibody
- HBsAg
Hepatitis B surface antigen
- HCV
Hepatitis C virus
- HIV
Human immunodeficiency virus
- INR
International normalization rate
- LDH
Lactate dehydrogenase
- NR
Not reported
- p-ANCA
Perinuclear antineutrophil cytoplasmic antibodies
- P/E
Physical examination
- PAP
Pulmonary alveolar proteinosis
- PAS
Periodic acid–Schiff stain
- PPD
Purified protein derivative
- PT
Prothrombin time
- RBC
Red blood cells
- RBG
Random blood glucose
- RF
Rheumatoid factor
- rhGM-CSF
Recombinant human granulocyte-macrophage colony-stimulating factor
- SOB
Shortness of breath
- WBC
White blood cells
- WLL
Whole lung lavage
Authors' contributions
AMS was the pulmonology consultant and head of the team who took care of the patient. RAA and MAA were the senior residents in the pulmonology and general internal medicine teams, respectively, who took care of the patient. AMH and NDA were the junior residents in the pulmonology and general internal medicine teams, respectively, who took care of the patient. RNB performed the histopathological examination of the bronchoalveolar lavage fluid and shared the microscopic pictures. AMS and RAA drafted the manuscript. All authors read and approved the final manuscript.
Funding
There was no funding for this report.
Availability of data and materials
Data are available from the corresponding author upon reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient’s father for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reference
- 1.Kumar A, Abdelmalak B, Inoue Y, Culver DA. Pulmonary alveolar proteinosis in adults: pathophysiology and clinical approach. Lancet Respir Med. 2018;6(7):554–565. doi: 10.1016/S2213-2600(18)30043-2. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki T, Trapnell BC. Pulmonary alveolar proteinosis syndrome. Clin Chest Med. 2016;37(3):431–440. doi: 10.1016/j.ccm.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135(2):223–235. doi: 10.1016/j.clim.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCarthy C, Avetisyan R, Carey BC, Chalk C, Trapnell BC. Prevalence and healthcare burden of pulmonary alveolar proteinosis. Orphanet J Rare Dis. 2018;13(1):129. doi: 10.1186/s13023-018-0846-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki T, Sakagami T, Young LR, Carey BC, Wood RE, Luisetti M, et al. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. Am J Respir Crit Care Med. 2010;182(10):1292–1304. doi: 10.1164/rccm.201002-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177(7):752–762. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstein LS, Kavuru MS, Curtis-McCarthy P, Christie HA, Farver C, Stoller JK. Pulmonary alveolar proteinosis: clinical features and outcomes. Chest. 1998;114(5):1357–1362. doi: 10.1378/chest.114.5.1357. [DOI] [PubMed] [Google Scholar]
- 8.Nogee LM, III FSC, Hamvas A. Diffuse Lung Diseases in Childhood. In: Kliegman R. M., Stan B. F., III J. W. S. G., Schor N. F., editors. Nelson Textbook of Pediatrics. 2. 20 ed. Philadelphia, PA: Elsevier, Inc.; 2016. p. 2116-20.
- 9.Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258(23):1123–1142. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 10.Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, et al. Pulmonary alveolar proteinosis. Nat Rev Dis Primers. 2019;5(1):16. doi: 10.1038/s41572-019-0066-3. [DOI] [PubMed] [Google Scholar]
- 11.Hamvas A, Cole FS, Nogee LM. Genetic disorders of surfactant proteins. Neonatology. 2007;91(4):311–317. doi: 10.1159/000101347. [DOI] [PubMed] [Google Scholar]
- 12.Ishii H, Tazawa R, Kaneko C, Saraya T, Inoue Y, Hamano E, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J. 2011;37(2):465–468. doi: 10.1183/09031936.00092910. [DOI] [PubMed] [Google Scholar]
- 13.Griese M. Pulmonary alveolar proteinosis: a comprehensive clinical perspective. Pediatrics. 2017;140:2. doi: 10.1542/peds.2017-0610. [DOI] [PubMed] [Google Scholar]
- 14.Wang BM, Stern EJ, Schmidt RA, Pierson DJ. Diagnosing pulmonary alveolar proteinosis. A review and an update. Chest. 1997;111(2):460–6. doi: 10.1378/chest.111.2.460. [DOI] [PubMed] [Google Scholar]
- 15.McReynolds LJ, Calvo KR, Holland SM. Germline GATA2 mutation and bone marrow failure. Hematol Oncol Clin North Am. 2018;32(4):713–728. doi: 10.1016/j.hoc.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809–821. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collin M, Dickinson R, Bigley V. Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol. 2015;169(2):173–187. doi: 10.1111/bjh.13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu AP, McReynolds LJ, Holland SM. GATA2 deficiency. Curr Opin Allergy Clin Immunol. 2015;15(1):104–109. doi: 10.1097/ACI.0000000000000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mir MA, Kochuparambil ST, Abraham RS, Rodriguez V, Howard M, Hsu AP, et al. Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations. Cancer Med. 2015;4(4):490–499. doi: 10.1002/cam4.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–97. doi: 10.1182/blood-2015-09-669937. [DOI] [PubMed] [Google Scholar]
- 22.Kitamura T, Uchida K, Tanaka N, Tsuchiya T, Watanabe J, Yamada Y, et al. Serological diagnosis of idiopathic pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2000;162(2 Pt 1):658–662. doi: 10.1164/ajrccm.162.2.9910032. [DOI] [PubMed] [Google Scholar]
- 23.Uchida K, Nakata K, Carey B, Chalk C, Suzuki T, Sakagami T, et al. Standardized serum GM-CSF autoantibody testing for the routine clinical diagnosis of autoimmune pulmonary alveolar proteinosis. J Immunol Methods. 2014;402(1–2):57–70. doi: 10.1016/j.jim.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shivji S, Amin R, Putra J. Bronchoalveolar lavage findings in a pediatric patient with primary pulmonary alveolar proteinosis. Int J Surg Pathol. 2020;28(3):294–295. doi: 10.1177/1066896919870598. [DOI] [PubMed] [Google Scholar]
- 25.Meka SG, Mohr M, Nair GB, Salman BA. Autoimmune pulmonary alveolar proteinosis mimicking Mycoplasma pneumonia in an adolescent. Respir Med Case Rep. 2020;30:101100. doi: 10.1016/j.rmcr.2020.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feld L, Jennings J, Fiorino EK, Harris M. Pulmonary alveolar proteinosis: a case of profound hypoxemia in a previously healthy teenager. Pediatr Emerg Care. 2019 doi: 10.1097/PEC.0000000000001820. [DOI] [PubMed] [Google Scholar]
- 27.Sirin Kose S, Asilsoy S, Uzuner N, Karaman O, Ozer E, Anal O. Pulmonary alveolar proteinosis in hereditary and autoimmune forms with 2 cases. Pediatr Emerg Care. 2018 doi: 10.1097/PEC.0000000000001536. [DOI] [PubMed] [Google Scholar]
- 28.Gajewska ME, Sritharan SS, Santoni-Rugiu E, Bendstrup EM. Autoimmune pulmonary alveolar proteinosis in an adolescent successfully treated with inhaled rhGM-CSF (molgramostim) Respir Med Case Rep. 2018;23:167–169. doi: 10.1016/j.rmcr.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trukalj M, Perica M, Ferencic Z, Erceg D, Navratil M, Redzepi G, et al. Successful treatment of autoimmune pulmonary alveolar proteinosis in a pediatric patient. Am J Case Rep. 2016;17:641-5. 10.12659/ajcr.897868 [DOI] [PMC free article] [PubMed]
- 30.Sideris GA, Josephson M. Pulmonary alveolar proteinosis and Niemann Pick disease type B: an unexpected combination. Respir Med Case Rep. 2016;19:37–39. doi: 10.1016/j.rmcr.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strickler A, Boza ML, Koppmann A, Gonzalez S. Autoimmune pulmonary proteinosis in a Chilean teenager, a rare aetiology of interstitial lung disease. BMJ Case Rep. 2014 doi: 10.1136/bcr-2012-006987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiBlasi RM, Crotwell D, Geiduscheck JM, Richardson P, Smith A, Salyer JW, et al. Therapeutic bilateral lung lavage in a child with pulmonary alveolar proteinosis. Pediatr Crit Care Med. 2010;11(3):e28–31. doi: 10.1097/PCC.0b013e3181b809d6. [DOI] [PubMed] [Google Scholar]
- 33.Robinson TE, Trapnell BC, Goris ML, Quittell LM, Cornfield DN. Quantitative analysis of longitudinal response to aerosolized granulocyte-macrophage colony-stimulating factor in two adolescents with autoimmune pulmonary alveolar proteinosis. Chest. 2009;135(3):842–848. doi: 10.1378/chest.08-1317. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto H, Yamaguchi E, Agata H, Kandatsu N, Komatsu T, Kawai S, et al. A combination therapy of whole lung lavage and GM-CSF inhalation in pulmonary alveolar proteinosis. Pediatr Pulmonol. 2008;43(8):828–830. doi: 10.1002/ppul.20856. [DOI] [PubMed] [Google Scholar]
- 35.Price A, Manson D, Cutz E, Dell S. Pulmonary alveolar proteinosis associated with anti-GM-CSF antibodies in a child: successful treatment with inhaled GM-CSF. Pediatr Pulmonol. 2006;41(4):367–370. doi: 10.1002/ppul.20347. [DOI] [PubMed] [Google Scholar]
- 36.Latzin P, Tredano M, Wust Y, de Blic J, Nicolai T, Bewig B, et al. Anti-GM-CSF antibodies in paediatric pulmonary alveolar proteinosis. Thorax. 2005;60(1):39–44. doi: 10.1136/thx.2004.021329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang T, Lazar CA, Fishbein MC, Lynch JP., 3rd Pulmonary alveolar proteinosis. Semin Respir Crit Care Med. 2012;33(5):498–508. doi: 10.1055/s-0032-1325160. [DOI] [PubMed] [Google Scholar]
- 38.Martin RJ, Rogers RM, Myers NM. PUlmonary alveolar proteinosis: shunt fraction and lactic acid dehydrogenase concentration as aids to diagnosis. Am Rev Respir Dis. 1978;117(6):1059–62. doi: 10.1164/arrd.1978.117.6.1059. [DOI] [PubMed] [Google Scholar]
- 39.Seymour JF, Doyle IR, Nakata K, Presneill JJ, Schoch OD, Hamano E, et al. Relationship of anti-GM-CSF antibody concentration, surfactant protein A and B levels, and serum LDH to pulmonary parameters and response to GM-CSF therapy in patients with idiopathic alveolar proteinosis. Thorax. 2003;58(3):252–257. doi: 10.1136/thorax.58.3.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johkoh T, Itoh H, Müller NL, Ichikado K, Nakamura H, Ikezoe J, et al. Crazy-paving appearance at thin-section CT: spectrum of disease and pathologic findings. Radiology. 1999;211(1):155–160. doi: 10.1148/radiology.211.1.r99ap10155. [DOI] [PubMed] [Google Scholar]
- 41.Maimon N, Paul N, Downey GP. Progressive dyspnea associated with a crazy-paving appearance on a chest computed tomography scan. Can Respir J. 2006;13(5):269–271. doi: 10.1155/2006/203830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishii H, Trapnell BC, Tazawa R, Inoue Y, Akira M, Kogure Y, et al. Comparative study of high-resolution CT findings between autoimmune and secondary pulmonary alveolar proteinosis. Chest. 2009;136(5):1348–1355. doi: 10.1378/chest.09-0097. [DOI] [PubMed] [Google Scholar]
- 43.Mahut B, Delacourt C, Scheinmann P, de Blic J, Mani TM, Fournet JC, et al. Pulmonary alveolar proteinosis: experience with eight pediatric cases and a review. Pediatrics. 1996;97(1):117–22. [PubMed] [Google Scholar]
- 44.Mazyck EM, Bonner JT, Herd HM, Symbas PN. Pulmonary lavage for childhood pulmonary alveolar proteinosis. J Pediatr. 1972;80(5):839–42. doi: 10.1016/s0022-3476(72)80143-4. [DOI] [PubMed] [Google Scholar]
- 45.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166(2):215–235. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 46.Zhang HT, Wang C, Wang CY, Fang SC, Xu B, Zhang YM. Efficacy of whole-lung lavage in treatment of pulmonary alveolar proteinosis. Am J Ther. 2016;23(6):e1671–e1679. doi: 10.1097/MJT.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 47.Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest. 2009;136(6):1678–1681. doi: 10.1378/chest.09-2295. [DOI] [PubMed] [Google Scholar]
- 48.Beccaria M, Luisetti M, Rodi G, Corsico A, Zoia MC, Colato S, et al. Long-term durable benefit after whole lung lavage in pulmonary alveolar proteinosis. Eur Respir J. 2004;23(4):526–531. doi: 10.1183/09031936.04.00102704. [DOI] [PubMed] [Google Scholar]
- 49.Trapnell BC, Inoue Y, Bonella F, Morgan C, Jouneau S, Bendstrup E, et al. Inhaled molgramostim therapy in autoimmune pulmonary alveolar proteinosis. N Engl J Med. 2020 doi: 10.1056/NEJMoa1913590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reed JA, Ikegami M, Cianciolo ER, Lu W, Cho PS, Hull W, et al. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol. 1999;276(4):L556–L563. doi: 10.1152/ajplung.1999.276.4.L556. [DOI] [PubMed] [Google Scholar]
- 51.Tazawa R, Ueda T, Abe M, Tatsumi K, Eda R, Kondoh S, et al. Inhaled GM-CSF for pulmonary alveolar proteinosis. N Engl J Med. 2019;381(10):923–932. doi: 10.1056/NEJMoa1816216. [DOI] [PubMed] [Google Scholar]
- 52.Khan A, Agarwal R, Aggarwal AN. Effectiveness of granulocyte-macrophage colony-stimulating factor therapy in autoimmune pulmonary alveolar proteinosis: a meta-analysis of observational studies. Chest. 2012;141(5):1273–1283. doi: 10.1378/chest.11-0951. [DOI] [PubMed] [Google Scholar]
- 53.Ohkouchi S, Akasaka K, Ichiwata T, Hisata S, Iijima H, Takada T, et al. Sequential granulocyte-macrophage colony-stimulating factor inhalation after whole-lung lavage for pulmonary alveolar proteinosis. A report of five intractable cases. Ann Am Thorac Soc. 2017;14(8):1298–304. doi: 10.1513/AnnalsATS.201611-892BC. [DOI] [PubMed] [Google Scholar]
- 54.Sheng G, Chen P, Wei Y, Chu J, Cao X, Zhang HL. Better approach for autoimmune pulmonary alveolar proteinosis treatment: inhaled or subcutaneous granulocyte-macrophage colony-stimulating factor: a meta-analyses. Respir Res. 2018;19(1):163. doi: 10.1186/s12931-018-0862-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2010;181(12):1345–1354. doi: 10.1164/rccm.200906-0978OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kavuru MS, Malur A, Marshall I, Barna BP, Meziane M, Huizar I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J. 2011;38(6):1361–1367. doi: 10.1183/09031936.00197710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soyez B, Borie R, Menard C, Cadranel J, Chavez L, Cottin V, et al. Rituximab for auto-immune alveolar proteinosis, a real life cohort study. Respir Res. 2018;19(1):74. doi: 10.1186/s12931-018-0780-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonfield TL, Kavuru MS, Thomassen MJ. Anti-GM-CSF titer predicts response to GM-CSF therapy in pulmonary alveolar proteinosis. Clin Immunol. 2002;105(3):342–350. doi: 10.1006/clim.2002.5301. [DOI] [PubMed] [Google Scholar]
- 59.Luisetti M, Rodi G, Perotti C, Campo I, Mariani F, Pozzi E, et al. Plasmapheresis for treatment of pulmonary alveolar proteinosis. Eur Respir J. 2009;33(5):1220–1222. doi: 10.1183/09031936.00097508. [DOI] [PubMed] [Google Scholar]
- 60.Akasaka K, Tanaka T, Kitamura N, Ohkouchi S, Tazawa R, Takada T, et al. Outcome of corticosteroid administration in autoimmune pulmonary alveolar proteinosis: a retrospective cohort study. BMC Pulm Med. 2015;15:88. doi: 10.1186/s12890-015-0085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the corresponding author upon reasonable request.