

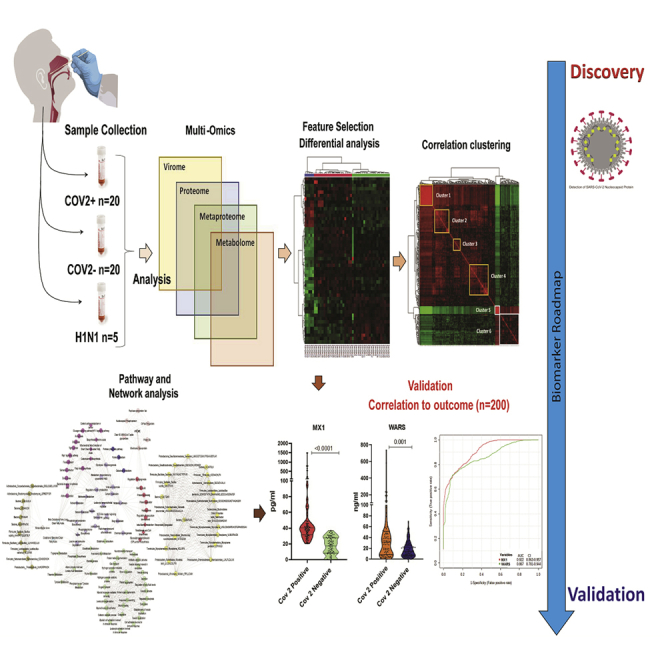
Summary
Rapid diagnosis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection still remains a major challenge. A multi-omic approach was adopted to analyze the respiratory specimens of 20 SARS-CoV-2-positive, 20 negative and 15 H1N1 pdm 2009 positive cases. Increased basal level of MX1 (MX dynamin-like GTPase 1) and WARS (tryptophan-tRNA ligase) correlated with SARS-CoV-2 infection and its outcome. These markers were further validated in 200 suspects. MX1>30pg/ml and WARS>25ng/ml segregated virus positives [AUC = 94% CI: (0.91–0.97)] and severe patients [AUC>0.85%]. Our results documented significant increase in immune activation; metabolic reprograming and decrease in oxygen transport, wound healing and others linked proteins and metabolites in patients with coronavirus disease 2019 (COVID-19). Multi-omics profiling correlated with viremia and segregated asymptomatic patients with COVID-19. Additionally, we identified increased respiratory pathogens (Burkholderiales, Klebsiella pneumonia) and decreased lactobacillus salivarius (FDR<0.05) in COVID-19 specimens. In conclusion, increased basal MX1 and WARS levels correlates with SARS-CoV-2 infection and could aid in the identification of patient's predisposed to higher severity.
Subject areas: Molecular biology, Virology
Graphical abstract
Highlights
-
•
Multi-omics analysis of respiratory specimen is distinct in SARS-CoV-2 infection
-
•
Change in oropharyngeal metaproteome correlates with SARS-CoV-2 infection
-
•
Multi-omics analysis reveals various process regulated by SARS-CoV-2 infection
-
•
Increased basal MX1 and WARS could segregate patients with severe and positive SARS-CoV-2
Molecular biology; Virology
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes coronavirus disease 2019 (COVID-19). Spread of the virus is severe, and within few months more than 5.69 million were infected; mortality was seen in more than 356 thousand patients and counting. The global recovery rate of COVID-19 is nearly 41% and the mortality rate is close to 7% (Ghinai et al., 2020; Guan et al., 2020; Li et al., 2007). About 80% of the patients infected with SARS-CoV-2 are asymptomatic or display mild symptoms and has good prognosis (Mehta et al., 2020; Thevarajan et al., 2020). However about 20% of patients who suffer from respiratory distress require immediate access to specialized intensive care; otherwise, they may die rapidly (Mehta et al., 2020; Murthy et al., 2020; Thevarajan et al., 2020; Wu and McGoogan, 2020). Therefore, it is vital to understand the pathogenesis of SARS-CoV-2 infection and identify patients who are positive, asymptomatic, or patients who are predisposed to COVID-19-associated severe respiratory distress. Thus, employment of new approaches which could characterize baseline molecular determinants associated with SARS-CoV-2 infection and outcome becomes critical.
Currently oropharyngeal/nasopharyngeal swab (respiratory specimen) is used for real time-polymerase chain reaction (RT-PCR) based detection of viral genetic material and is the gold standard of COVID-19 detection; clinical sensitivity ranging from 66% to 80% (Udugama et al., 2020). However, the respiratory specimen is a mixture of viral, host molecules (proteins, metabolites), and microbiome which could be indicative of viral presence and could be utilized as a surrogate for screening purpose. Additionally, the respiratory specimen could work as a liquid biopsy and provide an insight into the pathophysiology of COVID-19 disease. Methods with higher sensitivity and resolution than RT-PCR are needed to improve the management of the pandemic. Recently, Shen et al. (Shen et al., 2020) used high-resolution mass spectrometry-based proteomics and metabolomics in serum specimens to provide insights into the pathophysiology of COVID-19. However, detailed characterization of the respiratory specimen in terms of proteomics, metabolomics and metaproteomics needs to be analyzed for improving viral diagnostics and host related biomarkers linked to predisposition to severe outcome of disease.
In this pilot study, we hypothesized that SARS-CoV-2 induces characteristic molecular changes that can be detected in the respiratory specimen of COVID-19-positive patients. These molecular changes could be distinct and help segregate SARS-CoV-2-positive patients and provide pathophysiological insights and help in characterization of patients predisposed to severe outcome. To test this hypothesis, global proteome, metaproteome, and metabolome analysis was performed on the respiratory specimens followed by an in-depth analysis of the regulatory networks. The integrated analysis identified correlation clusters predictive of viral pathogenesis and promising targets for COVID-19 outcome. Targets identified in our derivative cohort were validated in 200 COVID-19 suspected specimens, and the sensitivity/specificity was recorded for COVID-19 infection and outcome (severity, symptomatic) prediction. We also analyzed the association of combined oropharyngeal and nasopharyngeal microbiome (metaproteome) with SARS-CoV-2 infection and outcome determination.
Results
Proteome profile of the respiratory specimen could stratify COVID-19-positive patients
We aimed to identify biomolecules having diagnostic value and outcome prediction value for SARS-CoV-2 infection (Figure 1A). Label-free quantitative proteomics was performed and SARS-CoV-2 proteins or linked proteins, host proteins, and bacterial peptides were identified in the respiratory specimen of COVID-19-positive, COVID-19-negative, and H1N1 pdm 2009 patients.
Figure 1.

Quantitative virome profile of the respiratory specimen
(A) Work flow for COVID-19 biomarker identification (discovery phase): Differentially regulated viral/host proteins, metabolites and bacterial peptides were identified which was subjected to random forest and AUROC analysis for identification of candidate for validation. Correlation clustering and pathway analysis provided insight on the pathophysiology of SARS-COV2 infection. Finally the identified candidate indicators were validated in 200 COVID suspect for SARS-CoV-2 infection and outcome prediction.
(B) Log normalized abundance of 6 viral and 1 viral associated proteins identified in the respiratory specimen of COVID-19 positive as compared to COVID-19 negative patients. ∗∗∗ signifies p < 0.05.
(C) Principle component analysis score plot showing segregation of COVID-19 positive patients from COVID-19 negative based on the identified Viral and associated proteins and Random forest analysis showing the mean decrease in accuracy of the viral and associated proteins (Red = upregulated and Green = downregulated) in COVID-19-positive as compared to COVID-19-negative patients.
(D) Partial least square discriminant analysis showing clear segregation of COVID-19-positive patients (Red dots) from COVID-19-negative (Green dots) and influenza A H1N1 pdm 2009 pdm 2009 positive patients (Blue dots) based on host proteomic evaluation and heatmap, hierarchical cluster analysis of top 40 proteins (p < 0.05) capable of segregating COVID-19-positive (orange bar) patients from COVID-19-negative (green bar) or influenza A H1N1 pdm 2009-positive patients (blue bar) (Red = upregulated, Green = downregulated and black = unregulated).
(E) Random forest analysis showing the mean decrease in accuracy of the proteins (Red = upregulated and Green = downregulated and yellow = unchanged) in COVID-19-positive compared to COVID-19-negative or Influenza A H1N1 pdm 2009 pdm 2009-positive patients and relative abundance (Log normalized) for MX1 and WARS showing significant increase in MX1 and WARS levels (p value∗∗ = p < 0.05, ∗∗∗<0.01).
(F) Joint AUROC analysis of MX1 and WARS documenting an AUC = 0.96 CI (0.82–1) p < 0.05 along with prediction class probability score plot showing segregation of CoV2 positive and CoV2 negative.
(G) KEGG pathway analysis of 184 proteins up regulated in COVID-19-positive respiratory specimen as compared to COVID-19-negative or Influenza A H1N1 pdm 2009 pdm 2009-positive cases (FC > 2; p < 0.05, FDR>0.05). Darker nodes are more significantly enriched gene sets. Bigger nodes represent larger gene sets. Thicker edges represent more overlapped genes. Yellow edge show viral linked network.
(H) KEGG pathway analysis of 60 proteins downregulated in COVID-19-positive respiratory specimen as compared to COVID-19-negative or Influenza A H1N1 pdm 2009 pdm 2009-positive cases (FC > 2; p < 0.05, FDR>0.05). Darker nodes are more significantly enriched gene sets. Bigger nodes represent larger gene sets. Thicker edges represent more overlapped genes. Three clusters of edges can be identified: (A) gas transport and oxygen transport cluster (B) wound healing and inflammation cluster and (C) vesicle transport cluster. Yellow edge show wound healing linked network.
(I) Principle component analysis (PCA) showing clear segregation of COVID-19-positive patients based on metaproteins estimations and hierarchical cluster analysis of the metaproteins (p < 0.05) show clear segregation of COVID-19-positive patients (Red bar) from COVID-19-negative (Green bar) or Influenza A H1N1 pdm 2009-positive patients (Blue bar). (Dark brown = upregulated, Blue = downregulated and white = unregulated).
(J) Sunburst plot representative of microbial population difference (phylum: order: family) in COVID-19-positive respiratory specimen as compared to COVID-19 negative or Influenza A H1N1 pdm 2009-positive cases. Percent distribution of identified peptide is provided in each sunburst plot.
Respiratory virus (SARS-CoV-2) or linked proteome analysis
We identified significant increase in 6 SARS-CoV-2 proteins along with ACE2 in the respiratory specimen of COVID-19-positive patients compared to negative patients (p < 0.05, Figure 1B). Principle component analysis (PCA) and unsupervised clustering analysis segregated patients with COVID-19 (Figures 1C and S1, and Table S1). Mean decrease in the accuracy was highest for ACE2 followed by viral proteins (Figures 1C and S1) suggesting that quantitation of these viral or viral linked protein could be used for early identification of SARS-CoV-2 infection.
Respiratory proteome (host) analysis
Analysis of the host proteome (Figure S2) identified 1256 proteins, of them 132 were differentially expressed (DEP; 116 up- and 16 downregulated) in COVID-19 positive compared to negative patients (FC > 2, p < 0.05, Table S1, Figure S4). Additionally, 166 differentially expressed host proteomes (DEPs) (117 upregulated and 49 downregulated) were identified in COVID-19-positive patients compared to H1N1 pdm 2009 patients (FC > 2, p < 0.05, Table S1, Figure S4). Partial least square discriminant analysis and unsupervised hierarchical clustering analysis (Figures 1D and S3) segregated COVID-19-positive patients. Amongst the identified DEPs, mean decrease in the accuracy (random forest; 1000 trees) was highest for MX1 (MX dynamin-like GTPase 1) and WARS (tryptophan-tRNA ligase) making them the most important proteins for segregating COVID-19-positive patients from negative or H1N1 pdm 2009 patients (Figure 1E). The diagnostic efficiency of MX1 was 0.895 (0.746–1) and WARS was 0.948 (0.85–1) for COVID-19-positive detection (p < 0.05; Figure S8). Combined diagnostic efficiency of MX1 and WARS was 0.96 (0.826–1) and showed clear segregation of COVID-19-positive from negative patients (Figure 1F).
Interestingly, COVID-19-positive patients showed significant increase in viral-associated interferon signaling, immune activation including monocyte, and neutrophil activation (IL-2, IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor signaling), IL-17, and nucleotide-binding oligomerization (NOD)-like receptor signaling, bacterial invasion of epithelial cells, fluid shear stress, and glucose metabolism (Figure 1G, KEGG; Figures S5 and S6), whereas oxygen transport, wound healing, regulation of body fluids, and other pathways were decreased (Figure 1H, KEGG, FDR<0.05, Figure S7). Together these findings showed that COVID-19-positive patients have virus-mediated hyperimmune activation, deregulated oxygen transport, increased fluid shear stress, and glucose metabolism. Furthermore, proteins such as MX1 and WARS (tryptophan-tRNA ligase) are regulated by SARS-CoV-2 infection and could be validated for a probable indicator of COVID-19 infection and outcome prediction.
Respiratory meta-proteome analysis
Often SARS-CoV-2 infection precedes bacterial co-infection and is linked with longer duration and more severe infection (Bengoechea and Bamford, 2020; Gu and Korteweg, 2007). SARS-CoV-2 is also known to modulate the gut microbiota and is associated to immune cell activation and severe outcome (Dhar and Mohanty, 2020). We hypothesized that change in naso-pharyngeal/oropharyngeal microbiome (likely source of lung microbiome) due to SARS-CoV-2 infection could be reflective of altered pathogenesis and help in identification of SARS-CoV-2-positive infection and outcome. To explore this, metaproteome analysis was performed in the respiratory specimen of the study group (Figure S9). PCA along with unsupervised clustering analysis segregated COVID-19-positive from other groups (Figure 1I). At the phylum level, proteobacteria and Tenericutes were increased, corroborating to an increase in alpha diversity seen in COVID-19-positive respiratory specimen (Figures 1J and S10, p < 0.05). Linear discriminating analysis showed significant increase in bacterial peptides linked to proteobacteria, firmicutes, and tenericutes in COVID-19-positive respiratory specimen compared to H1N1 pdm 2009 or negative (p < 0.05; Figure S11, Table S3). Amongst the differentially expressed metaproteins (DEMPs), mean decrease in the accuracy was highest for Bacillus subtilis (Variable 82), and Burkholderiales making them the most important bacterial lowest common ancestor (LCA) which could segregate COVID-19-positive from negative or H1N1 pdm 2009 cases (AUROC>0.99; p < 0.05, Figure S12 and 13). Interestingly, functionality assessment of COVID-19-positive microbiome showed significant increase in bacterial enzyme linked to pantothenate and CoA biosynthesis in actinobacteria, aminoacyl-trna biosynthesis, and terpenoid backbone biosynthesis in firmicutes and amino-nucleotide sugar metabolism, peptidoglycan biosynthesis, fatty acid biosynthesis, glycerophospholipid metabolism, and energy metabolism in proteobacteria (Figure S14). Together, these results suggest that SARS-CoV-2 infection modulates the oropharyngeal microbiome and associated function which correlates with COVID-19 pathophysiology and could aid in segregation of SARS-CoV-2-positive patients.
Metabolic phenotype of the respiratory specimen is predictive of SARS-CoV-2 infection
SARS-CoV-2 infection could change the metabolic phenotype of respiratory specimen; recently, a distinctive plasma metabolomic profile associated with SARS-CoV-2 infection and severity is also reported (Shen et al., 2020). We evaluated and compared the respiratory metabolome in COVID-19 positive, negative, and H1N1 pdm 2009 patients (Figure S15). Metabolomic analysis identified 106 differentially expressed metabolites (DEMs) in COVID-19 positive patients (53up- and 53 downregulated) compared to negative (Figure 3A, FC > 1.5, P < 0.05, Figure S16, Table S2). In addition, 274 DEMs were identified when COVID-19 positive patients were compared to H1N1 pdm 2009 patients (Figure 2A, FC > 1.5, P < 0.05, Figure S16, Table S2). Partial least square discriminant analysis (PLS-DA), followed by unsupervised clustering analysis clearly segregated COVID-19 positive from negative or H1N1 pdm 2009 cases (Figures 2B, 2C, and S17). Amongst the DEMs, mean decrease in the accuracy was highest for N-acetylserotonin (C00978) and azelaic acid (C08261) making them the most important metabolites segregating COVID-19 positive patients (Figure 2D). N-acetylserotonin and azelaic acid showed a combined diagnostic efficiency of 0.987 (0.98–1) for SARS-CoV-2 positive segregation from negatives (Figure 2E p < 0.05; Figure S18). Interestingly COVID-19 positive patients showed significant increase in metabolic pathways linked to biosynthesis of unsaturated fatty acids, glycerophospholipid metabolism, ubiquinone/terpenoid-quinone biosynthesis, aminoacyl-tRNA biosynthesis and amino acid metabolism including phenylalanine, tyrosine and tryptophan biosynthesis (Figure 2F). whereas, pathways linked to thiamine metabolism, one carbon pool by folate, vitamin B6 metabolism, riboflavin metabolism and steroid biosynthesis were decreased (Figure 2G). Together these results suggest that respiratory specimen of COVID-19 positive are rich in oxidative and inflammatory metabolites including tryptophan and derivative, histidine, fatty acids, glycerolipid and have significantly lower levels of anti-inflammatory steroids or vitamins. Further, metabolites such as N-acetylserotonin (C00978) and azelaic acid (C08261) could segregate COVID-19 positive patients from the negative population and warrant validation in larger cohort.
Figure 3.

Respiratory multi-omics cross correlation delineate pathways linked to SARS-CoV2 persistence and pathogenesis
(A) Schematic representation of the integration analysis performed in this study. DEVP, differently expressed viral/associated proteins; DEP, differently expressed host proteins; DEMP, differently expressed host metaproteins; DEM, differently expressed metabolites were cross correlated to identify viral regulatory network.
(B) Principle component analysis (PCA) showing clear segregation of COVID-19-positive patients from COVID-19-negative based on the multi-omics profile.
(C) Global cross correlation analysis identified 6 clusters (4 upregulated marked in yellow and 2 down regulated marked in white). Details of cluster 1 are shown adjacent which show viral protein along with bacterial peptides, proteins, and metabolites correlation with each other. Each correlation is represented with a dot where red dot = r2>0.8, p < 0.05 and pink dot = r2>0.5, p < 0.05, respectively.
(D) Mean cluster intensity is shown cluster 1 and cluster 5 are significantly different in COVID-19-positive compared to COVID-19-negative patients (p < 0.05).
(E) Global correlation map is shown for the six cluster identified; cluster1: RED, cluster2: pink, cluster3: blue, cluster4: purple, cluster5: light blue, cluster6: light green. Viral proteins are shown in light pink, and bacterial peptide is shown in yellow (upregulated) and olive green (downregulated). The most important cluster1 and cluster5 are highlighted. Connecting line are representing correlation r2>0.5 p < 0.05, respectively.
Figure 2.

Metabolic phenotype of the respiratory specimen is predictive of SARS-CoV2 infection
(A) Volcano plot showing differentially expressed metabolites in COVID-19-positive vs. COVID-19-negative respiratory specimen and COVID-19-positive vs. Influenza A H1N1 pdm 2009-positive (Pink dots are significant [p < 0.05, FC > 1.5]).
(B) Partial least square discriminant analysis showing clear segregation of COVID-19-positive patients (Red dots) from COVID-19-negative (Green dots) and influenza A H1N1 pdm 2009-positive cases patients (Blue dots) based on metabolomic estimations.
(C) Hierarchical cluster (Heatmap) analysis of the metabolites identified in the study (p < 0.05) show clear segregation of COVID-19-positive patients (Red bar) from COVID-19-negative (Green bar) or Influenza A H1N1 pdm 2009-positive cases patients (Blue bar; Red = upregulated, Green = downregulated and black = unregulated).
(D) Random forest analysis showing the mean decrease in accuracy of the metabolites (Red = upregulated and Green = downregulated and yellow = unchanged) in COVID-19-positive as compared to COVID-19-negative or influenza A H1N1 pdm 2009-positive patients.
(E) Joint AUROC analysis of N-acetylserotonin (C00978) and azelaic acid (C08261) and documenting an AUC = 0.987 CI (0.98–1) p < 0.05 along with prediction class probability score plot showing segregation of CoV2 positive and CoV2 negative.
(F) Pathway and metabolite set enrichment analysis (KEGG) for the upregulated metabolites (FC > 1.5, p < 0.05) in COVID-19-positive respiratory specimen as compared to COVID-19-negative or influenza A H1N1 pdm 2009-positive specimen.
(G) Pathway and metabolite set enrichment analysis (KEGG) for the downregulated metabolites (FC > 1.5, p < 0.05) in COVID-19-positive respiratory specimen as compared to COVID-19-negative or Influenza A H1N1 pdm 2009-positive specimen.
Multi-omics integration analysis delineates pathways linked to SARS-CoV-2 pathogenesis
Next we hypothesized that expression alteration of the viral proteome should reflect an analogs change in the host proteome, metabolome and metaproteome (microbiome). For example, an increase in viral proteome or bacterial proteome should recapitulate a corresponding increase in proteins or metabolites associated pathways and members of known pathways should co-cluster and provide insight of COVID-19 pathogenesis. To explore this, global cross correlation and hierarchal clustering analysis were performed for differentially expressed viral proteome (DEVP: 7), differentially expressed proteins (DEP: 132), differentially expressed metaproteins (DEMP: 33), and DEM: 106) in COVID-19-positive patient compared with COVID-19-negative (Figure 3 A). PCA recapitulates the distinction between COVID-19-positive and negative patients (Figure 3B). Correlation analysis followed by hierarchical clustering (r2>0.5, p < 0.05) showed that viral proteins significantly correlates with bacterial and host proteins or metabolites and identified 6 clusters (Figure 3C). Mean intensity of cluster 1 and cluster 5 was significantly altered in COVID-19 positives (p < 0.05, Figure 3D). Pathway analysis for the proteins/metabolites linked with each cluster (Figure 3E) showed that increase in the viral proteome: cluster 1 was associated with increase in metaproteome (Bacterial taxa), proteins/metabolites linked to viral life cycle, regulation of angiogenesis, vasculature development, protein processing and maturation, beta-alanine metabolism, pantothenate and CoA biosynthesis and glycosylphosphatidylinositol (GPI) anchor biosynthesis (r2 > 0.5, p < 0.05, Figure 3E, Table S4). In addition, decrease in the bacterial taxa: cluster 5 was linked with significant decrease in oxidative phosphorylation, thermogenesis, beta oxidation of fatty acids, and arachidonic acid metabolism (r2>0.5, p<0.05, Figure 3E, Table S4). This analysis identifies the underlying metabolic variations associated with CoV-2 infection. Over all, these results establish a linear and direct relationship between viral proteome, bacterial metaproteome, host proteome, and metabolome in COVID-19-positive respiratory specimen.
Respiratory multi-omics correlates with viremia and shows increase in monocyte, platelet and immune activation and segregate non-symptomatic COVID-19 patients
High viral load inversely relates to the CT values (Zou et al., 2020). On correlation of respiratory multi-omics data with SARS-CoV-2 viremia, a total of 54 proteins and/or metabolites were identified. Proteins and metabolites associated to energy metabolism (pentose phosphate pathway, glycolysis), glycerophospholipid metabolism, purine metabolism and inflammation (IL-17 signaling pathway) showed inverse correlation with the CT values, and these were increased with increase in viremia (R2 > 0.5, p < 0.05, Figure S19, Table S5).
Analysis of respiratory immune imprints by enriching DEPs on the blood transcription module (BTM) space (Li et al., 2017) showed increase in immune activation generic cluster, enriched in monocytes, cell cycle and transcription and platelet activation module I and II in COVID-19 positives (>3 genes, Figure S20, Table S6). These modules directly correlated with the metabolites linked to arginine biosynthesis, cysteine and methionine metabolism, and aminoacyl-tRNA biosynthesis (Table S6). Correlation of immune (monocyte and platelet) clusters with arginine metabolism again highlights activated state of these cells in COVID-19 respiratory specimen.
Next, we explored the efficacy of the multi-omic profile in COVID-19 asymptomatic identification. Of the 271 (DEP, DEMP, and DEM), a total of 20 variables could segregate COVID-19 asymptomatics (Figure S21). PLS-DA along with unsupervised clustering analysis showed clear distinction between symptomatic and asymptomatic patients (Figure S21). The diagnostic efficiency of AKR1A1, COPE, PSME2 and C02571 (L-acetyl carnitine) was highest for asymptomatic, whereas symptomatic patients showed significant increase in Klebsiella pneumoniae, CTBP2 (C-terminal-binding protein 2), and 4-alpha-methyl-5-alpha-cholest-7-en-3-one (Figure S22). Together, these results show that multi-omics profile of COVID-19-positive respiratory specimen correlates with viremia provide an insight on immune activation and could also aid in the identification of asymptomatic patients but warrants validation in larger cohort.
Multi-omics analysis of the respiratory specimen validates MX1 and WARS as candidate indicator of SARS-CoV-2 infection and outcome
Validation
Multi-omics analysis identified a panel of molecules (Table 1) which could segregate COVID-19 positives. Of them, expression of MX1 and WARS (tryptophan-tRNA ligase) was evaluated in a larger cohort of 200 COVID-19 suspects. COVID-19 positives showed significant increase in MX1 and WARS expression as compared to COVID-19 negatives (p < 0.05, Figure 4A). The diagnostic efficiency of MX1 was 92% (AUROC = 0.92 (0.863–0.957)) and WARS was 86% (AUROC = 0.867(0.781–0.944)) for segregation of COVID-19 positivity (p < 0.05, Figure 4B). MX1 independently had predictive accuracy of 83% (1000 permutation) for SARS-CoV-2 infection (Figure 4C, Figure S23). MX1>30pg/ml sensitivity (82.5% CI (73.7%–89.3%)) and specificity (82.6% (73.76%–89.3%)) in combination with WARS>25ng/ml sensitivity (72.3% CI (65.7%–82.3%)) and specificity (76.6% (66.6%–85.3%)) showed a diagnostic efficiency of 94% (AUROC = 0.948 CI (0.911–0.977)) and a predictive accuracy of 86% in segregating COVID-19 (Figures 4D, 4E, and S24). Results of our analysis clearly showed that baseline levels of MX1 and WARS could also segregate severe SARS-CoV-2 cases (n = 10) from non-severe cases (AUC>0.75 p < 0.05) and symptomatic (n = 38) from asymptomatic (AUC>0.70, p < 0.05; Figure 7F) and correlated significantly with the increase in respiratory rate and organ failure index (SOFA score; p < 0.05, r > 0.5).Further levels of MX1 and WARS significantly correlated with IL-6, VEGFA, immune interferon gamma, and MIF levels in patients with COVID-19 (Figure S27). On validation of MX1, WARS levels in a separate cohort (test cohort of 200 patients; CoV2+ve = 100 patients and CoV2-ve = 100 patients) show that MX1 was able to segregate CoV2+ve patients and severe patients with an AUC = 0.939 and WARS with AUC = 0.78. Together, they were able to document a joint AUROC level of AUC = 0.869 which showed significance under 2000 bootstrapping experiment (Figures S27–S29). Together, these results potentiate that basal level of MX1 and WARS could aid in early identification of SARS-CoV-2-positive patients and help in the prediction of outcome in such patients.
Table 1.
Biomarker panel identified from multi-omics analysis
| SRNO | Proteins | Mean decrease accuracy | AUC | p-val. (FDR corrected) | Log 2 FC (COV2+ vs. COV2-) |
|---|---|---|---|---|---|
| 1 | MX1 | 0.008728 | 0.885 | 2.64E-09 | 6.038 |
| 2 | WARS | 0.007161 | 0.94 | 2.57E-08 | 4.658 |
| 3 | PFN1 | 0.003191 | 0.885 | 0.0002 | 2.778 |
| 4 | CPSF6 | 0.003086 | 0.8125 | 0.000185 | -4.375 |
| 5 | S100P | 0.002588 | 0.735 | 0.000496 | 2.453 |
| 6 | TFF3 | 0.00258 | 0.8725 | 4.75E-05 | 5.049 |
| 7 | SERPINC1 | 0.002424 | 0.7375 | 0.014789 | 0.834 |
| 8 | HBD | 0.002374 | 0.835 | 3.82E-05 | -5.107 |
| 9 | HEBP2 | 0.002326 | 0.7375 | 2.63E-05 | 5.385 |
| 10 | SRP14 | 0.00227 | 0.7725 | 0.000863 | 4.106 |
| 11 | PGK1 | 0.002233 | 0.9175 | 5.55E-08 | 2.961 |
| 12 | S100A12 | 0.00223 | 0.82 | 0.000301 | 2.089 |
| 13 | HSP90AB1 | 0.002114 | 0.8625 | 9.15E-06 | 1.964 |
| 14 | GDI2 | 0.00206 | 0.8825 | 6.41E-06 | 2.950 |
| 15 | ARHGAP1 | 0.002021 | 0.795 | 0.000285 | 3.691 |
| 16 | BPIFB1 | 0.001944 | 0.86 | 7.43E-05 | 1.518 |
| 17 | RUVBL1 | 0.001919 | 0.7725 | 0.000568 | 3.433 |
| 18 | HNRNPA1 | 0.001867 | 0.79 | 0.001001 | 1.585 |
| 19 | OPRPN | 0.001803 | 0.78 | 0.004863 | -2.078 |
| 20 | ACTN4 | 0.001755 | 0.9275 | 4.08E-06 | 3.116 |
| 21 | FARSA | 0.001703 | 0.8025 | 0.000251 | 3.813 |
| 22 | DCD | 0.001617 | 0.6675 | 0.032855 | 2.365 |
| 23 | IDH1 | 0.001564 | 0.74 | 0.00246 | 2.748 |
| 24 | PSME2 | 0.001474 | 0.7425 | 0.022482 | 1.186 |
| 25 | CAPZB | 0.001452 | 0.79 | 0.001087 | 2.737 |
| 26 | TYMP | 0.001372 | 0.7875 | 0.010334 | 1.540 |
| 27 | PKM | 0.001337 | 0.8525 | 9.5E-05 | 2.385 |
| 28 | EEF1A1 | 0.001288 | 0.78 | 0.001064 | 2.096 |
| 29 | MYL6 | 0.001211 | 0.805 | 0.000254 | -3.703 |
| 30 | S100A6 | 0.001197 | 0.8325 | 9.01E-05 | 3.629 |
| Metabolites | Mean decrease accuracy | AUC | p-val. (FDR corrected) | Log 2 FC (COV2+ vs. COV2-) | |
|---|---|---|---|---|---|
| 1 | C00978 | 0.021992 | 0.99 | 2.89E-07 | -2.096 |
| 2 | C14774 | 0.017813 | 0.985 | 4.36E-11 | -1.129 |
| 3 | CE4969 | 0.009625 | 0.9475 | 4.03E-05 | -0.944 |
| 4 | C08261 | 0.009547 | 0.9075 | 1.03E-07 | 2.758 |
| 5 | C01747 | 0.009199 | 0.855 | 0.001579 | -0.848 |
| 6 | C04540 | 0.008258 | 0.9125 | 0.001682 | -1.632 |
| 7 | C00135 | 0.008031 | 0.855 | 9.54E-05 | 2.310 |
| 8 | HMDB0011719 | 0.007559 | 0.82 | 7.39E-05 | 1.470 |
| 9 | C00864 | 0.007174 | 0.9 | 4.66E-07 | 2.104 |
| 10 | C00555 | 0.006792 | 0.8625 | 0.000207 | -1.355 |
| 11 | C04500 | 0.006586 | 0.855 | 1.67E-05 | -2.552 |
| 12 | C06135 | 0.006539 | 0.885 | 8.24E-05 | -1.280 |
| 13 | CE2176 | 0.006415 | 0.8025 | 0.000304 | 1.198 |
| 14 | C03017 | 0.006253 | 0.8525 | 0.000253 | 1.873 |
| 15 | C04277 | 0.006133 | 0.85 | 0.00947 | -0.857 |
| 16 | C06186 | 0.005733 | 0.8375 | 0.000273 | 1.041 |
| 17 | C14875 | 0.005561 | 0.835 | 0.013234 | -0.478 |
| 18 | tymsf | 0.005265 | 0.82 | 0.004302 | 1.827 |
| 19 | C01194 | 0.005201 | 0.8825 | 9E-06 | 1.694 |
| 20 | prgnlones | 0.004341 | 0.755 | 0.003275 | -0.781 |
| 21 | C03764 | 0.003972 | 0.84 | 0.000269 | -0.966 |
| 22 | C06078 | 0.003862 | 0.805 | 0.006341 | -1.373 |
| 23 | C19670 | 0.003857 | 0.8775 | 6.61E-05 | 1.918 |
| 24 | C03758 | 0.003796 | 0.73 | 0.006803 | 1.549 |
| 25 | C01879 | 0.003592 | 0.86 | 8.34E-05 | -1.148 |
| 26 | C06536 | 0.003494 | 0.83 | 0.000431 | 1.059 |
| 27 | C03210 | 0.003324 | 0.8375 | 0.000301 | -1.135 |
| 28 | C16121 | 0.003144 | 0.8275 | 0.000837 | -0.764 |
| 29 | C04453 | 0.003088 | 0.7875 | 0.006481 | -0.930 |
| 30 | C01789 | 0.002887 | 0.685 | 0.002895 | -0.839 |
| Metaproteins | Mean decrease accuracy | AUC | p-val. (FDR corrected) | Log 2 FC (COV2+ vs. COV2-) | |
|---|---|---|---|---|---|
| 1 | Proteobacteria:Burkholderiales:Burkholderia:GEGGEHGDGGADTAAQNGDR | 0.033543 | 1 | 4.46E-29 | 19.093 |
| 2 | Actinobacteria:Streptomycetales:Streptomyces:LGPMDFYGR | 0.027812 | 1 | 5.28E-38 | -20.577 |
| 3 | Firmicutes:Bacillales:Lysinibacillus:LLLHVAGELALK | 0.026731 | 1 | 2.42E-38 | -20.390 |
| 4 | Actinobacteria:Corynebacteriales:Corynebacteriales:GIGLLQADLLVVNK | 0.02583 | 1 | 4.92E-39 | -21.465 |
| 5 | Firmicutes:Lactobacillales:Lactobacillus salivarius:VELSPIIIK | 0.025604 | 1 | 2.9E-38 | -23.767 |
| 6 | ::Bacteria:IQVLTQAR | 0.025283 | 1 | 2.45E-33 | 17.135 |
| 7 | ::Bacteria:ALQATGLK | 0.025233 | 1 | 4.33E-30 | 17.344 |
| 8 | ::Bacteria:IALIGRPNVGK | 0.024892 | 1 | 3.11E-41 | -18.958 |
| 9 | Proteobacteria:Desulfuromonadales:Geobacter:LIIAGGGTGGHLFPGIAIADEFLAR | 0.024735 | 1 | 2.09E-25 | 22.290 |
| 10 | ::Bacteria:LAALGLSLK | 0.024362 | 1 | 7.76E-37 | -19.295 |
| 11 | Firmicutes:Bacillales:Bacillus subtilis:AAANPVTQQQPDILP | 0.02414 | 1 | 1.25E-67 | -32.198 |
| 12 | ::Bacteria:YSTAGAR | 0.022161 | 0.994 | 1.32E-35 | -19.192 |
| 13 | Proteobacteria:Desulfobacterales:Desulfatibacillum:DQEGQDVLIFIDNIFR | 0.022079 | 0.994 | 2.93E-36 | 16.925 |
| 14 | Firmicutes:Bacillales:Bacillus subtilis:QINLTITVIK | 0.021523 | 0.994 | 4.42E-27 | 21.421 |
| 15 | ::Bacteria:TEDMLPILEK | 0.020414 | 0.994 | 3.16E-20 | 25.728 |
| 16 | Proteobacteria:Vibrionales:Aliivibrio:RPLLLGIAK | 0.018362 | 0.994 | 9.09E-28 | 19.575 |
| 17 | Proteobacteria:Enterobacterales:Klebsiella pneumoniae:VAVLXAAGGIQALALLLK | 0.017897 | 0.982 | 2.98E-26 | 18.305 |
| 18 | Firmicutes:Natranaerobiales:Natranaerobius:IEEAGGDVEIK | 0.017875 | 0.982 | 4.61E-20 | -21.371 |
| 19 | Firmicutes:Lactobacillales:Lactobacillus plantarum:GIDHGMTATALDAAAAMIAELGDGQVASGMVIGR | 0.01782 | 0.982 | 4.08E-30 | 28.118 |
| 20 | Proteobacteria:Pasteurellales:[Mannheimia] succiniciproducens:SVTDQDSK | 0.01747 | 0.982 | 1.64E-42 | 17.170 |
| 21 | Proteobacteria::Proteobacteria:LAVAGRPNVGK | 0.017045 | 0.975 | 2.6E-38 | -17.955 |
| 22 | Tenericutes:Mycoplasmatales:Mycoplasma:NQLIRPK | 0.016163 | 0.975 | 1.28E-26 | 24.527 |
| 23 | Tenericutes:Mycoplasmatales:Mycoplasma genitalium:IEPNINLK | 0.016142 | 0.975 | 4.79E-21 | 19.168 |
| 24 | Proteobacteria:Rickettsiales:Rickettsia bellii:ILIILSIILCSLFTK | 0.015536 | 0.975 | 2.03E-36 | 20.833 |
| 25 | Bacteroidetes:Bacteroidetes Order II. Incertae sedis:Salinibacter ruber:EVGCCSIESMNDAR | 0.015518 | 0.962 | 1.32E-35 | 19.985 |
| 26 | Tenericutes:Mycoplasmatales:Mycoplasma:VIGILDSNSNPDAVDFGIPANDDSAK | 0.014336 | 0.961 | 5.69E-28 | 19.027 |
| 27 | Firmicutes::Firmicutes:ADDNAEHLFK | 0.01373 | 0.958 | 3.09E-27 | 17.031 |
| 28 | Proteobacteria:Enterobacterales:Enterobacterales:LLNLPLQVLVK | 0.012895 | 0.956 | 1.53E-31 | 19.306 |
| 29 | Firmicutes:Bacillales:Bacillales:VAVTAGASTPTPIVK | 0.01284 | 0.954 | 7.75E-32 | 17.649 |
| 30 | Tenericutes:Mycoplasmatales:Ureaplasma:VLIIGKPNVGK | 0.010885 | 0.951 | 5.48E-31 | 18.380 |
| 31 | Tenericutes:Mycoplasmatales:Mycoplasma penetrans:DFNCNNVVLVLNK | 0.005724 | 0.89 | 0.000116 | 2.817 |
Figure 4.

Estimation of MX1 and WARS as candidate indicators for SARS-CoV-2 infection and outcome
(A) Quantitative assessment of MX1 and WARS in the respiratory specimen of 200 COVID-19 specimen show significant increase in MX1 and WARS levels in COVID-19 positive as compared to COVID-19 negative (FC > 2, p < 0.05)
.(B) Multivariate area under the receiver operating curve analysis show significant AUC of MX1 = 0.922 CI (0.863–0.957) and WARS = 0.867 CI (0.781–0.944).
(C) Prediction class probability for MX1 alone show clear segregation of COVID-19-positive from COVID-19-negative patients with a predictive accuracy of 84%.
(D) Area under the receiver operating curve analysis for MX1 and WARS together show AUC = 0.948 CI (0.911–0.977) p < 0.05.
(E) Prediction class probability for MX1 and WARS together show clear segregation of COVID-19-positive from COVID-19-negative patients with a predictive accuracy of 86%.
(F) Quantitative assessment of MX1 and WARS in the respiratory specimen show significant increase in MX1 and WARS levels in severe COVID-19 (n = 10) as compared to non-severe COVID-19 (n = 90, FC > 2, p < 0.05). The right panel show multivariate area under the receiver operating curve analysis of MX1 and WARS for the assessment of severity and symptomatic patients in COVID-19.
Discussion
In the present study, the molecular profile of the respiratory specimen was analyzed for the characterization of baseline molecular determinants which could be used as putative candidate for SARS-CoV-2 infection and outcome. This was complemented with global proteome (viral and host), metaproteome, and metabolome analysis of the respiratory specimens followed by analysis of the regulatory network. Multi-omics integrated analysis helped us in the characterization of a panel of biomolecules capable of stratifying COVID-19-positive patients but also provided insight on the pathogenesis of SARS-CoV-2 infection. Of the panel, we validated the expression of MX1 and WARS (tryptophan-tRNA ligase) in the respiratory specimen of 200 COVID-19 suspects and showed that increase in their expression corresponds to SARS-CoV-2 infection positivity. It showed a significant increase in severity and also correlated with known plasma predictors of COVID-19. These molecules could be used as a putative candidate for SARS-CoV-2 infection monitoring or modulation/therapeutic targets.
SARS-CoV-2 is known to mediate infection by entry into the cell via the ACE2 receptor (Li et al., 2003). ACE2 receptor is significantly expressed in the lung, kidney, blood vessels, and also in the mucosa of the oral cavity; this could explain the incidence of pneumonia and bronchitis seen in COVID-19-positive patients (Ciaglia et al., 2020). Results of our study show that 6 of 29 viral encoded proteins (nucleocaspid phosphoproteins, chain b spike glycoprotein, membrane glycoprotein, orf1ab polyprotein, replicas polyprotein 1ab, protein 9b) were significantly increased in COVID-19-positive patients as compared to COVID-19-negative patients. These results show that viral protein quantitation using mass spectrometry could aid in SARS-CoV-2 infection and outcome prediction. Interestingly, respiratory specimen of COVID-19-positive patient showed significant increase in the level of ACE 2 receptor. This increase in the ACE2 receptor level in the respiratory specimen could be attributed to a probable increase in ADAM17 activity (Ciaglia et al., 2020) or increase worn out bronchial epithelial cells in the respiratory specimen, although these observation warrants further evaluations.
Deep analysis of the respiratory specimen (complex mixture of biomolecules) not only can aid in the identification of a panel of indicators for SARS-CoV-2 infection but also could provide insight on the pathophysiology of the patients. Thus respiratory specimen could be classified as a liquid biopsy which on detailed analysis could provide clues for diagnostic/prognostic or therapeutic implication. Keeping this in mind, we adopted a multi-omics approach to characterize baseline molecular determinants associated with SARS-CoV-2 infection and outcome.
Results of our study segregate COVID-19-positive patients from COVID-19-negative or influenza A H1N1 pdm 2009 positive patients based on the respiratory specimen proteome. We were able to identify a panel of proteomic/metabolomic or metaproteomic signatures which could be used for screening of SARS-COV2 infection and segregation of COVID-19-positive patients. Panel proposed in the study comprises proteins linked to interferon activation, viral carcinogenesis, neutrophil and monocyte activation, and others and metabolites linked to glycerophospholipid metabolism, histidine metabolism, and inositol phosphate metabolism known to be linked to viral infection (Metzner et al., 2008; Thaker et al., 2019).
SARS-CoV-2 infection leads to severe immune activation which is followed by acute respiratory distress (Astuti and Ysrafil, 2020), concordant to the pathogenesis, proteome analysis of the respiratory specimen of COVID-19-positive patients showed significant increase in proteins linked to viral life cycle and carcinogenesis, immune activation including neutrophil, monocyte activation and degranulation, IL-17 signaling, NOD-like receptor signaling, leukocyte transendothelial migration, and antigen presentation which are known to be increase with the increase in SARS-CoV-2 infection (Li et al., 2020). Viral infection are also known for metabolic reprograming of host (Thaker et al., 2019) and proteome analysis of the respiratory specimen showed that there is significant increase in proteins associated to glucose metabolism suggesting that SARS-CoV-2 induces energy metabolism (Figure S25). Interestingly, we found increase in proteins linked to fluid shear stress and bacterial invasion of the epithelial cells in the respiratory specimen. This suggests that COVID-19-positive patients may have secondary bacterial infections and associated comorbidities which need further evaluation. Decrease in the oxygen caring capacity is a hall mark feature of SARS-CoV-2 infection which leads to acute respiratory distress syndrome (Geier and Geier, 2020), and respiratory specimen of COVID-19-positive patients showed concordant decrease in proteins linked to gas transport particularly oxygen transport (heamoglobin binding, haptoglobin binding), hydrogen peroxide, and cofactor catabolic/metabolic process. Decrease in the oxygen transport proteins was linked to an associated decrease in wound healing, regulation of body fluid levels and vesicular transport seen in the respiratory specimen of COVID-19-positive patients. These disturbances can lead to uncontrolled production of inflammatory mediators, contributing to a state of persistent injury in COVID-19-positive patients. Together, these observations suggest that the respiratory specimen could provide an insight into the pathophysiology (virus mediated hyper immune activation and decrease in oxygen transport and wound healing) of SARS-CoV-2 infection which could be explored for prognosis and therapeutic intervention.
Concordant to the proteomic analysis, results of the metabolome analysis showed that respiratory specimen of COVID-19 patients is enriched for metabolites linked to unsaturated fatty acids and glycerophospholipid metabolism involved in the early development of virus (Schoggins and Randall, 2013). Furthermore, there was significant increase in ubiquinone/terpenoid-quinone biosynthesis, aminoacyl-tRNA biosynthesis along with amino acid metabolism, whereas metabolites linked to vitamin metabolism and steroid metabolism was significantly decreased in COVID-19 respiratory specimen. These results suggest that respiratory specimen of COVID-19-positive patients is rich in oxidative and inflammatory metabolite and has significantly lower levels of anti-inflammatory steroids or vitamins.
Results of the respiratory proteomics show significant increase in bacterial invasion of the epithelial cells. Further bacterial co-infection aggravates viral infection and increase the duration of the disease (Gu and Korteweg, 2007). In addition, alteration of microbiome (gut) is linked to immune activation in SARS-CoV2 infection (Dhar and Mohanty, 2020). As oropharyngeal microbiome is reflective of lung microbiome (Dickson and Huffnagle, 2015) and SARS-COV2 infection could modulate it. Change in the oropharyngeal microbiome could be reflective of altered pathogenesis in COVID-19. This prompted us to perform metaproteome (microbiome) analysis of the respiratory specimen in COVID-19-positive, negative, and H1N1 pdm 2009 positive patients. Metaproteome analysis of COVID-19-positive respiratory specimen showed significant increase in Bacteroidetes Order II. Incertae sedis, Bacillales, Burkholderiales, Klebsiella-pneumonia, Pasteurellales, and other bacterial species known to be increased in lung and upper respiratory pathogenesis such as severe asthma, COPD, or SARS (Wu and Segal, 2017) and (Gu and Korteweg, 2007). Interestingly, Lactobacillus salivarius known as a potent antiallergic and probiotic was significantly reduced in COVID-19-positive patients (Li et al., 2010). Functionality assessment based on the respiratory metaproteome showed that proteobacteria: enzyme encode pathways such as fatty acid biosynthesis, glycerolipid metabolism, amino acid, energy metabolism, and oxidative phosphorylation and firmicutes or tenericutes: enzyme encodes pathways aminoacyl-trna biosynthesis and terpenoid backbone biosynthesis show similarity with the respiratory metabolome enriched pathways, suggesting to the involvement of bacteria in the metabolic regulation of SARS-CoV-2 infection. Together, these results suggest that SARS-CoV-2 infection modulates the respiratory microbiome and functionality thus providing destabilized microenvironment which may predispose patients to respiratory distress such as allergic cough, fever, and others.
Viral infection induces angiogenesis (Cerimele et al., 2003) and is known to increase proteins processing/maturation and GPI anchor biosynthesis (Metzner et al., 2008). Results of the global cross correlation analysis were concordant and showed that the viral proteome share a direct correlation with bacterial metaproteome and proteins or metabolites linked to viral life cycle, regulation of angiogenesis, vasculature development, protein processing/maturation, beta-alanine metabolism, pantothenate and CoA biosynthesis, and GPI anchor biosynthesis. Results of the multi-omics integration also show that with the increase in SARS-CoV-2 viremia, there is significant increase in energy metabolism (pentose phosphate pathway, glycolysis), glycerophospholipid metabolism, purine metabolism and inflammation (IL-17 signaling pathway) in COVID-19-positive patients; these observation correlates with classical features of virus replication, metabolic regulation, and inflammation (Thaker et al., 2019). Recently, Shen et al. (Shen et al., 2020) documented dysregulated lipid metabolism, increase in acute phase reactant, immune activation (macrophage, platelets) in patients with severe COVID-19. Our results was able to recapitulate the findings and showed that respiratory specimen of COVID-19-positive patients have significant increase in modules associated to immune activation generic cluster, enriched in monocytes and platelet activation module I and II. We additionally showed that these modules show direct correlation with the metabolites linked to arginine biosynthesis, cysteine, and methionine metabolism and aminoacyl-tRNA biosynthesis. Arginine metabolism is a classical feature of inflammatory macrophages and increase in arginine biosynthesis in monocytes suggests inflammatory activation of monocytes (Rath et al., 2014). Interestingly, multi-omics analysis was also able to segregated COVID-19 asymptomatic from the symptomatic. Molecules such as AKR1A1 (aldo-keto reductase family 1 member A), COPE (coatomer subunit epsilon), and C02571 (L-acetylcarnitine) were asymptomatic-specific, whereas symptomatic COVID-19 patients showed significant increase in Klebsiella pneumoniae, CTBP2 (C-terminal-binding protein 2), and 4-alpha-methyl-5-alpha-cholest-7-en-3-one suggesting that the multi-omics profile could also identify asymptomatic (silent carrier of infection) which could mediate an exponential increase in the infection rate.
Finally, molecular signatures (Table 1) capable of segregating COVID-19-positive patients. We validated MX1 and WARS (tryptophan-tRNA ligase) in 200 COVID-19 suspects. MX1 is an effector anti-viral protein which modulate the type-I interferon-mediated inflammatory response in lungs (Makris et al., 2017). MX1 is activated in response to novel viruses for which the body has no immune defense (antibodies) (Haller et al., 2015). SARS-COV2 as a novel virus mediates cellular entry via ACE2 receptor or via membrane fusion (cleavage of the spike glycoprotein by TMPRSS2 [type II transmembrane serine protease]). This increases expression of both MX1 and TMPRSS2 via interferon regulatory factor 1 (Panda et al., 2019). Increase in MX1 (chemoattractant) mediates infiltration of immune cells (neutrophil, monocyte) and induces interferon response (Haller et al., 2015). Increase in site-specific interferon response increases secretion of WARS (housekeeping enzyme) which activates monocytes to macrophage via TLR2-TLR4 pathway (Jin, 2019) followed by other immune cell activation which may lead to cytokine storm seen in patients with COVID-19. Thus, over expression of MX1 and WARS in response to SARS-COV2 infections in the respiratory specimen serves as attractive molecular targets and were cherry picked for evaluation of diagnostic potential in 200 COVID-19 suspects. Results of our study showed that MX1>30pg/ml and WARS>25ng/ml in the respiratory specimen was able to segregate COVID-19-positive from COVID-19-negative patients with a combined diagnostic efficiency of 94% and a predictive accuracy of 86%. Further results of our study also show that MX1 and WARS also have the potential to stratify severe SARS-CoV-2 from non-severe and symptomatic SARS-CoV-2 from non-symptomatic 85%. These results show a validated assessment that baseline level of MX1 and WARS could aid in identification of SARS-CoV-2-positive patients and help in segregating patients predisposed to differed outcome.
Many previous published proteomics studies (Coombs et al., 2010; Dove et al., 2012; Vester et al., 2009) has already highlighted significant increase in levels of the Mx proteins (interferon-induced key components of the host cell defense), postinfection of human influenza A virus in a cell culture setting. These observations were corroborated in our study; further, we additionally show that both human influenza A virus and SARS-CoV-2 modulates several global pathways including cell cycle regulation, carbohydrate metabolism, lipid metabolism, and others suggesting that the proteomic regulation of both human influenza A virus and SARS-CoV-2 are similar which could be one of the reason for similarity in there clinical presentation. Thus studies which compare different influenza virus and SARS-CoV-2 in terms of host proteomic and metabolomic response is warranted and should be performed in large cohort of patients.
Recently, Juan Bizzotto et al. (Bizzotto et al., 2020) showed that the expression of MX1 and MX2 are higher in patients with COVID-19 which is concordant to what we show in our study. In addition, they show that both MX1 and MX2 expression increases significantly with viral load and was seen to be reduced with age, which they pointed out to be linked with the severity of illness. Finally, they concluded MX1 as a solid responder to SARS-CoV-2 infection, and these results are concordant to our results.
Further as compared with human influenza A virus, the SARS-CoV-2 infection is much severe and induces significantly higher expression of MX-1, immune interferon gamma, and WARS (which works as an antiviral cytokine and also in activating the innate immune system) (Jin, 2019). Results of our study potentiates that MX-1 in combination with WARS give better diagnostic ability for SARS-CoV-2-positive patients as compared to SARS-CoV-2-negative patients.
Limitations of study
Owing to biosafety constrains in SARS-CoV-2 infection, large number of samples were not collected nor analyzed in the study. Only previously archived (left over) respiratory specimen of the patients coming for COVID-19 testing at the Institute of Liver and Biliary Sciences New Delhi were analyzed in the study. Detailed demographic profile and severity indices of the patients could not be collected as only archived samples were used for analysis. Parallel reaction monitoring-based assessment of viral proteins could help in absolute quantitation of viral proteins but was not performed in the study. We also want to emphasize that due to on-going pandemic and lockdown situation in most of the country, our sample size for control group on other respiratory virus was not there; future prospective studies can be done to evaluate the role of these respiratory proteins in the other respiratory viral infections also. We performed absolute quantitation of two best indicators of COVID-19 positivity. However, molecular targets such as metabolites, metaproteins, and viral proteins amongst the diagnostic panel for COVID-19 segregation warrant further validation. The multi-omics analysis performed in the study can be used for comparative analysis and to identify commonly altered proteomic and metabolic patterns and functional pathways during SARS-CoV-2 infection among diverse populations from different geographic locations.
In conclusion, our study presents a multi-omics investigation of respiratory specimen from COVID-19 positive and controls. Our data provides a systemic overview of the host response induced by SARS-COV2 infection in the respiratory specimen. Baseline levels of MX1 and WARS (tryptophan-tRNA ligase) could be utilized for early identification of SARS-COV2 infection, its prognosis, and outcome prediction and could be used for mass screening of patients with COVID-19. Systemic overview of the respiratory specimen could provide useful prognostic and therapeutic indications in the ongoing battle against the COVID-19 pandemic.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Discovery Cohort: Serum samples from 20 COVID-19 positive patients and 20 COVID-19 negative patients and 05 H1N1 pdm 2009 subjects in discovery cohort. Validation: Targets were validated on 200 COVID-19 patients. | ILBS, Hospital | (Table S7) in this Manuscript. |
| Proteomics | COVID-19 positive, negative and H1N1 Samples | Figure 1, Tables S1 and S3, Figures S1–S14 |
| Meta-proteomics | COVID-19 positive, negative and H1N1 Samples | Figure 1, Table S1 and S3, Figures S1–S14 |
| Metabolomics | COVID-19 positive, negative and H1N1 Samples | Figure 2, Table S2, Figures S15–S18 |
| Multi-omics integration | COVID-19 positive, negative and H1N1 Samples | Figure 3, Table S4 |
| Chemicals, peptides, and recombinant proteins | ||
| Triethylammonium bicarbonate buffer (TEAB) | Thermo Fisher Scientific | Cat # PI90114 |
| Urea | Thermo Fisher Scientific | Cat # 29,700 |
| Tris (2-carboxyethyl) phosphine (TCEP) | Thermo Fisher Scientific | Cat #T2556 |
| Iodoacetamide (IAA) | Sigma-Aldrich | Cat #I6125 |
| Dithiothreitol | Sigma-Aldrich | Cat #D0632 |
| Trypsin | Promega | Cat #V5111 |
| Acetone | JT Baker | Cat # 9006–03 |
| Water | Thermo Fisher Scientific | Cat #W6-4 |
| Acetonitrile | JT Baker | Cat # 14650359 |
| Formic acid (FA) | Sigma-Aldrich | Cat #F0507 |
| Ammonium hydroxide solution | Sigma-Aldrich | Cat # 221228 |
| Methanol | Sigma-Aldrich | Cat # 34,860 |
| C18 Spin Columns | Thermo Fisher Scientific | Cat # 89,870 |
| Bradford Total protein assay kit | Thermo Fisher Scientific | Cat # 22,662 |
| ELISA/Antibodies | ||
| MX1 ELISA | Cusabio | Cat: No; csb-El015249HU |
| WARS ELISA | Elabscience | Cat: No; E-EL-H1874 |
| Software and Algorithms | ||
| Xcalibur | Thermo Fisher Scientific | Cat # OPTON-30965 |
| Proteome Discoverer Version 2.4.1.15 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industial/mass spectrometry/liquidchromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| Compound Discoverer Software 3.1 | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30925#/OPTON-30925 |
| Metaboanalyst 4.0 | Webserver | https://www.metaboanalyst.ca/MetaboAnalyst/home.xhtml |
| R version 3.5.2 | R Project | https://www.r-project.org |
| Cytoscape | United States (Java) | https://cytoscape.org/ |
| Unipept | Webserver | https://unipept.ugent.be/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Jaswinder Singh Maras (jassi2param@gmail.com).
Material availability
The study did not generate any materials.
Data and code availability
The processed data is provided as supplemental information, and the raw data for the manuscript is available on request to the lead contact Dr. Jaswinder Singh Maras (jassi2param@gmail.com).
Experimental mode and subject details
Sample/patient selection
A cross sectional study was planned, and previously archived (left over) respiratory specimens (combined oropharyngeal and nasopharyngeal swabs in viral transport media) of the patients coming for COVID-19 testing at the Institute of Liver and Biliary Sciences New Delhi, were selected for the study. Multi-omics analysis was performed on respiratory specimen of RT-PCR proven COVID-19 positive, negative (n = 20) or H1N1 pdm 2009 patients. Fourteen of the 20 COVID-19 positive patients showed classical symptoms including fever, sore throat, cough and breathlessness, whereas 6 patients were asymptomatic yet positive for COVID-19 and were included in the study.
Discovery: For the discovery phase 150 ul of the respiratory specimen from COVID-19 positive patients (n = 20), COVID-19 negative patients (n = 20) and respiratory diseases control Influenza A H1N1 pdm 2009 pdm 2009 positive samples (n = 5) were subjected to proteomics (SARS-CoV-2 virus proteins and linked proteins and host proteins), metaproteomics (bacterial peptide) and metabolomics evaluations (Table S7). Baseline demographic profiles were recorded and baseline samples were used for multi-omics evaluation and correlation to disease pathogenesis. In brief, for proteomics and metaproteomics application, 4% SDS was added to 100 ul of the respiratory specimen, kept at 95°C for 15 min (viral deactivation). Total proteins in the respiratory specimen were precipitated by adding 6× cold acetone followed by centrifugation at 13,000 rpm for 10 min. The precipitated proteins were re-dissolved in ammonium bicarbonate buffer (pH-7) and were subjected to LC MS/MS evaluation of virome, host proteome and metaproteome detailed below. The left over 50 ul of the respiratory specimen was subjected to methanol precipitation followed by total metabolome evaluation as detailed below.
Validation: An annotated set of 200 COVID-19 suspect patients (100 COVID-19 positive and 100 COVID-19 negative; Table S7) was taken to validate the most significant indicators of SARS-CoV-2 infection and outcome in the discovery phase. The sensitivity/specificity and predictive accuracy was analyzed. Of the 100 positives a total of 10 SARS-CoV-2 patients were severe and 38 showed symptoms and were analyzed for outcome prediction analysis.
Proteomics of the respiratory specimen
Total proteins were isolated from the respiratory specimen of the study groups. 100 ug equivalent protein was subjected to reduction, alkylation followed by digestion for 16–20 hr at 37°C using sequencing-grade modified trypsin: proteins (1ug:20ug w/w). The samples were desalted and subjected to LC-MS/MS analysis. The peptides were eluted by a 3–95% gradient of buffer B (aqueous 80% acetonitrile in 0.1% formic acid) at a flow rate of 300 nL/min for about 60 min on a 25-cm analytical C18 column (C18, 3 μm, 100 Å) The peptides were ionized by nano-electrospray and subsequent tandem mass spectrometry (MS/MS) on a Q-ExactiveTM Plus (Thermo Fisher Scientific, San Jose, CA, United States). The peptides were analyzed using a mass spectrometer with the collision-induced dissociation mode with the electrospray voltage was 2.3 kV. Analysis on the orbitrap was performed with full scan MS spectra with a resolution of 70,000 from m/z 350 to 1800.The MS/MS data was analyzed by Proteome Discoverer (version 2.0, Thermo Fisher Scientific, Waltham, MA, United States) using the uniprot COVID-19 database (https://covid-19.uniprot.org) containing 14 SARS2, 15 CVHSA and 12 human protein for virome and virus linked proteins detection. The MS/MS data was reanalyzed by Proteome Discoverer (version 2.0, Thermo Fisher Scientific, Waltham, MA, United States) using the uniprot homo sapiens (Human) database (UP000005640) and human proteome with Mascot algorithm (Mascot 2.4, Matrix Science). Significant proteins were identified at (p < 0.05) and q values (p < 0.05). The threshold of false discovery rate was kept at 0.01. The identified proteins were subjected to standard statistical analysis and network and pathway analysis.
Metabolomics of the respiratory specimen
Metabolites were extracted from the 50ul of respiratory specimen using organic phase extraction protocol (Boudah et al., 2014). To 50 ul of respiratory specimen, 500 ul methanol was added and kept overnight at −20° for protein precipitation. The samples were centrifuged at 13.3∗1000 rpm for 10 min and the supernatant was dried under vacuum. The dried samples were reconstituted with (5:95:5) 5% acetonitrile: 95% water: 5% internal standards at known concentrations, for reverse-phase chromatography by using C18 column (Thermo Scientific25003102130: 3 μm, 2.1 mm, 100 mm) using ultra-high performance liquid chromatographic system followed by high-resolution mass spectrometry (MS) (Boudah et al., 2014). Metabolite features were extracted using compound discoverer 3.0 (Thermo) (Basu et al., 2017). Annotation of the features was performed using mass list searches (Basu et al., 2017; Boudah et al., 2014) mzCloud (www.mzcloud.org) and mummichog (Li et al., 2017). Identified and annotated features were subjected to log normalization and pareto-scaling using metaboanalyst 4.0 (http://metaboanalyst.ca.) server (Chong et al., 2018) and into SIMCA P12 software (Umetrix, Sweden) for multivariate projection analyses, such as principal component analysis and partial least square discriminant analysis. Pathway enrichment patterns were analyzed using Metaboanalyst 4.0 (Chong et al., 2018).
Metaproteome (microbiome) analysis of the respiratory specimens
Proteins were isolated from the respiratory specimen of the study groups. The isolated proteins were reduced, alkylated and digested using trypsin followed by mass spectrometry analysis similar to that stated in respiratory specimen proteome analysis section. The MS/MS data was acquired and analyzed by Proteome Discoverer (version 2.0, Thermo Fisher Scientific, Waltham, MA, United States) using the bacterial/fungal sequence (UniprotSwP_20170609, with sequences 467231 and MG_BG_UPSP with sequences 2019194). This was cross validated using Mascot algorithm (Mascot 2.4, Matrix Science) specifically for all possible microbial species. In brief, significant peptide groups were identified at (p < 0.05) and q values (p < 0.05) and the false discovery rate at 0.01. Only rank-1 peptides with Peptide Sequence Match (PSM) > 3 were subjected to biodiversity and functional analysis using unipept (Zhang et al., 2018). Peptides mapping to eukaryotic, fungal and viral database were rejected and only bacterial species associated peptides were segregated and were subjected to statistical, functional and biodiversity analysis (Zhang et al., 2018).
Global cross correlation, clustering, and integration analysis
Differentially expressed viral proteome (DEVP), differentially expressed host proteome (DEPs), differentially expressed metabolites (DEMs) and differentially expressed metaproteins (DEMP) were identified and subjected to cross correlation and clustering analysis (r2>0.5,p < 0.05). Significant and prominent clusters were identified and subjected to pathway analysis; for proteins (enricher/KEGG) and metabolites (KEGG/metaboanalyst). This was followed by developing a global cross correlation map between the virome, metaproteome and the pathways linked to the proteins and metabolites using cytoscape (https://cytoscape.org/) (Shannon et al., 2003).
ELISA quantitation: MX1 (Cat: No; csb-El015249HU) with a detection limit of 15.6 pg/mL-1000 pg/mL, sensitivity 3.9 pg/mL and WARS (Cat: No; E-EL-H1874) with a detection range 0.78–50 ng/mL and sensitivity of 0.47 ng/mL were used for validation as per manufacture protocol.
Statistical analyses
Results are shown as mean and standard deviation unless indicated otherwise. Statically analysis was performed using Graph Pad Prism v6, SPSS V20 and p values of <0.05 using Benjamini & Hochberg correction was considered statistically significant. Unpaired (two-tail) Student's t test, Mann- Whitney U test were performed for comparison of two groups. For comparison among more than two groups, one-way analysis of variance, Kruskal-Wallis test was performed. All correlations were performed using Spearman correlation analysis and R2 > 0.5, p < 0.05 was considered as statically significant. For the multi-omics analysis, features with over 80% missing values in particular group were neglected further feature with less than 80% missing values in particular group were imputed with the minimal value of the feature in the assigned group. Data for virome, proteome, metabolome and metaproteome analysis was log normalized and subjected to Perato scaling this was followed by calculation of Log2 Fold change for each pair of comparing group. Differentially expressed virome, proteome, metabolome or metaproteome were identified at p < 0.05 and fold change >±1.5 FC. From the discovery cohort important viral proteins, host proteins, metabolites and metaproteins were selected for validation based on random forest analysis and area under the curve (AUROC) analysis (AUROC>0.8,p < 0.05). Prediction class probability and predictive accuracy (for most important variables) was calculated in the discovery cohort and cross validated in the validation cohort.
Acknowledgments
We would like to acknowledge Science and Engineering Research Board Department of Science and Technology government of INDIA, for providing funds for the building of the manuscript. The work was supported from project DST (DST-SERB) (EMR/2016/004829).
Author contributions
J.S.M. conceptualized the work. Samples for the analysis were provided by E.G., R.A., and S.R. Sample processing and acquisition was performed by J.S.M., S.S., and A.B. Analysis was performed by J.S.M. Prof. Sarin provided critical inputs for the manuscript. The manuscript was read and approved by all authors.
Declaration of interests
There is no conflict of interest from any of the authors included in the manuscript.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102823.
Contributor Information
Jaswinder Singh Maras, Email: jassi2param@gmail.com.
Shiv K. Sarin, Email: sksarin@ilbs.in, shivsarin@gmail.com.
Supplemental information
References
- Astuti I., Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes Metab. Syndr. 2020;14:407–412. doi: 10.1016/j.dsx.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S., Duren W., Evans C.R., Burant C.F., Michailidis G., Karnovsky A. Sparse network modeling and metscape-based visualization metods for the analysis of large-scale metabolomics data. Bioinformatics. 2017;33:1545–1553. doi: 10.1093/bioinformatics/btx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoechea J.A., Bamford C.G.G. SARS-CoV-2, bacterial co-infections, and AMR: the deadly trio in COVID-19? EMBO Mol. Med. 2020;12:e12560. doi: 10.15252/emmm.202012560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizzotto J., Sanchis P., Abbate M., Lage-Vickers S., Lavignolle R., Toro A., Olszevicki S., Sabater A., Cascardo F., Vazquez E. SARS-CoV-2 infection boosts MX1 antiviral effector in COVID-19 patients. iScience. 2020;23:101585. doi: 10.1016/j.isci.2020.101585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudah S., Olivier M.F., Aros-Calt S., Oliveira L., Fenaille F., Tabet J.C., Junot C. Annotation of the human serum metabolome by coupling three liquid chromatography methods to high-resolution mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014;966:34–47. doi: 10.1016/j.jchromb.2014.04.025. [DOI] [PubMed] [Google Scholar]
- Cerimele F., Brown L.F., Bravo F., Ihler G.M., Kouadio P., Arbiser J.L. Infectious angiogenesis: bartonella bacilliformis infection results in endothelial production of angiopoetin-2 and epidermal production of vascular endothelial growth factor. Am. J. Pathol. 2003;163:1321–1327. doi: 10.1016/S0002-9440(10)63491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D.S., Xia J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018;46:W486–W494. doi: 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaglia E., Vecchione C., Puca A.A. COVID-19 infection and circulating ACE2 levels: protective role in women and children. Front Pediatr. 2020;8:206. doi: 10.3389/fped.2020.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs K.M., Berard A., Xu W., Krokhin O., Meng X., Cortens J.P., Kobasa D., Wilkins J., Brown E.G. Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J. Virol. 2010;84:10888–10906. doi: 10.1128/JVI.00431-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar D., Mohanty A. Gut microbiota and Covid-19- possible link and implications. Virus Res. 2020;285:198018. doi: 10.1016/j.virusres.2020.198018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson R.P., Huffnagle G.B. The lung microbiome: new principles for respiratory bacteriology in health and disease. PLoS Pathog. 2015;11:e1004923. doi: 10.1371/journal.ppat.1004923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove B.K., Surtees R., Bean T.J., Munday D., Wise H.M., Digard P., Carroll M.W., Ajuh P., Barr J.N., Hiscox J.A. A quantitative proteomic analysis of lung epithelial (A549) cells infected with 2009 pandemic influenza A virus using stable isotope labelling with amino acids in cell culture. Proteomics. 2012;12:1431–1436. doi: 10.1002/pmic.201100470. [DOI] [PubMed] [Google Scholar]
- Geier M.R., Geier D.A. Respiratory conditions in coronavirus disease 2019 (COVID-19): important considerations regarding novel treatment strategies to reduce mortality. Med. Hypotheses. 2020;140:109760. doi: 10.1016/j.mehy.2020.109760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghinai I., McPherson T.D., Hunter J.C., Kirking H.L., Christiansen D., Joshi K., Rubin R., Morales-Estrada S., Black S.R., Pacilli M. First known person-to-person transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the USA. Lancet. 2020;395:1137–1144. doi: 10.1016/S0140-6736(20)30607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J., Korteweg C. Pathology and pathogenesis of severe acute respiratory syndrome. Am. J. Pathol. 2007;170:1136–1147. doi: 10.2353/ajpath.2007.061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller O., Staeheli P., Schwemmle M., Kochs G. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015;23:154–163. doi: 10.1016/j.tim.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Jin M. Unique roles of tryptophanyl-tRNA synthetase in immune control and its therapeutic implications. Exp. Mol. Med. 2019;51:1–10. doi: 10.1038/s12276-018-0196-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.Y., Lin H.C., Hsueh K.C., Wu S.F., Fang S.H. Oral administration of Lactobacillus salivarius inhibits the allergic airway response in mice. Can. J. Microbiol. 2010;56:373–379. doi: 10.1139/w10-024. [DOI] [PubMed] [Google Scholar]
- Li G., Fan Y., Lai Y., Han T., Li Z., Zhou P., Pan P., Wang W., Hu D., Liu X. Coronavirus infections and immune responses. J. Med. Virol. 2020;92:424–432. doi: 10.1002/jmv.25685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.M., Li Y.G., Yamate M., Li S.M., Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect. 2007;9:96–102. doi: 10.1016/j.micinf.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Sullivan N.L., Rouphael N., Yu T., Banton S., Maddur M.S., McCausland M., Chiu C., Canniff J., Dubey S. Metabolic phenotypes of response to vaccination in humans. Cell. 2017;169:862–877 e817. doi: 10.1016/j.cell.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris S., Paulsen M., Johansson C. Type I interferons as regulators of lung inflammation. Front Immunol. 2017;8:259. doi: 10.3389/fimmu.2017.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., Hlh Across Speciality Collaboration U.K. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner C., Salmons B., Gunzburg W.H., Dangerfield J.A. Rafts, anchors and viruses--a role for glycosylphosphatidylinositol anchored proteins in the modification of enveloped viruses and viral vectors. Virology. 2008;382:125–131. doi: 10.1016/j.virol.2008.09.014. [DOI] [PubMed] [Google Scholar]
- Murthy S., Gomersall C.D., Fowler R.A. Care for critically ill patients with COVID-19. JAMA. 2020;323:1499–1500. doi: 10.1001/jama.2020.3633. [DOI] [PubMed] [Google Scholar]
- Panda D., Gjinaj E., Bachu M., Squire E., Novatt H., Ozato K., Rabin R.L. IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front Immunol. 2019;10:1019. doi: 10.3389/fimmu.2019.01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath M., Muller I., Kropf P., Closs E.I., Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Randall G. Lipids in innate antiviral defense. Cell Host Microbe. 2013;14:379–385. doi: 10.1016/j.chom.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker S.K., Ch'ng J., Christofk H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019;17:59. doi: 10.1186/s12915-019-0678-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevarajan I., Nguyen T.H.O., Koutsakos M., Druce J., Caly L., van de Sandt C.E., Jia X., Nicholson S., Catton M., Cowie B. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat. Med. 2020;26:453–455. doi: 10.1038/s41591-020-0819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugama B., Kadhiresan P., Kozlowski H.N., Malekjahani A., Osborne M., Li V.Y.C., Chen H., Mubareka S., Gubbay J.B., Chan W.C.W. Diagnosing COVID-19: the disease and tools for detection. ACS Nano. 2020;14:3822–3835. doi: 10.1021/acsnano.0c02624. [DOI] [PubMed] [Google Scholar]
- Vester D., Rapp E., Gade D., Genzel Y., Reichl U. Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics. 2009;9:3316–3327. doi: 10.1002/pmic.200800893. [DOI] [PubMed] [Google Scholar]
- Wu B.G., Segal L.N. Lung microbiota and its impact on the mucosal immune phenotype. Microbiol. Spectr. 2017;5:1–28. doi: 10.1128/microbiolspec.BAD-0005-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., McGoogan J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- Zhang X., Deeke S.A., Ning Z., Starr A.E., Butcher J., Li J., Mayne J., Cheng K., Liao B., Li L. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 2018;9:2873. doi: 10.1038/s41467-018-05357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L., Ruan F., Huang M., Liang L., Huang H., Hong Z., Yu J., Kang M., Song Y., Xia J. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 2020;382:1177–1179. doi: 10.1056/NEJMc2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The processed data is provided as supplemental information, and the raw data for the manuscript is available on request to the lead contact Dr. Jaswinder Singh Maras (jassi2param@gmail.com).