

Abstract
Background:
Amphetamine analogs with a 3,4-methylenedioxy ring-substitution are among the most popular illicit drugs of abuse, exerting stimulant and entactogenic effects. Enzymatic N-demethylation or opening of the 3,4-methylenedioxy ring via O-demethylenation gives rise to metabolites that may be pharmacologically active. Indeed, previous studies in rats show that specific metabolites of 3,4-methylenedioxymethamphetmine (MDMA), 3,4-methylenedioxymethcathinone (methylone), and 3,4-methylenedioxypyrovalerone (MDPV) can interact with monoaminergic systems.
Aim:
Interactions of metabolites of MDMA, methylone, and MDPV with human monoaminergic systems were assessed.
Methods:
The ability of parent drugs and their metabolites to inhibit uptake of tritiated norepinephrine, dopamine, and serotonin (5-HT) was assessed in human embryonic kidney 293 cells transfected with human monoamine transporters. Binding affinities and functional activity at monoamine transporters and various receptor subtypes were also determined.
Results:
MDMA and methylone displayed greater potency to inhibit norepinephrine uptake as compared to their effects on dopamine and 5-HT uptake. N-demethylation of MDMA failed to alter uptake inhibition profiles, whereas N-demethylation of methylone decreased overall transporter inhibition potencies. O-demethylenation of MDMA, methylone, and MDPV resulted in catechol metabolites that maintained norepinephrine and dopamine uptake inhibition potencies, but markedly reduced activity at 5-HT uptake. O-methylation of the catechol metabolites significantly decreased norepinephrine uptake inhibition, resulting in metabolites lacking significant stimulant properties.
Conclusions:
Several metabolites of MDMA, methylone, and MDPV interact with human transporters and receptors at pharmacologically relevant concentrations. In particular, N-demethylated metabolites of MDMA and methylone circulate in unconjugated form and could contribute to the in vivo activity of the parent compounds in human users.
Keywords: MDMA, methylone, MDPV, metabolite, transporter
Introduction
3,4-Methylenedioxymethamphetmine (MDMA, “ecstasy”) is a popular drug of abuse which exerts stimulant and entactogenic effects by evoking release and inhibiting uptake of presynaptic norepinephrine, 5-HT, and dopamine (Rickli et al., 2015a; Del Bello et al., 2015). The enzymatic biotransformation of MDMA includes two main pathways: 1) N-demethylation to form 3,4-methylenedioxyamphetamine (MDA) and 2) O-demethylenation to form catechol metabolites 3,4-dihydroxymethamphetamine (HHMA) and 3,4-dihydroxyamphetamine (HHA) (Kreth et al., 2000; Meyer et al., 2008; de la Torre et al., 2004; Segura et al., 2005). The latter metabolites may contribute to MDMA-induced adverse clinical effects, since the cytotoxicity of highly reactive catechols is well established (Carmo et al., 2006; Antolino-Lobo et al., 2011). Additionally, the 4-hydroxy-3-methoxy metabolites of MDMA exhibit increased potency to stimulate vasopressin secretion, which could increase the risk of fatal hyponatremia (Forsling et al., 2002; Fallon et al., 2002).
Previous investigations in rats show that subcutaneous injection of MDMA or its N-demethylated metabolite MDA (1–10 mg/kg) increases blood pressure, heart rate, and locomotor activity (Schindler et al., 2014). By contrast, injection of the O-demethylenated catechol metabolites HHMA and HHA (1–10 mg/kg) induces potent sympathomimetic effects on the cardiovascular system but fails to affect locomotor activity, suggesting these more polar compounds do not readily cross the blood-brain-barrier and target norepinephrine transporters or receptors in the periphery (Schindler et al., 2014; Mueller et al., 2009). Escobedo and colleagues demonstrated that HHMA can be detected in mouse brain after intraperitoneal administration of high doses of HHMA (30 mg/kg) but not MDMA (30 mg/kg) (Escobedo et al., 2005), which indicates that catechol metabolites can cross the blood-brain-barrier under certain circumstances.
The synthetic cathinones 3,4-methylenedioxymethcathinone (methylone) and 3,4-methylenedioxypyrovalerone (MDPV) share the methylenedioxy ring-substitution with MDMA but their pharmacological effects are different. Methylone is a transporter substrate like MDMA (Baumann et al., 2012; Simmler et al., 2013) but it inhibits dopamine uptake more potently than 5-HT uptake in transfected cells (Simmler et al., 2013; Eshleman et al., 2013). MDPV is a potent blocker of the norepinephrine transporter (NET) and dopamine transporter (DAT) devoid of substrate activity, similar to the mechanism of action for methylphenidate (Luethi et al., 2018a; Eshleman et al., 2013; Baumann et al., 2013; Simmler et al., 2013) but with increased potency and toxicity. Importantly, the metabolism pattern of methylone and MDPV is similar to MDMA, giving rise to potentially active metabolites (Baumann et al., 2012; Schindler et al., 2014; de la Torre et al., 2004). In the present study, we wished to assess the potential clinical relevance of in vivo and in vitro studies in rats (Schindler et al., 2014; Elmore et al., 2017; Anizan et al., 2016) by examining the interactions of metabolites of MDMA, methylone and MDPV with human monoamine transporters and receptors, and rat and mouse trace amine-associated receptor 1 (TAAR1).
2. Methods
2.1. Drugs/Test substances
Mazindol, MDA HCl, MDMA HCl, MDPV HCl, methylone HCl, and fluoxetine HCl were purchased from Lipomed (Arlesheim, Switzerland). Nisoxetine HCl was obtained from Sigma Aldrich (Buchs, Switzerland). 3,4-Dihydroxyamphetamine (HHA) HCl, 3,4-dihydroxymethamphetamine (HHMA) fumarate, 4-hydroxy-3-methyoxyamphetamine (HMA) fumarate, and 4-hydroxy-3-methoxymethamphetamine (HMMA) HCl were synthesized and analysed for purity at the Research Triangle Institute (RTI, Durham, NC, USA) (Schindler et al., 2014). 3,4-Dihydroxymethcathinone (HHMC) HBr, 4-hydroxy-3-methyoxymethcathinone (HMMC) HCl, and 3,4-methylenedioxycathinone (MDC) HCl, were synthesized as described in (Ellefsen et al., 2015); 3,4-dihydroxypyrovalerone (HHPV) HBr and 4-hydroxy-3-methyoxypyrovalerone (HMPV) HCl were synthesized as described in (Anizan et al., 2014). All drugs were used as racemic mixtures. Radiolabeled [3H]norepinephrine (13.1 Ci/mmol) and [3H]dopamine (30.0 Ci/mmol) were attained from Perkin-Elmer (Schwerzenbach, Switzerland). Radiolabeled [3H]5-HT (80 Ci/mmol) was obtained from Anawa (Zurich, Switzerland).
2.2. Monoamine uptake transporter inhibition
Human embryonic kidney (HEK) 293 cells (Invitrogen, Zug, Switzerland) stably overexpressing the human NET, DAT, or 5-HT transporter (SERT) were used to investigate the inhibition of monoamine uptake as previously described (Luethi et al., 2018b). Briefly, cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Life Technologies, Zug, Switzerland) containing 10% fetal bovine serum (FBS; Gibco) and upon 70–90% confluency, the cells were detached and resuspended (3 × 106 cells/ml) in Krebs-Ringer Bicarbonate Buffer (KRB; Sigma-Aldrich, Buchs, Switzerland). For [3H]dopamine uptake experiments, the uptake buffer was additionally supplied with 0.2 mg/ml of L-ascorbic acid (Sigma Aldrich). To 100 μl of cell suspension, 25 μl of uptake buffer containing test compounds (at concentrations of 1 nM to 900 μM), transporter-specific inhibitors (10 μM nisoxetine for NET, 10 μM mazindol for DAT, and 10 μM fluoxetine for SERT), or vehicle control were added in a round bottom 96-well plate. After 10 min shaking on a rotary shaker at 450 rotations per min at room temperature, 50 μl of radiolabelled [3H]norepinephrine, [3H]dopamine, or [3H]5-HT dissolved in uptake buffer was added for an additional 10 min to initiate uptake transport. Thereafter, 100 μl of the suspension was transferred into microcentrifuge tubes containing 50 μl of 3 M KOH and 200 μl of a 1:1 mixture of silicone oil type AR 200 and type AR 20 (Sigma-Aldrich). The tubes were centrifuged for 3 min (13,200 rotations per min) and frozen in liquid nitrogen. The cell pellet was then cut into 6 ml scintillation vials (Perkin-Elmer) containing lysis buffer (1% NP-40, 5 mM EDTA, 50 mM NaCl, 0.05 M TRIS HCl). The vials were shaken for 1 h, filled with 4.5 ml of scintillation fluid (Ultimagold, Perkin Elmer), and subsequently, the uptake was quantified using a scintillation counter (Packard Tri-Carb Liquid Scintillation Counter 1900 TR). Uptake in the presence of the selective inhibitors was subtracted to determine specific uptake. A summary of cell culture and assay conditions for the monoamine reuptake inhibition assays is given in Supplemental Table S1.
2.3. Receptor and transporter binding
Receptor and transporter binding affinities were determined as previously described in detail for each receptor and transporter (Luethi et al., 2018c). Briefly, cell membrane preparations were derived from various cell lines (Supplemental Table S2) and overexpressed the respective monoamine receptors or transporters (human genes, with the exception of rat and mouse genes for TAAR1). The membrane preparations were incubated with radiolabeled selective ligands at concentrations equal to Kd, and ligand displacement by the compounds was measured. The difference between total binding (binding buffer alone) and nonspecific binding (in the presence of specific competitors) was determined to be specific binding. The following radioligands and competitors, respectively, were used: 2.9 nM N-methyl-[3H]nisoxetine and 10 μM indatraline (NET), 3.3 nM [3H]WIN35,428 and 10 μM indatraline (DAT), 1.5 nM [3H]citalopram and 10 μM indatraline (SERT), 0.90 nM [3H]8-hydroxy-2-(di-n-propylamine)tetralin and 10 μM pindolol (5-HT1A receptor), 0.40 nM [3H]ketanserin and 10 μM spiperone (5-HT2A receptor), 1.4 nM [3H]mesulgerine and 10 μM mianserin (5-HT2C receptor), 0.11 nM [3H]prazosin and 10 μM chlorpromazine (α1 adrenergic receptor), 2 nM [3H]rauwolscine and 10 μM phentolamine (α2 adrenergic receptor), 1.2 nM [3H]spiperone and 10 μM spiperone (dopamine D2 receptors), and 3.5 nM (rat TAAR1) or 2.4 nM (mouse TAAR1) [3H]RO5166017 and 10 μM RO5166017 (rat and mouse TAAR1). IC50 values were assessed by calculating nonlinear regression curves for a one-site model using three independent 10-point concentration-response curves. Ki values were determined by the Cheng-Prusoff equation. A summary of cell culture and assay conditions for the radioligand binding assays is given in Supplemental Table S2.
2.4. Activity at the 5-HT2B receptor
Human embryonic kidney (HEK) 293 cells expressing the human 5-HT2B receptor were incubated in growth medium at a density of 50,000 cells per well at 37 °C in poly-D-lysine-coated 96-well plates overnight. Thereafter, the growth medium was removed by snap inversion, and 200 μl of no wash dye (FLIPR calcium 6 assay kit Cat # R8191; Molecular Devices, Sunnyvale, CA, USA) were added to each well. The plates were incubated for 2 h at 37 °C. Thereafter, the plates were placed into a FLIPR, and 50 μl of the test drugs diluted in assay buffer were added to each well online. The increase in fluorescence was measured for 51 s, and EC50 values were derived from the concentration-response curves using nonlinear regression. A summary of cell culture and assay conditions for the 5-HT2B receptor activation assay is given in Supplemental Table S3.
2.5. Functional activity at the human TAAR1
Activity at the human TAAR1 was assessed as previously described in detail (Luethi et al., 2018c). Recombinant HEK 293 cells that expressed the human TAAR1 were harvested and pelleted by centrifugation at 900 rotations per min for 3 min at room temperature. Thereafter, the supernatant was removed and the cell pellet was resuspended in fresh culture medium. The cells were plated into 96-well plates (100 μl, containing 80,000 cells per well) and incubated for 20 h at 37 °C. The cell culture medium was removed and 50 μl PBS (without Ca2+ and Mg2+) was added. The PBS was removed by snap inversion, 90 μl of KRB containing 1 mM IBMX was added, and the plates were incubated for 60 min at 37 °C. All of the compounds were tested at a broad concentration range (300 pM – 30 μM) in duplicate and a standard curve (0.13 nM – 10 μM cAMP) was included on each plate. Additionally, a reference plate containing RO5256390, β-phenylethylamine and p-tyramine was included in each experiment. Compound solution, β-phenylethylamine (as maximal response), or basal control were added at a volume of 30 μl, and the cells were incubated for 40 min at 37 °C. Finally, the cells were lysed with 50 μl of detection mix solution containing Ru-cAMP Alexa700 anti-cAMP antibody and lysis buffer for 120 min at room temperature under heavy shaking; fluorescence was then measured. A summary of cell culture and assay conditions for the human TAAR1 activation assay is given in Supplemental Table S3.
2.6. Data analysis.
Prism software (version 7.0a, GraphPad, San Diego, CA, USA) was used for calculations. Monoamine uptake data were fitted by nonlinear regression to variable-slope sigmoidal dose-response curves and IC50 values and 95% confidence intervals were derived from 3 to 5 individual 11-point inhibition curves. The DAT/SERT ratio is expressed as 1/DAT IC50 : 1/SERT IC50. For radioligand binding assays, logistic regression was used to calculate IC50 values derived from 3 10-point curves. No observed affinity within a concentration range indicates that there was no binding at the highest tested concentration as well as no binding at 9 different lower concentrations. Unpaired two-tailed Student’s t-test was used to compare individual Ki values and P values < 0.05 were considered to be statistically significant. EC50 values for 5-HT2B receptor activation were determined using nonlinear regression concentration-response curves. The maximal activity at the receptors was calculated relative to 5-HT activity, which was defined as 100%. The activity at the human TAAR1 was measured using a NanoScan (IOM reader; 456 nm excitation wavelength; 630 and 700 nm emission wavelengths). The FRET signal was calculated as the following: FRET (700 nM) −P × Å~ FRET (630 nM), where P = Ru (700 nM) / Ru (630 nM).
3. Results
3.1. Monoamine transporter inhibition
The uptake inhibition curves for MDMA, methylone, MDPV, and their respective metabolites are shown in Figures 1–3, with corresponding IC50 values and DAT/SERT inhibition ratios listed in Table 1. Numbers in parentheses indicate the number of individual 11-point curves (NET/DAT/SERT): MDMA (3/3/3), MDA (3/3/3), HHMA (4/–/5), HHA (3/–/3), HMMA (3/3/4), HMA (3/4/3) methylone (3/3/3), MDC (4/3/3), HHMC (3/–/3), HMMC (3/4/4), MDPV (3/3/3), HHPV (3/4/3), HMPV (3/3/4). No sigmoidal uptake curves could be plotted for dopamine uptake inhibition in the cases of HHA, HHMA, and HHMC but the curve intercepts between 0 and 100% uptake were included in Figures 1–3 for comparison; the full non-sigmoidal curves of three of individual 11-point curves are shown in Supplemental Figure S1.
Figure 1. Metabolism of MDMA and corresponding monoamine uptake inhibition curves.
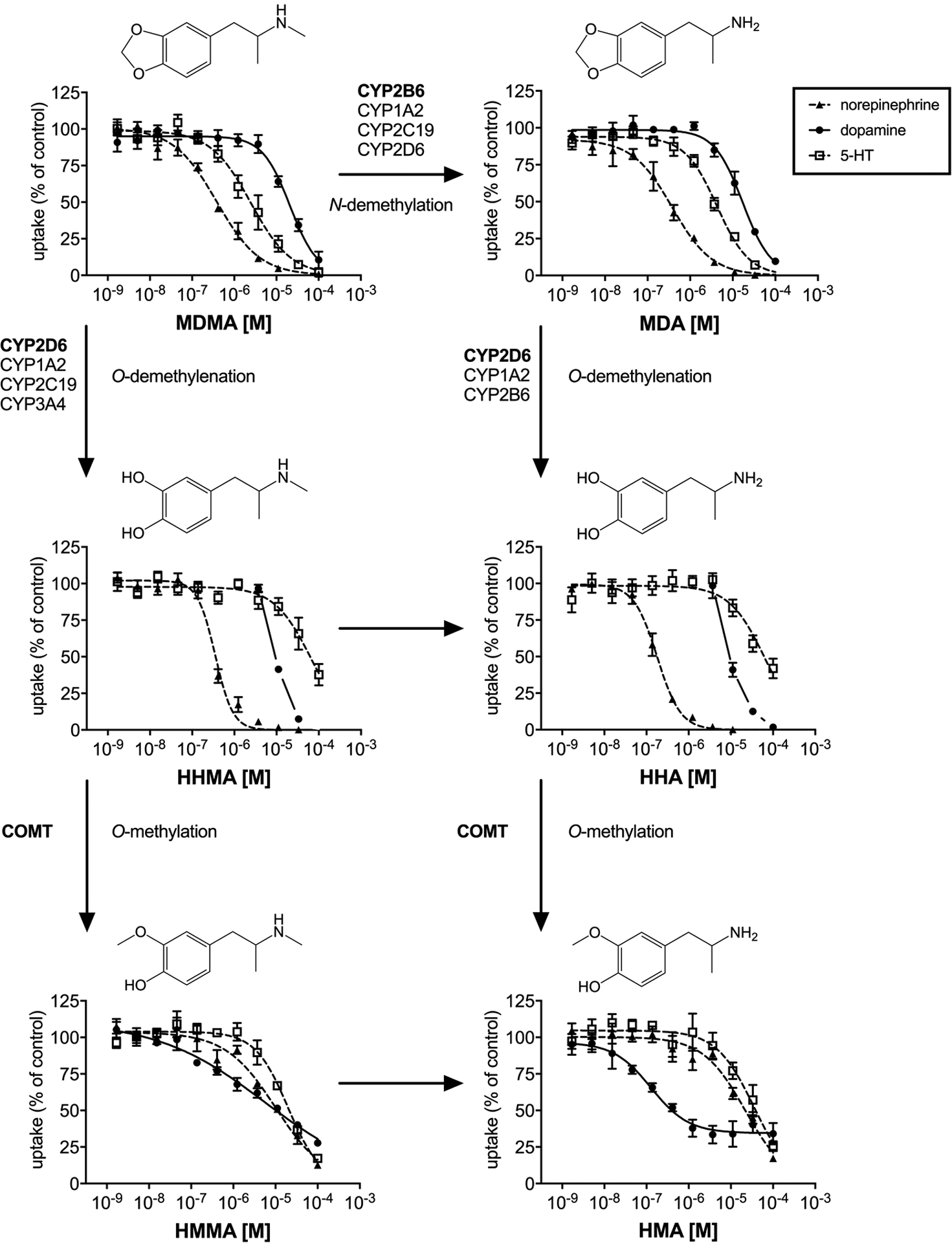
Major (bold) and minor enzymes involved in MDMA metabolism as described in (Meyer et al., 2008; Kreth et al., 2000). Monoamine uptake curves were fitted by non-linear regression and the data are presented as the mean±S.E.M. For HHMA and HHA no IC50 value was calculated for DAT, due to the lack of sigmoidal shape of the uptake curves. For these compounds, the DAT uptake curve intercept between 0 and 100% is shown in the figure for comparison and full curves are shown in Supplemental Figure S1.
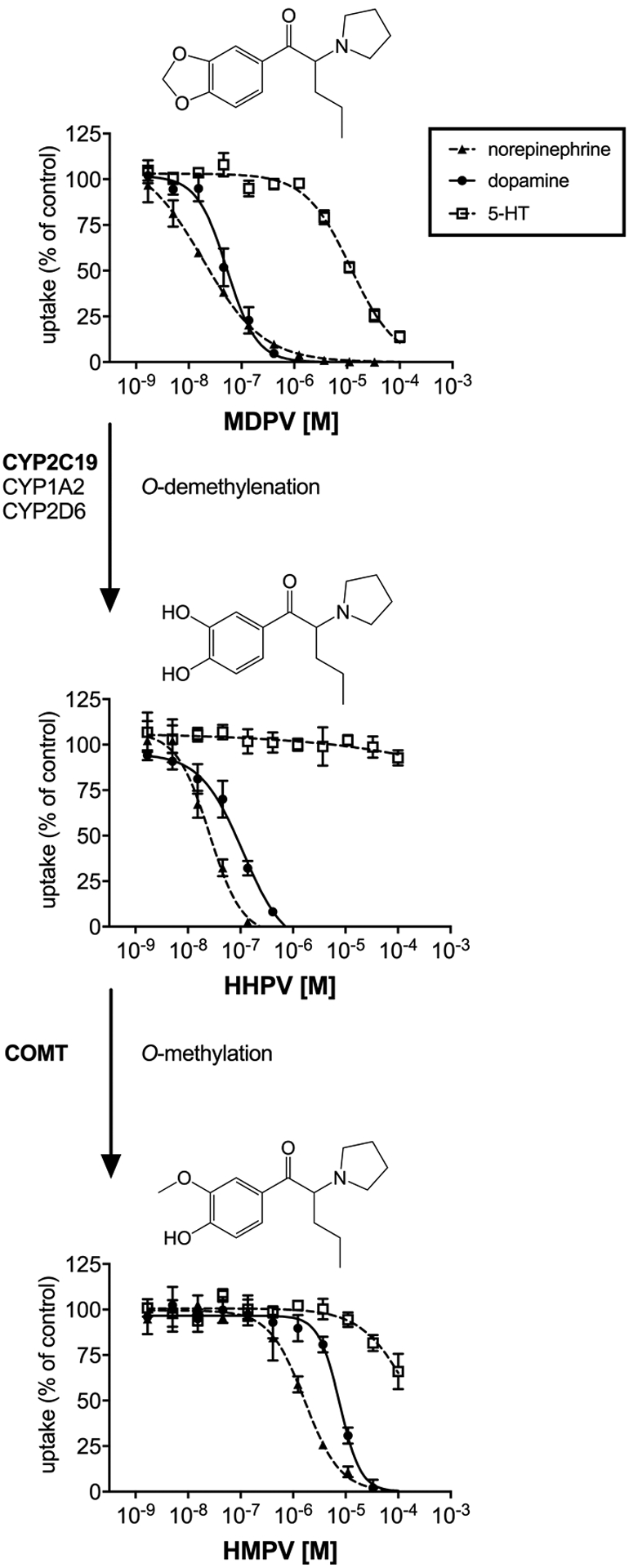
Figure 3. Metabolism of MDPV and corresponding monoamine uptake inhibition curves.
Major (bold) and minor enzymes involved in methylone metabolism as described in (Meyer et al., 2010). Monoamine uptake curves were fitted by non-linear regression and the data are presented as the mean±S.E.M.
Table 1.
Monoamine uptake inhibition.
| NET | DAT | SERT | DAT/SERT ratio | |
|---|---|---|---|---|
| IC50 [μM] | IC50 [μM] | IC50 [μM] | ||
| MDMA | 0.38 (0.28–0.52) | 21 (17–26) | 2.5 (1.7–3.6) | 0.12 (0.07–0.21) |
| HHMA | 0.35 (0.30–0.41) | 9.8a | 65 (46–94) | |
| HMMA | 10 (7–15) | 5.6 (2.8–11.4) | 20 (16–25) | 3.6 (1.4–8.9) |
| MDA | 0.38 (0.27–0.54) | 17 (14–21) | 4.3 (3.5–5.3) | 0.25 (0.17–0.38) |
| HHA | 0.18 (0.15–0.21) | 9.6a | 63 (43–93) | |
| HMA | 22 (16–30) | 0.13 (0.06–0.26)b | 35 (25–50) | 270 (96–830) |
| Methylone | 0.56 (0.41–0.76) | 6.6 (5.5–7.9) | 18 (15–21) | 2.7 (1.9–3.8) |
| HHMC | 0.78 (0.58–1.05) | 25a | >100 | |
| HMMC | 30 (20–3) | 0.34 (0.16–0.71)b | >100 | |
| MDC | 2.3 (1.6–3.2) | 34 (27–42) | 13 (10–16) | 0.38 (0.24–0.59) |
| MDPV | 0.018 (0.010–0.033) | 0.053 (0.040–0.070) | 12 (10–14) | 226 (143–350) |
| HHPV | 0.024 (0.018–0.031) | 0.092 (0.067–0.126) | >100 | |
| HMPV | 1.7 (1.2–2.2) | 7.7 (6.2–9.6) | >100 |
Values are means and 95% confidence intervals. DAT/SERT ratio = 1/DAT IC50 : 1/SERT IC50.
no IC50 value could be calculated as the curve did not display a sigmoidal shape; given concentrations represent 50% uptake inhibition.
partial uptake inhibition.
MDMA potently inhibited norepinephrine uptake (IC50 = 0.38 μM) and had a distinct preference to inhibit 5-HT (IC50 = 2.5 μM) vs. dopamine (IC50 = 21 μM) uptake. The N-demethylation of MDMA did not alter potency to inhibit norepinephrine and barely influenced its selectivity for 5-HT (IC50 = 4.3 μM) vs. dopamine (IC50 = 17 μM) uptake inhibition. The catechol metabolites of MDMA and MDA that are formed by O-demethylenation maintained potent norepinephrine uptake inhibition with IC50 values of 0.35 and 0.18 μM for HHMA and HHA, respectively. However, both catechol metabolites displayed a substantial decrease in inhibition of 5-HT uptake (IC50 of 63–65 μM) compared to the parent compounds. As no IC50 values could be calculated for HHMA and HHA for dopamine uptake inhibition, the intercepts of the dopamine uptake curves and 50% uptake inhibition values were determined as estimates of their transporter inhibition potencies. For these metabolites, 50% dopamine uptake inhibition was reached at 9.6–9.8 μM.
O-methylation of the catechol metabolites decreased the potency to inhibit norepinephrine uptake, which resulted in IC50 values of 10 and 22 μM for HMMA and HMA, respectively. Dopamine uptake inhibition for HMMA (IC50 = 5.6 μM) and HMA (IC50 = 0.13 μM) was significantly increased compared to MDMA and MDA (confidence intervals of the IC50 values do not overlap). However, HMA only partially inhibited dopamine uptake, with a maximum of 66% transporter inhibition (34% dopamine uptake).
Similar to MDMA, the β-keto analog methylone potently inhibited norepinephrine uptake (IC50 = 0.58 μM). Unlike MDMA, methylone had higher preference to inhibit dopamine (IC50 = 6.6 μM) vs. 5-HT (IC50 = 18 μM) uptake. MDC, the metabolite formed by N-demethylation of methylone, was more selective for 5-HT vs. dopamine inhibition but showed lower overall potency for all transporters. As observed for MDMA metabolites, the O-demethylenated catechol metabolite HHMC had substantially decreased 5-HT uptake inhibition potency (IC50 > 100 μM) without significant change in norepinephrine uptake inhibition (IC50 = 0.78 μM). HMMC, which is formed by O-methylation of HHMC, had decreased the potency to inhibit norepinephrine uptake (IC50 = 30 μM) while altering DAT function at lower concentrations (IC50 = 0.34 μM) compared to the parent compound. Like HMA, HMMC only partially (26%) inhibited dopamine uptake transport (74% dopamine uptake).
MDPV inhibited NET and DAT with high potency (IC50 = 0.018 and 0.053 μM, respectively) but had much lower potency at SERT (IC50 = 12 μM). The catechol metabolite HHPV potently inhibited NET and DAT as well (IC50 = 0.024 and 0.092 μM, respectively) but was devoid of SERT inhibition activity (IC50 > 100 μM). O-methylation of HHPV resulted in substantially decreased NET and DAT inhibition potency (IC50 = 1.7 and 7.7 μM, respectively) for the corresponding hydroxy-methoxy metabolite HMPV.
3.2. Monoamine receptor and transporter binding affinities
The binding affinities and activation potencies of MDMA, methylone, MDPV, and their metabolites at monoamine transporters and receptors are shown in Table 2 (n=3). MDMA did not bind to any monoamine transporters in the investigated concentration range. However, the catechol metabolites HHMA and HHA displayed sub-micromolar affinity at the NET and low micromolar affinity at the DAT. MDMA and MDA bound to adrenergic α1 and α2 receptors (Ki of 2.6–8.8 μM) whereas the catechol metabolites HHMA and HHA bound to α2 receptors only (Ki of 1.3 and 2.0 μM, respectively). Neither MDMA nor any of its metabolites bound to dopaminergic D2 receptors. MDMA and MDA showed affinity at serotonergic 5-HT1A, 5-HT2A, 5-HT2C receptors in the range of 3–11 μM. The remaining metabolites of MDMA were devoid of any binding to serotonergic receptors with the exception of HHA that bound to the 5-HT2A receptor (Ki = 9.2 μM). MDMA and its metabolites showed high affinity to both rat and mouse TAAR1 in the range of 0.1–3.1 μM, with the exception of HHMA that did not bind to the mouse TAAR1 in the investigated concentration range (Ki > 4.4 μM).
Table 2.
Monoamine transporter and receptor binding.
| NET | DAT | SERT | D2 | α1A | α2A | 5-HT1A | 5-HT2A | 5-HT2c | TAAR1rat | TAAR1mouse | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | Ki [μM] | |
| MDMA | >8.7 | >8.5 | >7.5 | >13 | 6.9±1.2 | 4.6±0.1 | 11±2 | 4.6±1.1 | 4.4±0.8 | 0.25±0.01 | 3.1±0.7 |
| HHMA | 0.94±0.14 | 1.9±0.0 | >7.4 | >13 | >9.5 | 1.3±0.1 | >18 | >12 | >5.1 | 3.0±0.3 | >4.4 |
| HMMA | >8.8 | >8.5 | >7.4 | >13 | >9.5 | >4.7 | >18 | >12 | >5.1 | 0.25±0.04 | 0.45±0.05 |
| MDA | >8.8 | >8.5 | >7.4 | >13 | 8.8±0.6 | 2.6±0.1 | 9.0±0.8 | 3.2±0.8 | 4.8±0.4 | 0.22±0.03 | 0.18±0.03 |
| HHA | 0.79±0.30 | 2.9±0.3 | >7.4 | >13 | >9.5 | 2.0±0.4 | >18 | 9.2±1.7 | >5.1 | 1.9±0.4 | 1.4±0.2 |
| HMA | >8.8 | >8.5 | >7.4 | >13 | >9.5 | >4.7 | >18 | >12 | >5.1 | 0.15±0.02 | 0.05±0.02 |
| Methylone | >8.7 | 2.3±0.1 | >7.5 | >13 | >9.5 | >5.0 | >17 | >12 | >14 | >5.0 | >4.7 |
| HHMC | 4.0±1.0 | 7.1 ±0.3 | >7.4 | >13 | >9.5 | >4.7 | >18 | >12 | >5.1 | 2.2±0.1 | >4.4 |
| HMMC | 4.2±1.0 | >8.5 | >7.4 | >13 | >9.5 | >4.7 | >18 | >12 | >5.1 | >5.2 | >4.4 |
| MDC | >8.8 | >8.5 | >7.4 | >13 | >9.5 | >4.7 | >17 | >12 | >5.1 | 4.8±0.5 | 3.5±0.3 |
| MDPV | 0.10±0.01 | 0.011±0.001 | 2.2±0.1 | >13 | >9.5 | >5.0 | 12±2 | >12 | 2.1 ±0.2 | >5.0 | >4.7 |
| HHPV | 0.11±0.01 | 0.023±0.003 | >7.4 | >13 | >9.5 | >4.7 | 4.3±1.6 | >12 | >5.1 | >5.2 | >4.4 |
| HMPV | 6.2±0.7 | 2.0±0.2 | >7.4 | >13 | >9.5 | >4.7 | >17 | >12 | >5.1 | >5.2 | >4.4 |
Ki values are given as mean±SD.
Methylone bound to the DAT (Ki = 2.3 μM) but not to the NET or SERT in the investigated concentration range. The N-demethylated metabolite MDC showed no affinity to any monoamine transporter. The dihydroxy and hydroxy-methoxy metabolites HHMC and HMMC both bound to the NET with Ki of 4 μM and HHMC additionally bound to the DAT (Ki = 7.1 μM). Methylone and its metabolites did not bind to any adrenergic, dopaminergic, or serotonergic receptors in the investigated concentration range. Binding to the rat TAAR1 was observed for MDC and HHMC in the range of 2–5 μM, and binding to the mouse TAAR1 was observed for MDC only (Ki = 3.5 μM).
MDPV displayed high affinity at the NET and DAT (Ki = 0.10 and 0.01 μM, respectively) with less potent affinity at the SERT (Ki = 2.2 μM). The dihydroxy metabolite HHPV maintained potent NET inhibition potency (Ki = 0.11 μM, respectively) but displayed a slight but significantly lower DAT inhibition (0.02 μM). The hydroxy-methoxy metabolite HMPV bound to the NET and DAT significantly less potently (Ki = 6.2 and 2.0 μM, respectively) compared to MDPV and HHPV. HHPV and HMPV did not bind to the SERT in the investigated concentration range (Ki >7.4 μM). MDPV and its metabolites did not bind to adrenergic or dopaminergic receptors in the investigated concentration range. Moderate affinity at serotonergic receptors was observed for MDPV at the 5-HT1A and 5-HT2C receptor (Ki = 12 and 2 μM, respectively) and for HHPV at the 5-HT1A receptor only (Ki = 4.3 μM). MDPV and its metabolite did not bind to rat or mouse TAAR1 in the investigated concentration range.
3.3. Activity at the serotonergic 5-HT2B receptor and human TAAR1
No binding affinity at the 5-HT2B receptor was examined; however, to assess for possible 5-HT2B interactions, the activation potency for this receptor subtype was determined (n=3). Of all the parent compounds and metabolites, MDA was the only compound to partially activate the 5-HT2B receptor with EC50 of 0.20 ± 0.01 μM and activation efficacy of 51 ± 7%. The remaining drugs did not activate the 5-HT2B receptor at concentrations up to 10 μM. Activation of the human TAAR1 was assessed for compounds with considerable affinity (Ki < 2 μM) for either rat or mouse TAAR1. HMA was the only compound to partially activate human TAAR1 with EC50 of 10.4 ± 0.2 μM and activation efficacy of 51 ± 12%.
4. Discussion
4.1. Pharmacological effects of MDMA metabolites
The metabolism of MDMA in humans consists of two pathways: 1) N-demethylation to form MDA and 2) O-demethylenation to form HHMA and HHA (de la Torre et al., 2004; Kreth et al., 2000; Meyer et al., 2008; de la Torre et al., 2012; Schmid et al., 2016). MDA formation is primarily mediated by cytochrome P450 (CYP) 2B6 with contributions from CYP1A2, CYP2C19, and CYP2D6 (Kreth et al., 2000; Meyer et al., 2008; Vizeli et al., 2017; Schmid et al., 2016). CYP2D6 is the primary enzyme involved in O-demethylenation of MDMA and MDA, with contributions from CYP1A2, CYP2C19, and CYP3A4 (Meyer et al., 2008; Kreth et al., 2000; Schmid et al., 2016; Vizeli et al., 2017). The demethylenated catechol metabolites HHMA and HHA are subsequently O-methylated by catechol-O-methyltransferase (COMT) to yield HMMA and HMA (de la Torre et al., 2004; de la Torre et al., 2012). A study in healthy human subjects revealed Tmax values of 2.2–2.6 h for MDMA, 4.6–5.9 h for MDA, and 2.7–5.0 h for HMMA after oral administration of different MDMA doses (Schmid et al., 2016). Cmax levels of MDA accounted for 4–8% of the Cmax of MDMA. The Cmax of HMMA strongly depended on CYP2D6 activity and ranged from 9–48% of the Cmax of MDMA; however, the Cmax of unconjugated HMMA was only about 1–3% of the Cmax of MDMA, suggesting a high proportion of conjugated HMMA (Schmid et al., 2016). It is noteworthy that the biotransformation of MDMA is generally similar between rats and humans, with the notable exception that rats metabolize the parent compound at a much faster rate (Baumann et al., 2009; Concheiro et al., 2014).
In the current study, both MDMA and MDA displayed their most potent effects at norepinephrine uptake inhibition, along with an entactogenic pharmacological profile, which is expressed as more potent effects at 5-HT vs. dopamine uptake inhibition (Simmler et al., 2013; Liechti, 2015). MDA showed activity at the serotonergic 5-HT2B receptor. The affinity of MDA and MDMA at the 5-HT2A receptor did not significantly differ, but MDA has previously been shown to activate the 5-HT2A receptor about 10-fold more potently compared to MDMA (Rickli et al., 2015b). However, a recent study showed that the hallucinogenic potency of psychedelic drugs correlates with their 5-HT2A and 5-HT2C receptor binding affinities but not with their 5-HT2A receptor functional activities. This intriguing observation may be explained by higher sensitivity of the ligand-binding assay or inherent limitations of the calcium mobilization assay to provide a valid index of in vivo receptor subtype activity (Luethi and Liechti, 2018). Taken together, MDA might exert mild psychedelic effects that are slightly more pronounced than MDMA, but an increase in such effects may not be noticeable over the time course after MDMA use, as only a small fraction of the parent (<10%) is metabolized to MDA.
Our findings show that O-demethylenation resulted in more pronounced dopaminergic activity of the metabolites, which is not surprising given the similarities in chemical structures for the hydroxy metabolites and dopamine. The catechol metabolites HHMA and HHA maintained the NET inhibition potency of their precursors MDMA and MDA, respectively. Furthermore, the corresponding catechol metabolites displayed substantial binding affinity to both NET and DAT. In contrast to the overall reduced potency at monoamine transporters, the hydroxy-methoxy metabolites showed high affinity to rodent TAAR1. However, HMA was the only compound that partially activated human TAAR1 in this study, with only moderate potency (EC50 of 10 μM). Further research is needed to decipher the effect of MDMA metabolites on TAAR1.
The catechol and hydroxy-methoxy metabolites of MDMA are almost exclusively found in conjugated form as sulfates (HHMA and partially HMMA) or glucuronides (HMMA) in plasma and urine (Schmid et al., 2016; Segura et al., 2001; de la Torre et al., 2004; Steuer et al., 2015). Thus, despite the rather modest amount of MDA generated from biotransformation of MDMA, it is the most relevant contributor to the pharmacological effects among all MDMA metabolites. Transporter inhibition data and animal studies (Schindler et al., 2014) suggest that the fraction of unconjugated catechol metabolites could contribute to cardiovascular effects of MDMA while a substantial contribution of the hydroxy-methoxy metabolites seems unlikely. The anomalous increase of dopamine uptake to more than 200% as observed for HHMA and HHA has been reported before for unlabeled dopamine and other DAT substrates (Henry et al., 2018), but the underlying mechanisms responsible for this phenomenon are not yet understood and more research is needed to provide better insight. It is worth mentioning that Escubedo et al. (Escubedo et al., 2011) reported that HHMA induces wash-resistant inhibition of dopamine uptake in rat brain tissue suggesting that dihydroxy metabolites might induce long-term changes in DAT structure and function related to cytotoxic effects. Furthermore, various mechanistic studies suggest that MDMA metabolites are in fact responsible for the neurotoxic effects related to MDMA use (Moratalla et al., 2017). Besides metabolism in the liver, CYP enzymes located near drug targets in the brain may affect local metabolism of centrally acting drugs (Miksys and Tyndale, 2002). Therefore, metabolite concentrations in the brain may differ from the measured plasma levels and potential effects of the metabolites cannot solely be derived from transporter and receptor interaction studies.
4.2. Pharmacological effects of methylone metabolites
In vitro studies with human liver microsomes demonstrate that methylone is N-demethylated to form MDC and O-demethylenated to form HHMC, analogous to the metabolism of MDMA. These biotransformations mainly involve the actions of CYP2D6 with minor contributions from other CYP enzymes (Pedersen et al., 2013). HHMC is further metabolized by COMT to yield HMMC and to a lesser extent to 3-hydroxy-4-methoxymethcathinone (Pedersen et al., 2013). Following a 5 mg/kg injection of methylone in rats, about 3% of total methylone was excreted in urine in unchanged form within 48 h post-dosing, whereas the amount of MDC and HMMC accounted for about 2% and 26%, respectively (Kamata et al., 2006). Additionally, 5% of the HMMC isomer 3-hydroxy-4-methoxy-methcathinone was formed. However, > 80% of the hydroxy-methoxy metabolites of methylone were excreted as conjugates (Kamata et al., 2006). In the same study, a single human urinary sample of a patient admitted to an emergency department after ingestion of an unknown amount of methylone powder was analyzed. In accordance with the rat data, the analysis revealed that HMMC was the most abundant metabolite with MDC and 3-hydroxy-4-methoxy-methcathinone being minor metabolites (Kamata et al., 2006). HMMC was the most abundant metabolite also in rat plasma following subcutaneous injections of methylone, reaching 12–22% of the parent compound; HHMC and MDC were detected in amounts of 10–13% of injected methylone (Elmore et al., 2017). Cmax was reached after 15 min for methylone, after 30–45 min for MDC, after 60–70 min for HHMC, and after 90–120 min for HMMC (Elmore et al., 2017). In another study, maximum brain and serum concentrations of methylone were reached 30 min after subcutaneous injection with approximately five times higher brain vs. serum concentrations (Stefkova et al., 2017). Serum levels of MDC peaked 30 min later than those of methylone and reached about 20% of the methylone levels (Stefkova et al., 2017). In that study, HMMC was found to be the second most abundant metabolite in serum after MDC. However, the precise amount of HMMC could not be quantified. Lopez-Arnau and colleagues reported Tmax values of 0.5 and 1 h for 15 mg/kg and 30 mg/kg oral doses, respectively (Lopez-Arnau et al., 2013). The similar Tmax values observed after subcutaneous injection and oral administration suggests fast absorption of the drug. Different observations have been made regarding the linearity of methylone pharmacokinetics, with one study suggesting linear pharmacokinetics after oral administration (Lopez-Arnau et al., 2013) and another suggesting non-linear pharmacokinetics after subcutaneous administration (Elmore et al., 2017).
In the current study, methylone was a potent inhibitor of norepinephrine uptake and to a lesser extent an inhibitor of dopamine and 5-HT uptake. The N-demethylated metabolite MDC exerted similar 5-HT uptake inhibition potency as methylone but much weaker potency at norepinephrine and dopamine uptake inhibition, suggesting weaker psychotropic effects compared to the parent compound (Luethi and Liechti, 2018), which is in accordance with animal studies (Elmore et al., 2017). The partial dopamine inhibition by HHMC at sub-micromolar concentrations may not be sufficient to produce discernable dopaminergic effects over the course of time after methylone intake. In fact, neither HHMC nor HMMC increase basal dialysate dopamine concentrations when administered intravenously to rats (Elmore et al., 2017). Like methylone (Baumann et al., 2012), MDC and HHMC are known transporter substrates (Elmore et al., 2017). Such substrate-type activity has been previously observed for phase I metabolites of the transporter substrate 4-methylmethcathinone (mephedrone) (Mayer et al., 2016). The binding of methylone to DAT and of HHMC to both DAT and NET suggests that these compounds may adopt both substrate-type and inhibitory binding modes, as has been described before for certain MDMA analogs (Sandtner et al., 2016). As observed for the catechol metabolites of MDMA and other hDAT substrates (Henry et al., 2018), HHMC caused a more than 200% increase in dopamine uptake at some concentrations.
4.3. Pharmacological effects of MDPV metabolites
The main metabolic pathway of MDPV biotransformation is O-demethylenation by CYP2D6 to form HHPV, followed by O-methylation to yield HMPV (Meyer et al., 2010; Strano-Rossi et al., 2010). Meyer and colleagues found HMPV to be the most abundant metabolite in human and rat urine (Meyer et al., 2010). Following subcutaneous injections of 0.5–2 mg/kg MDPV to rats, Tmax was reached after 13–19 min (Anizan et al., 2016). In that study, HMPV was the most abundant metabolite, with maximal concentrations reaching 53–61% of the parent compound and Tmax of 189–206 min; HHPV was detected at maximal concentrations of 12–19% of MDPV and Tmax of 206–257 min (Anizan et al., 2016). MDPV and its metabolites displayed linear pharmacokinetics and both metabolites are mainly present as conjugates (Anizan et al., 2014; Meyer et al., 2010; Strano-Rossi et al., 2010). Anizan and colleagues did not observe a correlation between concentrations of the hydroxylated MDPV metabolites and horizontal locomotor activity or stereotypy in rats; the authors therefore hypothesized that the metabolites either do not cross the blood-brain-barrier or potentially exert an inhibitory effect on locomotor activity (Anizan et al., 2016).
The results of the current study confirm that MDPV and HHPV are both highly potent NET and DAT inhibitors (Baumann et al., 2017; Meltzer et al., 2006). In contrast to the entactogen MDMA, the high selectivity for dopaminergic vs. serotonergic activity indicates stronger reinforcing properties and a higher addictive liability for MDPV (Liechti, 2015). The radioligand binding data indicate that both metabolites of MDPV are devoid of any transporter substrate-activity, as observed for the parent compound (Rickli et al., 2015a; Baumann et al., 2013). However, previous studies in rats indicate that HHPV may not significantly contribute to stimulant effects due to low blood-brain-barrier permeability and high proportion of formed conjugates (Schindler et al., 2016). Consistent with this hypothesis, microdialysis studies in conscious rats show that intravenous administration of HHPV fails to alter locomotor activity or substantially increase extracellular dopamine concentrations in the nucleus accumbens (Baumann et al., 2017). Unlike the catechol metabolites of MDMA and methylone, HHPV displayed a sigmoidal uptake curve and did not cause and increase in dopamine uptake more than 100%, which may be explained by the lack of substrate activity for this metabolite.
5. Conclusion
Metabolites of the 3,4-methylenedioxy ring-substituted stimulants MDMA, methylone, and MDPV interact with monoamine transporters. The N-demethylation of MDMA only slightly changes the monoamine uptake inhibition profile but potentially increases the 5-HT2A receptor-mediated psychedelic properties of the formed metabolite MDA. By contrast, N-demethylation of methylone substantially decreases the norepinephrine and dopamine uptake inhibition potencies of the corresponding metabolite MDC. The O-demethylenated catechol metabolites of all substances maintained the norepinephrine uptake inhibition potency of the parent compounds but showed marked reductions in 5-HT uptake inhibition potency. O-methylation of the catechol metabolites significantly decreased the inhibition potency across all monoamine transporters. The hydroxy-methoxy metabolites of MDMA and methylone displayed rather uncommon dopamine uptake inhibition curves, expressed by an unusual flat slope for HMMA and only partial uptake inhibition for HMA and HMMC. The DAT inhibition curve of the hydroxy-methoxy metabolite of MDPV displayed a common sigmoidal shape, which suggests that the irregularities in shape for DAT uptake curves are caused by the substrate properties of the respective metabolites. The catechol metabolites of MDMA, methylone, and MDPV could potentially contribute to cardiovascular effects in humans, due to potent norepinephrine uptake inhibition. However, pharmacokinetic data from human and rat studies show a high proportion of conjugates for the catechol and hydroxy-methoxy metabolites, suggesting only minor contribution of these metabolites to in vivo pharmacological effects. On the other hand, N-demethylated metabolites MDA and MDC are found in unconjugated form at pharmacologically relevant amounts. The effects of MDA are expected to contribute more to the pharmacological effects compared to MDC, as the monoamine inhibition potency of the former is similar to the parent compound. Placebo-controlled clinical studies are needed to gain better insight into the pharmacokinetics of methylone and MDPV in humans and therefore a clearer interpretation of the results of the current study.
Supplementary Material
Figure 2. Metabolism of methylone and corresponding monoamine uptake inhibition curves.
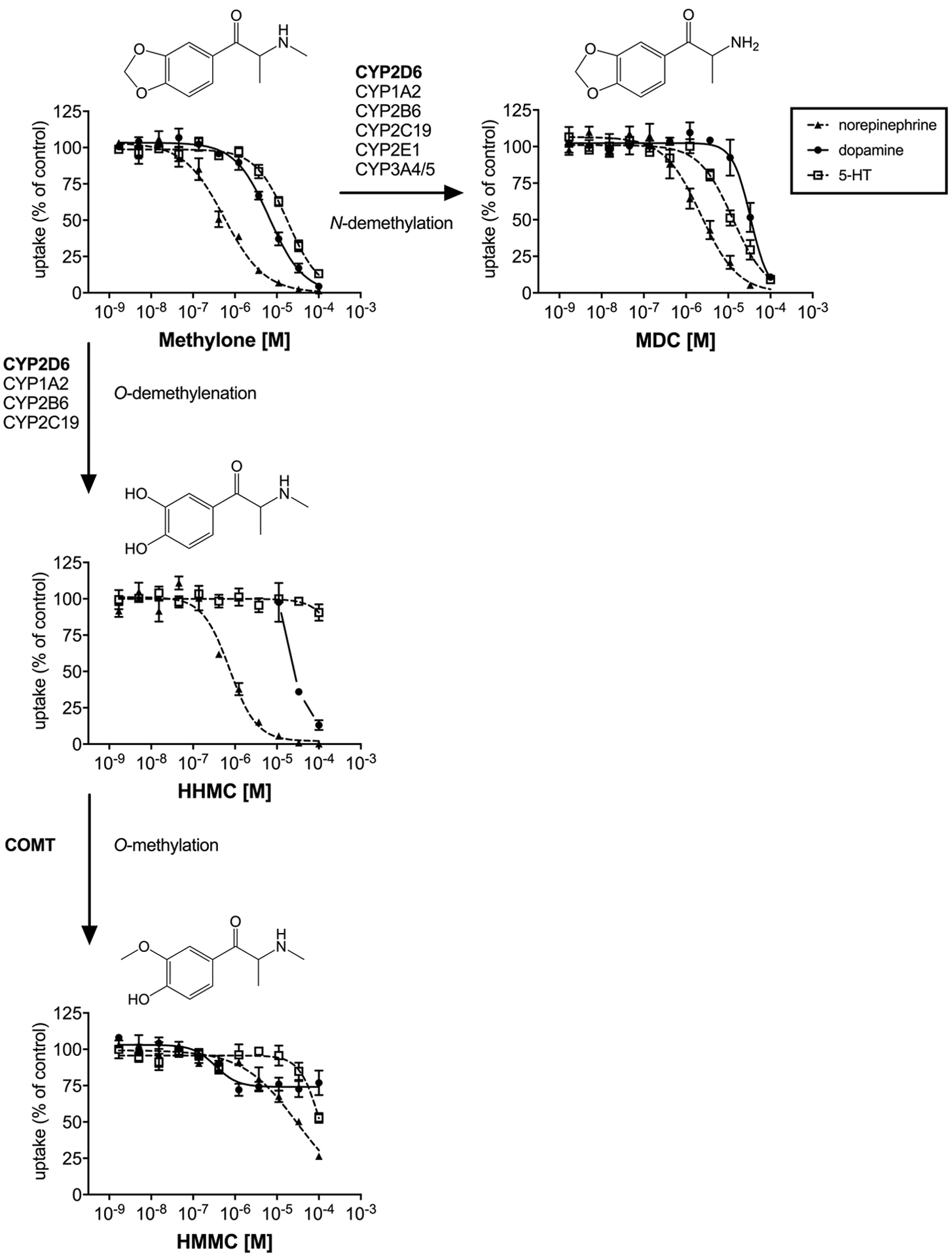
Major (bold) and minor enzymes involved in methylone metabolism as described in (Pedersen et al., 2013). Monoamine uptake curves were fitted by non-linear regression and the data are presented as the mean±S.E.M. No DAT IC50 value was calculated for HHMC, due to the lack of sigmoidal shape of the uptake curve. The DAT uptake curve intercept between 0 and 100% for HHMC is shown in the figure for comparison and the full curve is shown in Supplemental Figure S1.
Acknowledgements
This work was supported by the Federal Office of Public Health (grant no. 16.921318) and the NIH Intramural Research Programs of the National Institute on Drug Abuse (NIDA) and the National Institute of Alcohol Abuse and Alcoholism (NIAAA). The authors thank Sylvie Chaboz and Danièle Buchy for technical assistance.
Footnotes
Declaration of conflicting interests
M.C.H. is an employee of F. Hoffmann-La Roche. The other authors do not have any conflicts of interest to declare for this work.
References
- Anizan S, Concheiro M, Lehner KR, et al. (2016) Linear pharmacokinetics of 3,4-methylenedioxypyrovalerone (MDPV) and its metabolites in the rat: relationship to pharmacodynamic effects. Addict Biol 21: 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anizan S, Ellefsen K, Concheiro M, et al. (2014) 3,4-Methylenedioxypyrovalerone (MDPV) and metabolites quantification in human and rat plasma by liquid chromatography-high resolution mass spectrometry. Anal Chim Acta 827: 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolino-Lobo I, Meulenbelt J, Molendijk J, et al. (2011) Induction of glutathione synthesis and conjugation by 3,4-methylenedioxymethamphetamine (MDMA) and 3,4-dihydroxymethamphetamine (HHMA) in human and rat liver cells, including the protective role of some antioxidants. Toxicology 289: 175–184. [DOI] [PubMed] [Google Scholar]
- Baumann MH, Ayestas MA Jr., Partilla JS, et al. (2012) The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology 37: 1192–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Bukhari MO, Lehner KR, et al. (2017) Neuropharmacology of 3,4-methylenedioxypyrovalerone (MDPV), its metabolites, and related analogs. Curr Top Behav Neurosci 32: 93–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Partilla JS, Lehner KR, et al. (2013) Powerful cocaine-like actions of 3,4-methylenedioxypyrovalerone (MDPV), a principal constituent of psychoactive ‘bath salts’ products. Neuropsychopharmacology 38: 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Zolkowska D, Kim I, et al. (2009) Effects of dose and route of administration on pharmacokinetics of (+ or −)−3,4-methylenedioxymethamphetamine in the rat. Drug Metab Dispos 37: 2163–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo H, Brulport M, Hermes M, et al. (2006) Influence of CYP2D6 polymorphism on 3,4-methylenedioxymethamphetamine (‘ecstasy’) cytotoxicity. Pharmacogenet Genomics 16: 789–799. [DOI] [PubMed] [Google Scholar]
- Concheiro M, Baumann MH, Scheidweiler KB, et al. (2014) Nonlinear pharmacokinetics of (+/−)3,4-methylenedioxymethamphetamine (MDMA) and its pharmacodynamic consequences in the rat. Drug Metab Dispos 42: 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre R, Farre M, Roset PN, et al. (2004) Human Pharmacology of MDMA. Therapeutic Drug Monitoring 26: 137–144. [DOI] [PubMed] [Google Scholar]
- de la Torre R, Yubero-Lahoz S, Pardo-Lozano R, et al. (2012) MDMA, methamphetamine, and CYP2D6 pharmacogenetics: what is clinically relevant? Front Genet 3: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bello F, Sakloth F, Partilla JS, et al. (2015) Ethylenedioxy homologs of N-methyl-(3,4-methylenedioxyphenyl)-2-aminopropane (MDMA) and its corresponding cathinone analog methylenedioxymethcathinone: Interactions with transporters for serotonin, dopamine, and norepinephrine. Bioorg Med Chem 23: 5574–5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellefsen KN, Concheiro M, Suzuki M, et al. (2015) Quantification of methylone and metabolites in rat and human plasma by liquid chromatography-tandem mass spectrometry. Forensic Toxicology 33: 202–212. [Google Scholar]
- Elmore JS, Dillon-Carter O, Partilla JS, et al. (2017) Pharmacokinetic profiles and pharmacodynamic effects for methylone and its metabolites in rats. Neuropsychopharmacology 42: 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobedo I, O’Shea E, Orio L, et al. (2005) A comparative study on the acute and long-term effects of MDMA and 3,4-dihydroxymethamphetamine (HHMA) on brain monoamine levels after i.p. or striatal administration in mice. Br J Pharmacol 144: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escubedo E, Abad S, Torres I, et al. (2011) Comparative neurochemical profile of 3,4-methylenedioxymethamphetamine and its metabolite alpha-methyldopamine on key targets of MDMA neurotoxicity. Neurochem Int 58: 92–101. [DOI] [PubMed] [Google Scholar]
- Eshleman AJ, Wolfrum KM, Hatfield MG, et al. (2013) Substituted methcathinones differ in transporter and receptor interactions. Biochem Pharmacol 85: 1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JK, Shah D, Kicman AT, et al. (2002) Action of MDMA (ecstasy) and its metabolites on arginine vasopressin release. Ann N Y Acad Sci 965: 399–409. [DOI] [PubMed] [Google Scholar]
- Forsling ML, Fallon JK, Shah D, et al. (2002) The effect of 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’) and its metabolites on neurohypophysial hormone release from the isolated rat hypothalamus. Br J Pharmacol 135: 649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry LK, Allen MD, Shetty M, et al. (2018) Elucidation of self-mediated enhancement of dopamine transport by the dopamine transporter which can be modulated by extracellular gate and N-terminal residues. Experimental Biology 2018. San Diego, CA. [Google Scholar]
- Kamata HT, Shima N, Zaitsu K, et al. (2006) Metabolism of the recently encountered designer drug, methylone, in humans and rats. Xenobiotica 36: 709–723. [DOI] [PubMed] [Google Scholar]
- Kreth K, Kovar K, Schwab M, et al. (2000) Identification of the human cytochromes P450 involved in the oxidative metabolism of “Ecstasy”-related designer drugs. Biochem Pharmacol 59: 1563–1571. [DOI] [PubMed] [Google Scholar]
- Liechti ME. (2015) Novel psychoactive substances (designer drugs): overview and pharmacology of modulators of monoamine signaling. Swiss Med Wkly 145: w14043. [DOI] [PubMed] [Google Scholar]
- Lopez-Arnau R, Martinez-Clemente J, Carbo M, et al. (2013) An integrated pharmacokinetic and pharmacodynamic study of a new drug of abuse, methylone, a synthetic cathinone sold as “bath salts”. Prog Neuropsychopharmacol Biol Psychiatry 45: 64–72. [DOI] [PubMed] [Google Scholar]
- Luethi D, Kaeser PJ, Brandt SD, et al. (2018a) Pharmacological profile of methylphenidate-based designer drugs. Neuropharmacology 134: 133–140. [DOI] [PubMed] [Google Scholar]
- Luethi D, Kolaczynska KE, Docci L, et al. (2018b) Pharmacological profile of mephedrone analogs and related new psychoactive substances. Neuropharmacology 134: 4–12. [DOI] [PubMed] [Google Scholar]
- Luethi D and Liechti ME. (2018) Monoamine transporter and receptor interaction profiles in vitro predict reported human doses of novel psychoactive stimulants and psychedelics. Int J Neuropsychopharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luethi D, Trachsel D, Hoener MC, et al. (2018c) Monoamine receptor interaction profiles of 4-thio-substituted phenethylamines (2C-T drugs). Neuropharmacology 134: 141–148. [DOI] [PubMed] [Google Scholar]
- Mayer FP, Wimmer L, Dillon-Carter O, et al. (2016) Phase I metabolites of mephedrone display biological activity as substrates at monoamine transporters. Br J Pharmacol 173: 2657–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer PC, Butler D, Deschamps JR, et al. (2006) 1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (Pyrovalerone) analogues: a promising class of monoamine uptake inhibitors. J Med Chem 49: 1420–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Du P, Schuster F, et al. (2010) Studies on the metabolism of the alpha-pyrrolidinophenone designer drug methylenedioxy-pyrovalerone (MDPV) in rat and human urine and human liver microsomes using GC-MS and LC-high-resolution MS and its detectability in urine by GC-MS. J Mass Spectrom 45: 1426–1442. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Peters FT and Maurer HH. (2008) The role of human hepatic cytochrome P450 isozymes in the metabolism of racemic 3,4-methylenedioxy-methamphetamine and its enantiomers. Drug Metab Dispos 36: 2345–2354. [DOI] [PubMed] [Google Scholar]
- Miksys SL and Tyndale RF. (2002) Drug-metabolizing cytochrome P450s in the brain. J Psychiatry Neurosci 27: 406–415. [PMC free article] [PubMed] [Google Scholar]
- Moratalla R, Khairnar A, Simola N, et al. (2017) Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog Neurobiol 155: 149–170. [DOI] [PubMed] [Google Scholar]
- Mueller M, Yuan J, Felim A, et al. (2009) Further studies on the role of metabolites in (+/−)−3,4-methylenedioxymethamphetamine-induced serotonergic neurotoxicity. Drug Metab Dispos 37: 2079–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen AJ, Petersen TH and Linnet K. (2013) In vitro metabolism and pharmacokinetic studies on methylone. Drug Metab Dispos 41: 1247–1255. [DOI] [PubMed] [Google Scholar]
- Rickli A, Hoener MC and Liechti ME. (2015a) Monoamine transporter and receptor interaction profiles of novel psychoactive substances: para-halogenated amphetamines and pyrovalerone cathinones. Eur Neuropsychopharmacol 25: 365–376. [DOI] [PubMed] [Google Scholar]
- Rickli A, Kopf S, Hoener MC, et al. (2015b) Pharmacological profile of novel psychoactive benzofurans. Br J Pharmacol 172: 3412–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandtner W, Stockner T, Hasenhuetl PS, et al. (2016) Binding mode selection determines the action of ecstasy homologs at monoamine transporters. Mol Pharmacol 89: 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler CW, Thorndike EB, Blough BE, et al. (2014) Effects of 3,4-methylenedioxymethamphetamine (MDMA) and its main metabolites on cardiovascular function in conscious rats. Br J Pharmacol 171: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler CW, Thorndike EB, Suzuki M, et al. (2016) Pharmacological mechanisms underlying the cardiovascular effects of the “bath salt” constituent 3,4-methylenedioxypyrovalerone (MDPV). Br J Pharmacol 173: 3492–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid Y, Vizeli P, Hysek CM, et al. (2016) CYP2D6 function moderates the pharmacokinetics and pharmacodynamics of 3,4-methylene-dioxymethamphetamine in a controlled study in healthy individuals. Pharmacogenet Genomics 26: 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura M, Farre M, Pichini S, et al. (2005) Contribution of cytochrome P450 2D6 to 3,4-methylenedioxymethamphetamine disposition in humans: use of paroxetine as a metabolic inhibitor probe. Clin Pharmacokinet 44: 649–660. [DOI] [PubMed] [Google Scholar]
- Segura M, Ortuno J, Farre M, et al. (2001) 3,4-Dihydroxymethamphetamine (HHMA). A major in vivo 3,4-methylenedioxymethamphetamine (MDMA) metabolite in humans. Chem Res Toxicol 14: 1203–1208. [DOI] [PubMed] [Google Scholar]
- Simmler LD, Buser TA, Donzelli M, et al. (2013) Pharmacological characterization of designer cathinones in vitro. Br J Pharmacol 168: 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefkova K, Zidkova M, Horsley RR, et al. (2017) Pharmacokinetic, ambulatory, and hyperthermic effects of 3,4-methylenedioxy-N-methylcathinone (methylone) in rats. Front Psychiatry 8: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuer AE, Schmidhauser C, Schmid Y, et al. (2015) Chiral plasma pharmacokinetics of 3,4-methylenedioxymethamphetamine and its phase I and II metabolites following controlled administration to humans. Drug Metab Dispos 43: 1864–1871. [DOI] [PubMed] [Google Scholar]
- Strano-Rossi S, Cadwallader AB, de la Torre X, et al. (2010) Toxicological determination and in vitro metabolism of the designer drug methylenedioxypyrovalerone (MDPV) by gas chromatography/mass spectrometry and liquid chromatography/quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 24: 2706–2714. [DOI] [PubMed] [Google Scholar]
- Vizeli P, Schmid Y, Prestin K, et al. (2017) Pharmacogenetics of ecstasy: CYP1A2, CYP2C19, and CYP2B6 polymorphisms moderate pharmacokinetics of MDMA in healthy subjects. Eur Neuropsychopharmacol 27: 232–238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.