

Abstract
Interruptions or alterations in the B cell development pathway can lead to primary B cell immunodeficiency with resultant absence or diminished immunoglobulin production. While the most common cause of congenital agammaglobulinemia is X-linked agammaglobulinemia (XLA), accounting for approximately 85% of cases, other genetic forms of agammaglobulinemia have been identified. Early recognition and diagnosis of these conditions are pivotal for improved outcomes and prevention of sequelae and complications. The diagnosis of XLA is often delayed, and can be missed if patient has a mild phenotype. The lack of correlation between phenotype and genotype in this condition makes management and predicting outcomes quite difficult. In contrast, while less common, autosomal recessive forms of agammaglobulinemia present at younger ages and with typically more severe clinical features resulting in an earlier diagnosis. Some diagnostic innovations, such as KREC level measurements and serum BCMA measurements, may aid in facilitating an earlier identification of agammaglobulinemia leading to prompt treatment. Earlier diagnosis may improve the overall health of patients with XLA.
Keywords: Agammaglobulinemia, X-linked agammaglobulinemia, Immunoglobulin replacement therapy, Bruton’s tyrosine kinase, Common variable immunodeficiency
Introduction
B cell development, from hematopoietic stem cell to plasma cell, occurs in the bone marrow as a result of signals derived from the bone marrow, the surrounding environment, and through interaction with antigen and T cells [1]. Different interruptions or alterations in this complex pathway can lead to maturational arrest in the early stages of B cell development resulting in humoral immunodeficiency with decreased or absent antibody production [1]. Hypogammaglobulinemia or agammaglobulinemia are seen in a heterogenous set of diseases broadly termed primary B cell immunodeficiencies [2]. The immunophenotype and clinical presentation of these conditions vary largely among patients even when the same genetic alteration is detected, which highlights the multifactorial nature of these diseases [2]. Patients present with recurrent bacterial infections, typically S. pneumoniae and/or H. influenza, as well as other pathogens, which are specific to certain conditions, such as enterovirus and Giardia with X-linked agammaglobulinemia (XLA) [2].
The most common genetic cause of agammaglobulinemia is XLA, which comprises about 85% of cases of agammaglobulinemia [3]. Additionally, several genetic mutations causing autosomal recessive agammaglobulinemia have been described, as well as three gene mutations resulting in an autosomal dominant form of agammaglobulinemia [3, 4]. Formation of the pre-B cell receptor complex (pre-BCR) on the pre-B cell surface is necessary for survival and maturation of these cells. A genetic defect affecting development of the pre- BCR complex can halt B cell development and lead to primary B cell immunodeficiency [5]. According to the European Society for Immunodeficiencies (ESID), agammaglobulinemia is defined by recurrent infections starting before the age of 5 years, < 500 mg/dL of IgG and < 2 standard deviations for normal levels of IgA and IgM, and < 2% circulating B cells [6].
The aim of this article is to review the genetic defects and pathways that result in absent production of immunoglobulin, which includes XLA, autosomal recessive, and autosomal dominant agammaglobulinemia. We will cover the common manifestations of disease and sequelae of disease, as well as diagnosis and treatments currently available. Relevant to present day, we will also briefly cover what has been reported to date regarding agammaglobulinemia/hypogammaglobulinemia and Sars-CoV-2.
X-Linked Agammaglobulinemia
BTK
The most common genetic disorder resulting in maturational arrest of B cell development is X-linked agammaglobulinemia (XLA). The incidence of XLA ranges from 1:100,000 to 1:200,000. This disorder results from a mutation in Bruton’s tyrosine kinase (BTK) [1]. BTK is a cytoplasmic tyrosine kinase expressed mainly in hematopoietic cells. As a member of the Tec kinase family, BTK is activated by a src kinase, and then further activated through autophosphorylation. With activation, it is then able to bind to BLNK, a scaffolding protein resulting in PLCy2 phosphorylation and calcium influx [2]. There have been an excess of 600 mutations in BTK identified, with the majority of mutations involving 1–4 base pairs [2]. Of note, there have been larger deletions reported in 3–5% of individuals involving BTK and extending through the adjacent or linked genes TIMM8A and TAF7L, which results in both XLA and deafness-dystonia-optic neuropathy syndrome [7]. An early study by Plebani et. al, of 73 patients with XLA reported that missense mutations accounted for 55.6% of BTK mutations in this cohort, followed by small deletions (15.9%), premature stop codon (14.3%), splice site mutations (7.9%), and small insertions (6.3%) [8]. A more recent study, which included 168 XLA patients, reported missense mutations accounting for the most common mutation type in BTK (49%) followed insertion or deletions (18%), nonsense mutations (17%), splice site mutations (12%), and large deletions (4%) [9].
The BTK protein is composed of 5 structural domains (pleckstrin homology domain (PH), Tec homology domain (TH), src homology domain 3 (SH3), and Src homology domain 2 (SH2), and the SH1 domain which is the domain responsible for the kinase activity [6]. A study of 122 XLA patients, from countries in Eastern and Central Europe, found the most common type of mutation reported to be missense. It was also noted that the majority of mutations in BTK identified in this cohort occurred in the SH1 domain (45.3%). The other domains were also found with mutations, although less frequently, with the SH2 domain accounting for 22.3%, SH3 domain at 7.3%, and the TH domain 6.3%. Analysis of this cohort found 98 different mutations, of which 46 were novel [10].
Clinical Manifestations
The clinical manifestations of XLA include recurrent, pyogenic, bacterial infections in young boys, and near complete absence of all isotypes of immunoglobulins [1]. The common pathogens in XLA include Streptococcus pneumoniae, Staphylococcus spp., Pseudomonas spp., and Haemophilus Influenzae. (Tables 1 and 2 include some of the pathogens that have been isolated in patients with XLA.)
Table 1.
Organisms that have been identified in patients with XLA
| Bacteria | Virus | Fungus | Parasite |
|---|---|---|---|
|
Branhamella catharralis Campylobacter spp Escherichia coli Haemophilus spp Helicobacter spp Klebsiella pneumoniae Mycobacterium spp Mycoplasma Pseudomonas spp Salmonella Shigella spp Staphylococcus spp Streptococcus pneumoniae |
Adenovirus Enterovirus Hepatitis C Measles Rotavirus |
Pneumocystis jirovecii Candida spp |
Giardia lamblia Blastocystis hominis |
Table 2.
Pathogens identified in some XLA by infection type. Tables 1 and 2
adapted from: Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality, X-linked agammaglobulinemia report on a U Registry of 201 patients
| Infection type | Pathogens identified |
|---|---|
| Pneumonia |
Haemophilus influenzae, type b Haemophilus parainfluenzae Haemophilus parahemolytica Klebsiella pneumoniae Measles Pneumocystis jirovecii Pseudomonas spp Mycobacterium avium Staphylococcus spp Streptococcus pneumoniae |
| Diarrhea |
Campylobacter fetus Clostridium difficile Enterovirus Giardia lamblia Helicobacter pylori Rotavirus Salmonella spp Shigella spp |
| Meningitis/encephalitis |
Adenovirus Enterovirus Haemophilus influenzae Streptococcus pneumoniae |
| Sepsis |
Campylobacter fetus Helicobacter cinaidi Haemophilus influenza, type b Pseudomonas spp Salmonella spp Streptococcus pneumoniae |
| Hepatitis | Hepatitis C |
| Cellulitis | Staphylococcus Aureus |
| Arthritis | Mycoplasma |
The disease, however, appears to have clinical heterogeneity, in that some patients present with later onset of disease and higher than expected levels of immunoglobulins [1]. This heterogeneity may be due to which domain of BTK is affected. A study of 201 XLA patients in a US registry found infection (85%) accounted for the most frequently reported presentation with positive family history (41%) and neutropenia (11%) accounting for other initial clinical presentations. Patients with a family history of XLA had an earlier age of diagnosis on average (2.59 years mean age) when compared to patients without a family history of XLA (5.37 years mean age) [11]. Of concern is that, despite family history, the age of diagnosis was above 2 years.
Another study of 174 Chinese patients with XLA found 127 distinct mutations in BTK among this cohort, of which 45 were novel mutations. The majority of mutations were found to be missense mutations (38.1%) as reported in other studies with the majority of mutations occurring in the SH1 (50.79%) domain [12].
Infections
In a recent study of the USIDNET registry, of 226 patients with XLA and known BTK mutation, infections of the ear, nose, and throat occurred the most frequently (77.8%) followed by respiratory infections (72.5%). Common offending organisms were Influenza B (11.5%), Hemophilus influenzae (11%), Staphylococcus aureus (10.6%), Streptococcus pneumoniae (8.4%), Pseudomonas aeruginosa (6.1%), and Giardia (4.4%) [13]. A study of the Italian Primary Immunodeficiency Network Registry (IPINet) reported the most frequently reported infections occurred in the respiratory tract, gastrointestinal tract, and integumentary system. Although a pathogen could not always be identified, this study found Hemophilus Influenzae (53.6%) and Streptococcus pneumoniae (17.9%) to be the most commonly isolated microorganisms from sputum cultures [9]. Other pathogens isolated, in order from most to least common, included Pseudomonas, Staphylococcus, Klebsiella, Branhamella catharralis, and Pneumocystis jirovecii [9]. Gastrointestinal infections were most commonly caused by Giardia lamblia (48.7%), and infections of the skin were most commonly due to Staphylococcus aureus (75.8%) [9].
Phenotypic Heterogeneity
The genotype–phenotype correlation in XLA has been difficult to study due to the number of different mutations in BTK. There are also no set criteria to measure symptom severity objectively. In one study, mild mutations in BTK (defined as amino acid substitutions or splice defects in conserved, non-invariant, or consensus sequences) were associated with older age of diagnosis and higher number of peripheral B cells when compared to severe mutations in BTK (premature stop codons, frameshift mutations, complex mutations, large deletions, and splice defects at invariant sites). A higher level of serum IgM was associated with mild mutations. These findings suggested that amino acid substitution may result in proteins that have some function, although they are unstable [2]. Another study found mild mutations in BTK were associated with older age of diagnosis, increased IgG and IgA levels, and decreased hospitalizations prior to immunoglobulin replacement therapy initiation. However, there were some exceptions to this observation, as there were cases of severe symptoms with “mild” mutations, and mild phenotypes seen with mutations resulting in absent BTK expression [14]. In another study by Conley et al., of 82 patients with XLA the average age of diagnosis was 35 months of age for patients with sporadic mutations [15].
Delay in diagnosis and atypical clinical features have been reported in several patients with BTK mutations in the non-kinase domains [6]. In a study of 5 XLA patients with delayed and atypical presentation of XLA, mutations in BTK were found in the PH or SH2 domains [6]. Another report aiming to compare BTK mutations in XLA patients with peripheral B cells present did not find a correlation with the type or location of the BTK mutation and the presence of peripheral B cells [16].
There have been case reports further demonstrating this phenotypic heterogeneity. For example, in one case report, a man with a diagnosis of CVID and Crohn’s disease was evaluated for BTK mutation after his grandson was diagnosed with XLA. The grandfather presented the same mutation in BTK, but had vastly different clinical manifestations and was diagnosed, much later in his adult life, incorrectly, with CVID [17].
One study of 174 XLA patients in China found patients with severe genotype mutations (defined in this study as a frameshift or nonsense mutation resulting in a truncated protein) had a statistically significant earlier age of diagnosis when compared to the group defined as having a mild genotype mutation [12].
Diagnosis
The majority of children with BTK mutation will have some serum immunoglobulin that can be measured, and some B cells detected in the periphery, although this measurement tends to decrease with increasing age. For this reason, XLA has been described as a “leaky” B cell immunodeficiency in some patients [2]. B cell differentiation is blocked at the pro- to pre-B cell stage and partially blocked at the pre-mature to immature B cell stage [1]. The majority of CD19 + cells in the bone marrow of XLA patients will show pro-B cells (CD19 + , CD34 + TdT expression, no cytoplasmic mu heavy chain), and the minority will be pre-B cells ((< 10%) CD19 + , CD34-, TdT, cytoplasmic mu heavy chain expression) with an even smaller population of cells, which represent a stage between the pro- and the pre- B cell. These cells express CD19 + , CD34 + , TdT, and cytoplasmic mu heavy chain [2].
Criteria for a diagnosis of XLA reported by Winkelstein et. al can be fulfilled with (a) a mutation in the BTK gene and/or defective BTK protein expression or (b) family history of XLA in a maternally related male relative or (c) < 2% peripheral B cells and hypogammaglobulinemia [11].
The diagnostic criteria for XLA are illustrated in Figs. 1 and 2.
Fig. 1.

Criteria for probable diagnosis of agammaglobulinemia in patients with no genetic diagnosis (
adapted from https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria)
Fig. 2.
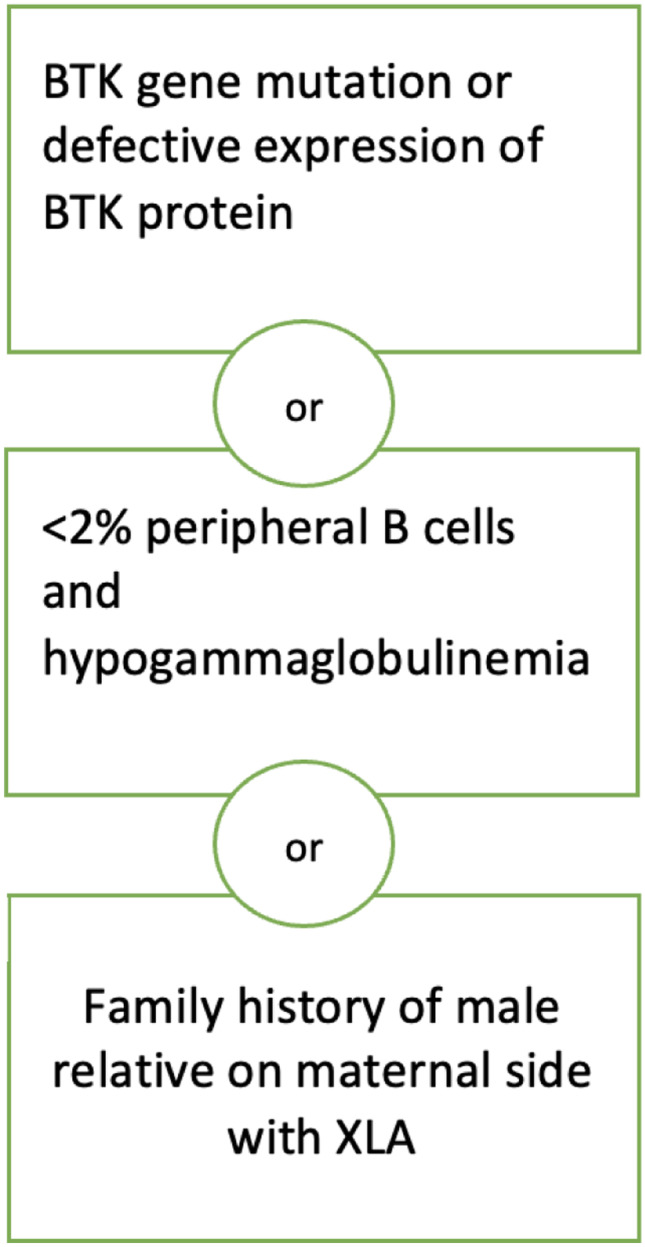
Diagnostic criteria for X-linked agammaglobulinemia.
Adapted from Winkelstein, Jerry A. MD; Marino, Mary C. MLS; Lederman, Howard M. MD, PhD; Jones, Stacie M. MD; Sullivan, Kathleen MD, PhD; Burks, A. Wesley MD; Conley, Mary Ellen MD; Cunningham-Rundles, Charlotte MD, PhD; Ochs, Hans D. MD X-linked agammaglobulinemia, report on a United States Registry of 201 Patients Medicine: July 2006—Volume 85—Issue 4—p 193–202 https://doi.org/10.1097/01.md.0000229482.27398.ad
Genetic testing for BTK pathogenic variants may be obtained via several methods. There are tests available specifically testing for the single BTK gene and protein analysis. Chromosomal microarray can identify large deletions of the BTK protein. Gene panels are available which include BTK and other possible genes involved with agammaglobulinemia and/or autosomal recessive causes of agammaglobulinemia. If these tests are negative, but there is a high index of suspicion for XLA or agammaglobulinemia due to patient characteristics, more in-depth testing may be carried out in the form of exome sequencing or genome sequencing [7].
Survival
A large multicenter study from different countries reported survival rates at 20 years of age as low as 22%, while other countries reported survival above 70%. This study, which surveyed 40 centers around the world, aimed to describe the different clinical manifestations, diagnostic approaches, and management of XLA patients globally. The most common cause of death reported was chronic or acute lung disease in 41% of patients. Thirty-four percent of patients had more than 24 months delay in diagnosis, and 39% of centers reported lack of genetic studies as a diagnostic challenge. A common concern reported among centers was lack of access to immunoglobulin replacement products presenting a challenge to managing patients with XLA. Additionally, many different complications were reported which highlights the heterogeneity of inflammatory, infectious, and autoimmune complications, which can be seen in patients with XLA. Overall, the majority of centers reported a good survival rate [3].
A study of 201 patients in a US registry found the most common cause of death in this cohort to be disseminated enteroviral infection (35%), followed by chronic lung disease (25%), then hepatitis (15%). Additionally, there were 2 deaths secondary to treatments including one from hepatitis C, which was acquired from IVIG contaminated with hepatitis C virus, and another death attributed to stem cell transplant [11].
A recent study of 168 XLA patients in the Italian Primary Immunodeficiency Network Registry (IPINet) found a survival rate of 92.7% at 43 years of age, significantly reduced compared to the survival rate of healthy controls. Chronic lung disease (CLD) was diagnosed in 51.8% of patients, and 13.1% of these patients had evidence of CLD at the time of diagnosis. The rate of malignancy diagnosed at follow up was 3.7%. The survival rate was further reduced in patients with a diagnosis of chronic lung disease (90.5% survival rate) [9]. A study of 36 XLA patients in India, over the course of two decades, found the mean survival to be 11.4 years of age with a 20% mortality rate [18].
Autosomal Recessive Agammaglobulinemia
Autosomal recessive agammaglobulinemia comprise rare causes of agammaglobulinemia, compared to XLA. Thus far, mutations have been described in mu heavy chain, Lambda 5, Ig alpha, Ig beta, BLNK, PI3K genes, and TCF3. These conditions tend to be more severe and present earlier than cases of XLA. Some patients with early B cell developmental defects have yet to have a genetic cause identified, although with advances in diagnostic and genetic testing, this is improving [19].
Mu Heavy Chain Deficiency
Several cases of mu heavy chain deficiency have been reported, resulting in a clinical phenotype similar to patients with BTK mutation. The patients with mu heavy chain deletion, however, tend to present at younger ages (mean age: 11 months versus 35 months in BTK mutation) and with more severe infections. Specifically, these patients present more frequently with enteroviral infections and sepsis secondary to Pseudomonas [20]. Mu heavy chain deletion is responsible as the cause for approximately 5% of agammaglobulinemia [21]. Thirty percent of the mutations of the mu heavy chain gene have been found to affect the mu heavy constant region. Thirty percent of patients with mu heavy chain mutation are also found to have neutropenia [20]. The majority of mutations result from large gene deletions, while a minority are caused by point mutations. While mutations in BTK may cause a partial interruption in B cell development that can lead to a “leaky” phenotype, mutations in the mu heavy chain gene results in a complete block, which inhibits B-cell maturation from the pro- to pre- B cell stage [21]. There has been one case of a patient with both gene deletion and missense mutation resulting in a compound heterozygous alteration of the mu heavy chain gene. The patient presented with severe infection at 7 months of age (peritonitis, pseudomonas bacteremia, E. coli urinary tract infection, neutropenia, and hypogammaglobulinemia) [21].
Lambda 5 Deficiency
The IGLL1 gene encodes the lamba-5 and the VpreB chains, which are both components of the pre-B cell receptor complex. Lambda 5 and the VpreB chain are together known as the surrogate light chain. Few cases of IGLL1 gene mutations have been reported. One published case consisted of a 2-month-old female who presented with bilateral upper lobe pneumonia. Immunodeficiency evaluation showed absent B cells with undetectable IgA and IgM and an IgG level of 329 mg/dL. Chromosomal microarray identified a homozygous missense mutation in exon 3 of the IGLL1 gene [5]. Another patient presented with recurrent otitis media at 2 months old and immunodeficiency evaluation revealed undetectable CD19 + B cells as well as hypogammaglobulinemia. This male patient was found to have mutations in exon 1 and exon 5 of lambda5/14.1 gene. Analysis of peripheral lymphocytes in this patient demonstrated 0.06% expressed CD19 with low intensity expression of surface IgM and CD38 dim, which is seen in normal B cells. Bone marrow analysis was consistent with an arrest in pro-B to pre-B cell differentiation with 85% of the few CD19 + cells expressing CD34 [22].
Ig Alpha Deficiency
Ig alpha, along with Ig beta, is a transmembrane protein heterodimer responsible for signal transduction in the B cell receptor complex [23]. Ig alpha and Ig beta form a disulfide bond complex and function to assist the mu heavy chain to the surface of the cell [24]. A 2-year-old female patient from Turkey with agammaglobulinemia has been reported with a homozygous splice defect in exon 3 of the Ig alpha gene. Analysis of peripheral lymphocytes showed < 0.01% CD19 + B cells and bone marrow showed more than 75% of CD19 + expressed CD34, which indicated a block in differentiation from pro-B to pre-B cell [23]. There have been 5 patients with mutations in Ig alpha reported, and all mutations have been reported to involve a region upstream of the transmembrane domain [25]. In another case, a 6-month-old female, who presented with human herpes virus 8 (HHV8) and John Cunningham (JC) viral encephalitis, with immune evaluation pertinent for hypogammaglobulinemia and absent peripheral B cells, was found to have a novel homozygous mutation in CD79a resulting in a premature stop codon [25]. A recent publication describes a 16-year-old male with agammaglobulinemia secondary to mutation in CD79a, diagnosed at 2 years of age, who developed Campylobacter jejuni spondylodiscitis, although he was on immunoglobulin replacement therapy and prophylactic antibiotics. This patient received immunoglobulin therapy with IgM supplementation, which was found to aid in complement activation and increase bacterial killing [26].
Ig Beta Deficiency
The first patient reported with Ig beta deficiency was a 15-year-old female with a long-standing history of recurrent bronchitis, which began at 5 months of age, and two episodes of pneumonia. She was diagnosed with hypogammaglobulinemia at 15 months of age [24]. Although peripheral CD19 + B cells were very low, they were not completely absent (0.08%). This patient was identified as having a homozygous mutation of codon 137 in the Ig beta gene, neighboring the amino acid residue required for disulfide bonding between the Ig alpha and Ig beta proteins [24]. The mutation in this patient resulted in a “leaky” phenotype, as mutant Ig beta in vitro was found to form some disulfide bonds with Ig alpha and deliver the mu heavy chain to the cell surface. This was demonstrated by the finding of a small amount of peripheral B cells in the circulation being IgM dim [24].
A 20-year-old Italian male with history of pneumonia, Salmonella enteritis, and chronic sinusitis was found to have a mutation in exon 3 of the Ig beta gene resulting in complete lack of Ig beta expression. Immunologic phenotype consisted of severe hypogammaglobulinemia, for which he had been on replacement since the age of 8 months and absent peripheral B cells [27].
In another report, a new, homozygous, null mutation in codon 13 of the Ig beta gene was found in a 15-month-old female patient with a history of recurrent upper respiratory infections, fever, ecthyma, and severe neutropenia (10 cells/mm3). Immune evaluation was positive for absence of peripheral CD19 + cells and hypogammaglobulinemia (IgG < 35 mg/dL). Parents were consanguineous and both harbored the same heterozygous mutation [28].
BLNK Deficiency
BLNK, a scaffolding protein that is phosphorylated after B cell receptor cross-linking, plays a role in B cell development. In one published report, a patient with recurrent otitis beginning at 8 months of age, two episodes of pneumonia, undetectable serum immunoglobulins and < 1% peripheral B cells, were found to have BLNK deficiency. The patient demonstrated absence of pre-B cells indicating an important role for BLNK in the pro- to pre-B cell transition [29]. It is also suspected that the older brother of this patient, who passed away from neutropenia and pseudomonal sepsis, likely had BLNK deficiency [2]. There have been 5 patients with BLNK deficiency described as of this paper’s writing, although some have been cited as non-published observations [2, 29–31]. In one published report, a patient with agammaglobulinemia, no circulating B cells, and homozygous BLNK mutation was found to have a block in the pre-BI to pre-BII stage of B cell maturation [30].
PIK3R1 and PIK3CD Mutation
The PI3K family of kinases is involved in signal transduction, ultimately resulting in production of PIP3 and subsequent membrane recruitment and activation of AKT [32]. The PI3K kinases play several other roles in the cell including signaling for growth, metabolism, survival, and more. There have been several cases of malignancies and immune dysregulation and few cases of immunodeficiency reported with overactivation or underactivation of the PI3K pathway [33]. Specifically, underactivation of this pathway leads to a primarily humoral defect with near absent B cells, hypogammaglobulinemia, recurrent sinopulmonary infections, colitis, and/or autoimmunity, while T cell numbers are normal.
Class 1A PI3Ks play an important role in immunology. The PIK3R1 gene encodes p85α, p55α, and p50α, which are classified as regulatory subunits. One regulatory subunit will form a heterodimer with a catalytic subunit p110a, p110b, or PI3K delta. The PI3K delta enzyme complex is composed of p85 alpha and p110 delta, and p110 delta is encoded by the PIK3CD gene [33].
There have been 3 cases of agammaglobulinemia reported with PIK3R1 gene mutation all reported with absent B cells, elevated T cells, and agammaglobulinemia [32, 34]. The first patient described, diagnosed at 19 years of age, presented at 3.5 months old with neutropenia, interstitial pneumonia, and gastroenteritis. Later in life, she developed erythema nodosum, inflammatory colitis, and recurrent bacteremia secondary to campylobacter. Initial immune evaluation revealed hypogammaglobulinemia and absent B cells (< 1% CD19 + B cells) in the periphery and bone marrow. Analysis of bone marrow showed < 0.1% of pro-B cells, despite normal numbers of early B cell precursors, indicating near absence of cells arising from B cell lineage. The patient was found to have absent p85alpha due to a premature stop codon in the PIK3R1 gene [34]. In murine studies, lack of p85 alpha has a phenotype consisting of decreased B cells, increased IL12 production from dendritic cells, and hyper-responsiveness to insulin [34].
In another published report of premature stop codon in the PIK3R gene, a 10-month-old female displayed mucosal bleeding secondary to thrombocytopenia, neutropenia, pan-hypogammaglobulinemia, and absence of B cells. Her clinical phenotype consisted of recurrent upper respiratory infections, oral thrush, and prolonged diarrhea on two occasions. After a male sibling was born, he was clinically diagnosed as having autosomal recessive agammaglobulinemia and found to have absent B cells on flow cytometry and neutropenia. Genetic testing was only obtained in the female patient [32].
There are an additional 6 cases of homozygous PIK3CD gene mutation resulting in LOF of PIK3 delta enzyme complex. These patients also displayed hypogammaglobulinemia or agammaglobulinemia as well as decreased B cells [34].
SLC39A7 Mutation
In a study by Anzilotti et. al, 6 individuals were identified as having hypomorphic mutations inSLC39A7, a gene encoding the protein ZIP7, which is responsible for transportation of zinc from the endoplasmic reticulum to the cytoplasm. These patients had absent B cells, agammaglobulinemia, and early infections. A subset of patients had blistering dermatoses, failure to thrive, and thrombocytopenia. These two patients with severe phenotype were cured after hematopoietic stem cell transplant. Analysis of B cell development found an arrest from the late pre-B stage to the immature B stage. These abnormal immature cells had intact expression of the genes for Rag and IL7R, but not BAFFR and CD20, which are usually expressed at the immature B cell stage [35].
Autosomal Dominant Agammaglobulinemia
LRRC8 Deficiency
In one report, a 17-year-old female with agammaglobulinemia, absent peripheral B cells, abnormal facial features, and normal BTK expression was found to have a balanced translocation t(9;20)(q33.2;q12) on karyotype analysis. This translocation resulted in the truncation of the LRRC8 gene, which when transferred into mouse stem cells, results in inhibition of B cell maturation with arrest at the pro-B cell stage of development. The mice showed decrease in B cells and T cell proportions; however, the patient had normal number of T cells. It is hypothesized that the truncated LRRC8 gene leads to a dominant-negative effect, as normal gene is expressed from the other allele and results in transcription of a normal LRRC8 protein [36].
TOP2B Mutation (Hoffman Syndrome)
Autosomal dominant mutations in TOP2B, a gene coding for a type II topoisomerase, has been reported in patients with recurrent infections with encapsulated organisms, absent B cells, and agammaglobulinemia, in addition to facial dysmorphism and limb abnormalities [37].
Autosomal Dominant and Autosomal Recessive Form
TCF3 Mutation
A study of 4 patients with agammaglobulinemia, decreased B cells in the periphery, were found to have mutations in the gene TCF3 (E2A) which encodes two transcription factors, E12 and E47. The small number of B cells that were present showed absent B cell receptor, and increased CD19 expression. In all 4 patients, the de novo mutation was expressed in E47 and resulted in an abnormal protein (E555K). This abnormal protein caused a dominant-negative effect when it was associated with wild-type E47 binding to DNA. Additionally, this mutation resulted in maturational arrest of B cells prior to differentiation into the pro-B cell [38]. For reference, XLA patients have a defect in the pro-B cell to pre-B cell stage, which is the subsequent stage [6]. A recent case report describes a novel mutation in TCF3 resulting in an autosomal recessive form of agammaglobulinemia with recurrent pneumonia, chronic diarrhea, and failure to thrive in a 9-year-old female [39].
Unique Features
Neutropenia
Neutropenia has been seen in certain primary immunodeficiencies. Neutropenia has been suspected to be secondary to the concurrent severe infection at the time of presentation in some cases, such as with XLA. Ten to 25% of XLA patients are found to have neutropenia [40]. Neutropenia has been noted as a common feature in XLA. It has also been seen in some cases with ARA due to mu heavy chain mutation, BLNK deficiency, Ig alpha deficiency, and in one patient with Ig beta deficiency. The 3 patients reported with homozygous mutations in PIK3R1 gene all had neutropenia as a clinical feature. The neutropenia was present early on in the disease course in these patients, improved with passing time [34].
Enterovirus Infection
Enterovirus infection in non-immunocompromised patients can present as asymptomatic or mild disease. In immunocompromised patients, this infection can lead to more devastating complications, such as meningoencephalitis. The rate of enteroviral infections is higher in XLA patients compared to other primary immunodeficiencies. Chronic meningoencephalitis due to enterovirus commonly presents as progressive neurological symptoms, including gradual loss of cognitive and motor milestones, with death commonly resulting after 2 years. The diagnosis requires a high index of suspicion as CSF studies can be normal in many cases. Currently, there are no FDA approved antiviral medications for enterovirus infection. Treatment consists of high-dose IVIG and intrathecal immunoglobulin, but success is not common [40].
Colitis
In an article based on a survey of patients with XLA and USIDNET registry data by Hernandez-Trujillo et al., 4% of patients reported a formal diagnosis of Crohn’s disease had been given to them, although many more patients reported chronic diarrhea (21%) and abdominal pain (17%) as on-going symptoms. When compared to data from the USIDNET registry of XLA patients, 3% of patients had a diagnosis of colitis or enteritis, while 7% had abdominal pain and 9% had chronic diarrhea [41].
In another more recent study of the USIDNET registry, 35% of patients with XLA were reported to have some form of gastrointestinal symptoms, with diarrhea being the most commonly reported. Ten percent of patients had a diagnosis of inflammatory bowel disease or enteritis. The study reported a case of a 23-month-old male with XLA who had a maternal grandfather with a diagnosis of Crohn’s disease (diagnosed at 35 years old) and CVID (diagnosed at 48 years old). He was evaluated for BTK mutation after his grandson was diagnosed with XLA, and was found to have the same missense mutation, and subsequently making his diagnosis XLA and not CVID. This study also raised the point that IVIG has been emerging as a possible therapy for Crohn’s disease, which poses the question: could IVIG therapy in XLA patients be preventing development of inflammatory bowel disease in some patients? [23].
Autoimmunity
XLA has been associated with inflammatory and autoimmune conditions, although it is thought to occur much less in XLA when compared to other primary immunodeficiencies. In a survey of 128 XLA patients, 69% reported at least one symptom of an inflammatory process [41]. Twenty percent reported joint pain, and 11% reported swelling of a joint; however, only 7% reported a formal diagnosis of arthritis. Cytopenias were also reported, but the cause of the cytopenia (autoimmune versus infectious) was not specified. In the USIDNET registry of XLA patients, 16% of patients with XLA had a diagnosis of arthritis, 5% with hypothyroidism or goiter, and 1% had vasculitis [41].
Diagnosis
Agammaglobulinemia should be considered when there is a history of recurrent sinopulmonary infections prior to the age of 5 years, a severe bacterial infection such as meningitis or sepsis, and/or a lack of lymphoid tissue on physical exam. However, clinical diagnosis alone of XLA is difficult due to phenotypic heterogeneity and de novo presentations [7]. Laboratory measurements that suggest a diagnosis of XLA include significantly reduced immunoglobulin levels with IgG usually < 200 mg/dL and decreased peripheral B cells (< 1%). Antibody titers showing lack of response to childhood vaccines may also be helpful. A minority of individuals are also found to be severely neutropenic; however, this is usually in conjunction with a severe infection at the time of diagnosis.
Family history is also important to obtain when suspecting a diagnosis of XLA; however, de novo presentation occur in 33% of patients, and autosomal recessive cases of agammaglobulinemia will show a different inheritance pattern (Table 3) [7]. Examination of peripheral monocytes via flow cytometry or western blot for BTK protein expression can be done prior to more expensive genetic testing. The added benefit of flow cytometry is the ability to identify carriers [1].
Table 3.
Summary of mutations identified as causing agammaglobulinemia
| Mutation | Inheritance pattern |
|---|---|
| BTK (85%) | X-linked |
| Mu-heavy chain | AR |
| Lambda 5 | AR |
| Ig alpha | AR |
| Ig beta | AR |
| PIK3 | AR |
| BLNK | AR |
| TCF3 | AR and AD |
| LRRC8 | AD |
In a patient with a positive family history of XLA, genetic testing for pathogenic hemizygous BTK variant can be obtained by single gene testing, via a genetic panel including multiple suspect genes. In cases where the patient displays the clinical phenotype of XLA, but no mutation in BTK is identified, genome sequencing or exome sequencing should be contemplated [7].
Kappa-deleting recombination excision circle (KREC) levels can be used to make the diagnosis of XLA shortly after birth. KRECs are formed during the process of V(D)J recombination of B lymphocytes. One report found KREC measurement using PCR on Guthrie card samples identified patients with XLA [42]. Another report commented on 13 patients, 12 with XLA, and one patient with ARA, and found KREC levels were low or undetectable at birth, and remained low or undetectable overtime [43]. Utilization of KREC level on newborn screening could facilitate earlier diagnosis, not only of XLA, but of autosomal recessive agammaglobulinemia as well. Earlier diagnosis can decrease morbidity and mortality by preventing severe infections or complications, by prompt treatment with immunoglobulin replacement therapy and prophylactic antibiotics [43]. A study of 306 patients, 30 with XLA, 5 with non-XLA hypogammaglobulinemia (no BTK mutation with < 2% peripheral B cells), 133 healthy neonates, 138 healthy controls aged 1 month to 35 years old, aimed to measure levels of both coding joint KRECs and signal joint KRECs. All healthy patients were positive for both coding joint KRECs and signal joint KRECs, while all XLA and non-XLA patients were negative for signal joint KRECs and all, but one, were negative for coding joint KRECs, again indicating this could be a valid neonatal screening tool for B cell defects [44].
A TNF receptor on the surface of plasma cells, which is elevated in multiple myeloma, known as B-cell maturation antigen (BCMA), promotes cell survival when ligand interaction occurs. In a study of patients with primary antibody deficiency (PAD), levels of serum BCMA (sBCMA) were measured in an attempt to characterize severity among primary antibody deficiency patients. This paper defined severe PAD as patients with a diagnosis of CVID, XLA, or serum IgG < 600 mg/dL, and found sBCMA levels to be significantly reduced in these patients, when compared to normal patients and patients with mild forms of PAD (selective IgA deficiency, IgG deficiency). There was a 97% positive predictive value for the diagnosis of XLA when the serum BCMA level was < 15 ng/mL. These findings suggest that although more studies are needed, sBCMA measurements may aid in the diagnosis of severe PAD and determine which patients need to be started on immunoglobulin replacement therapy [45].
Management
The treatment for agammaglobulinemia is immunoglobulin replacement therapy, in order to prevent infection in these patients. In the USA, immunoglobulin products are composed of donated plasma from 10 to 20,000 donors, which is then tested for pathogens and processed to eliminate any potential viruses and prions [46]. Immunoglobulin replacement is available in several preparations and can be administered intravenously or subcutaneously. With intravenous immunoglobulin replacement, the patient is infused every 3–4 weeks. Subcutaneous infusions can be offered daily to weekly or biweekly to monthly depending on the product [7]. When administered intravenously, there is a higher IgG level peak on day one compared to the peaks obtained with subcutaneous (and facilitated subcutaneous) infusions. Studies have shown a rate of 3–4 infections per patient-year, while on immunoglobulin replacement therapy [46]. There is no set dose required or ideal trough for prevention of infection. It has been shown that different patients do clinically well with different trough levels, and therefore, this should be individualized based on patient’s clinical response [46].
Prophylactic antibiotics can also be used to prevent infection, although there are no current published guidelines. Prophylactic antibiotics may be considered, particularly, if patients continue to have infections despite immunoglobulin replacement therapy at adequate doses. If a patient develops an infection, they require prompt treatment with antibiotics for a longer course, in some patients double the time, than a non-affected patient [7]. Monitoring for disease sequelae includes consideration of regular chest X-rays and sinus imaging [7]. Ideally, patient should receive a baseline pulmonary function test early on in diagnosis with periodic tests to follow lung function over time [7].
Agammaglobulinemia and Stem Cell Transplant
Stem cell transplant has been proposed in the past as a possible cure for XLA; however, there are many possible risks and complications associated with allogeneic stem cell transplant, which has limited this as an option, due to current accepted treatments available. There have been few cases reported of XLA patients receiving stem cell transplant with different conditioning regimens with varying results. The first 6 cases of XLA patients treated with allogeneic stem cell transplant, without utilization of a pre-conditioning regimen, did not result in engraftment in any of the 6 patients [47].
One case has been reported of a 13-year-old male with XLA and newly diagnosed acute myeloid leukemia (AML) who underwent allogeneic stem cell transplant with myeloablative conditioning and total body irradiation as treatment for his AML. One year after transplant, the patient had BTK gene analysis, which no longer showed his prior BTK mutation, and subsequent normal BTK expression in monocytes and B cells [48].
Another XLA patient, at 25 years of age, developed pre-B acute lymphocytic leukemia (ALL) with dominant negative TP53 mutation. As part of his treatment for ALL, he received myeloablative conditioning therapy, followed by allogeneic stem cell transplant. At 14 months post-transplant, the patient had normal circulating B cells; normal levels of immunoglobulins A, G, and M; and appropriate antibody response to vaccination with Prevnar-13 [49].
In another report, a 28 year-old-male with XLA and history of severe potentially fatal infections, despite IVIG replacement therapy, received allogeneic stem cell transplant with reduced intensity conditioning with successful immune reconstitution of CD19 + B cells, IgA and IgM, and increasing IgG levels after IVIG discontinuation [50].
Agammaglobulinemia and Lung Transplant
Bronchiectasis and chronic lung disease is a known complication of XLA which may progress to require lung transplantation. One series of 6 patients with XLA and end-stage bronchiectasis received lung transplantation in a single center. The results showed 4 out of the 6 patients developed repeated pulmonary sepsis and chronic lung allograft dysfunction in the long term [51]. This demonstrates the great risks associated with lung transplant in these patients.
Agammaglobulinemia and COVID-19
With the emergence of the Sars-CoV-2 pandemic in 2019, it stands to reason that immunodeficient patients would be of particular concern. There have been few cases reported of patients with agammaglobulinemia and CVID with COVID-19 infection. In a recent letter to the editor, Quinti et al. described 7 patients, one with XLA, one with autosomal recessive agammaglobulinemia, and five patients with CVID with COVID. The authors reported mild clinical courses in the two patients with agammaglobulinemia, and severe disease in the patients with CVID and one fatality [52]. In another case report, two patients with XLA in Italy developed interstitial pneumonia and lymphopenia secondary to SARS-CoV-2 infection, and both patients were able to recover without mechanical ventilation or intensive care treatment. The authors hypothesize that T cell response plays a more important role in fighting infection with SARS-CoV-2 than B cell response [53]. Another recent report of a 26-year-old male with XLA, currently off of IVIG for the past 2 years, developed COVID-19 infection and was treated with convalescent plasma, which resulted in recovery [54]. This patient presented with fever and chills for 1 week, and had labs remarkable for transaminitis, elevated lactate dehydrogenase, sedimentation rate (ESR), c-reactive protein (CRP), ferritin, and low d-dimer [54]. A 39-year-old male with XLA receiving monthly IVIG developed COVID-19 infection and had a complicated course of prolonged hospitalization, persistent fever, and bilateral pneumonia. Despite aggressive treatment with IVIG, hydroxychloroquine, azithromycin, and ceftriaxone, symptoms persisted. On the 23rd day of hospitalization, he received convalescent plasma and was afebrile after 24 h and negative for SARS-Cov-2 PCR at 48 h [55].
The evolution of SARS-Cov-2 infection to acute respiratory distress syndrome (ARDS) in many patients is thought to be secondary to a cytokine storm induced by the virus. Patients with COVID-19 have been shown to have elevation of inflammatory cytokines and chemokines, even more elevated in those patients requiring intensive care treatment. Another study of COVID-19 and patients with inborn errors of immunity was conducted via a survey of multiple immunodeficiency societies worldwide. Ninety-six patients with immunodeficiency were included in the study with 56% having primary antibody deficiency. Eight patients in the study had agammaglobulinemia (6 with XLA and two with AR agammaglobulinemia) and 29 patients with CVID. Findings showed many of the patients with COVID-19, and humoral immunodeficiency had mild or asymptomatic presentations. Eleven of the patients with CVID had mild disease presentation and four patients expired [56].
The hypothesis that has been proposed surrounding mild cases of COVID-19 with XLA patients is possible decrease in IL-6 production, therefore decreased chance of cytokine storm, in patients with BTK deficiency [56].
Another study of patients with primary immunodeficiency at Mount Sinai Hospital described their cohort of patients with immunodeficiency and clinical course with COVID-19 infection. Of the cohort, three patients had a diagnosis of XLA, 9 had CVID, one had hypogammaglobulinemia, and three had other primary immunodeficiencies. All the patients with XLA received convalescent plasma and recovered from the illness, and none necessitated mechanical ventilation. Two CVID patients and the one patient with hypogammaglobulinemia died. The patients with XLA had lower levels of inflammatory markers, including IL-6, compared to patients with CVID [57]. Early studies have shown BTK activation in monocytes is increased in severe cases of COVID-19 and utilization of BTK inhibitors reduces inflammatory markers [58]. In one study, use of the BTK inhibitor acalabrutinib in 19 patients with severe COVID infection showed decrease in inflammatory markers (Il-6, CRP) and decrease in oxygen requirement with extubation of 50% of patients on mechanical ventilation [58].
Conclusion/Summary
Congenital agammaglobulinemia results from a mutation in BTK about 85% of the time. Autosomal recessive forms of agammaglobulinemia have been reported, and mutations have been identified in genes encoding components of the pre-BCR. Mutations have been identified in genes encoding the mu heavy chain, Ig alpha and beta, BLNK, lambda 5, PIK3R1, PIK3CD, and LRRC8, as well as an autosomal dominant form of agammaglobulinemia from mutation in TCF3. Early identification and diagnosis of agammaglobulinemia is important in decreasing morbidity and mortality, as swift initiation of immunoglobulin replacement may help to prevent sequelae, such as life-threatening infection. The lack of phenotype and genotype correlation in BTK mutations make prediction of clinical course and identification of patients only mildly affected difficult. Diagnosis continues to be delayed in patients, even in those with family history of XLA. New research suggests newborn screening for B cell disorders may help in accelerating the diagnosis of this disorder involving measurements of KRECs, as is seen with TREC measurements, currently performed to rule out severe combined immunodeficiency on newborn screening in the USA. Furthermore, research in measurements of serum BCMA may aid in the management of primary antibody deficiencies by identifying severe cases that need immunoglobulin replacement immediately, including XLA. Management of agammaglobulinemia consists of life-long immunoglobulin replacement therapy. While widely used, there are no current guidelines regarding the use of prophylactic antibiotics. Further studies are needed to identify if prophylactic antibiotics in XLA have a role in improving infection prevention, when used concurrently with immunoglobulin replacement. A curative option continues to be needed for these patients. This would help to streamline the current practices among physicians managing these immunodeficiencies. Lastly, the current global pandemic with Sars-CoV-2 raised initial concerns about patients with primary immunodeficiencies. While more formal studies are needed, observations and case reports have shown several patients with XLA presenting with a milder form of COVID-19 when compared to patients with other primary immunodeficiencies.
Declarations
Conflict of Interest
Vivian Hernandez-Trujillo reports conflict with Ad board for Takeda and CSL; Speaker for Takeda; Consultant for Takeda. Melissa Cardenas-Morales does not have any conflicts of interest to disclose.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gaspar HB, Conley ME. Early B cell defects. Clin Exp Immunol. 2000;119(3):383–389. doi: 10.1046/j.1365-2249.2000.01192.x.PMID:10691907;PMCID:PMC1905597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, Coustan-Smith E, Howard V, Campana D. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227. doi: 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- 3.El-Sayed ZA, Abramova I, Aldave JC, Al-Herz W, Bezrodnik L, Boukari R, Bousfiha AA, Cancrini C, Condino-Neto A, Dbaibo G, Derfalvi B, Dogu F, Edgar JDM, Eley B, El-Owaidy RH, Espinosa-Padilla SE, Galal N, Haerynck F, Hanna-Wakim R, Hossny E, Ikinciogullari A, Kamal E, Kanegane H, Kechout N, Lau YL, Morio T, Moschese V, Neves JF, Ouederni M, Paganelli R, Paris K, Pignata C, Plebani A, Qamar FN, Qureshi S, Radhakrishnan N, Rezaei N, Rosario N, Routes J, Sanchez B, Sediva A, Seppanen MR, Serrano EG, Shcherbina A, Singh S, Siniah S, Spadaro G, Tang M, Vinet AM, Volokha A, Sullivan KE. X-linked agammaglobulinemia (XLA):Phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ J. 2019;12(3):100018. doi: 10.1016/j.waojou.2019.100018.PMID:30937141;PMCID:PMC6439403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hernandez-Trujillo, VP (2021) Agammaglobulinemia. In: UpToDate, Post, TW (Ed), UpToDate, Waltham, MA
- 5.Gemayel KT, Litman GW, Sriaroon P. Autosomal recessive agammaglobulinemia associated with an IGLL1 gene missense mutation. Ann Allergy Asthma Immunol. 2016;117(4):439–441. doi: 10.1016/j.anai.2016.07.038. [DOI] [PubMed] [Google Scholar]
- 6.Carrillo-Tapia E, García-García E, Herrera-González NE, Yamazaki-Nakashimada MA, Staines-Boone AT, Segura-Mendez NH, Scheffler-Mendoza SC, O Farrill-Romanillos P, Gonzalez-Serrano ME, Rodriguez-Alba JC, Santos-Argumedo L, Berron-Ruiz L, Sanchez-Flores A, López-Herrera G (2018) Delayed diagnosis in X-linked agammaglobulinemia and its relationship to the occurrence of mutations in BTK non-kinase domains. Expert Rev Clin Immunol 14(1):83–93. 10.1080/1744666X.2018.1413349. Epub 2017 Dec 11. PMID: 29202590 [DOI] [PubMed]
- 7.Smith CIE, Berglöf A. X-Linked Agammaglobulinemia (2001) [Updated 2016 Aug 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021
- 8.Plebani A, Soresina A, Rondelli R, Amato GM, Azzari C, Cardinale F, Cazzola G, Consolini R, De Mattia D, Dell'Erba G, Duse M, Fiorini M, Martino S, Martire B, Masi M, Monafo V, Moschese V, Notarangelo LD, Orlandi P, Panei P, Pession A, Pietrogrande MC, Pignata C, Quinti I, Ragno V, Rossi P, Sciotto A (2002) Stabile A; Italian Pediatric Group for XLA-AIEOP. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol 104(3):221–30. 10.1006/clim.2002.5241. PMID: 12217331 [DOI] [PubMed]
- 9.Lougaris V, Soresina A, Baronio M, Montin D, Martino S, Signa S, Volpi S, Zecca M, Marinoni M, Baselli LA, Dellepiane RM, Carrabba M, Fabio G, Putti MC, Cinetto F, Lunardi C, Gazzurelli L, Benvenuto A, Bertolini P, Conti F, Consolini R, Ricci S, Azzari C, Leonardi L, Duse M, Pulvirenti F, Milito C, Quinti I, Cancrini C, Finocchi A, Moschese V, Cirillo E, Crescenzi L, Spadaro G, Marasco C, Vacca A, Cardinale F, Martire B, Trizzino A, Licciardello M, Cossu F, Di Matteo G, Badolato R, Ferrari S, Giliani S, Pession A, Ugazio A, Pignata C, Plebani A. Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J Allergy Clin Immunol. 2020;146(2):429–437. doi: 10.1016/j.jaci.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Tóth B, Volokha A, Mihas A, Pac M, Bernatowska E, Kondratenko I, Polyakov A, Erdos M, Pasic S, Bataneant M, Szaflarska A, Mironska K, Richter D, Stavrik K, Avcin T, Márton G, Nagy K, Dérfalvi B, Szolnoky M, Kalmár A, Belevtsev M, Guseva M, Rugina A, Kriván G, Timár L, Nyul Z, Mosdósi B, Kareva L, Peova S, Chernyshova L, Gherghina I, Serban M, Conley ME, Notarangelo LD, Smith CI, van Dongen J, van der Burg M, Maródi L. Genetic and demographic features of X-linked agammaglobulinemia in Eastern and Central Europe: a cohort study. Mol Immunol. 2009;46(10):2140–2146. doi: 10.1016/j.molimm.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, Conley ME, Cunningham-Rundles C, Ochs HD. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore) 2006;85(4):193–202. doi: 10.1097/01.md.0000229482.27398.ad. [DOI] [PubMed] [Google Scholar]
- 12.Chen XF, Wang WF, Zhang YD, Zhao W, Wu J, Chen TX. Clinical characteristics and genetic profiles of 174 patients with X-linked agammaglobulinemia: Report from Shanghai, China (2000–2015) Medicine (Baltimore) 2016;95(32):e4544. doi: 10.1097/MD.0000000000004544.PMID:27512878;PMCID:PMC4985333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groth D, Wright H, Marsh R, Fuleihan R, Cunningham-Rundles C, Sullivan K, Feuille E (2020) X-linked agammaglobulinemia: infection frequencies in 226 patients from the USIDNET Registry. J Allergy Clin Immunol 145(2):AB80. ISSN 0091–6749. 10.1016/j.jaci.2019.12.676. (https://www.sciencedirect.com/science/article/pii/S0091674919323784)
- 14.López-Granados E, Pérez de Diego R, Ferreira Cerdán A, Fontán Casariego G, García Rodríguez MC (2005) A genotype-phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia. J Allergy Clin Immunol 116(3):690–7. 10.1016/j.jaci.2005.04.043. PMID: 16159644 [DOI] [PubMed]
- 15.Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr. 2002;141(4):566–571. doi: 10.1067/mpd.2002.127711. [DOI] [PubMed] [Google Scholar]
- 16.Tao L, Boyd M, Gonye G, Malone B, Schwaber J. BTK mutations in patients with X-linked agammaglobulinemia: lack of correlation between presence of peripheral B lymphocytes and specific mutations. Hum Mutat. 2000;16(6):528–529. doi: 10.1002/1098-1004(200012)16:6<528::AID-HUMU12>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 17.Barmettler S, Otani IM, Minhas J, Abraham RS, Chang Y, Dorsey MJ, Ballas ZK, Bonilla FA, Ochs HD, Walter JE (2017 ) Gastrointestinal manifestations in X-linked agammaglobulinemia. J Clin Immunol 37(3):287–294. 10.1007/s10875-017-0374-x. Epub 2017 Feb 24. PMID: 28236219; PMCID: PMC5414010 [DOI] [PMC free article] [PubMed]
- 18.Singh S, Rawat A, Suri D, Gupta A, Garg R, Saikia B, Minz RW, Sehgal S, Chan KW, Lau YL, Kamae C, Honma K, Nakagawa N, Imai K, Nonoyama S, Oshima K, Mitsuiki N, Ohara O. X-linked agammaglobulinemia: twenty years of single-center experience from North West India. Ann Allergy Asthma Immunol. 2016;117(4):405–411. doi: 10.1016/j.anai.2016.07.044. [DOI] [PubMed] [Google Scholar]
- 19.Conley ME, Broides A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T, Shurtleff SA. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005;203:216–234. doi: 10.1111/j.0105-2896.2005.00233.x. [DOI] [PubMed] [Google Scholar]
- 20.Smith T, Cunningham-Rundles C (2019) Primary B-cell immunodeficiencies. Hum Immunol 80(6):351–362. 10.1016/j.humimm.2018.10.015. Epub 2018 Oct 22. PMID: 30359632; PMCID: PMC7395616 [DOI] [PMC free article] [PubMed]
- 21.Silva P, Justicia A, Regueiro A, Fariña S, Couselo JM, Loidi L. Autosomal recessive agammaglobulinemia due to defect in μ heavy chain caused by a novel mutation in the IGHM gene. Genes Immun. 2017;18(3):197–199. doi: 10.1038/gene.2017.14. [DOI] [PubMed] [Google Scholar]
- 22.Minegishi Y, Coustan-Smith E, Wang YH, Cooper MD, Campana D, Conley ME (1998) Mutations in the human lambda5/14.1 gene result in B cell deficiency and agammaglobulinemia. J Exp Med 187(1):71–77. 10.1084/jem.187.1.71 [DOI] [PMC free article] [PubMed]
- 23.Minegishi Y, Coustan-Smith E, Rapalus L, Ersoy F, Campana D, Conley ME. Mutations in Igalpha (CD79a) result in a complete block in B-cell development. J Clin Invest. 1999;104(8):1115–1121. doi: 10.1172/JCI7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobbs AK, Yang T, Farmer D, Kager L, Parolini O, Conley ME. Cutting edge: a hypomorphic mutation in Igbeta (CD79b) in a patient with immunodeficiency and a leaky defect in B cell development. J Immunol. 2007;179(4):2055–2059. doi: 10.4049/jimmunol.179.4.2055. [DOI] [PubMed] [Google Scholar]
- 25.Khalili A, Plebani A, Vitali M, Abolhassani H, Lougaris V, Mirminachi B, Rezaei N, Aghamohammadi A. Autosomal recessive agammaglobulinemia: a novel non-sense mutation in CD79a. J Clin Immunol. 2014;34(2):138–141. doi: 10.1007/s10875-014-9989-3. [DOI] [PubMed] [Google Scholar]
- 26.Langereis JD, Henriet SS, Kuipers S, Weemaes CMR, van der Burg M, de Jonge MI, van der Flier M (2018) IgM augments complement bactericidal activity with serum from a patient with a novel CD79a mutation. J Clin Immunol 38(2):185–192. 10.1007/s10875-017-0474-7. Epub 2018 Jan 15. PMID: 29335801; PMCID: PMC5840230 [DOI] [PMC free article] [PubMed]
- 27.Ferrari S, Lougaris V, Caraffi S, et al. Mutations of the Igbeta gene cause agammaglobulinemia in man. J Exp Med. 2007;204(9):2047–2051. doi: 10.1084/jem.20070264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lougaris V, Vitali M, Baronio M, Moratto D, Tampella G, Biasini A, Badolato R, Plebani A. Autosomal recessive agammaglobulinemia: the third case of Igβ deficiency due to a novel non-sense mutation. J Clin Immunol. 2014;34(4):425–427. doi: 10.1007/s10875-014-0033-4. [DOI] [PubMed] [Google Scholar]
- 29.Minegishi Y, Rohrer J, Coustan-Smith E, Lederman HM, Pappu R, Campana D, Chan AC, Conley ME. An essential role for BLNK in human B cell development. Science. 1999;286:1954–1957. doi: 10.1126/science.286.5446.1954. [DOI] [PubMed] [Google Scholar]
- 30.Lagresle-Peyrou C, Millili M, Luce S, Boned A, Sadek H, Rouiller J, Frange P, Cros G, Cavazzana M, André-Schmutz I, Schiff C. The BLNK adaptor protein has a nonredundant role in human B-cell differentiation. J Allergy Clin Immunol. 2014;134(1):145–154. doi: 10.1016/j.jaci.2013.12.1083. [DOI] [PubMed] [Google Scholar]
- 31.van Zelm MC, Geertsema C, Nieuwenhuis N, de Ridder D, Conley ME, et al. Gross deletions involving IGHM, BTK, or Artemis: a model for genomic lesions mediated by transposable elements. Am J Hum Genet. 2008;82:320–332. doi: 10.1016/j.ajhg.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang P, Upton JEM, Barton-Forbes MA, Salvadori MI, Clynick MP, Price AK, Goobie SL. Autosomal recessive agammaglobulinemia due to a homozygous mutation in PIK3R1. J Clin Immunol. 2018;38(1):88–95. doi: 10.1007/s10875-017-0462-y. [DOI] [PubMed] [Google Scholar]
- 33.Nunes-Santos CJ, Uzel G, Rosenzweig SD. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol. 2019;143(5):1676–1687. doi: 10.1016/j.jaci.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 34.Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, Smith AM, Perez EE, Murray PJ (2012) Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med 209(3):463–70. 10.1084/jem.20112533. Epub 2012 Feb 20. PMID: 22351933; PMCID: PMC3302225 [DOI] [PMC free article] [PubMed]
- 35.Anzilotti C, Swan DJ, Boisson B, Deobagkar-Lele M, Oliveira C, Chabosseau P, Engelhardt KR, Xu X, Chen R, Alvarez L, Berlinguer-Palmini R, Bull KR, Cawthorne E, Cribbs AP, Crockford TL, Dang TS, Fearn A, Fenech EJ, de Jong SJ, Lagerholm BC, Ma CS, Sims D, van den Berg B, Xu Y, Cant AJ, Kleiner G, Leahy TR, de la Morena MT, Puck JM, Shapiro RS, van der Burg M, Chapman JR, Christianson JC, Davies B, McGrath JA, Przyborski S, Santibanez Koref M, Tangye SG, Werner A, Rutter GA, Padilla-Parra S, Casanova JL, Cornall RJ, Conley ME, Hambleton S (2019) An essential role for the Zn2+ transporter ZIP7 in B cell development. Nat Immunol 20(3):350–361. 10.1038/s41590-018-0295-8. Epub 2019 Feb 4. PMID: 30718914; PMCID: PMC6561116. [DOI] [PMC free article] [PubMed]
- 36.Sawada A, Takihara Y, Kim JY, Matsuda-Hashii Y, Tokimasa S, Fujisaki H, Kubota K, Endo H, Onodera T, Ohta H, Ozono K, Hara J. A congenital mutation of the novel gene LRRC8 causes agammaglobulinemia in humans. J Clin Invest. 2003;112(11):1707–1713. doi: 10.1172/JCI18937.PMID:14660746;PMCID:PMC281644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broderick L, Yost S, Li D, et al. Mutations in topoisomerase IIβ result in a B cell immunodeficiency. Nat Commun. 2019;10:3644. doi: 10.1038/s41467-019-11570-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boisson B, Wang YD, Bosompem A, Ma CS, Lim A, Kochetkov T, Tangye SG, Casanova JL, Conley ME. A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR(-) B cells. J Clin Invest. 2013;123(11):4781–4785. doi: 10.1172/JCI71927.PMID:24216514;PMCID:PMC3809807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qureshi S, Sheikh MDA, Qamar FN. Autosomal recessive agammaglobulinemia—first case with a novel TCF3 mutation from Pakistan. Clin Immunol. 2019;198:100–101. doi: 10.1016/j.clim.2018.07.016. [DOI] [PubMed] [Google Scholar]
- 40.Bearden D, Collett M, Quan PL, Costa-Carvalho BT, Sullivan KE (2016) Enteroviruses in X-linked agammaglobulinemia: update on epidemiology and therapy. J Allergy Clin Immunol Pract 4(6):1059–1065. 10.1016/j.jaip.2015.12.015. Epub 2016 Feb 13. PMID: 26883540 [DOI] [PubMed]
- 41.Hernandez-Trujillo VP, Scalchunes C, Cunningham-Rundles C, et al. Autoimmunity and inflammation in X-linked agammaglobulinemia. J Clin Immunol. 2014;34(6):627–632. doi: 10.1007/s10875-014-0056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borte S, von Döbeln U, Fasth A, Wang N, Janzi M, Winiarski J, Sack U, Pan-Hammarström Q, Borte M, Hammarström L. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood. 2012;119(11):2552–2555. doi: 10.1182/blood-2011-08-371021. [DOI] [PubMed] [Google Scholar]
- 43.King J, Borte S, Brodszki N, von Döbeln U, Smith CIE, Hammarström L. Kappa-deleting recombination excision circle levels remain low or undetectable throughout life in patients with X-linked agammaglobulinemia. Pediatr Allergy Immunol. 2018;29(4):453–456. doi: 10.1111/pai.12893. [DOI] [PubMed] [Google Scholar]
- 44.Nakagawa N, Imai K, Kanegane H, Sato H, Yamada M, Kondoh K, Okada S, Kobayashi M, Agematsu K, Takada H, Mitsuiki N, Oshima K, Ohara O, Suri D, Rawat A, Singh S, Pan-Hammarström Q, Hammarström L, Reichenbach J, Seger R, Ariga T, Hara T, Miyawaki T, Nonoyama S. Quantification of κ-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J Allergy Clin Immunol. 2011;128(1):223–225.e2. doi: 10.1016/j.jaci.2011.01.052. [DOI] [PubMed] [Google Scholar]
- 45.Maglione PJ, Ko HM, Tokuyama M, Gyimesi G, Soof C, Li M, Sanchez E, Chen H, Radigan L, Berenson J, Cunningham-Rundles C (2020) Serum B-cell maturation antigen (BCMA) levels differentiate primary antibody deficiencies. J Allergy Clin Immunol Pract 8(1):283–291.e1. 10.1016/j.jaip.2019.08.012. Epub 2019 Aug 17. PMID: 31430592; PMCID: PMC6980522 [DOI] [PMC free article] [PubMed]
- 46.Wasserman RL. Personalized therapy: immunoglobulin replacement for antibody deficiency. Immunol Allergy Clin North Am. 2019;39(1):95–111. doi: 10.1016/j.iac.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Howard V, Myers LA, Williams DA, Wheeler G, Turner EV, Cunningham JM, Conley ME. Stem cell transplants for patients with X-linked agammaglobulinemia. Clin Immunol. 2003;107(2):98–102. doi: 10.1016/s1521-6616(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 48.Abu-Arja RF, Chernin LR, Abusin G, Auletta J, Cabral L, Egler R, Ochs HD, Torgerson TR, Lopez-Guisa J, Hostoffer RW, Tcheurekdjian H, Cooke KR (2015) Successful hematopoietic cell transplantation in a patient with X-linked agammaglobulinemia and acute myeloid leukemia. Pediatr Blood Cancer 62(9):1674–6. doi: 10.1002/pbc.25554. Epub 2015 Apr 20. PMID: 25900577; PMCID: PMC4876715 [DOI] [PMC free article] [PubMed]
- 49.van Zelm MC, Pumar M, Shuttleworth P, Aui PM, Smart JM, Grigg A, Bosco JJ. Functional antibody responses following allogeneic stem cell transplantation for TP53 mutant pre-B-ALL in a patient with X-linked agammaglobulinemia. Front Immunol. 2019;26(10):895. doi: 10.3389/fimmu.2019.00895.PMID:31105705;PMCID:PMC6498405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikegame K, Imai K, Yamashita M, Hoshino A, Kanegane H, Morio T, Kaida K, Inoue T, Soma T, Tamaki H, Okada M, Ogawa H. Allogeneic stem cell transplantation for X-linked agammaglobulinemia using reduced intensity conditioning as a model of the reconstitution of humoral immunity. J Hematol Oncol. 2016;13(9):9. doi: 10.1186/s13045-016-0240-y.PMID:26873735;PMCID:PMC4752762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barnes S, Kotecha S, Douglass JA, Paul E, Hore-Lacy F, Stirling R, Snell GI, Westall GP (2015) Evolving practice: X-linked agammaglobulinemia and lung transplantation. Am J Transplant 15(4):1110–1113. 10.1111/ajt.13084. Epub 2015 Mar 3. Erratum in: Am J Transplant 15(8):2278. Hore-Lacey, F [corrected to Hore-Lacy, F]. PMID: 25736826 [DOI] [PubMed]
- 52.Quinti I, Lougaris V, Milito C, Cinetto F, Pecoraro A, Mezzaroma I, Mastroianni CM, Turriziani O, Bondioni MP, Filippini M, Soresina A, Spadaro G, Agostini C, Carsetti R, Plebani A (2020) A possible role for B cells in COVID-19? Lesson from patients with agammaglobulinemia. J Allergy Clin Immunol 146(1):211–213.e4. 10.1016/j.jaci.2020.04.013. Epub 2020 Apr 22. PMID: 32333914; PMCID: PMC7175894 [DOI] [PMC free article] [PubMed]
- 53.Soresina A, Moratto D, Chiarini M, et al. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr Allergy Immunol. 2020;31(5):565–569. doi: 10.1111/pai.13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hovey JG, Tolbert D, Howell D (2020) Bruton's Agammaglobulinemia and COVID-19. Cureus 12(11):e11701. Published 2020 Nov 25. 10.7759/cureus.11701 [DOI] [PMC free article] [PubMed]
- 55.Mira E, Yarce OA, Ortega C, et al. Rapid recovery of a SARS-CoV-2-infected X-linked agammaglobulinemia patient after infusion of COVID-19 convalescent plasma. J Allergy Clin Immunol Pract. 2020;8(8):2793–2795. doi: 10.1016/j.jaip.2020.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyts I, Bucciol G, Quinti I, Neven B, Fischer A, Seoane E, Lopez-Granados E, Gianelli C, Robles-Marhuenda A, Jeandel PY, Paillard C, Sankaran VG, Demirdag YY, Lougaris V, Aiuti A, Plebani A, Milito C, Dalm VA, Guevara-Hoyer K, Sánchez-Ramón S, Bezrodnik L, Barzaghi F, Gonzalez-Granado LI, Hayman GR, Uzel G, Mendonça LO, Agostini C, Spadaro G, Badolato R, Soresina A, Vermeulen F, Bosteels C, Lambrecht BN, Keller M, Mustillo PJ, Abraham RS, Gupta S, Ozen A, Karakoc-Aydiner E, Baris S, Freeman AF, Yamazaki-Nakashimada M, Scheffler-Mendoza S, Espinosa-Padilla S, Gennery AR, Jolles S, Espinosa Y, Poli MC, Fieschi C, Hauck F, Cunningham-Rundles C, Mahlaoui N; IUIS Committee of Inborn Errors of Immunity, Warnatz K, Sullivan KE, Tangye SG (2021) Coronavirus disease 2019 in patients with inborn errors of immunity: An international study. J Allergy Clin Immunol 147(2):520–531. 10.1016/j.jaci.2020.09.010. Epub 2020 Sep 24. PMID: 32980424; PMCID: PMC7832563 [DOI] [PMC free article] [PubMed]
- 57.Ho HE, Mathew S, Peluso MJ, Cunningham-Rundles C (2021) Clinical outcomes and features of COVID-19 in patients with primary immunodeficiencies in New York City. J Allergy Clin Immunol Pract 9(1):490–493.e2. 10.1016/j.jaip.2020.09.052. Epub 2020 Oct 8. PMID: 33039649; PMCID: PMC7543763 [DOI] [PMC free article] [PubMed]
- 58.Roschewski M, Lionakis MS, Sharman JP et al (2020) Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci Immunol 5(48):eabd0110. 10.1126/sciimmunol.abd0110 [DOI] [PMC free article] [PubMed]