

SUMMARY
The bioactive sphingolipid metabolites ceramide and sphingosine-1-phosphate (S1P) are a recent addition to the lipids accumulated in obesity and have emerged as important molecular players in metabolic diseases. Here we summarize evidence that dysregulation of sphingolipid metabolism correlates with pathogenesis of metabolic diseases in humans. This review discusses the current understanding of how ceramide regulates signaling and metabolic pathways to exacerbate metabolic diseases and the Janus faces for its further metabolite S1P, the kinases that produce it, and the multifaceted and at times opposing actions of S1P receptors in various tissues. Gaps and limitations in current knowledge are highlighted together with the need to further decipher the full array of their actions in tissue dysfunction underlying metabolic pathologies, pointing out prospects to move this young field of research toward the development of effective therapeutics.
Sphingolipid metabolites, particularly ceramide and sphingosine-1-phosphate, have emerged as important mediators in the development of metabolic diseases. In this review, Green et al. discuss the multifacted, tissue-specific roles of these sphingolipid metabolites and how their dysregulation contributes to the pathogenesis of metabolic diseases.
INTRODUCTION
Obesity is one of the most threatening health scourges of our time, increasing the risk of numerous diseases and complicating many others. There are currently nearly 1000 FDA registered clinical trials investigating pharmacological, surgical, and / or behavioral interventions for obesity and its complications. Writers from antiquity viewed obesity as an undesirable condition, and its association with disease has long been recognized. It is remarkable that despite tremendous research effort, the physiologic and molecular mechanisms by which excess adiposity promotes disease remain murky. Obesity increases the risk for development of numerous metabolic diseases, including type 2 diabetes (T2D), liver disease, and cardiovascular diseases (CVD). We urgently need a better understanding of the physiological and molecular mechanisms linking obesity to metabolic imbalance, and thus, to metabolic diseases and their consequences. This is undoubtedly a multi-factorial process involving myriad endogenous and exogenous factors, and the complex interplay between many tissues including adipose, skeletal muscle, liver, pancreas, vasculature, and others. Research over the past decade has shed light on roles for alterations in lipid metabolism and, more recently, bioactive sphingolipids, in linking obesity to the development of metabolic diseases. In this review, we discuss the roles of the bioactive sphingolipid metabolites ceramide and sphingosine-1-phosphate (S1P) in metabolic regulation of the pathological sequelae of obesity. The advent of new technologies to measure these sphingolipid metabolites in cells and tissues has revealed significant alterations that correlate with pathological conditions. We also highlight new mechanistic insights gained from in vivo studies on modulation of the synthesis and degradation of ceramide and S1P that begin to define them as important mediators in metabolic diseases.
BASICS OF SPHINGOLIPID METABOLISM
Sphingolipids are ubiquitous and essential components of all eukaryotic membranes (Hannun and Obeid, 2018). Their biosynthesis at the endoplasmic reticulum begins with condensation of an amino acid, most commonly serine, with fatty acids, typically palmitate, activated by reaction with CoA (fatty acyl-CoA), and catalyzed by serine palmitoyl transferase (SPT), an enzyme complex composed of two large subunits, encoded by Sptlc1 and either Sptlc2 or −3, and a small regulatory subunit (Figure 1). A family of 3 ORMDL proteins sense ceramide levels and feed-back inhibit SPT to regulate nutrients entering the sphingolipid pool. The transient intermediate, 3-ketodihydrosphingosine, is rapidly reduced to dihydrosphingosine that is then N-acylated by one of six ceramide synthases (CerS1–6) with a saturated or monounsaturated fatty acid of 14–26 carbons, forming dihydroceramides. Dihydroceramide desaturase (DES1) then introduces a 4–5 trans double bond to yield ceramide, a bioactive metabolic signaling lipid that accumulates in obesity (Chaurasia et al., 2019). Ceramide is the central component of all complex sphingolipids including sphingomyelin and glycosphingolipids, which are generated in the Golgi. Internalized membrane complex sphingolipids reach the lysosomal compartment, where they are sequentially degraded by acidic hydrolases to ceramide and then by ceramidase to sphingosine. Sphingosine can be recycled in the salvage pathway to ceramide or phosphorylated by sphingosine kinases (SphK1 and SphK2) to form sphingosine-1-phosphate (S1P). S1P can then be irreversibly cleaved by ER-localized S1P lyase (SGPL1) in the only exit pathway for total sphingolipid degradation, or dephosphorylated by S1P phosphatases for further reutilization of sphingosine to ceramide. S1P can also be exported out of cells by specific transporters to activate five specific G protein-coupled receptors (GPCRs), termed S1PR1–5, that mediate many of its known functions (Spiegel and Milstien, 2011). Note that generation of sphingosine occurs only through the degradation of ceramide, which has important ramifications as, similar to ceramide, sphingosine and S1P are potent signaling molecules in their own right. Indeed, as described below, ceramide and S1P predominantly have opposing effects, making the ratio of these two molecules crucial to understanding their pathological roles, and making the enzymes that interconvert these two molecules critical for the regulation of cell fate. Moreover, many diseases including obesity and T2D perturb biosynthesis and tissue levels of both ceramides and S1P, and these sphingolipids play multiple roles in the context of various metabolic diseases.
Figure 1. Spatial distribution and compartmentalization of sphingolipid metabolism.
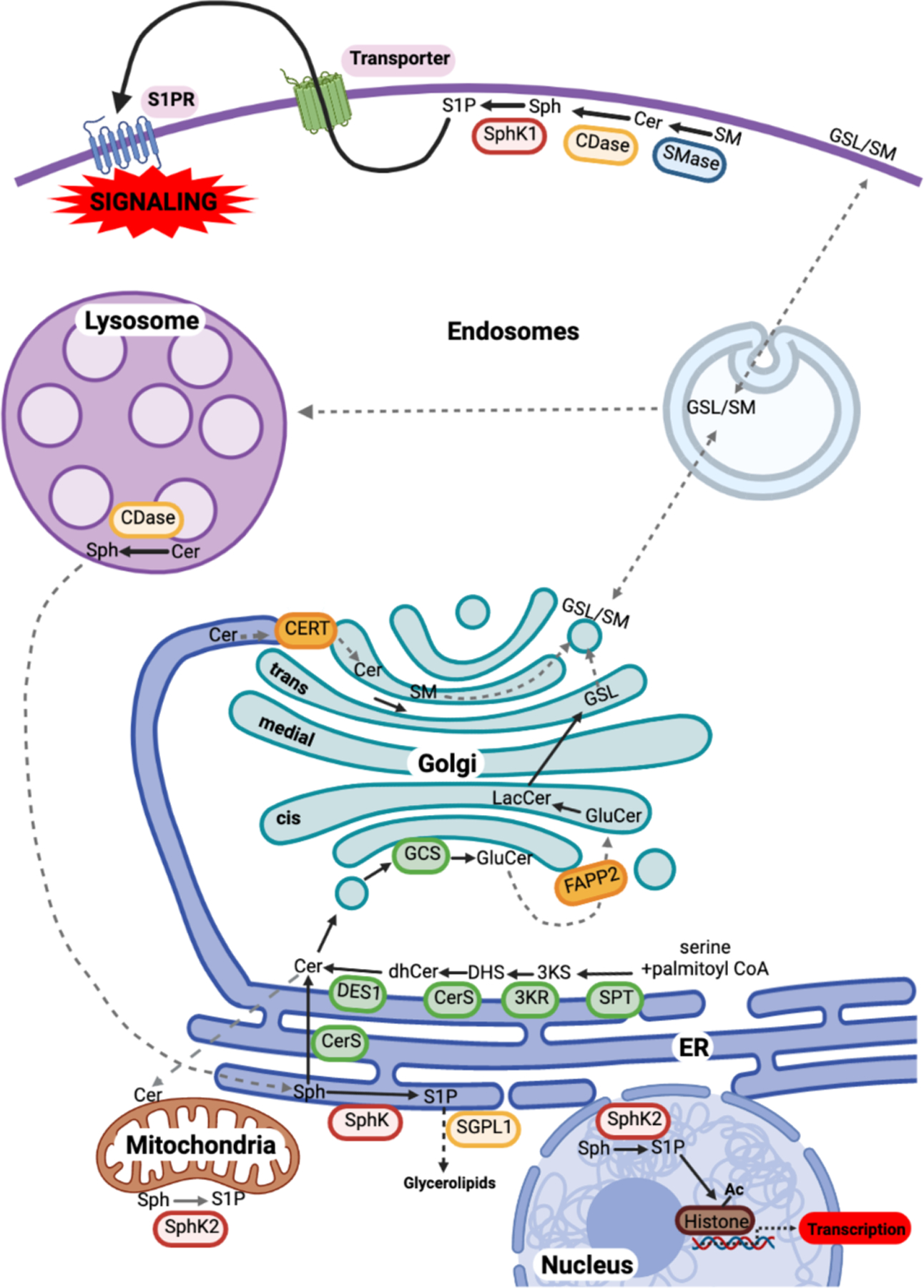
De novo sphingolipid synthesis begins at the ER as described in the text, leading to the formation of ceramide (Cer). Cer is transported to the Golgi either by the ceramide transfer protein CERT at ER-trans-Golgi contact sites for the formation of sphingomyelin (SM), or by vesicular transport to the cis-Golgi. There, Cer is glucosylated to GluCer, which is then trafficked further in the Golgi via FAPP2 to form LacCer, and sequentially glycosylated to form complex glycosphingolipids (GSLs). Cer may also be translocated to mitochondria at membrane contact sites with the ER. At the plasma membrane, in a signal-mediated process, sphingomyelinase (SMase), ceramidase (CDase), and sphingosine kinases (SphK) produce the bioactive metabolites Cer, sphingosine (Sph) and sphingosine-1-phosphate (S1P), respectively. S1P is then transported outside of cells and acts in a paracrine or autocrine fashion via S1P receptors (S1PRs) to initiate myriad signaling pathways. Plasma membrane sphingolipids are internalized by the endocytic pathway to the lysosome for degradation to Sph, which can be trafficked by unknown mechanisms to the ER. In the ER, Sph can either be recycled back to Cer for reutilization or degraded after phosphorylation by SphKs and cleavage by S1P lyase (SGPL1). S1P produced by SphK2 in mitochondria interacts with the electron transport chain, and in the nucleus, it regulates histone acetylation.
Color code: Red box, enzymes; orange box, lipid binding and transport proteins; blue arrows, vesicular transport steps; red arrows, protein-mediated lipid transport step; dashed arrows, unknown transport step(s).
LIPOTOXICITY AS A LINK BETWEEN OBESITY AND DISEASE
A combination of events can arise in obesity leading to metabolic syndrome, T2D and other pathological conditions, including chronic inflammation, lipotoxicity, endocrine perturbation, and modifications to gut microbiota. How obesity precipitates these, however, is not completely understood.
A role for adipose tissue expansion has emerged as a process critical to metabolic homeostasis (Kusminski et al., 2016). However, the capacity for adipocyte lipid storage is not infinite, and therefore excess energy consumption can push adipocytes beyond their potential for lipid storage. This can instigate inflammation, hypoxia, and fibrosis, processes that derail critical endocrine and lipolytic functions of adipose tissue (Crewe et al., 2017). Thus, dysfunction of adipose tissue in obese humans is associated with increased risk for diseases, such as T2D, dyslipidemia, nonalcoholic fatty liver disease (NAFLD), hypertension, and CVD (Kusminski et al., 2016). However, pathways driving healthy vs. unhealthy adipose tissue expansion remain to be unequivocally identified.
As the primary function of insulin in adipocytes is to suppress lipolysis, insulin resistant adipocytes continually release free or non-esterified fatty acids (FFA) into the circulation, where they in turn overwhelm muscle, liver, heart, and other peripheral organs and tissues. However, viewing obesity as a direct cause of insulin resistance via increased circulating FFA may be an oversimplification. Nevertheless, T2D patients indisputably display continually elevated FFA. Additionally, T2D gives rise to numerous disruptions in lipid and lipoprotein handling including the elevation of circulating triacylglycerols, leading to oversupply of FFA in organs expressing endothelial lipoprotein lipase, including heart and skeletal muscle (Wang et al., 2017b). The surfeit of FFA increases mitochondrial β-oxidation and subsequent oxidative stress and increases storage as triacylglycerols in lipid droplets. Moreover, increased mitochondrial damage may reduce the capacity to clear FFA via β-oxidation, leading to a vicious cycle of intracellular lipid accumulation. Lipotoxicity may also arise from cell-autonomous perturbances to FFA metabolism. For example, in certain pathophysiological conditions, e.g. hypoxia, FFA oxidation is also attenuated, shunting FFA into the aberrant production of lipid mediators including ceramides and diacylcglycerols (Petersen and Shulman, 2017). These deleterious lipid metabolites regulate intracellular pathways that impact insulin action, inflammation and metabolism. Palmitate, the most abundant circulating saturated fatty acid in humans, increases production of ceramide as it is a substrate for both SPT and CerS (Figure 1), and upregulates transcription of important sphingolipid biosynthetic enzymes including DES1 (Hu et al., 2011) and SphK1 (Ross et al., 2013). Because of the abundant changes that occur in metabolic diseases, it is unlikely that the mechanisms by which sphingolipid metabolism is perturbed arise merely directly from fatty acid oversupply; oxidative stress, inflammatory signaling, endocrine signaling, dysbiosis, and other routes likely contribute in ways yet to be discovered.
ELEVATED CERAMIDES AS A CAUSE OF METABOLIC DISEASES?
Increased ceramide correlates with pathogenesis of metabolic diseases
Profiling studies in high fat-fed mice, ob/ob mice, and lipid-infused rats, as well as in other animal models of metabolic syndrome support a correlation between increased ceramide and obesity and/or insulin resistance (Frangioudakis et al., 2010; Holland et al., 2007). Unbiased analyses of large metabolomics and lipidomics data sets from patients have also indicated associations between serum and tissue levels of ceramides and/or dihydroceramides and comorbidities of obesity, steatosis, T2D, non-alcoholic steatohepatitis (NASH), and major adverse cardiac events (Anroedh et al., 2018; Havulinna et al., 2016; Lemaitre et al., 2018; Luukkonen et al., 2016; Wigger et al., 2017). The importance of ceramide in human skeletal muscle was initially controversial due to conflicting reports on whether obesity and/or T2D increase intramuscular ceramides, and whether interventions that improve insulin sensitivity (e.g., diet-induced weight loss, exercise, etc.) also lower ceramides (Petersen and Jurczak, 2016; Skovbro et al., 2008). However, recent evidence points to subcellular localization and/or specific pools of distinct species of ceramide, rather than their total mass per se, in disease processes. For example, ceramides in the mitochondria, ER, and nucleus were inversely correlated with insulin signaling while lipids in the cytosolic fraction showed no relationship (Perreault et al., 2018). Moreover, mass spectrometry technological advances since the 1990s have enabled a more complex view of ceramide as not one bulk substance but rather a family of chemically and biologically distinct ceramide species. Particular insight has been gained from comparison and contrast of long chain (C16–C20) vs. very long chain (C22–26) ceramides, which are produced by acyl chain-specific ceramide synthases, CerS1–6. The ceramides that most tightly correlate with insulin resistance and hepatic steatosis are long chain species, C16:0 or C18:0 ceramides produced predominantly by CerS6 in adipose and liver tissue (Turpin et al., 2014) and CerS1 in muscle (Bergman et al., 2016). Therefore, some of the conflicting conclusions might be resolved by taking into account subcellular localization and/or specific ceramide species. Together, these studies make a strong case that long chain vs. very long chain ceramides serve distinct functions; however, it is also possible that characteristics of CerS beyond their catalytic output, such as subcellular localization, induction under high fat feeding, etc., may also contribute to the different biological effects observed.
Intriguingly, circulating C16, C18, and C24:1 ceramides are not only associated with heart failure, but are also predictive of major adverse cardiac events (Havulinna et al., 2016; Wang et al., 2017a). Patients with heart failure with preserved ejection fraction, a condition strongly correlated with diabetes, demonstrated specific elevations in circulating C16:0 ceramide (Javaheri et al., 2020). Moreover, the ratio of C16:0/C24:0 ceramides in circulation from the Framingham Heart study was predictive of heart failure (Nwabuo et al., 2019). This ceramide ratio also provides predictive information about CVD and mortality in the general population years prior to the onset of disease and may reflect sphingolipid remodeling in cardiac tissues (Peterson et al., 2018). Diastolic relaxation in T2D patients treated with fenofibrate also correlated with this ratio (Peterson et al., 2020). The connection between plasma ceramides and metabolic dysfunction and associated underlying CVD suggests that ceramide could be a useful diagnostic biomarker of adverse cardiac incident risks (Havulinna et al., 2016). These studies also suggest that specific ceramide species likely regulate distinct (patho)physiological processes. Interventions that decrease the C16:0/C24:0 ceramide ratio might lead to better disease outcomes.
In addition to serving as biomarkers, experimental evidence suggests that specific ceramides also play mechanistic roles in heart pathophysiology in diabetes, including diabetic cardiomyopathy. Studies in both genetic and diet-based mouse models of liptoxic cardiomyopathy have shown that inhibiting sphingolipid biosynthesis, either genetically and/or pharmacologically, attenuates cardiac hypertrophy and loss of function (Park et al., 2008; Russo et al., 2012). Moreover, altering sphingolipid metabolism by overexpression of acid ceramidase attenuated cell death and inflammatory response after myocardial infarction (Hadas et al., 2020). Further mechanistic work suggested that distinct CerS isoforms mediated distinct outcomes, with macroautophagy stimulated by CerS5, and mitochondrial damage caused by upregulation of CerS2 (Law et al., 2018). Translational significance of these studies in cell culture and animal models gain support from human studies demonstrating altered regulation of SPT subunits and CerS, coupled to concomitant alterations in ceramide in humans with heart failure (Ji et al., 2017). This is further supported by studies showing that these changes are reversed upon unloading in patients after placement of a left-ventricular assist device (Chokshi et al., 2012). The reader is also referred to excellent recent reviews on the contribution of sphingolipid metabolites to CVD associated with obesity (Kovilakath et al., 2020; Summers, 2018).
While the roles of ceramides in promoting metabolic disease have been a major focus, evidence also exists for beneficial effects of specific ceramide species. In models of type 1 diabetes and HFD-induced obesity, C24:1 ceramide is reduced in liver, heart, and plasma (Keppley et al., 2020). Restoration of liver C24:1 ceramide, by dietary supplementation reduced body weight, improved glucose tolerance and insulin sensitivity, and increased FA oxidation in HFD fed mice (Keppley et al., 2020). Similarly, elevating liver C18:1 ceramide in mice fed a western diet by deletion of alkaline ceramidase 3 (Acer3), which is up-regulated in NASH, decreased the severity of NASH by reducing oxidative stress (Wang et al., 2020). Together, these findings emphasize the necessity to consider changes in specific ceramide species, ceramide backbones, and ceramide metabolites, and their subcellular localizations in the context of metabolic dysfunction.
Mass spectrometry approaches have also been used to examine changes in S1P in metabolic diseases. Elevations in plasma S1P are a feature of both human and rodent obesity and correlate with metabolic abnormalities such as adiposity and insulin resistance (Geng et al., 2015; Kowalski et al., 2013). However, it was reported that plasma S1P and ApoM, a minor HDL apolipoprotein and carrier for S1P, are inversely associated with mortality in African Americans with T2D who are at higher risk for CVD (Liu et al., 2019). Thus, it is not surprising that an accumulating body of evidence is emerging implicating disrupted bioactive sphingolipid signaling as both an underlying cause and a link between metabolic disease and its pathological sequelae.
Regulation of ceramide levels in metabolic diseases
Although initially increased supplies of the substrates palmitate and serine were assumed to be the major cause of increased ceramide in obesity, it has become apparent that many other factors including inflammation, oxidative stress, hormonal cues and the microbiome influence ceramide synthesis and degradation in metabolic disorders. This topic will only be briefly discussed as it has been reviewed recently (Summers et al., 2019). Saturated fatty acids stimulate toll-like receptor 4 (TLR4) signaling leading to transcriptional activation of ceramide biosynthetic genes, including Sptlc2 and specific CerS isoforms in a NF-κB-dependent manner (Holland et al., 2011a; Hu et al., 2009; Hu et al., 2011) (Figure 2). Activation of intestinal hypoxia-inducible factor 2α (HIF-2α) during obesity contributes to hepatic steatosis by increasing ceramide levels mainly due to degradation of complex sphingolipids in the salvage pathway (Xie et al., 2017). In obese mice, a bile acid agonist produced in the liver stimulates the farnesoid X receptor (FXR) in the ileum, resulting in increased production of ceramides (Jiang et al., 2015a). Moreover, administration of a bile acid that is not hydrolyzed by gut bacterial bile salt hydrolase and is an intestine-selective FXR inhibitor reduced biosynthesis of ceramides resulting in decreased steatosis and increased adipose beiging (Jiang et al., 2015b) (Figure 2). While gut microbiota composition is altered in obese patients and affects the pathophysiology of obesity (Ley et al., 2006), few studies have characterized the effects of bacteria-derived sphingolipids on metabolic disease. Early studies reported that sphingomyelin and other sphingolipids are effectively digested in the intestine (Schmelz et al., 1994). However, sphingolipids can also be generated by Bacteroides, the gut commensal bacteria that have SPT allowing them to produce odd-chain length sphingoid bases that can affect inflammation in the colon (An et al., 2014). Furthermore, sphingolipids produced by gut bacteria enter host sphingolipid metabolic pathways increasing hepatic ceramide levels that influence insulin resistance (Johnson et al., 2020). Nevertheless, how specific bacteria-derived sphingolipid species effect whole body metabolism remains unclear.
Figure 2. Ceramide and S1P are critical links in the development of metabolic disease.
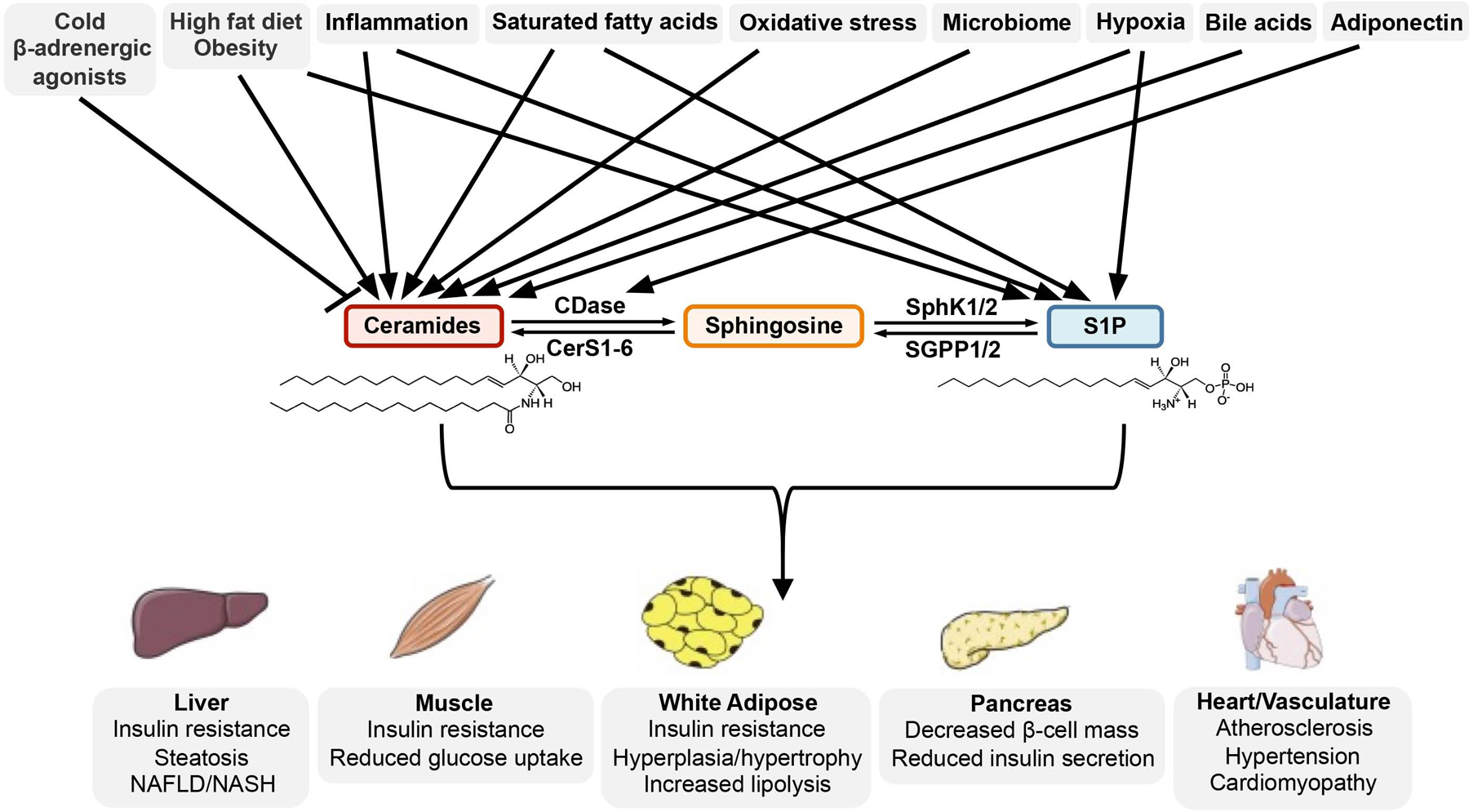
Multiple factors associated with increased susceptibility to metabolic dysfunction influence the synthesis of ceramides and S1P. Subsequent derangements in ceramide/S1P levels have significant consequences to glucose and lipid homeostasis in metabolic tissues and the heart and vasculature systems.
Levels of sphingolipid metabolites can also be regulated by the insulin-sensitizing and cardioprotective adipokine, adiponectin. Stimulation of the adiponectin receptors AdipoR1 or AdipoR2 in vivo decreased liver ceramides by enhancing their intrinsic ceramidase activity that was suggested to contribute to the anti-diabetic action of adiponectin (Holland et al., 2011b; Holland et al., 2017). Based on crystal structures (Tanabe et al., 2020; Vasiliauskaite-Brooks et al., 2017), there is still debate on whether adiponectin receptors are bona fide ceramidases and further studies are required to fully characterize their hydrolase activities and substrate specificities. Moreover, ceramidase activity not only degrades ceramide but also generates sphingosine that can be converted to pro-survival S1P, known to prevent apoptosis of pancreatic β-cells and cardiomyocytes and to exert an anti-diabetic effect. It will be important to test whether any ceramidase-dependent actions of the adiponectin receptor also require SphKs and/or S1PRs signaling, a possibility previously suggested (Holland et al., 2011b), but not yet rigorously tested.
Intervening in ceramide biosynthesis
Glucose homeostasis is regulated primarily through insulin action and is the net result of complex interplay between pancreas, the producer of insulin, liver, the main producer of glucose through gluconeogenesis, adipose tissue, the major supplier of fatty acids to liver, and skeletal muscle, the major depot for final glucose disposal. Ceramide accumulation in these tissues has been implicated in the impairment of many metabolic processes that underlie T2D and diabetic complications (Figure 2). Ceramide accumulation contributes to pancreatic β-cell apoptosis and decline of their functions. C16:0 ceramide in liver reduces glucose metabolism and insulin sensitivity and promotes hepatic steatosis (Turpin et al., 2014). Its excessive accumulation induces adipose tissue lipid storage, inflammation, and reduced thermogenesis capacity, leading to dysfunction that underlies lipotoxic cardiomyopathy (Law et al., 2018; Park et al., 2008). In skeletal muscle, accumulation of C18:0 ceramide contributes to the development of obesity-associated insulin resistance (Turpin-Nolan et al., 2019).
Numerous studies have shown that pharmacological reduction of elevated ceramides suppresses or even prevents progression of metabolic diseases in rodents (Chaurasia and Summers, 2020). The potent SPT inhibitor myriocin that has frequently been used to block de novo ceramide synthesis was shown to improve insulin resistance and hepatic steatosis, and to protect from atherosclerosis and cardiomyopathy in several animal models (Frangioudakis et al., 2010; Holland et al., 2007). Indeed, in diet-induced obese mice adipocyte-specific ablation of Sptlc2, one of the subunits of SPT, reduced adipose ceramide levels and fat mass, while increasing adipose browning, mitochondrial activity, and insulin sensitivity (Chaurasia et al., 2016). However, others reported that deletion of either Sptlc1 or Sptlc2 in adipose tissue impaired adipose differentiation and elicited a lipodystrophic phenotype (Alexaki et al., 2017; Lee et al., 2017). Other approaches are needed to determine whether the discrepancy could be due to different roles of ceramide in pre-adipocytes and mature adipocytes as the CRE-recombinase expression in the deletion strains showed developmentally distinct patterns of expression (Chaurasia et al., 2020).
Fenretinide, an inhibitor of desaturase 1 (DES1), which converts dihydroceramides to ceramides (Figure 1), resolved insulin resistance and hepatic steatosis (Bikman et al., 2012). Although Des1 homozygous knockout mice are not viable, the heterozygous knockout reduced ceramide levels in multiple tissues and improved insulin sensitivity (Holland et al., 2007). The lethality of Des1 homozygous deletion was recently overcome by generation of inducible Des1 knockout mice (Chaurasia et al., 2019). Global as well as liver and adipose-specific Des1 deletion in adult mice protected from obesogenic diet-induced hepatic steatosis and insulin resistance (Chaurasia et al., 2019). Taken together these studies demonstrate that elevated de novo ceramide synthesis is a key event in the development of metabolic disease and significant protection is achieved by inhibition of its generation. This provides compelling evidence for the development of potent pharmacological inhibitors of key enzymes in de novo sphingolipid biosynthesis.
On the other hand, elevation of dihydroceramides by depletion or inhibition of DES1 may also contribute to the metabolic benefits observed. While dihydroceramides have previously been considered merely biologically inert ceramide precursors, more recent studies reveal that, among other activities, dihydroceramides promote autophagy (Young and Wang, 2018), a process that is emerging as largely beneficial in the context of metabolic disease. Moreover, only ceramides, but not dihydroceramides, induce apoptosis and insulin resistance (Chaurasia and Summers, 2015). Furthermore, dihydroceramide-related metabolites including dihydrosphingosine and its phosphorylated derivative dihydrosphingosine-1-phosphate have also been shown to have distinct biological activities. Though these metabolites have not historically gained much research attention, the striking phenotype of DES1-deficient mice may warrant such studies.
MECHANISMS LINKING CERAMIDES TO THE DEVELOPMENT OF METABOLIC DISEASES
Although abundant evidence links ceramide elevation to development of metabolic diseases, the mechanisms involved remain understudied. It is now well accepted that in most types of tissue, ceramide induces insulin resistance due to suppression of insulin-stimulated AKT, a key serine/threonine kinase that regulates gluconeogenesis in the liver and glucose uptake in adipose and muscle tissue (Chavez et al., 2003; Holland et al., 2007) (Figure 3A–C). Many detailed mechanistic studies that followed found that inhibition of AKT by ceramide is due to activation of two independent effectors, protein phosphatase 2A (PP2A) and protein kinase Cζ (PKCζ). Activation of PP2A causes dephosphorylation at T308 that inactivates AKT (Stratford et al., 2004). Interestingly, ceramide only activates atypical PKC isoforms such as PKCζ that lack the calcium-sensitive C2 domain and contain a C1 domain that binds phosphatidylinositol-3,4,5-triphosphates (PIP3) and ceramides (Hannun and Obeid, 2008). PKCζ activated by ceramide phosphorylates threonine 34 in the PH domain of AKT, preventing PIP3 binding and inhibiting AKT translocation and subsequent activation in response to insulin (Powell et al., 2003; Stratford et al., 2004). In liver, PKCζ also likely mediates the effect of ceramide on expression of both sterol regulatory element binding transcription factor 1c (Srebp1c) (Jiang et al., 2015a), a key regulator of triglycerides, and the fatty acid translocase CD36 that facilitates uptake of fatty acids and enhances their esterification (Chaurasia et al., 2019; Xia et al., 2015). In addition, PP2A also inhibits hormone-sensitive lipase (HSL), slowing lipolysis in adipose tissue (Chaurasia et al., 2019) (Figure 3B). A recent study provided direct evidence that hepatic C16:0 ceramide specifically formed by CerS6, but not by CerS5, binds to mitochondrial fission factor (MFF) to promote mitochondrial fission, causing impairment of mitochondrial function and insulin resistance in obesity (Hammerschmidt et al., 2019). C16:0 ceramide may also directly inhibit Complex II and IV activity of mitochondrial electron transport (Zigdon et al., 2013) by yet unknown mechanisms thereby suppressing β-oxidation in liver and adipose tissue and increasing accumulation of excessive triglycerides in lipid droplets (Raichur et al., 2014; Turpin et al., 2014) (Figure 3B).
Figure 3. Ceramide modulation and tissue-specific effects on cellular metabolism.
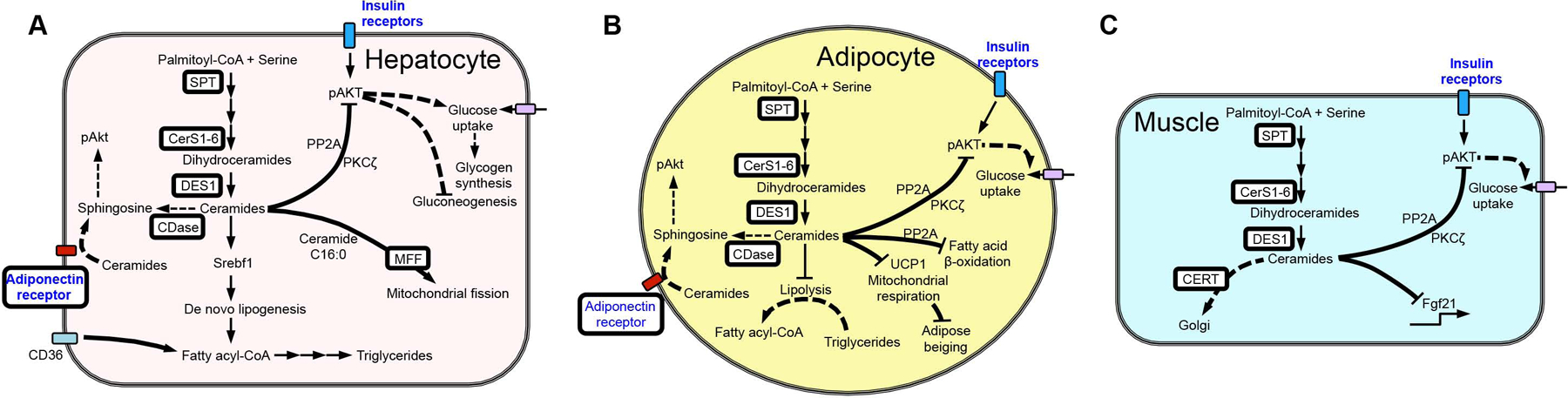
Ceramides generated during the progression of metabolic disease activate protein phosphatase 2A (PP2A) and protein kinase Cζ (PKCζ), which inhibit AKT activation and reduce insulin signaling and glucose uptake. (A) Hepatic ceramides also reduce mitochondrial function and increase lipid accumulation. (B) Adipose tissue ceramides are also linked to decreased fatty acid β-oxidation and lipolysis. (C) In muscle, excess ceramides inhibit expression of the myokine FGF21, a key regulator of glucose metabolism. White boxes highlight the sphingolipid biosynthetic enzymes shown to affect metabolic outcomes when altered in vivo.
The most well studied genetic risk factor for the development of NAFLD is a mutant variant of the patatin-like phospholipase domain-containing 3 (PNPLA3I148M) (Romeo et al., 2008). Interestingly, a recent study demonstrated that overexpression of this mutant promotes NASH by metabolic reprogramming, causing increased triglycerides and ceramides. It was suggested that ceramide activates STAT3 in hepatocytes and consequent downstream inflammatory pathways drive stellate cell fibrogenic activity (Banini et al., 2020). However, hepatic stellate cells express higher levels of PNPLA3 than hepatocytes (Bruschi et al., 2017) and the clinical significance of its overexpression is not clear.
Recently it was shown that ceramides interact directly and specifically with the voltage-dependent anion channel VDAC2, a mitochondrial platform for BAX/BAK translocation and an effector of ceramide-mediated apoptosis (Dadsena et al., 2019). However, it is still not known whether this mechanism contributes to apoptosis of pancreatic β-cells (Shimabukuro et al., 1998) or cardiomyocytes (Law et al., 2018) that underlie diabetes and cardiomyopathy, respectively.
Inducible overexpression of adiponectin receptors to enhance ceramidase activity (Holland et al., 2017) as well as adipose-specific overexpression of acid ceramidase that reduced ceramide-activated PKCζ, increased adipose glucose uptake while also reducing hepatic steatosis (Xia et al., 2015). This emphasizes the importance of cross-talk between liver and adipose sphingolipids to regulate systemic glucose metabolism. Aberrant ceramide accumulation in adipocytes induced SOCS-3 and inhibited UCP3 expression suggesting a role for ceramide in weight gain, energy expenditure and thermogenesis (Yang et al., 2009). Indeed, cold or β-adrenergic agonists reduced adipose ceramides whereas adipocyte-specific inhibition of ceramide synthesis induced adipose beiging by enhancing expression of the uncoupling protein UCP1 and the transcriptional co-activator PGC-1α (Chaurasia et al., 2016). Conversely, ceramide treatment of adipocytes suppressed mitochondrial respiration and expression of several genes important for browning including Ucp1 and Ppargc1a, thereby impairing beige fat thermogenesis (Jiang et al., 2015b).
In muscle, overexpression of the ceramide transporter (CERT) that diverts ceramides from the ER to the Golgi for synthesis of sphingomyelin improved insulin sensitivity in mice by enhancing insulin-mediated activation of AKT, glycogen synthase kinase 3 beta (GSK3β), and extracellular signal-regulated kinases (ERK1/2) (Bandet et al., 2018). Intriguingly, CerS1 and its product C18:0 ceramide are increased in skeletal muscle of obese mice. Furthermore, muscle-specific CerS1 deletion, but not deficiency of C16:0 ceramide-producing CerS5 and CerS6, improved glucose tolerance likely by increasing expression of Fgf21 that acts as an insulin sensitizer to overcome peripheral insulin resistance (Turpin-Nolan et al., 2019) (Figure 3C). However, although a selective CerS1 inhibitor enhanced fat oxidation in muscle, it did not improve insulin sensitivity (Turner et al., 2018). Further studies are needed to better understand the different effects between genetic inactivation of Cers1 compared to its pharmacological inhibition.
THE JANUS FACES OF SPHINGOSINE KINASES AND S1P IN METABOLIC DISEASES
While there is overwhelming evidence that ceramide functions in myriad ways to exacerbate metabolic diseases, roles for its further metabolite S1P and the SphKs that produce it are more nuanced. Initially it was suggested that S1P has beneficial roles in cell functions that pertain to metabolic disease. Whereas earlier studies of lipotoxicity implicated ceramide in β-cell death (Shimabukuro et al., 1998), genetic ablation of Sphk1 predisposed diet-induced obese mice to the onset of diabetes by promoting pancreatic β-cell death, and treatment with S1P bolstered resistance to palmitate-induced cell death (Qi et al., 2013). Moreover, expression of dominant-negative SphK1 promoted palmitate-induced cell death in pancreatic β-cells. These data support a beneficial role of SphK1 in protection from lipotoxicity-induced pancreatic dysfunction. Moreover, in contrast to ceramides that antagonize insulin function, SphKs have been reported in earlier studies to promote insulin signaling in some tissues. For example, injection of SphK1-expressing adenovirus improved glucose tolerance in diabetic KK/Ay mice, increased glycogen storage, and reduced triglycerides that correlated with activation of hepatic AKT and GSK3β signaling (Ma et al., 2007). Likewise, transgenic mice globally overexpressing SphK1 have improved glucose tolerance and muscle insulin sensitivity and reduced ceramide levels, as well as activation of c-Jun N-terminal kinase (JNK), a serine threonine kinase associated with insulin resistance (Bruce et al., 2012). Indeed, skeletal muscle and whole body insulin resistance were improved in HFD-fed SphK1-overexpressing mice. However, in contrast, SphK1 expression was found to be elevated specifically in adipose tissue of HFD-mice and human T2D patients. Moreover, SphK1 deletion or its pharmacological inhibition improved systemic insulin sensitivity of obese mice (Wang et al., 2014), and protected from HFD-induced hepatic inflammation, NASH (Geng et al., 2015) and hepatosteatosis, likely due to reduced activation of PPARγ (Chen et al., 2016). Similarly, tissue-specific opposing functions for SphK2 in metabolic diseases have been reported. On one hand, overexpression of SphK2 in the liver ameliorated glucose intolerance and insulin resistance in diet-induced obese mice (Lee et al., 2015). On the other hand, deletion of SphK2 preserved insulin production and prevented progression of diabetes by protecting pancreatic β-cells against lipoapoptosis (Song et al., 2019).
What is the explanation for the seemingly opposing actions of SphKs?
First, expression of SphKs in various tissues can have different functions. For example, it was suggested that global SphK2−/− mice are protected from age-related obesity due to increased adipocyte-produced adiponectin, which has glucose- and lipid-lowering effects, and the adipocyte triglyceride lipase (ATGL), which catalyzes the rate-limiting step of lipolysis (Ravichandran et al., 2019). Conversely, exacerbated insulin resistance and glucose intolerance in hepatocyte-specific SphK2 knockout obese mice was due to increase of its substrate sphingosine that mediated inhibition of the hepatic PI3 kinase/AKT signaling pathway (Aji et al., 2020) (Figure 4A). However, it is still not clear how sphingosine inhibits PI3K and further studies might reveal a new aspect of hepatic insulin signaling regulation. Furthermore, in contrast to global loss of SphK1 that ameliorates steatosis and adipocyte pro-inflammatory responses (Wang et al., 2014), adipocyte-specific SphK1 deletion exacerbated glucose intolerance and adipocyte hypertrophy, and impaired lipolysis (Anderson et al., 2020) (Figure 4B). Although the molecular mechanisms by which SphK1 regulates adipocyte function are not clear, it seems that SphK1 has an essential homeostatic role in adipocytes that can protect from obesity-associated pathology (Anderson et al., 2020). Together, these studies emphasize the need for better understanding of the complex inter-organ communication and tissue-specific roles of SphKs and S1P signaling involved in the pathogenesis of metabolic disease.
Figure 4. S1P/S1PR signaling modulation in metabolic tissues.
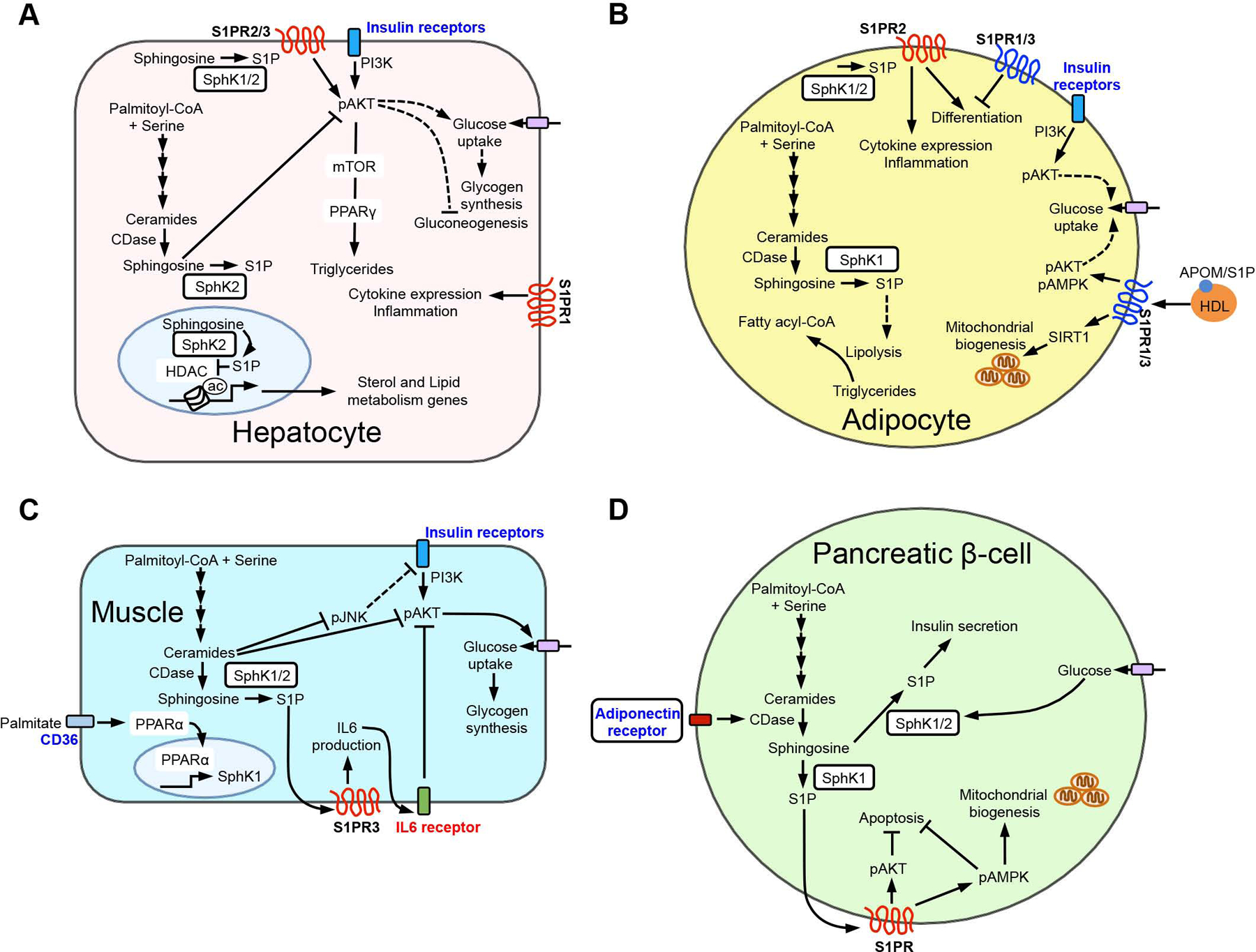
Regulation of S1P/S1PR signaling exhibits tissue-specific metabolic outcomes. (A) S1P generated in liver hepatocytes can activate AKT to enhance insulin signaling and triglyceride storage as well as regulate sterol, lipid, and inflammatory gene expression. (B) S1P and S1P/S1PR signaling modulates adipocytes inflammation, differentiation, and pathways affecting lipolysis, glucose uptake, and mitochondrial biogenesis. (C) In muscles, S1P/S1PR affects insulin signaling in part through IL6 generation and receptor activation to block AKT activity. (D) In pancreatic β-cells, S1P enhances insulin secretion, and activation of S1PRs suppresses apoptosis and regulates mitochondrial biogenesis.
HOW DO SPHKS AND S1P AFFECT METABOLIC DISEASES?
SphKs are key enzymes that regulate levels of several bioactive sphingolipid metabolites. As mentioned above (Figure 1), ceramide is hydrolyzed by ceramidases to sphingosine, which undergoes phosphorylation by SphKs to form S1P. One critical aspect of this phosphorylation is that S1P can be irreversibly degraded, thus reducing total sphingosine and ceramide burden. Therefore, one function of SphKs in metabolic disease may be the removal of excess ceramide. However, S1P is also a potent signaling molecule in its own right. Intracellularly generated S1P can be transported out of cells by several ATP-binding cassette transporters and the major facilitator superfamily member SPNS2, where it acts in an autocrine or paracrine fashion as a ligand for S1PR1–5, that mediate signaling in numerous cell types. S1PR1–3 are ubiquitously expressed whereas, S1PR4 is predominantly expressed in the immune system and S1PR5 in the central nervous system. These receptors are associated with different profiles of Gα subunits. For example, S1PR1 couples solely to Gi/Go; S1PR2 couples to Gs, Gq/G11, G12/G13; S1PR3 couples to Gi/Go, Gq/G11, G12/G13; S1PR4 and S1PR5 couple to Gi/Go and G12/G13 (Spiegel and Milstien, 2003). Because all cells express some flavor of these receptors, which are coupled to distinct G proteins that activate effectors, S1P signaling is involved in myriad physiological and pathophysiological functions (for review see (Spiegel and Milstien, 2003)). Thus, many drugs have been developed that target S1P/S1PR signaling axes, some of which are already in the clinic. This has allowed for more targeted studies into the roles of S1P signaling which has revealed that SphKs, S1P, and S1PRs have primarily adaptive but also maladaptive roles in metabolic disease.
S1P also has intracellular targets (Maceyka and Spiegel, 2014). In contrast to SphK1, which is localized in the cytosol and translocates upon activation to the plasma membrane, SphK2 is also present in several intracellular compartments, including nuclei and mitochondria depending on cell type (Maceyka and Spiegel, 2014). The relevance of its function in these compartments to metabolic syndrome has not yet been extensively studied. S1P formed in the nucleus by SphK2 inhibited histone deacetylases HDAC1/2 (Hait et al., 2009), causing an increase in acetylation of histones and upregulation of hepatic genes encoding nuclear receptors and enzymes involved in lipid metabolism (Nagahashi et al., 2015) (Figure 4A). In this regard, the pro-drug FTY720/Fingolimod that is phosphorylated in vivo by SphK2 protects from insulin resistance and hepatosteatosis by reducing muscle ceramides and CD36 expression (Bruce et al., 2013), and in part by inhibition of HDAC and attenuating fatty acid synthase expression (Rohrbach et al., 2019).
S1PRS SIGNALING IN METABOLIC TISSUES
Despite these beneficial roles, SphKs and S1P have deleterious functions likely due to their known effects in immune cell trafficking and proinflammatory signaling (Maceyka and Spiegel, 2014). Saturated fatty acid overload upregulates SphK1 in livers from mice and in humans with NASH. S1P in turn activates S1PR1 signaling in hepatocytes leading to NF-κB activation, elevated cytokine/chemokine production, and immune cell infiltration (Geng et al., 2015). Others have shown that binding of S1P to S1PR2/3 in hepatocytes also regulates expression of peroxisome proliferator-activated receptor gamma (PPARγ) through the AKT-mTOR pathway, which promotes lipogenesis and lipid storage (Chen et al., 2016) (Figure 4A). It has been suggested that S1P synthesized in hepatocytes in response to excess palmitate is released, leading to paracrine activation of hepatic stellate cells by binding to S1PR3 (Al Fadel et al., 2016), and also increases migration of myofibroblasts into the damaged areas in a S1PR1/3-dependent manner (Li et al., 2011). These initiate fibrosis and further studies are needed to determine whether altered S1P metabolism in fibrotic liver regulates NASH disease progression.
Of note, palmitate upregulates SphK1 in skeletal muscles by activation of the key transcription factor PPARα involved in regulating lipid catabolism. S1P then stimulates S1PR3 to induce the pro-inflammatory cytokine IL-6 and the autocrine loop of IL-6 signaling leading to insulin resistance (Ross et al., 2013) (Figure 4C). Furthermore, it was suggested that adiponectin stimulates ceramidase activity of AdipoR1/2 to produce sphingosine and subsequently S1P (Holland et al., 2011b) that can be secreted and activate S1PRs to protect skeletal muscle cells from palmitate-induced ROS production and cell death (Botta et al., 2020). Similarly, activation of S1PR by this adiponectin-S1P autocrine axis led to stimulation of AMPK and AKT, thus suppressing β-cell apoptosis and enhancing their survival (Holland et al., 2011b) (Figure 4D). Unfortunately, the S1PR involved in this adiponectin-S1P autocrine-axis has not yet been identified.
The role of the SphK/S1P/S1PR axis in adipose tissue is also complicated. Increased SphK activity can initially remove harmful ceramide. However, depletion of adipocyte SphK1 also caused adipocyte hypertrophy and decreased insulin sensitivity (Anderson et al., 2020). The S1P formed can also increase pro-inflammatory cytokine secretion (Wang et al., 2014) and its binding to S1PR2 recruits circulating pro-inflammatory M1-macrophages that can lead to adipose dysfunction and insulin resistance (Kitada et al., 2016). Moreover, whereas S1PR1/3 inhibit, S1PR2 activates preadipocyte differentiation, important for adipogenesis (Kitada et al., 2016) (Figure 4B). The biological functions of each S1PR need to be validated in vivo as the majority of studies were carried out using cell lines that do not fully mimic adipose or skeletal muscle tissues nor their potential communication with the liver.
THE APOLIPOPROTEIN M AND S1P AXIS IN METABOLIC DISEASE
High levels of S1P are present in blood, much of which is carried by HDL-associated apolipoprotein M (ApoM) secreted by the liver (Maceyka and Spiegel, 2014). In addition to the well-known function of HDL in clearance of excess cholesterol, HDL/ApoM-associated S1P signaling through S1PRs is responsible for some of the beneficial effects of HDL, such as attenuation of apoptosis and inflammation, and vasoprotection (Rohrbach et al., 2017). Recently it was suggested that ApoM/S1P attenuates the development of insulin resistance by stimulating AKT and AMPK, the main insulin signaling pathways in liver, adipose tissue, and skeletal muscle, through S1PR1 and/or S1PR3, and perhaps also by improving mitochondrial functions through upregulation of SIRT1 protein levels (Kurano et al., 2020) (Figure 4B). However, another report suggested that the ApoM/S1P axis directly affects S1PR1 endothelial barrier function in brown adipose tissue in a S1PR1-dependent manner to decrease browning and to slow triglycerides clearance, thus exacerbating diet-induced obesity (Christoffersen et al., 2018) (Figure 5A).
Figure 5. S1P/S1PR signaling in inflammation and metabolic dysregulation.
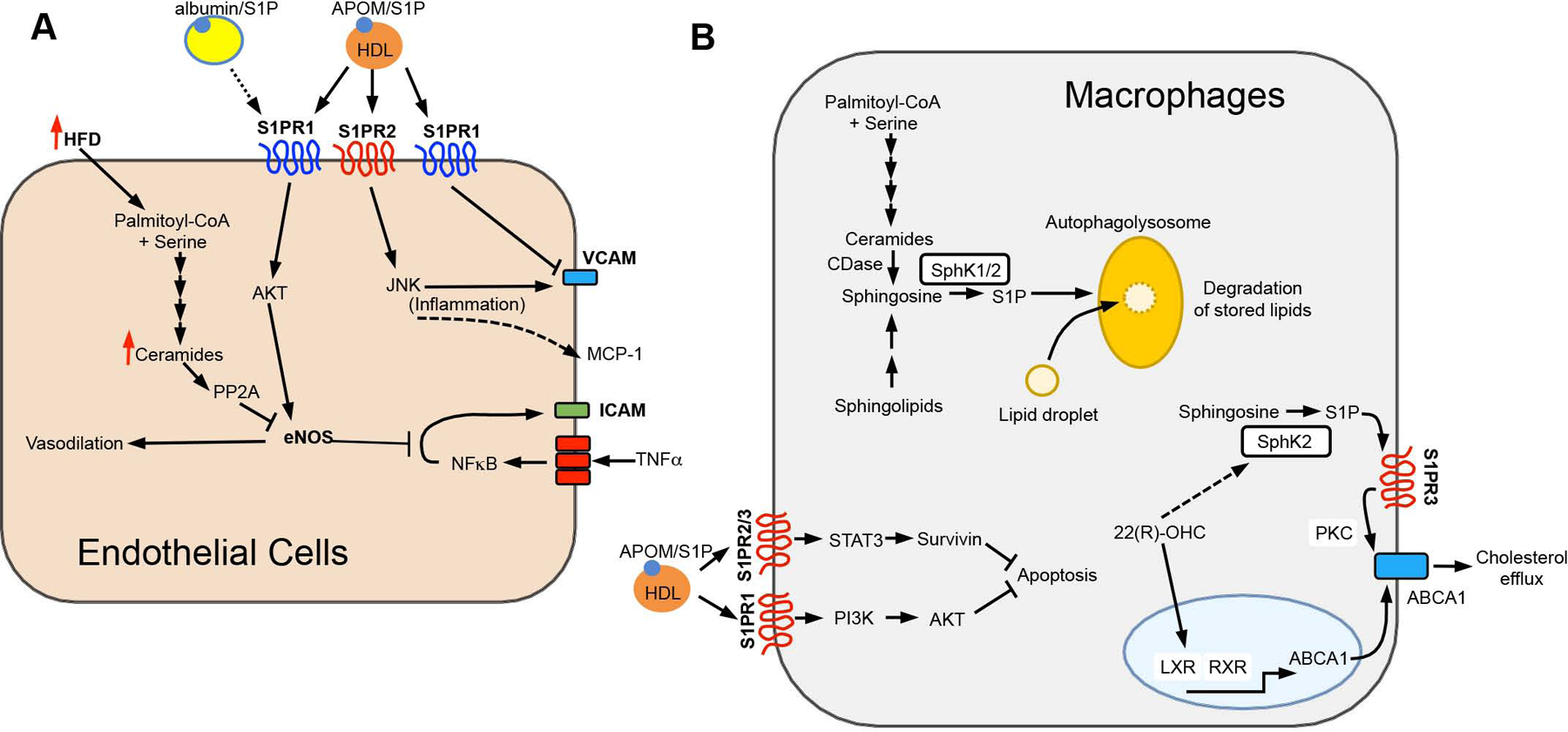
(A) Endothelial S1PRs influence inflammation and increase vasodilation through eNOS activation, counteracting effects mediated by ceramides. (B) In macrophages, intracellular S1P enhances degradation of stored lipids, while S1P/S1PR signals block apoptosis and increase ABCA1-mediated cholesterol export. White boxes highlight the sphingolipid biosynthetic enzymes shown to affect metabolic outcomes when altered in vivo.
S1PR signaling in endothelial cells and macrophages is also involved in metabolic disorders and T2D. In high fat diet-fed ApoE−/− mice, inhibition of S1PR2 reduced plaque formation and atherogenesis, likely through inhibition of JNK signaling, which promotes macrophage recruitment by inducing expression of the adhesion molecule VCAM-1 and monocyte chemoattractant protein-1 (MCP-1) in endothelial cells (Figure 5A) (Ganbaatar et al., 2020; Skoura et al., 2011). Conversely, activation of S1PR1 via ApoM-S1P limits endothelial inflammation and monocyte adhesion by inhibition of endothelial expression of VCAM-1 and E-selectin without affecting MCP-1 expression (Ruiz et al., 2017) (Figure 5A). As endothelial cell dysfunction can cause metabolic dysregulation, it is likely that these opposing mechanisms within endothelial cells function to maintain homeostasis and restore balance after inflammatory insults or injury. This raises the interesting question of how the balance between S1PR1 and S1PR2 activity is maintained. One intriguing facet of this balance is the carrier of S1P. It was shown that ApoM-S1P acts as a biased agonist that prolongs S1PR1 signaling leading to sustained S1PR1-PI3K-AKT-eNOS-dependent enhanced barrier function (Wilkerson et al., 2012), while albumin-S1P leads to rapid internalization and desensitization of S1PR1 (Galvani et al., 2015). Moreover, activation of S1PR1 by ApoM-S1P antagonized the cytokine-induced NF-κB pathway that increased ICAM-1 and VCAM-1 in endothelial cells (Galvani et al., 2015) (Figure 5A). Together, this suggests that endothelial S1PR1 protects the vessel wall from inflammation and atherosclerosis. A biased agonist of S1PR1, SAR247799, that mimicked ApoM-S1P effects in endothelial cells was recently developed (Poirier et al., 2020). In a clinical trial, SAR247799 treatment of patients with T2D reversed endothelial dysfunction without the side effects of other S1PR1 agonists, likely through restoring endothelial junctions and barrier function as no significant effects on lymphocyte trafficking were observed (Bergougnan et al., 2020).
Impaired vasodilation in hypertension is a serious complication of metabolic diseases. HDL-ApoM-S1P activates S1PR1 on endothelial cells, stimulates AKT-dependent phosphorylation and activation of endothelial nitric oxide synthase (eNOS) to produce NO, a critical regulator of vasorelaxation (Wilkerson et al., 2012) (Figure 5A). HDL from T2D patients had lower S1P and reduced ability to stimulate eNOS and suppress NF-κB-mediated immune responses (Vaisar et al., 2018). Similarly, HDL from non-diabetic metabolic syndrome patients had lower associated S1P and reduced ability to stimulate AKT and eNOS, which could be reversed with S1P enrichment (Denimal et al., 2017). Together, these studies suggest that decreased S1P may contribute to the impairment of HDL functionality and antiatherogenic properties in these patients. In contrast, increased TNFα vascular smooth muscle led to S1P-dependent augmented myogenic tone in a mouse model of diabetes, suggesting that S1P may also contribute to microvascular complications in diabetes (Sauve et al., 2016).
Macrophage apoptosis is a central feature of atherosclerotic plaque development. HDL-ApoM-S1P was also shown to inhibit macrophage cell death. (Feuerborn et al., 2017). Stimulation of S1PR2/3 enhanced JAK2 activity and phosphorylation and translocation of the master transcription factor STAT3 to the nucleus where it enhanced expression of survivin that suppressed ER stress and apoptosis by inhibiting caspase 3 activation (Feuerborn et al., 2017) (Figure 5B). The infiltration of cholesterol-accumulating macrophages in the vascular wall, another characteristic of atherosclerosis, was greatly exacerbated in SphK2 deficient ApoE−/− mice. It was shown that SphK2, but not SphK1, in macrophages was required for autophagosome- and lysosome-mediated catabolism of intracellular lipid droplets to prevent cholesterol accumulation and limit the development of atherosclerosis (Ishimaru et al., 2019). Whether SphK2 functions internally by regulating levels of sphingosine or ceramide to stimulate autophagic lipid degradation or externally via S1PR signaling remains to be elucidated. Evidence for S1P acting in an autocrine/paracrine role in macrophages originated from the finding that the nuclear transcription factor, liver X receptor (LXR), known to be activated by oxysterols, stimulated SphK and that S1P/S1PR3 signaling, perhaps through PKC, regulated ABCA1-mediated cholesterol efflux to lipid-poor HDL (Vaidya et al., 2019) (Figure 5B). Together, these results demonstrate that the complex roles of S1P depend on cell type, the effectors that S1PRs signal through, and the synthesis and degradation of S1P.
THE UNKNOWNS: CONCLUSIONS AND FUTURE PERSPECTIVES.
We have come a long way from the initial mindset that sphingolipid pools represent a sink for excess palmitate, resulting in aberrant biosynthesis of ceramide, which drives insulin resistance and metabolic diseases. However, a relic of this mindset is that catabolic pathways of sphingolipid metabolism have been largely unexplored. Although deficiency of acid sphingomyelinase that generates ceramide from sphingomyelin hydrolysis protects mice against adipocyte hypertrophy and diet-induced steatosis (Sydor et al., 2017), the mechanism of the protection has not been elucidated. Furthermore, while the major human neutral sphingomyelinase, SMPD3, was shown to be elevated in inflamed vs. healthy adipose tissue in obese women (Kolak et al., 2012), the link to metabolic disease is still unclear.
This review has focused on the signaling roles of ceramide and S1P, as these are the the best studied sphingolipid metabolites. However, it is possible that these metabolites, found to be altered by targeted lipidomics, are not the cause but a symptom of dysregulated sphingolipid metabolism that leads to the observed pathophysiologies. Moreover, many studies have found altered levels of other sphingolipid species, including glucosylceramides, sphingomyelins, and ganglioside GM3, in T2D, obesity, and other metabolic diseases (Drouin-Chartier et al., 2021; Ooi et al., 2021). An additional layer of complexity arises from the recent discovery of several atypical sphingolipids formed by alternative utilization by SPT of amino acids, such as alanine or shorter chain acyl-CoAs to form 1-deoxy or shorter-chain sphingoid bases, respectively (Kovilakath et al., 2020; Lone et al., 2019). Because the majority of studies that have revealed relationships between sphingolipids and metabolic diseases have relied on targeted lipidomics approaches, these minor atypical sphingolipids have been largely ignored. However, a recent study revealed association of 1-deoxy-sphingolipids that cannot be converted into complex sphingolipids or phosphorylated and terminally degraded by S1P lyase (Lone et al., 2019), with steatosis but not steatohepatitis or fibrosis in NAFLD (Weyler et al., 2020). Also, myristate-derived d16:0 ceramides generated by SPTLC3, a subunit of SPT that promotes utilization of alternative shorter fatty acyl-CoAs, were shown to increase in a mouse model of obesity and contribute to cardiomyocyte apoptosis (Russo et al., 2013). Additionally, genome-wide association analyses revealed loci of SPTLC3 associated with CVD risk in humans (Tabassum et al., 2019). Future studies are needed to address the roles of atypical sphingolipids and SPTLC3 and its products in metabolic diseases to define potential mechanistic functions. It will also be important to determine other genetic determinants that influence aberrant sphingolipid levels in patient populations compared to healthy controls.
There are still many open questions remaining. Although much is known about S1PRs and S1P intracellular targets, only a few direct targets of specific ceramides or dihydroceramide species have been identified and even much less is known in relation to their mechanisms of action in metabolic diseases. Uncovering additional molecular targets and mechanisms are central for understanding the functions of critical dihydroceramide/ceramide species. It is also still unclear whether specific dihydroceramide species are the key regulators of metabolic diseases or just biomarkers for increased de novo biosynthesis of ceramides whose inappropriate buildup triggers adverse effects. Moreover, tissue-specific roles of distinct ceramide species or S1P in disease etiology are still not fully delineated. The challenge for this field is that these bioactive sphingolipid metabolites are rapidly interconvertible and are intermediates in the biosynthesis of complex sphingolipids and thus assigning specific functions to distinct sphingolipid species is a daunting task. For example, depletion of a ceramide synthase isoform may not only impact those downstream dihydroceramide and ceramide pools, but also sphingomyelins and glycosphingolipids derived from them. In fact, in the context of gain- and loss-of-function studies targeting sphingolipid metabolic enzymes, identification of specific lipid mediators is impossible without taking into account potential impacts on the greater sphingolipidome.
Preclinical research use of mouse models and pharmacological tools together with sophisticated mass spectrometry methods to measure the sphingolipidome in human patients support the idea of development of new therapeutic approaches that reduce ceramide biosynthesis (e.g., by inhibiting DES1 or a specific CerS) or by targeting specific S1PRs to combat metabolic disease. Mechanistically, further tissue- and organelle-specific studies of ceramide, sphingosine, and S1P actions will be imperative for delineating how these bioactive sphingolipids affect disease development. Together, a better understanding of sphingolipid synthesis and degradation and how they are regulated by metabolic alterations holds great promise for the development of tractable therapeutic targets and approaches.
ACKNOWLEDGMENTS
The authors wish to thank Dr. Sheldon Milstien for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health (R01GM043880 to S.S. and R01HL151243 to L.A.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors have nothing to disclose.
REFERENCES
- Aji G, Huang Y, Ng ML, Wang W, Lan T, Li M, Li Y, Chen Q, Li R, Yan S, et al. (2020). Regulation of hepatic insulin signaling and glucose homeostasis by sphingosine kinase 2. Proc. Natl. Acad. Sci. USA 117, 24434–24442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Fadel F, Fayyaz S, Japtok L, and Kleuser B (2016). Involvement of Sphingosine 1-Phosphate in Palmitate-Induced Non-Alcoholic Fatty Liver Disease. Cell Physiol. Biochem 40, 1637–1645. [DOI] [PubMed] [Google Scholar]
- Alexaki A, Clarke BA, Gavrilova O, Ma Y, Zhu H, Ma X, Xu L, Tuymetova G, Larman BC, Allende ML, et al. (2017). De Novo Sphingolipid Biosynthesis Is Required for Adipocyte Survival and Metabolic Homeostasis. J. Biol. Chem 292, 3929–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, Lu X, Zeissig S, Blumberg RS, and Kasper DL (2014). Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AK, Lambert JM, Montefusco DJ, Tran BN, Roddy P, Holland WL, and Cowart LA (2020). Depletion of adipocyte sphingosine kinase 1 leads to cell hypertrophy, impaired lipolysis, and nonalcoholic fatty liver disease. J. Lipid Res 61, 1328–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anroedh S, Hilvo M, Akkerhuis KM, Kauhanen D, Koistinen K, Oemrawsingh R, Serruys P, van Geuns RJ, Boersma E, Laaksonen R, et al. (2018). Plasma concentrations of molecular lipid species predict long-term clinical outcome in coronary artery disease patients. J. Lipid Res 59, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandet CL, Mahfouz R, Veret J, Sotiropoulos A, Poirier M, Giussani P, Campana M, Philippe E, Blachnio-Zabielska A, Ballaire R, et al. (2018). Ceramide Transporter CERT Is Involved in Muscle Insulin Signaling Defects Under Lipotoxic Conditions. Diabetes 67, 1258–1271. [DOI] [PubMed] [Google Scholar]
- Banini BA, D PK, Cazanave S, Seneshaw M, Mirshahi F, Santhekadur PK, Wang L, Guan HP, Oseini A, Alonso C, et al. (2021). Identification of a metabolic, transcriptomic and molecular signature of PNPLA3-mediated acceleration of steatohepatitis. Hepatology 73,1290–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman BC, Brozinick JT, Strauss A, Bacon S, Kerege A, Bui HH, Sanders P, Siddall P, Wei T, Thomas MK, et al. (2016). Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia 59, 785–798. [DOI] [PubMed] [Google Scholar]
- Bergougnan L, Andersen G, Plum-Morschel L, Evaristi MF, Poirier B, Tardat A, Ermer M, Herbrand T, Arrubla J, Coester HV, et al. (2021). Endothelial-protective effects of a G-protein-biased sphingosine-1 phosphate receptor-1 agonist, SAR247799, in type-2 diabetes rats and a randomized placebo-controlled patient trial. Br. J. Clin. Pharmacol 87, 2303–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikman BT, Guan Y, Shui G, Siddique MM, Holland WL, Kim JY, Fabrias G, Wenk MR, and Summers SA (2012). Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J. Biol. Chem 287, 17426–17437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta A, Elizbaryan K, Tashakorinia P, Lam NH, and Sweeney G (2020). An adiponectin-S1P autocrine axis protects skeletal muscle cells from palmitate-induced cell death. Lipids Health Dis 19, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Risis S, Babb JR, Yang C, Kowalski GM, Selathurai A, Lee-Young RS, Weir JM, Yoshioka K, Takuwa Y, et al. (2012). Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 61, 3148–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Risis S, Babb JR, Yang C, Lee-Young RS, Henstridge DC, and Febbraio MA (2013). The sphingosine-1-phosphate analog FTY720 reduces muscle ceramide content and improves glucose tolerance in high fat-fed male mice. Endocrinology 154, 65–76. [DOI] [PubMed] [Google Scholar]
- Bruschi FV, Claudel T, Tardelli M, Caligiuri A, Stulnig TM, Marra F, and Trauner M (2017). The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 65, 1875–1890. [DOI] [PubMed] [Google Scholar]
- Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DL, Gopalan V, Prakash KN, Velan SS, Bulchand S, et al. (2016). Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metab 24, 820–834. [DOI] [PubMed] [Google Scholar]
- Chaurasia B, and Summers SA (2015). Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab 26, 538–550. [DOI] [PubMed] [Google Scholar]
- Chaurasia B, and Summers SA (2021). Ceramides in Metabolism: Key Lipotoxic Players. Annu. Rev. Physiol 83, 303–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, Wilkerson JL, Sweeney CR, Pereira RF, Sumida DH, et al. (2019). Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 365, 386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasia B, Ying L, Talbot CL, Maschek JA, Cox J, Schuchman EH, Hirabayashi Y, Holland WL, and Summers SA (2021). Ceramides are necessary and sufficient for diet-induced impairment of thermogenic adipocytes. Mol Metab 45, 101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, and Summers SA (2003). A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem 278, 10297–10303. [DOI] [PubMed] [Google Scholar]
- Chen J, Wang W, Qi Y, Kaczorowski D, McCaughan GW, Gamble JR, Don AS, Gao X, Vadas MA, and Xia P (2016). Deletion of sphingosine kinase 1 ameliorates hepatic steatosis in diet-induced obese mice: Role of PPARgamma. Biochim. Biophys. Acta 1861, 138–147.26615875 [Google Scholar]
- Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, et al. (2012). Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 125, 2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen C, Federspiel CK, Borup A, Christensen PM, Madsen AN, Heine M, Nielsen CH, Kjaer A, Holst B, Heeren J, et al. (2018). The Apolipoprotein M/S1P Axis Controls Triglyceride Metabolism and Brown Fat Activity. Cell Rep 22, 175–188. [DOI] [PubMed] [Google Scholar]
- Crewe C, An YA, and Scherer PE (2017). The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J. Clin. Invest 127, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadsena S, Bockelmann S, Mina JGM, Hassan DG, Korneev S, Razzera G, Jahn H, Niekamp P, Muller D, Schneider M, et al. (2019). Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun 10, 1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denimal D, Monier S, Brindisi MC, Petit JM, Bouillet B, Nguyen A, Demizieux L, Simoneau I, Pais de Barros JP, Verges B, et al. (2017). Impairment of the Ability of HDL From Patients With Metabolic Syndrome but Without Diabetes Mellitus to Activate eNOS: Correction by S1P Enrichment. Arterioscler Thromb Vasc. Biol 37, 804–811. [DOI] [PubMed] [Google Scholar]
- Drouin-Chartier JP, Hernandez-Alonso P, Guasch-Ferre M, Ruiz-Canela M, Li J, Wittenbecher C, Razquin C, Toledo E, Dennis C, Corella D, et al. (2021). Dairy consumption, plasma metabolites, and risk of type 2 diabetes. Am. J. Clin. Nutr in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerborn R, Becker S, Poti F, Nagel P, Brodde M, Schmidt H, Christoffersen C, Ceglarek U, Burkhardt R, and Nofer JR (2017). High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis 257, 29–37. [DOI] [PubMed] [Google Scholar]
- Frangioudakis G, Garrard J, Raddatz K, Nadler JL, Mitchell TW, and Schmitz-Peiffer C (2010). Saturated- and n-6 polyunsaturated-fat diets each induce ceramide accumulation in mouse skeletal muscle: reversal and improvement of glucose tolerance by lipid metabolism inhibitors. Endocrinology 151, 4187–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H, Dahlback B, Kono M, Proia RL, Smith JD, et al. (2015). HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal 8, ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganbaatar B, Fukuda D, Shinohara M, Yagi S, Kusunose K, Yamada H, Soeki T, Hirata KI, and Sata M (2021). Inhibition of S1P Receptor 2 Attenuates Endothelial Dysfunction and Inhibits Atherogenesis in Apolipoprotein E-Deficient Mice. J. Atheroscler. Thromb 28, 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng T, Sutter A, Harland MD, Law BA, Ross JS, Lewin D, Palanisamy A, Russo SB, Chavin KD, and Cowart LA (2015). SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes. J. Lipid Res 56, 2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadas Y, Vincek AS, Youssef E, Zak MM, Chepurko E, Sultana N, Sharkar MTK, Guo N, Komargodski R, Kurian AA, et al. (2020). Altering Sphingolipid Metabolism Attenuates Cell Death and Inflammatory Response After Myocardial Infarction. Circulation 141, 916–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, et al. (2009). Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, Sprenger HG, Awazawa M, Nicholls HT, Turpin-Nolan SM, et al. (2019). CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell 177, 1536–1552 e1523. [DOI] [PubMed] [Google Scholar]
- Hannun YA, and Obeid LM (2008). Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol 9, 139–150. [DOI] [PubMed] [Google Scholar]
- Hannun YA, and Obeid LM (2018). Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol 19, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havulinna AS, Sysi-Aho M, Hilvo M, Kauhanen D, Hurme R, Ekroos K, Salomaa V, and Laaksonen R (2016). Circulating Ceramides Predict Cardiovascular Outcomes in the Population-Based FINRISK 2002 Cohort. Arterioscler. Thromb. Vasc. Biol 36, 2424–2430. [DOI] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, et al. (2011a). Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest 121, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, et al. (2007). Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5, 167–179. [DOI] [PubMed] [Google Scholar]
- Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, et al. (2011b). Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med 17, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, Quittner-Strom E, Tippetts TS, Gordillo R, and Scherer PE (2017). Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol. Metab 6, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Bielawski J, Samad F, Merrill AH Jr., and Cowart LA (2009). Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J. Lipid Res 50, 1852–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Ross J, Geng T, Brice SE, and Cowart LA (2011). Differential regulation of dihydroceramide desaturase by palmitate versus monounsaturated fatty acids: implications for insulin resistance. J. Biol. Chem 286, 16596–16605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru K, Yoshioka K, Kano K, Kurano M, Saigusa D, Aoki J, Yatomi Y, Takuwa N, Okamoto Y, Proia RL, et al. (2019). Sphingosine kinase-2 prevents macrophage cholesterol accumulation and atherosclerosis by stimulating autophagic lipid degradation. Sci. Rep 9, 18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaheri A, Allegood JC, Cowart LA, and Chirinos JA (2020). Circulating Ceramide 16:0 in Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol 75, 2273–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji R, Akashi H, Drosatos K, Liao X, Jiang H, Kennel PJ, Brunjes DL, Castillero E, Zhang X, Deng LY, et al. (2017). Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight 2, e96203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, et al. (2015a). Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Invest 125, 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, et al. (2015b). Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun 6, 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EL, Heaver SL, Waters JL, Kim BI, Bretin A, Goodman AL, Gewirtz AT, Worgall TS, and Ley RE (2020). Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nat. Commun 11, 2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppley LJW, Walker SJ, Gademsey AN, Smith JP, Keller SR, Kester M, and Fox TE (2020). Nervonic acid limits weight gain in a mouse model of diet-induced obesity. FASEB J 34, 15314–15326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada Y, Kajita K, Taguchi K, Mori I, Yamauchi M, Ikeda T, Kawashima M, Asano M, Kajita T, Ishizuka T, et al. (2016). Blockade of Sphingosine 1-Phosphate Receptor 2 Signaling Attenuates High-Fat Diet-Induced Adipocyte Hypertrophy and Systemic Glucose Intolerance in Mice. Endocrinology 157, 1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolak M, Gertow J, Westerbacka J, Summers SA, Liska J, Franco-Cereceda A, Oresic M, Yki-Jarvinen H, Eriksson P, and Fisher RM (2012). Expression of ceramide-metabolising enzymes in subcutaneous and intra-abdominal human adipose tissue. Lipids Health Dis 11, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovilakath A, Jamil M, and Cowart LA (2020). Sphingolipids in the Heart: From Cradle to Grave. Front Endocrinol (Lausanne) 11, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski GM, Carey AL, Selathurai A, Kingwell BA, and Bruce CR (2013). Plasma sphingosine-1-phosphate is elevated in obesity. PLoS One 8, e72449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurano M, Tsukamoto K, Shimizu T, Kassai H, Nakao K, Aiba A, Hara M, and Yatomi Y (2020). Protection Against Insulin Resistance by Apolipoprotein M/Sphingosine-1-Phosphate. Diabetes 69, 867–881. [DOI] [PubMed] [Google Scholar]
- Kusminski CM, Bickel PE, and Scherer PE (2016). Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov 15, 639–660. [DOI] [PubMed] [Google Scholar]
- Law BA, Liao X, Moore KS, Southard A, Roddy P, Ji R, Szulc Z, Bielawska A, Schulze PC, and Cowart LA (2018). Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J 32, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Hong IK, Kim BR, Shim SM, Sung Lee J, Lee HY, Soo Choi C, Kim BK, and Park TS (2015). Activation of sphingosine kinase 2 by endoplasmic reticulum stress ameliorates hepatic steatosis and insulin resistance in mice. Hepatology 62, 135–146. [DOI] [PubMed] [Google Scholar]
- Lee SY, Lee HY, Song JH, Kim GT, Jeon S, Song YJ, Lee JS, Hur JH, Oh HH, Park SY, et al. (2017). Adipocyte-Specific Deficiency of De Novo Sphingolipid Biosynthesis Leads to Lipodystrophy and Insulin Resistance. Diabetes 66, 2596–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre RN, Yu C, Hoofnagle A, Hari N, Jensen PN, Fretts AM, Umans JG, Howard BV, Sitlani CM, Siscovick DS, et al. (2018). Circulating Sphingolipids, Insulin, HOMA-IR, and HOMA-B: The Strong Heart Family Study. Diabetes 67, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, and Gordon JI (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. [DOI] [PubMed] [Google Scholar]
- Li C, Zheng S, You H, Liu X, Lin M, Yang L, and Li L (2011). Sphingosine 1-phosphate (S1P)/S1P receptors are involved in human liver fibrosis by action on hepatic myofibroblasts motility. J. Hepatol 54, 1205–1213. [DOI] [PubMed] [Google Scholar]
- Liu M, Frej C, Langefeld CD, Divers J, Bowden DW, Carr JJ, Gebre AK, Xu J, Larsson B, Dahlback B, et al. (2019). Plasma apoM and S1P levels are inversely associated with mortality in African Americans with type 2 diabetes mellitus. J. Lipid Res 60, 1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone MA, Santos T, Alecu I, Silva LC, and Hornemann T (2019). 1-Deoxysphingolipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 512–521. [DOI] [PubMed] [Google Scholar]
- Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, Hyotylainen T, and Yki-Jarvinen H (2016). Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol 64, 1167–1175. [DOI] [PubMed] [Google Scholar]
- Ma MM, Chen JL, Wang GG, Wang H, Lu Y, Li JF, Yi J, Yuan YJ, Zhang QW, Mi J, et al. (2007). Sphingosine kinase 1 participates in insulin signalling and regulates glucose metabolism and homeostasis in KK/Ay diabetic mice. Diabetologia 50, 891–900. [DOI] [PubMed] [Google Scholar]
- Maceyka M, and Spiegel S (2014). Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahashi M, Takabe K, Liu R, Peng K, Wang X, Wang Y, Hait NC, Allegood JC, Yamada A, Aoyagi T, et al. (2015). Conjugated Bile Acid Activated S1P Receptor 2 Is a Key Regulator of Sphingosine Kinase 2 and Hepatic Gene Expression. Hepatology 61, 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwabuo CC, Duncan M, Xanthakis V, Peterson LR, Mitchell GF, McManus D, Cheng S, and Vasan RS (2019). Association of Circulating Ceramides With Cardiac Structure and Function in the Community: The Framingham Heart Study. J. Am. Heart Assoc 8, e013050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi GJ, Meikle PJ, Huynh K, Earnest A, Roberts SK, Kemp W, Parker BL, Brown W, Burton P, and Watt MJ (2021). Hepatic lipidomic remodeling in severe obesity manifests with steatosis and does not evolve with non-alcoholic steatohepatitis. J. Hepatol in press. [DOI] [PubMed] [Google Scholar]
- Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, et al. (2008). Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res 49, 2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault L, Newsom SA, Strauss A, Kerege A, Kahn DE, Harrison KA, Snell-Bergeon JK, Nemkov T, D’Alessandro A, Jackman MR, et al. (2018). Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 3, e96805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, and Jurczak MJ (2016). CrossTalk opposing view: Intramyocellular ceramide accumulation does not modulate insulin resistance. J. Physiol 594, 3171–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MC, and Shulman GI (2017). Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol. Sci 38, 649–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LR, Jiang X, Chen L, Goldberg AC, Farmer MS, Ory DS, and Schaffer JE (2020). Alterations in plasma triglycerides and ceramides: links with cardiac function in humans with type 2 diabetes. J. Lipid Res 61, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LR, Xanthakis V, Duncan MS, Gross S, Friedrich N, Volzke H, Felix SB, Jiang H, Sidhu R, Nauck M, et al. (2018). Ceramide Remodeling and Risk of Cardiovascular Events and Mortality. J. Am. Heart Assoc 7, e007931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier B, Briand V, Kadereit D, Schafer M, Wohlfart P, Philippo MC, Caillaud D, Gouraud L, Grailhe P, Bidouard JP, et al. (2020). A G protein-biased S1P1 agonist, SAR247799, protects endothelial cells without affecting lymphocyte numbers. Sci. Signal 13, eaax8050. [DOI] [PubMed] [Google Scholar]
- Powell DJ, Hajduch E, Kular G, and Hundal HS (2003). Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol. Cell Biol 23, 7794–7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Chen J, Lay A, Don A, Vadas M, and Xia P (2013). Loss of sphingosine kinase 1 predisposes to the onset of diabetes via promoting pancreatic beta-cell death in diet-induced obese mice. FASEB J 27, 4294–4304. [DOI] [PubMed] [Google Scholar]
- Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, Dogra S, Ohman MK, Takeda K, Sugii S, et al. (2014). CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 20, 687–695. [DOI] [PubMed] [Google Scholar]
- Ravichandran S, Finlin BS, Kern PA, and Ozcan S (2019). Sphk2(−/−) mice are protected from obesity and insulin resistance. Biochim Biophys Acta Mol. Basis Dis 1865, 570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach T, Maceyka M, and Spiegel S (2017). Sphingosine kinase and sphingosine-1-phosphate in liver pathobiology. Crit. Rev. Biochem. Mol. Biol 52, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach TD, Asgharpour A, Maczis MA, Montefusco D, Cowart LA, Bedossa P, Sanyal AJ, and Spiegel S (2019). FTY720/fingolimod decreases hepatic steatosis and expression of fatty acid synthase in diet-induced nonalcoholic fatty liver disease in mice. J. Lipid Res 60, 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, and Hobbs HH (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 40, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross JS, Hu W, Rosen B, Snider AJ, Obeid LM, and Cowart LA (2013). Sphingosine kinase 1 is regulated by peroxisome proliferator-activated receptor alpha in response to free fatty acids and is essential for skeletal muscle interleukin-6 production and signaling in diet-induced obesity. J. Biol. Chem 288, 22193–22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz M, Frej C, Holmer A, Guo LJ, Tran S, and Dahlback B (2017). High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler. Thromb. Vasc. Biol 37, 118–129. [DOI] [PubMed] [Google Scholar]
- Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, and Cowart LA (2012). Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J. Clin. Invest 122, 3919–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SB, Tidhar R, Futerman AH, and Cowart LA (2013). Myristate-derived d16:0 sphingolipids constitute a cardiac sphingolipid pool with distinct synthetic routes and functional properties. J. Biol. Chem 288, 13397–13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve M, Hui SK, Dinh DD, Foltz WD, Momen A, Nedospasov SA, Offermanns S, Husain M, Kroetsch JT, Lidington D, et al. (2016). Tumor Necrosis Factor/Sphingosine-1-Phosphate Signaling Augments Resistance Artery Myogenic Tone in Diabetes. Diabetes 65, 1916–1928. [DOI] [PubMed] [Google Scholar]
- Schmelz E, Crall K, Larocque R, Dillehay D, and Merrill AJ (1994). Uptake and metabolism of sphingolipids in isolated intestinal loops of mice. J. Nutr 124, 702–712. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Zhou YT, Levi M, and Unger RH (1998). Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 95, 2498–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoura A, Michaud J, Im DS, Thangada S, Xiong Y, Smith JD, and Hla T (2011). Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol 31, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovbro M, Baranowski M, Skov-Jensen C, Flint A, Dela F, Gorski J, and Helge JW (2008). Human skeletal muscle ceramide content is not a major factor in muscle insulin sensitivity. Diabetologia 51, 1253–1260. [DOI] [PubMed] [Google Scholar]
- Song Z, Wang W, Li N, Yan S, Rong K, Lan T, and Xia P (2019). Sphingosine kinase 2 promotes lipotoxicity in pancreatic beta-cells and the progression of diabetes. FASEB J 33, 3636–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, and Milstien S (2003). Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol 4, 397–407. [DOI] [PubMed] [Google Scholar]
- Spiegel S, and Milstien S (2011). The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol 11, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford S, Hoehn KL, Liu F, and Summers SA (2004). Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem 279, 36608–36615. [DOI] [PubMed] [Google Scholar]
- Summers SA (2018). Could Ceramides Become the New Cholesterol? Cell Metab 27, 276–280. [DOI] [PubMed] [Google Scholar]
- Summers SA, Chaurasia B, and Holland WL (2019). Metabolic Messengers: ceramides. Nat. Metab 1, 1051–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydor S, Sowa JP, Megger DA, Schlattjan M, Jafoui S, Wingerter L, Carpinteiro A, Baba HA, Bechmann LP, Sitek B, et al. (2017). Acid sphingomyelinase deficiency in Western diet-fed mice protects against adipocyte hypertrophy and diet-induced liver steatosis. Mol. Metab 6, 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabassum R, Ramo JT, Ripatti P, Koskela JT, Kurki M, Karjalainen J, Palta P, Hassan S, Nunez-Fontarnau J, Kiiskinen TTJ, et al. (2019). Genetic architecture of human plasma lipidome and its link to cardiovascular disease. Nat. Commun 10, 4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe H, Fujii Y, Okada-Iwabu M, Iwabu M, Kano K, Kawana H, Hato M, Nakamura Y, Terada T, Kimura-Someya T, et al. (2020). Human adiponectin receptor AdipoR1 assumes closed and open structures. Commun. Biol 3, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Lim XY, Toop HD, Osborne B, Brandon AE, Taylor EN, Fiveash CE, Govindaraju H, Teo JD, McEwen HP, et al. (2018). A selective inhibitor of ceramide synthase 1 reveals a novel role in fat metabolism. Nat. Commun 9, 3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, Mauer J, Xu E, Hammerschmidt P, Bronneke HS, et al. (2014). Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 20, 678–686. [DOI] [PubMed] [Google Scholar]
- Turpin-Nolan SM, Hammerschmidt P, Chen W, Jais A, Timper K, Awazawa M, Brodesser S, and Bruning JC (2019). CerS1-Derived C18:0 Ceramide in Skeletal Muscle Promotes Obesity-Induced Insulin Resistance. Cell Rep 26, 1–10 e17. [DOI] [PubMed] [Google Scholar]
- Vaidya M, Jentsch JA, Peters S, Keul P, Weske S, Graler MH, Mladenov E, Iliakis G, Heusch G, and Levkau B (2019). Regulation of ABCA1-mediated cholesterol efflux by sphingosine-1-phosphate signaling in macrophages. J. Lipid Res 60, 506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisar T, Couzens E, Hwang A, Russell M, Barlow CE, DeFina LF, Hoofnagle AN, and Kim F (2018). Type 2 diabetes is associated with loss of HDL endothelium protective functions. PLoS One 13, e0192616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliauskaite-Brooks I, Sounier R, Rochaix P, Bellot G, Fortier M, Hoh F, De Colibus L, Bechara C, Saied EM, Arenz C, et al. (2017). Structural insights into adiponectin receptors suggest ceramidase activity. Nature 544, 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Toledo E, Hruby A, Rosner BA, Willett WC, Sun Q, Razquin C, Zheng Y, Ruiz-Canela M, Guasch-Ferre M, et al. (2017a). Plasma Ceramides, Mediterranean Diet, and Incident Cardiovascular Disease in the PREDIMED Trial (Prevencion con Dieta Mediterranea). Circulation 135, 2028–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Badeanlou L, Bielawski J, Ciaraldi TP, and Samad F (2014). Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am. J. Physiol. Endocrinol. Metab 306, E756–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li C, Lin X, Sun H, Xu R, Li Q, Wei Y, Li Y, Qian J, Liu C, et al. (2020). Targeting alkaline ceramidase 3 alleviates the severity of nonalcoholic steatohepatitis by reducing oxidative stress. Cell Death. Dis 11, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X, Qiang X, Sun L, Gurley EC, Lai G, et al. (2017b). The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 65, 2005–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyler J, Verrijken A, Hornemann T, Vonghia L, Dirinck E, von Eckardstein A, Vanwolleghem T, Michielsen P, Peiffer F, Driessen A, et al. (2021). Association of 1-deoxy-sphingolipids with steatosis but not steatohepatitis nor fibrosis in non-alcoholic fatty liver disease. Acta Diabetol 58, 319–327. [DOI] [PubMed] [Google Scholar]
- Wigger L, Cruciani-Guglielmacci C, Nicolas A, Denom J, Fernandez N, Fumeron F, Marques-Vidal P, Ktorza A, Kramer W, Schulte A, et al. (2017). Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep 18, 2269–2279. [DOI] [PubMed] [Google Scholar]
- Wilkerson BA, Grass GD, Wing SB, Argraves WS, and Argraves KM (2012). Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: high density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J. Biol. Chem 287, 44645–44653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia JY, Holland WL, Kusminski CM, Sun K, Sharma AX, Pearson MJ, Sifuentes AJ, McDonald JG, Gordillo R, and Scherer PE (2015). Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab 22, 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Yagai T, Luo Y, Liang X, Chen T, Wang Q, Sun D, Zhao J, Ramakrishnan SK, Sun L, et al. (2017). Activation of intestinal hypoxia-inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat. Med 23, 1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, and Samad F (2009). Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab 297, E211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MM, and Wang HG (2018). Sphingolipids as Regulators of Autophagy and Endocytic Trafficking. Adv. Cancer Res 140, 27–60. [DOI] [PubMed] [Google Scholar]
- Zigdon H, Kogot-Levin A, Park JW, Goldschmidt R, Kelly S, Merrill AH Jr., Scherz A, Pewzner-Jung Y, Saada A, and Futerman AH (2013). Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J. Biol. Chem 288, 4947–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]