

Supplemental Digital Content is available in the text.
Keywords: bacterial killing, humans; complex II; macrophages; metabolism; mitochondrial respiration; monocytes; salt
Abstract
Background:
Dietary high salt (HS) is a leading risk factor for mortality and morbidity. Serum sodium transiently increases postprandially but can also accumulate at sites of inflammation affecting differentiation and function of innate and adaptive immune cells. Here, we focus on how changes in extracellular sodium, mimicking alterations in the circulation and tissues, affect the early metabolic, transcriptional, and functional adaption of human and murine mononuclear phagocytes.
Methods:
Using Seahorse technology, pulsed stable isotope-resolved metabolomics, and enzyme activity assays, we characterize the central carbon metabolism and mitochondrial function of human and murine mononuclear phagocytes under HS in vitro. HS as well as pharmacological uncoupling of the electron transport chain under normal salt is used to analyze mitochondrial function on immune cell activation and function (as determined by Escherichia coli killing and CD4+ T cell migration capacity). In 2 independent clinical studies, we analyze the effect of a HS diet during 2 weeks (URL: http://www.clinicaltrials.gov. Unique identifier: NCT02509962) and short-term salt challenge by a single meal (URL: http://www.clinicaltrials.gov. Unique identifier: NCT04175249) on mitochondrial function of human monocytes in vivo.
Results:
Extracellular sodium was taken up into the intracellular compartment, followed by the inhibition of mitochondrial respiration in murine and human macrophages. Mechanistically, HS reduces mitochondrial membrane potential, electron transport chain complex II activity, oxygen consumption, and ATP production independently of the polarization status of macrophages. Subsequently, cell activation is altered with improved bactericidal function in HS-treated M1-like macrophages and diminished CD4+ T cell migration in HS-treated M2-like macrophages. Pharmacological uncoupling of the electron transport chain under normal salt phenocopies HS-induced transcriptional changes and bactericidal function of human and murine mononuclear phagocytes. Clinically, also in vivo, rise in plasma sodium concentration within the physiological range reversibly reduces mitochondrial function in human monocytes. In both a 14-day and single meal HS challenge, healthy volunteers displayed a plasma sodium increase of  and
and  respectively, that correlated with decreased monocytic mitochondrial oxygen consumption.
respectively, that correlated with decreased monocytic mitochondrial oxygen consumption.
Conclusions:
Our data identify the disturbance of mitochondrial respiration as the initial step by which HS mechanistically influences immune cell function. Although these functional changes might help to resolve bacterial infections, a shift toward proinflammation could accelerate inflammatory cardiovascular disease.
Clinical Perspective.
What Is New?
High extracellular salt can disturb mitochondrial function of murine and human mononuclear phagocytes.
Mitochondrial function has a profound impact on immune cell activation and function.
Minimal changes in plasma sodium concentration on high dietary salt can rapidly and transiently inhibit mitochondrial function of circulating monocytes.
What Are the Clinical Implications?
High salt–induced shift toward proinflammatory macrophage activation can help fight off bacterial infections while worsening end-organ damage in the context of cardiovascular disease.
Salt sensitivity is typically related to blood pressure. Mitochondrial function analyses in monocytes and other immune cells on a high salt challenge could serve as test for salt sensitivity of immune cells.
A Western lifestyle is associated with an increased risk for hypertension and cardiovascular disease.1 In modern societies, the consumption of processed foods is on the rise. Unfortunately, they generally contain a high salt (HS) content, leading to a salt intake often greater than the recommendations of medical guidelines.2 High sodium (Na+) content of the so-called Western diet is linked to numerous pathologies,3 in particular to cardiometabolic diseases.4 On salt challenge, serum Na+ stays remarkably constant and only transiently changes by 2 to 4 mM shortly after a HS meal before returning to normal.5,6 In contrast, recent data demonstrate that Na+ can accumulate in various tissues and may even reach hypertonic concentrations in the interstitial fluid. Na+ magnetic resonance imaging measurements in humans and absolute Na+ quantification in experimental models demonstrate that both chronic HS intake and local inflammation can lead to tissue Na+ accumulation in skin and muscle.7,8 Furthermore, tissue Na+ accumulates in humans on aging,9 in patients with hypertension,9 primary hyperaldosteronism,10 heart failure,11 and end-stage renal disease,12 but also in noncardiovascular disease, such as autoimmune disease.13 Of note, tissue Na+ can be removed by sodium-glucose cotransporter 2 inhibition (SGLT2i) in patients with type 2 diabetes,14 by diuretics in patients with heart failure,11 and after surgery of aldosterone-producing tumors.10 Furthermore, macrophages (MΦ) possess a homeostatic function, by which they sense and regulate peripheral salt and water balance.15 Conversely, HS boosts in vitro and in vivo proinflammatory M1 MΦ8,16–19 and helper T type 17 cell20–23 responses, whereas it inhibits anti-inflammatory regulatory T cells23–25 and M2 MΦ.18,26 These findings indicate that Na+ is an important factor in tissue microenvironment, probably acting also at the cellular level of immune cells, and thereby impacts disease.
MΦ play a pivotal role in the pathogenesis of atherosclerosis, acute myocardial infarction, and heart failure, highlighting the importance of myeloid cells for cardiovascular health and cardiovascular disease.27 MΦ display a wide range of phenotypes. They sense signals from the environment, followed by profound energetic and transcriptional reprogramming.28 This reprogramming determines MΦ functions, such as immune responses, homeostatic tissue function, phagocytosis, and clearance of apoptotic cells, and defense against pathogens and malignancies.29 The programming of MΦ is directly linked to changes in central metabolic pathways. Mitochondria synthesize ATP by oxidative phosphorylation. For efficient oxidative phosphorylation, mitochondria create a membrane potential (ψ) across their inner membrane, determined by the proton (H+) gradient generated by the electron transport chain (ETC) to drive the synthesis of ATP.30 In vitro analyses of “classic” lipopolysaccharide (LPS)-activated macrophages (M[LPS]) have shown that the latter increase glycolysis and pentose phosphate pathways, and exhibit a truncated tricarboxylic acid cycle that effectively redirects their mitochondria from ATP to reactive oxygen species production.31,32 On the other hand, late-phase M2-like MΦ upregulate fatty acid oxidation and oxidative phosphorylation.33,34 These experiments are mostly performed 24 hours after activation and therefore reflect an advanced state of phenotypic adaptation. By contrast, little is known about early metabolic alterations that occur within the first hours of activation and environmental adaptation. Here we examine how changes in Na+, mimicking alterations in the circulation (+4 mM) and tissues (+40 mM), might affect the early metabolism of monocytes and MΦ, respectively. We hypothesize that mononuclear phagocytes challenged by an altered Na+ microenvironment immediately adapt their cellular metabolism, activation status, and function.
Methods
The authors declare that all supporting data and analytical methods are available within the article and its Data Supplement. All materials used for this study are listed with their respective sources in Table I in the Data Supplement. The data, analytical methods, and study materials that support the findings of this study are available from the corresponding authors on reasonable request.
Human Material
Human monocytes were derived from venous whole blood, drawn from healthy volunteers in accordance with the ethical standards of the institutional review board of the Charité-Universitätsmedizin Berlin (ethical approval EA2/046/17), and written informed consent was obtained from all participants before study entry. Subjects were recruited locally at the Experimental and Clinical Research Center Berlin-Buch. The long-term salt intervention study reevaluated here was previously performed in our laboratory (URL: http://www.clinicaltrials.gov. Unique identifier: NCT02509962). The short-term salt intervention study (pizza meal challenge) can be found at Clinical Trials (URL: http://www.clinicaltrials.gov. Unique identifier: NCT04175249).
Volunteers and study participants recruited during this study were between 24 and 56 years old. Study participants from the long-term salt intervention study (URL: http://www.clinicaltrials.gov. Unique identifier: NCT02509962) were all male. Study participants from the short-term salt intervention study (URL: http://www.clinicaltrials.gov. Unique identifier: NCT04175249), as well as volunteers for the in vitro +2 and +4 mM NaCl treatment, were equally distributed (10 male, 10 female). Volunteers for the human monocytes and human Escherichia coli killing studies were healthy donors.
Human peripheral blood mononucleated cells (PBMCs) used for functional killing experiments were drawn from healthy volunteers with the ethical approval 20-1707-101 from the institutional review board of the University of Regensburg. In addition, PBMCs were obtained from the peripheral blood of healthy subjects or isolated from buffy coats of healthy donors purchased from the Belgian Red Cross in compliance with institutional review board protocols (CME2019/042 and CME2016/629).
Isolation of PBMCs
Venous whole blood was drawn from healthy volunteers into K3-EDTA tubes. PBMCs were separated by density gradient centrifugation, using isotonic Biocoll solution (L6115, Biochrom by Merck) and SepMate tubes (85460, Stemcell Technologies). After hypotonic lysis of remaining erythrocytes with 155 mM NH4Cl (1145.0500, Merck), 10 mM NaHCO3 (017 K13779929, Merck), and 1 mM EDTA (E-4884, Sigma by Merck) in water, cells were washed and frozen in medium, supplemented with 10% (v/v) fetal bovine serum (S0615, Biochrom by Merck) and 10% (v/v) dimethyl sulfoxide (D4540, Sigma by Merck).
Monocyte Isolation Out of PBMCs
Monocyte isolation was performed using the Pan Monocyte Isolation Kit human (130-096-537, Miltenyi Biotec) and the MultiMACS Cell24 Separator Plus (Miltenyi Biotec) according to the manufacturer’s instructions. Purity of monocyte enrichment was determined by flow-cytometric analysis, using antihuman CD14 (130-110-579, Miltenyi Biotec; 12-0149-42, Invitrogen), antihuman CD16 (130-106-703, Miltenyi Biotec; 17-0168-42, Invitrogen), and the LIVE/DEAD Fixable Aqua Dead Cell Stain kit, for 405-nm excitation (L34966, Invitrogen by Thermo Fisher Scientific). Cells were analyzed using a FACSCanto II flow cytometer and FACSDiva software (BD). Data analysis was performed with FlowJo.
Generation of Human Macrophages
Monocytes were sorted from PBMCs by using the EasySep CD14 Positive Selection kit II (17858, Stemcell Technologies) according to the manufacturer’s protocol. Sorted monocytes were further differentiated in X-VIVO15 medium (BE02-060F, Lonza), supplemented with 10% FCS (S1400, Biowest) as well as 100 ng/mL macrophage colony-stimulating factor (300-25, Peprotech) for 7 days.
Generation of Murine BMDMs
Animal care and use followed the regulations of the German Animal Welfare Act. The procedures followed were approved by the local State authorities (X9009/18) or by Umweltamt der Stadt Regensburg and performed in accordance with institutional guidelines. Murine MΦ were generated from the bone marrow of male, 9- to 12-week-old, wild type C57BL/6J mice. Animals were purchased from Harlan Laboratories and Charles River and bred at the animal facility from the Max Delbrück Center, Berlin, and from the University of Regensburg, respectively. Mice were housed under standard light-dark cycled (12 h/12 h) and specific pathogen–free conditions. They were fed a normal chow diet (V1124-300, Ssniff Spezialdiäten GmbH) and tap water ad libitum. Bone marrow cells were differentiated for 9 to 10 days in DMEM (41966-029, Gibco by Thermo Fisher Scientific), supplemented with 10% (v/v) fetal bovine serum (S0615, Biochrom by Merck), 5% (v/v) adult horse serum (S-HEU03-I, Cell Concepts), 1% (v/v) nonessential amino acids (M7145, Sigma by Merck), 50 μM ß-mercaptoethanol (M3148, Sigma by Merck), and 20% (v/v) L929 cell conditioned medium in in-house–sealed, hydrophobic Teflon-coated bags (FT FEP 100C, DuPont/American Durafilm) at 37°C and 10% CO2. Purity of BMDMs was regularly determined by flow-cytometric analysis and consistently at ≥90% CD11b+ F4/80+ double-positive cells (553310, BD Biosciences and 48-4801-80, eBioscience by Thermo Fisher Scientific, respectively).
Macrophage Activation and Stimulation
BMDMs as well as human blood-derived monocytes were cultivated in DMEM (11966-025, Gibco by Thermo Fisher Scientific), supplemented with 10 mM glucose (X997.2, Carl Roth), 10% (v/v) fetal bovine serum (S0615, Biochrom by Merck), 1% (v/v) penicillin-streptomycin (15140-122, Gibco by Thermo Fisher Scientific), 1% (v/v) HEPES (15630-056, Gibco by Thermo Fisher Scientific), and 50 μM ß-mercaptoethanol (M3148, Sigma by Merck). BMDMs were activated with 10 ng/mL LPS (L3024, Sigma by Merck) or 10 ng/mL interleukin (IL) 4 (404-ML-010, R&D Systems by bio-techne) and 10 ng/mL IL13 (PMC0134, Invitrogen by Thermo Fisher Scientific). Human MΦ were activated with 100 ng/mL macrophage colony-stimulating factor (300-25, Peprotech), 100 ng/mL ultrapure LPS (tlrl-3pelps, Invivogen) and 20 ng/mL interferon (IFN)γ (300-02, Peprotech) or with 100 ng/mL macrophage colony-stimulating factor (300-25, Peprotech) and 20 ng/mL IL-4 (113040045, Immunotools). For the activation under HS conditions, respective concentrations of sodium chloride (NaCl, 9265.1, Carl Roth) were added to the medium. Pharmacological inhibition of mitochondrial function was performed with Oligomycin A (75351, Sigma by Merck), N5,N6-bis(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (BAM15, SML1760-5MG, Sigma by Merck), rotenone (R8875, Sigma by Merck) or antimycin A (A8674, Sigma by Merck) at concentrations of 10 µM, respectively. DMSO (D4540, Sigma by Merck) was included as solvent control if necessary. Dimethyl malonate (136441, Sigma by Merck) was used at a concentration of 10 mM.
Bacterial Infection
E. coli infection experiments are described in the Supplemental Methods in the Data Supplement.
Seahorse
Real-time measurement of oxygen consumption rate (OCR) was performed using the Seahorse XFe96 FluxPak (102416-100, Agilent) and the Seahorse XFe Analyzer (Agilent). BMDMs and human monocytes were cultivated (and stimulated) in XFe96 microplates as described under Macrophage Activation and Stimulation. Before the analysis, cells were washed with unbuffered, DMEM-formulated Seahorse base medium (102363-100, Agilent), supplemented with 10 mM glucose (X997.2, Carl Roth) and 2 mM L-glutamine (25030-081, Gibco by Thermo Fisher Scientific) and incubated for 30 to 60 minutes at 37°C, without CO2. For the analysis, cells were then loaded onto the Seahorse XFe Analyzer together with a precalibrated disposable cartridge. A mitochondrial stress test was performed. The assay was designed to repeat 5 cycles (of 3 min mixing followed by 3 min measuring) per phase. During the assay, 2 µM Oligomycin A (75351, Sigma by Merck), 1 µM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (C2920, Sigma by Merck), 1 µM rotenone (R8875, Sigma by Merck), and 1µM antimycin A (A8674, Sigma by Merck) were injected. After the assay, a nuclear staining with Hoechst (B2261, Sigma by Merck) was performed (10 µg/mL in cell culture medium for 45 minutes at 37°C). Cells were then washed with PBS and detached with 2 mM EDTA in PBS for 10 min on ice. Cell number was then determined on a BD LSRFortessa with high-throughput sampler (BD) using FACSDiva and FlowJo to normalize OCR to cell count per well. Human blood-derived monocytes were treated equally. However, as monocytes are nonadherent, cells needed to be centrifuged between washes (for 5 min at 300 g), and detaching was not necessary.
Pulsed Stable Isotope-Resolved Metabolomics
BMDMs were cultivated in 10-cm cell culture dishes and stimulated as indicated above. One hour before the stimulatory end point (ie, after 2 hours and 23 hours), medium was changed to U-13C-glucose (CLM-1396-PK, Cambridge Isotope Laboratories) or U-13C-glutamine (CLM-1822-H-PK, Cambridge Isotope Laboratories) containing DMEM (A14430-01, Gibco by Thermo Fisher Scientific), supplemented with 10% (v/v) fetal bovine serum (S0615, Biochrom by Merck), 1% (v/v) penicillin-streptomycin (15140-122, Gibco by Thermo Fisher Scientific), 1% (v/v) HEPES (15630-056, Gibco by Thermo Fisher Scientific), 50 μM ß-mercaptoethanol (M3148, Sigma by Merck), 10 mM glucose, and 2 mM glutamine (unlabeled or U-13C-labeled, depending on the experiment). After 1 hour of 13C-labeling (including the respective stimulants), cells were washed with a buffer containing 140 mM NaCl, 5 mM HEPES, and 4 mM KCl and lysed in methanol-chloroform-water (5:2:1 v/v/v), containing 2 µg/mL cinnamic acid (133760, Sigma by Merck) as internal control. For metabolite extraction, cell lysates were shaken for 60 min at 4°C and centrifuged for 15 min at 4°C and 10 000 g. Polar phase was collected and dried under vacuum. Extracts were resuspended in 20% methanol, shaken for 60 min at 4°C and polar phase was again dried under vacuum. Sample derivatization was performed as described elsewhere.35 In parallel to the samples, a set of Ident and Quant mixes were extracted, derivatized, and measured.35 These allow a more reliable identification of isomeric metabolites as well as quantification of 63 compounds, respectively.
The samples and Ident/Quant standards were injected in split mode (split 1:5, injection volume 1 µL) in a temperature-controlled injector (CAS4, Gerstel) with a baffled glass liner (Gerstel). The following temperature program was applied during sample injection: initial temperature of 80°C for 30 s followed by a ramp with 12°C/min to 120°C and a second ramp with 7°C/min to 300°C and final hold for 2 minutes. Gas chromatographic separation was performed on an Agilent 6890 N, equipped with a VF-5 ms column. Helium was used as carrier gas with a flow rate of 1.2 mL/min. Gas chromatography was performed with the following temperature gradient: 2-minute heating at 67.5°C, first temperature gradient with 5°C/min up to 120°C; subsequently, a second temperature increase of 7°C/min up to 200°C, 12°C/min up to 320°C and a hold of 6 minutes. The spectra were recorded in a mass range of 60 to 600 mass units with 10 spectra/s at a detector voltage of 1650 V. The gas chromatography–mass spectrometry chromatograms were initially preprocessed with ChromaTOF (Leco). Metabolite identification and quantification were performed with MAUI-SILVIA.36 Metabolite quantities were normalized to protein content (determined in parallel), the internal control cinnamic acid, and the sum of all peak areas. Data processing was performed with R (MTXQC).
Intracellular Na+ Quantification
Human monocytes (106 cells) were stimulated with HS for 60 minutes, harvested, and washed with iso-osmolal sucrose solution. The pellet was lysed, and total Na+ was determined by atomic absorption spectrometry (iCE 3000 Series, Thermo Scientific).37
Statistical Analyses
No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to the sample allocation during experiments and outcome assessment. Statistical analyses were performed with GraphPad Prism Version 8.3.0. Normality of the data were tested by the Kolmogorov-Smirnov test. Significance between 2 groups was analyzed by t test (when normally distributed), Mann-Whitney test (when nonnormally distributed) or Wilcoxon matched-pairs signed-rank test (for nonnormally distributed, matched data). In Na+ quantifications, outliers were identified with ROUT and excluded from analysis. For >2 groups with 1 variable only, 1-way ANOVA with a Tukey post hoc test (for normally distributed data) or Kruskal-Wallis test with a Dunn post hoc test (for nonnormally distributed data) were used. Matched data with more than 2 groups (or time points) were analyzed by Friedman test, and false discovery rate (FDR) correction was performed using the Benjamini-Hochberg procedure. The relationship between normal distributed data sets with single time point comparison was determined by Pearson correlation and linear regression analysis. Comparison between multiple time points was performed by linear mixed-effects model applied to rank-transformed between–time point difference data. Exact statistical tests used are described in the respective figure legends. Respective P values were depicted in the figures. After false discovery rate correction, q values were reported. For the statistical analysis of the gene expression data, the Mann-Whitney U test comparing M(LPS) and M(IL4+IL13) at each time point to untreated M0, as well as comparing normal salt (NS) to HS at each time point for each activation group, was performed. False discovery rate correction was done across genes using the Benjamini-Hochberg procedure.
Results
HS Inhibits Mitochondrial Respiration in Murine and Human Mononuclear Phagocytes
The importance of cellular metabolism for immune cell function in the advanced phase has been extensively studied.32,38,39 However, little is known about how murine and human MΦ adapt their cellular metabolism in the first hours after sensing a change in a microenvironment. Therefore, we performed real-time measurement of glycolysis and mitochondrial respiration after activating murine BMDMs (Figure 1A, Figure IA in the Data Supplement) for 3 hours under NS or HS (addition of 40 mM NaCl) conditions. It is interesting that although the extracellular acidification rate was not affected (Figure IB in the Data Supplement), basal and maximal OCR were decreased under HS (Figure 1B, Figure IC and ID in the Data Supplement). Accordingly, HS induced a significant reduction in ATP levels (Figure 1C) and ψ, determined by tetramethylrhodamine ethyl ester flow cytometry (Figure 1D, Figure IE in the Data Supplement). Recently, we showed that an additional 40 mM of extracellular Na+ increases intracellular Na+ within minutes by approximately 20%, accompanied by an enhanced antibacterial defense.17 To mimic the effect of raised intracellular Na+ and to demonstrate that Na+ directly affects mitochondrial function, we treated isolated mitochondria from unactivated BMDM with increasing concentrations of NaCl and found the ψ to decrease dose-dependently (Figure 1E). In line with our data, Ip and Medzhitov previously reported an inhibition of mitochondrial respiration in nonactivated MΦ under HS, although at much higher concentrations (100 mM).19 To verify our findings, we next sought to determine whether HS diminishes mitochondrial function in human mononuclear phagocytes. We treated freshly isolated CD14+ human monocytes (Figure IF in the Data Supplement) as well as monocyte-derived MΦ (Figure 1F) with HS. In human monocytes, HS led to a significant increase in intracellular Na+ content (Figure 1G). As for murine MΦ, we found significantly decreased OCR (Figure 1H and 1I), ATP levels (Figure 1J), and ψ (Figure 1K) compared with NS, independent of the activation status. Furthermore and in agreement with our previous findings,17 short-term HS challenge significantly accelerated E. coli killing in murine MΦ (Figure 1L) and human CD14+ monocytes (Figure 1M). We next tested whether HS would not only alter M1-like killing capacity but also affect M2-like MΦ functionality. As a surrogate assay, we measured T cell migration, which is promoted by M2-like MΦ function.40 Indeed, HS-treated M(IL-4) less potently promoted CD4+ T cell and CD4+ regulatory T cell migration (Figure 1N). Taken together, these data demonstrate that a change in the sodium microenvironment results in a rapid loss of mitochondrial function consistently in all M1- and M2-like murine and human MΦ, highlighting a species-overarching regulatory mechanism. The functional outcome, however, was different in M1- and M2-like MΦ.
Figure 1.
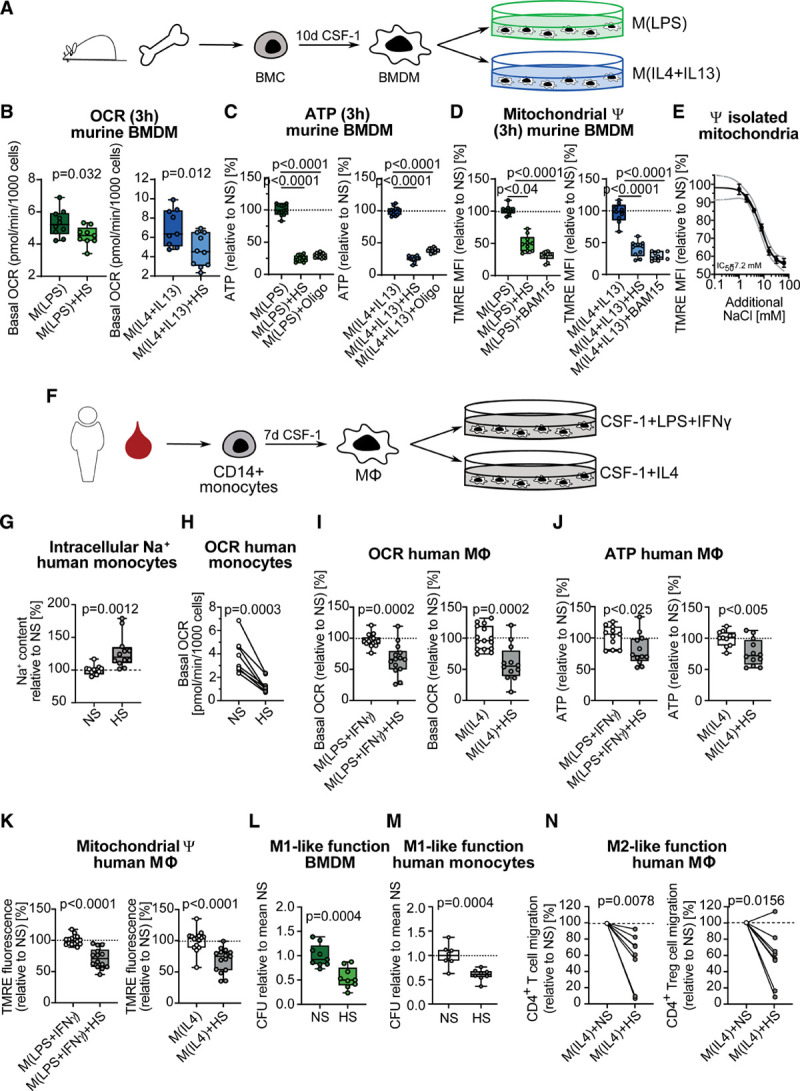
High salt inhibits mitochondrial respiration in murine and human mononuclear phagocytes. A, Pictogram of the generation and activation of murine bone marrow–derived macrophages (BMDMs). B through D, BMDMs were treated for 3 hours with lipopolysaccharide (LPS) or interleukin (IL) 4+IL13, under normal salt (NS) or high salt (HS) conditions. (B) Basal oxygen consumption rate (OCR) (pooled data n=10 from 3 independent experiments). Data are depicted as box and whisker with minimum to maximum. Significance was analyzed by unpaired, 2-tailed t test. C, Relative ATP content, with Oligomycin A (Oligo, 10 µM) treatment as positive control for ATP decrease (pooled data n=12 from 2 independent experiments). Data are depicted as box and whisker with minimum to maximum. Significance was analyzed by 1-way ANOVA with Tukey’s post hoc test. D, Relative TMRE mean fluorescence intensity (MFI), with mitochondrial uncoupler BAM15 (10 µM) as positive control for mitochondrial depolarization (pooled data n=10 from 2 independent experiments). Data are depicted as box and whisker with minimum to maximum. Significance was analyzed by Kruskal-Wallis test with Dunn’s post hoc test (LPS groups) or by one-way ANOVA with Tukey’s post hoc test (IL4+IL13 groups). E, Relative TMRE MFI in isolated unactivated BMDM mitochondria, treated with increasing concentrations of NaCl (serial dilutions from 64 mM to 1 mM NaCl) for 3 hours (n=4). F, Pictogram of the generation and activation of human monocyte-derived macrophages. G, Relative intracellular Na+ content in human monocytes after 1 hour of HS treatment (n=13). Significance was analyzed by unpaired, two-tailed t test. H, Basal OCR in freshly isolated human monocytes (n=8 male donors), treated for 3 hours with 40 mM of NaCl (HS). Data are shown as donor-paired mean and were analyzed by paired, 2-tailed t test. I through K, Human monocyte-derived macrophages (MΦ) activated for 6 hours with LPS+ interferon (IFN) γ or IL4, under NS or HS conditions. I, Basal OCR (n=14 for M[LPS+INFγ] NS and HS, n=15 for M[IL4] NS, and n=12 for M[IL4] HS). J, ATP level in relation to NS (n=11 for NS, n=12 for HS for both M[LPS+INFγ] and M[IL4], respectively). K, TMRE fluorescence intensity in relation to NS (n=14 for M[LPS+INFγ] NS and HS, n=15 for M[IL4] NS and HS). I through K, Data are shown as box and whisker with minimum to maximum and were analyzed by unpaired, 2-tailed t test. Relative E. coli colony-forming units in murine BMDMs (L) (pooled data n=9 from 3 independent experiments) and human monocytes (M) (n=8), treated under NS or HS conditions, infected with E. coli for 3 hours. Data are shown as box and whisker with minimum to maximum in relation to NS and were analyzed by unpaired, 2-tailed t test. N, Effect of human M2 macrophages treated with NS and HS on CD4+ T cell and CD4+ regulatory T cell (Treg) migration (n=8 each). Data are shown as donor-paired mean and were analyzed by Wilcoxon matched-pairs signed-rank 2-tailed test. ψ indicates mitochondrial membrane potential; CFU, colony-forming unit; HS, high salt; NS, normal salt; and TMRE, tetramethylrhodamine ethyl ester.
HS Inhibits Mitochondrial ETC at Complex II
To dissect the molecular mechanism underlying the HS-induced mitochondrial dysfunction, we performed pulsed stable isotope-resolved metabolomics41 in murine BMDM. Using pulsed isotope labeling with U-13C6-glucose or U-13C5-glutamine (Figure 2A), we analyzed the metabolic turnover within a defined time window during activation, in contrast to classic cumulative labeling experiments that cover the entire course of activation. We performed these analyses in the third and 24th hours of M(LPS) and M(IL4+IL13) activation, labeling during the last hour only (Figure IIA in the Data Supplement). Surprisingly, we found a significant reduction in the succinate to fumarate conversion under HS after only 3 hours of activation (Figure 2B, Figure IIB through IIE in the Data Supplement), which continued to persist at 24 hours (Figure 2C, Figure IIF through II-I in the Data Supplement). Of note, this finding was independent of the activating stimulus. Furthermore, glycolysis was only minimally induced after 3 hours of LPS-treatment compared with 24 hours, and in line with our previous extracellular acidification rate results, HS had no effect either, independent of LPS or IL4+IL13 treatment (Figure IIJ and IIK in the Data Supplement). During the oxidation of succinate to fumarate, reduced flavin adenine dinucleotide (FADH2) is formed and serves as a first electron acceptor. However, FADH2 is covalently bound to complex II. Therefore, we measured levels of nonprotein-bound flavin adenine dinucleotide (FAD) in murine M(LPS) and M(IL4+IL13) and found them to be decreased after 3 hours of HS (Figure 2D). Equally, HS led to a decrease of FAD in human monocytes (Figure 2E). Both decreased fumarate to succinate ratio, and lower levels of FAD released after electron transfer from FADH2 to ubiquinone suggest a complex II inhibition under HS. The importance of complex II activity in MΦ activation has been studied extensively.32,42 Citrate-derived itaconate has been shown to inhibit succinate dehydrogenase in MΦ and thereby regulate their function.42 However, itaconate was not induced by LPS with or without HS at 3 hours (Figure IIJ in the Data Supplement) and is thus not responsible for the HS-induced complex II inhibition. To test whether HS inhibits complex II activity directly and to avoid compensatory regulation during stimuli processing occurring in intact MΦ, we used a cell- and mitochondrion-independent assay. We isolated complex II from BMDMs and analyzed its activity under increasing concentrations of NaCl within the physiological dynamic range of the cytosol.17,37 Strikingly, mimicking the HS-induced intracellular rise of Na+-inhibited isolated complex II activity significantly, even at low Na+ concentrations (Figure 2F). Next, we studied the combined complex II+III activity in intact purified mitochondria on HS challenge. Although the assay determines complex III activity by measuring the reduction of oxidized cytochrome C, it depends on succinate oxidation to start the electron transfer reaction. Here, in intact mitochondria, HS dose-dependently inhibited complex II+III activity similar to the complex III inhibitor antimycin A (Figure 2G). In contrast, complexes I and IV were not affected by HS (Figure 2H and 2I). In addition, reduced nicotinamide adenine dinucleotide (NADH+H+) to oxidized nicotinamide adenine dinucleotide (NAD+) conversion at complex I remained intact after 3 hours of activation under HS conditions in both M(LPS) and M(IL4+IL13) MΦ (Figure 2J). Thus, our data indicate that HS specifically inhibits the ETC at complex II, without consecutive reverse electron flow to complex I and NAD+ to NADH conversion, but with subsequent disturbance in electron transfer to complex III. Of note, transmission electron microscopy and 3-dimensional reconstruction of mitochondrial networks of M(LPS) and M(IL4+IL13) MΦ showed no differences under NS and HS conditions after 3 hours of activation (Figure 3A–3D), suggesting that HS-induced complex II inhibition and subsequent energetic collapse were independent of large structural changes. It is interesting that increased intracellular Na+ levels not only occur on extracellular hypertonic conditions in immune cells17 but also occur under ischemic isotonic conditions, eg, in the heart43 and astrocytes. Furthermore, it has been reported earlier that on hypoxia, mitochondrial sodium calcium exchanger-dependent Na+ entry into the mitochondrial matrix leads to a reduction in inner mitochondrial membrane fluidity. Subsequently, coenzyme Q/ubiquinone diffusion from complex II to complex III is impaired and combined complex II+III activity diminished to a comparable extent as shown by us.44 We extent this concept by showing a specific inhibition of complex II on Na+ rise, a possibly compensatory mechanism to prevent electron oversaturation of the inner mitochondrial membrane on Na+-induced fluidity deficiency.
Figure 2.

High salt inhibits mitochondrial electron transport chain at complex II. A, Pictogram of the central carbon metabolism and ETC. Glucose-derived 13C-label is represented as red dots. Glutamine-derived 13C-label is represented as blue dots. Mitochondrial membrane potential is ψ. B through D, Bone marrow–derived macrophages (BMDMs) treated for 3 h (B and D) or 24 h (C) with lipopolysaccharide (LPS) or interleukin (IL) 4+IL13 under normal salt (NS) or high salt (HS) conditions. B and C, 13C-glutamine-derived 13C-fumarate to 13C-succinate ratio as surrogate for succinate dehydrogenase/complex II activity (n=5). Data are depicted as box and whisker with minimum to maximum. Significance was analyzed by unpaired 2-tailed t test. D, Relative (deproteinized) FAD content (n=4). Data are depicted as box and whisker with minimum to maximum. Significance was analyzed by unpaired 2-tailed t test. E, Relative (deproteinized) FAD content in freshly isolated monocytes (n=6 donors), cultivated for 3 hours under NS or HS conditions. Data are depicted in relation to the donor-paired NS control. Significance was analyzed by Wilcoxon matched-pairs signed-rank 2-tailed test. F, Relative complex II activity after purification of complex II from BMDMs under increasing concentrations of additional NaCl (0 mM to 4 mM) (n=3 or 4). Data are depicted as mean in relation to the NS control. Significance was analyzed by 1-way ANOVA with Tukey’s post hoc test. G, Relative complex II+III activity in bovine heart mitochondria under increasing concentrations of additional NaCl (0.0625 mM to 64 mM) or antimycin A (AA, 0.3438 nM to 352 nM) (n=3 per concentration, respectively). H, Relative complex IV activity in bovine heart mitochondria under increasing concentrations of additional NaCl (0.0625 mM to 64 mM) or potassium cyanide (KCN, 0.01 µM to 100 µM) (n=3 for NaCl and n=1 for KCN). Data are depicted as mean±SEM in relation to the NS control for G and H. I, Relative complex I activity after purification of complex I from BMDMs under increasing concentrations of additional NaCl (0 mM to 4 mM) (n=3). Data are depicted as mean in relation to the NS control. Significance was analyzed by 1-way ANOVA with Tukey’s post hoc test. J, NAD+ to NADH ratio as surrogate for complex I activity in BMDMs treated for 3 hours with LPS or IL4+IL13 under NS, HS, or NS+rotenone (10 µM) treatment (n=6). Data are depicted as box and whisker with minimum to maximum. Significance was analyzed by 1-way ANOVA with Tukey’s post hoc test. 2OG indicates 2-Oxoglutarate; ATP, Adenosine Triphosphate; Cit, Citrate; ETC, electron transport chain; FAD, Flavin Adenine Dinucleotide; Fh, Fumarate hydratase; Fum, Fumarate; Glc, Glucose; Gln, Glutamine; Glu, Glutamate; Glud1, Glutamate Dehydrogenase 1; H+, protons; Ita, Itaconate; Lac, Lactate; Ldha, Lactate Dehydrogenase A; Mal, Malate; NAD+, oxidized Nicotinamide Adenine Dinucleotide; NADH+H+, reduced Nicotinamide Adenine Dinucleotide; O2, oxygen; OAA, Oxaloacetate; Pcx, Pyruvate Carboxylase; Pdha, Pyruvate Dehydrogenase E1 Subunit Alpha 1; Pyr, Pyruvate; Sdha, Succinate Dehydrogenase complex, subunit A; and Suc, Succinate.
Figure 3.

Mitochondrial morphology is not affected by HS. A through D, Representative electron micrographs of M(lipopolysaccharide [LPS]) and M(interleukin [IL] 4+IL13) bone marrow–derived macrophages (BMDMs), activated for 3 hours under normal salt (NS) or high salt (HS) conditions, depicting an entire cell, a representative mitochondrion, and (in yellow) a 3-dimensional reconstruction of the mitochondrial network in a representative section of the cell.
Coupled ETC Is Crucial for Early MΦ Activation and Function
To highlight the importance of complex II inhibition and mitochondrial ψ depolarization on MΦ function, we evaluated MΦ activation on HS, as well as on dimethyl malonate, antimycin A, and BAM15 treatment, respectively, under NS conditions. Indeed, pharmacological inhibition of complex II with dimethyl malonate (Figure 4A), inhibition of complex III with antimycin A (Figure 4B), and mitochondrial depolarization with BAM15 (Figure 4C) phenocopied the HS effect on the expression profile of representative marker genes Nos2 and Retnla1 after only 3 hours. Functionally, mitochondrial uncoupling under NS conditions with BAM15 accelerated defenses against E. coli in murine MΦ and human monocytes similarly as under HS conditions (Figure 4D and 4E). Of note, BAM15 neither affected E. coli growth directly (Figure IIID in the Data Supplement), nor compromised cell viability, as determined by lactate dehydrogenase secretion (Figure IIIE and IIIF in the Data Supplement). It is interesting that, whereas after 3 hours of activation, HS inhibited the expression of several proinflammatory genes in mouse M(LPS+IFNγ) MΦ (Figure 4A–4C, Figure IIIA and IIIB in the Data Supplement), over time there was a switch toward enhanced proinflammatory signature at 24 hours (Figure 4F and 4H, Figure IIIA and IIIB in the Data Supplement) and nitric oxide production (Figure 4G), as described previously.8,17,37 Considering anti-inflammatory activation, HS reduced the overall early and late marker gene expression in mouse M(IL4+IL13) MΦ (Figure IIIA and IIIB in the Data Supplement), as reported previously for 24 hours.26 Similarly, 24 hours of HS boosted the expression of proinflammatory genes in LPS+IFNγ-treated human MΦ (Figure 4I), whereas it inhibited anti-inflammatory gene expression in IL4-treated human MΦ after 24 hours (Figure IIIC in the Data Supplement).
Figure 4.

Coupled electron transport chain is crucial for early macrophage activation and function. Relative expression of Nos2 and Retnla1 in untreated, lipopolysaccharide (LPS)- or interleukin (IL) 4+IL13-treated bone marrow–derived macrophages (BMDMs) activated for 3 hours under normal salt (NS) or high salt (HS) or NS+dimethyl malonate (DMM) (A) (pooled data n=16 from 2 independent experiments), NS+antimycin A (AA) (B) (pooled data n=8 from 2 independent experiments), or NS+BAM15 (C) (pooled data n=6 from 2 independent experiments). D and E, Relative Escherichia coli (E. coli) colony-forming units (CFUs) in BMDMs (D) (pooled data n=18 from 3 independent experiments) and freshly isolated human monocytes (E) (pooled data n=10 from 3 independent experiments), 3 hours after infection under NS, HS, or NS+BAM15 (10 µM) treatment. Relative gene expression of Nos2 (F) and Il6 (H), as well as nitrite in the supernatant (G) in untreated or LPS-treated BMDMs activated for 24 hours under NS or HS. F through H, Data (n=6 each) are depicted as box and whisker with minimum to maximum. Significance was analyzed by 1-way ANOVA with Tukey’s post hoc test (A through C, E through H) and Kruskal-Wallis test with Dunn’s post hoc test for D. I, Relative gene expression of Il6, Il8, Tnf, and Il23 in human macrophages activated for 24 hours with LPS+interferon (IFN)γ under NS or HS conditions (n=6 each for IL6, Il8, and Il23, n=5 each for Tnf). For I, data are shown as donor-paired mean and were analyzed by paired, 2-tailed t test (Il6, Il8, Il23) or Wilcoxon matched-pairs signed-rank 2-tailed test (Tnf).
Taken together, our data demonstrate that a change in the Na+ microenvironment results in a rapid loss of mitochondrial respiration. This mitochondrial dysfunction, in line with previous reports,45 has profound consequences on murine MΦ activation and function. We recently demonstrated that HS boosts Bnip3-expression.17 Bnip3 is linked to mitochondrial dysfunction46 and thus could contribute to our antibacterial phenotype.
High Dietary Salt Intake Transiently Inhibits Mitochondrial Respiration in Human Monocytes
In contrast to tissue MΦ, which might sense substantially higher local Na+ levels, especially in inflammatory areas,8 circulating monocytes are exposed to the more subtle Na+ changes in the bloodstream. To directly assess how physiologically relevant changes in plasma Na+ affect monocytic mitochondrial function, we analyzed plasma Na+ changes and oxidative respiration in monocytes from healthy humans challenged with 6 g of salt (administered as NaCl tablets) in addition to their accustomed diets over the course of 14 days, followed by a 14-day washout phase (NCT02509962). We found a significant increase in plasma Na+ levels during the HS intervention compared with baseline. Fourteen days after cessation of HS intake, plasma Na+ returned to baseline (Figure 5A). Of note, changes in basal OCR in human monocytes over the course of the study displayed a significant negative correlation with changes in plasma Na+ concentrations (Figure 5B). These findings were independent and not driven by differences in monocyte subpopulations, as these were unchanged during the HS intervention (Figure IVA and IVB in the Data Supplement). We thus analyzed in vitro whether the observed 2 to 4 mM increase in extracellular NaCl could also trigger mitochondrial dysfunction in human monocytes acutely. Strikingly, our measurements showed that mitochondrial function in monocytes obtained from healthy study participants was reduced, as indicated by significantly decreased OCR, ATP, and ψ levels, when treating them in vitro for 3 hours with only +2 and +4 mM of NaCl (Figure 5C–5E). All parameters showed a dose-dependent reduction independent of sex (Figure 5C–5E). We aimed to mimic these acute and relatively mild Na+ changes in vivo with a meal challenge, highly representative of a real-life situation. Here, 20 healthy donors (10 men and 10 women) ate a single pizza meal with almost 10 g of salt per serving (NCT04175249) (Figure 6A). Three hours after the meal challenge, plasma Na+ increased significantly by 1 to 6 mM, returning to baseline by 8 hours after the meal challenge (Figure 6B). It is striking that basal OCR (Figure 6C) and ψ (Figure 6E) decreased significantly 3 hours after the meal intake. One limitation related to our study design is the fact that we cannot fully exclude the caloric challenge as a potential confounder. However, the significant negative correlation of OCR and plasma Na+ differences between 3 hours after the meal and baseline speak against it (Figure 6D). These data demonstrate that dietary sodium intake has immediate effects on monocytic mitochondrial respiration, and this in turn might affect their activation and function, further highlighting the significance of dietary sodium intake on monocyte/macrophage immunometabolism and function in humans.
Figure 5.

Minimal sodium (Na+) changes inhibit monocytic mitochondrial function. A, Plasma Na+ (n=8) as well as (B) fold-change in monocytic basal oxygen consumption rate (OCR) in relation to difference in plasma Na+ compared with baseline (n=5 for day 3, n=4 for day 14, n=6 for day 28) from a chronic salt intervention study in male volunteers. C, Basal OCR, (D) ATP level, and (E) tetramethylrhodamine ethyl ester (TMRE) mean fluorescence intensity (MFI) in freshly isolated monocytes from 10 male and 10 female volunteers, treated in vitro for 3 hours with +2 mM and +4 mM NaCl. A, Significance of plasma Na+ was analyzed by Friedman test, and false discovery rate (FDR) correction was performed via Benjamini-Hochberg procedure. B, Relationship between the change in plasma Na+ and the change in basal OCR (of days 3, 14, and 28 compared with baseline) was determined by linear mixed-effects model applied to rank-transformed between–time point difference data, accounting for donor. C through E, Data were analyzed by Friedman test with Benjamini-Hochberg FDR correction. NS indicates normal salt; and TMRE, tetramethylrhodamine ethyl ester.
Figure 6.

Single meal inhibits monocytic mitochondrial function. A, Scheme of the study protocol with a picture of a representative pizza containing 9.8±0.3 g NaCl. Before, 3 hours and 8 hours after the meal challenge, monocytes were isolated from 10 male and 10 female volunteers (n=20 per time point). B, Plasma Na+, (C) monocytic basal oxygen consuption rate (OCR) in relation to baseline, and (E) TMREhigh monocytes in relation to baseline at baseline, 3 hours, and 8 hours after pizza. D, Relation between fold-change in monocytic basal OCR and difference in plasma Na+ as single time point comparison between 3 hours after pizza and baseline. B, C, and E, Data were analyzed by Friedman test with Benjamini-Hochberg false discovery rate (FDR) correction. D, Relationship between the change in plasma Na+ and the change in basal OCR (of 3 hours compared with baseline) was determined by Pearson correlation and linear regression analysis. TMRE indicates tetramethylrhodamine ethyl ester.
Discussion
One major finding of our study is that circulating monocytes respond to moderate changes in plasma Na+ within the reference range, whereas MΦ sense higher Na+ concentrations with subsequent changes on mitochondrial energetics, MΦ activation, and function. Furthermore, we have found a strong inhibition of complex II+III and ATP production in vitro under HS. Of note, HS actions on gene expression and bactericidal function could be phenocopied under NS using a mitochondrial uncoupler. Intracellular electrolyte and energy homeostasis are tightly regulated processes. From the physiological perspective, the early reduced mitochondrial ATP production induced by HS will provoke adaptive responses, by which the cell will reach again a more favorable energetic homeostasis. Our data provide the first evidence that disturbances in this critical phase might have long-term functional consequences. These findings raise the intriguing questions of whether and to what extent changes in intracellular Na+ affect oxidative phosphorylation and function of other cell types, such as dendritic, T, B, and intestinal cells or cardiomyocytes, equally subjective to dietary sodium changes. We did not investigate functional consequences in vivo. Thus, future HS intervention studies will be needed to identify whether immune and other cells modulate their function on different diets. Especially in the context of cardiovascular disease, functional downstream effects of HS will be of great interest, considering salt-sensitive and -insensitive patients. Therapy could in the future be accompanied by dietary recommendations, dependent on the personal responsiveness of cells on HS. Including diseased patients will therefore be of major interest in future studies, as well as the investigation if salt sensitivity is correlated or even partially caused by mitochondrial dysfunction in certain cell types.
Recently, we found that Slc8a1/NCX1 senses high extracellular Na+.37 Pharmacological and genetic inhibition of Slc8a1/NCX1 provided mechanistic insights on Na+ influx in mononuclear phagocyte cells,37 although we cannot exclude that other channels or mechanisms also promote the redistribution of Na+ from the extracellular to intracellular compartment. Interestingly, mitochondrial mitochondrial sodium calcium exchanger has been shown to be crucial for mitochondrial salinization on isotonic hypoxia,43 with subsequent deficiency in electron transfer from complex II to complex III and superoxide burst.44 Increased intracellular Na+ levels not only occur on extracellular hypertonic conditions in immune cells,17 but also occur under ischemic NS conditions, eg, in the heart.43,47 Excitation coupling of healthy cardiac myocytes consumes large amounts of ATP, which is replenished by mitochondrial oxidative phosphorylation.48 During cardiac ischemia, intracellular and mitochondrial Na+ levels increase along with a reduction in mitochondrial respiratory function with reduced ATP, which might be a reason for the imbalance of the energy demand and thus further contribute to heart failure.48 SGLT2i is a new promising class that reduces mortality in patients with cardiovascular risk.49 Experimentally, SGLT2i has direct cardiac effects by decreasing cardiac intracellular Na+ and Ca2+ and increasing mitochondrial Ca2+.50 Therefore, it is tempting to speculate that SGLT2i might also affect immunometabolism in other cell types including mononuclear phagocytes. Future studies will thus be needed to elucidate the molecular actions of SGLT2i in mitochondrial function.
In conclusion, the current data highlight a previously unappreciated interaction between dietary salt intake and mitochondrial energetics in mononuclear phagocytes. Our data suggest that early and transient disturbance of mitochondrial ETC with reduced ATP production may translate into long-term functional changes. Although these functional changes might help to resolve bacterial infections, a shift toward an inflammatory phenotype might accelerate cardiometabolic disease.
Acknowledgments
The authors thank Jana Czychi, Gabriele N’diaye, Juliane Anders, Ute Gerhardt, May-Britt Köhler, and Fardad Ramezani for assistance. S.G. led and conceived the project, designed and performed most experiments, and analyzed and interpreted the data. H.B. conducted the clinical study, and performed PBMC isolation and flow-cytometric analysis together with S.G. P.N. performed murine BMDM and human peripheral blood monocyte bacterial killing and growth experiments. R.W., D.S., and A.G. performed analyses of ATP, tetramethylrhodamine ethyl ester, gene expression, and T cell migration in human macrophages. C.Z. together with S.G. measured and analyzed pulsed stable isotope-resolved metabolomics experiments. T.B. and E.T. performed sectioned transmission electron microscopy imaging and 3-dimensional reconstruction. V.M.P. performed mitochondrial isolation and ETC complex assays together with S.G. L.K. performed peripheral blood monocyte bacterial killing and cell culture experiments in human monocytes for intracellular Na+ measurement. M.V. performed intracellular Na+ quantifications. A. Maifeld, A. Mähler, and N.W. performed the clinical study, which was reanalyzed by S.G. and H.B. S.K.F. performed statistical analyses. K.B., J.S., and R.D. gave major conceptual input. D.N.M., S.K., J.J., and M.K. supervised the experiments and interpreted the data. S.G. and D.N.M. wrote the article with key editing by S.K., H.B., and M.K. and further input from all authors.
Sources of Funding
S.G. was supported by the Bundesministerium für Bildung und Forschung funding MSTARS (Multimodal Clinical Mass Spectrometry to Target Treatment Resistance). D.N.M., H.B., N.W., and S.K.F. were supported by the Deutsche Forschungsgemeinschaft (German Research Foundation; Projektnummer 394046635 - SFB 1365). D.N.M. was supported by the Deutsches Zentrum für Herz-Kreislauf-Forschung (DZHK, 81Z0100106). J.J. received funding from the Deutsche Forschungsgemeinschaft (JA1993/6-1), Deutsche Forschungsgemeinschaft SFB 1350 grant (project No. 387509280, TPB5) and the Bavarian Ministry of Science and the Arts in the framework of the Bavarian Research Network „New Strategies Against Multi-Resistant Pathogens by Means of Digital Networking – bayresq.net.” M.K. and N.W. were supported by the European Research Council under the European Union’s Horizon 2020 research and innovation program (M.K.: 640116; N.W.: 852796). M.K. was further supported by a Strategic Action Plan for Limburg (Strategisch Actieplan voor Limburg in het Kwadraat, SALK) grant from the government of Flanders, Belgium, and by an Odysseus grant from the Research Foundation Flanders. N.W. is supported by a grant from the Corona-Stiftung. N.W. is participant in the Clinician Scientist Program funded by the Berlin Institute of Health. S.K. was supported by Impuls und Vernetzungsfond Aging and Metabolic Programming (AMPro, ZT-0026, Helmholtz Association).
Disclosures
None.
Supplemental Materials
Expanded Methods
Data Supplement Figures I–IV
Data Supplement Table I
Supplementary Material
Footnotes
Nonstandard Abbreviations and Acronyms
- ψ
- mitochondrial membrane potential
- BMDM
- bone marrow–derived macrophage
- ETC
- electron transport chain
- HS
- high salt
- MΦ
- macrophage
- NS
- normal salt
- OCR
- oxygen consumption rate
- PBMC
- peripheral blood mononuclear cell
H.B. and P.N. contributed equally.
M.K., J.J., S.K., and D.N.M. contributed equally.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCULATIONAHA.120.052788.
For Sources of Funding and Disclosures, see page 156.
Contributor Information
Sabrina Geisberger, Email: Sabrina.Geisberger@mdc-berlin.de.
Hendrik Bartolomaeus, Email: tbartolomaeus@evolution.uni-bonn.de.
Patrick Neubert, Email: Patrick.Neubert@klinik.uni-regensburg.de.
Ralf Willebrand, Email: ralf.dechend@charite.de.
Christin Zasada, Email: christin.zasada@mdc-berlin.de.
Thomas Bartolomaeus, Email: tbartolomaeus@evolution.uni-bonn.de.
Victoria McParland, Email: Victoria.McParland@mdc-berlin.de.
Dries Swinnen, Email: dries.swinnen@uhasselt.be.
Anneleen Geuzens, Email: anneleen.geuzens@uhasselt.be.
András Maifeld, Email: andras.maifeld@charite.de.
Luka Krampert, Email: Luka.Krampert@klinik.uni-regensburg.de.
Marion Vogl, Email: Marion.Vogl@klinik.uni-regensburg.de.
Anja Mähler, Email: anja.maehler@charite.de.
Nicola Wilck, Email: nicola.wilck@charite.de.
Lajos Markó, Email: lajos.marko@charite.de.
Ekin Tilic, Email: etilic@evolution.uni-bonn.de.
Sofia K. Forslund, Email: sofia.forslund@mdc-berlin.de.
Katrina J. Binger, Email: k.binger@latrobe.edu.au.
Johannes Stegbauer, Email: johannes.stegbauer@med.uni-duesseldorf.de.
Ralf Dechend, Email: ralf.dechend@charite.de.
Markus Kleinewietfeld, Email: markus.kleinewietfeld@uhasselt.be.
Jonathan Jantsch, Email: Jonathan.Jantsch@klinik.uni-regensburg.de.
References
- 1.Collaborators GBDD. Health effects of dietary risks in 195 countries, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2019;393:1958–1972. doi: 10.1016/S0140-6736(19)30041-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor J. 2013 ESH/ESC guidelines for the management of arterial hypertension. Eur Heart J. 2013;34:2108–2109 [PubMed] [Google Scholar]
- 3.Müller DN, Wilck N, Haase S, Kleinewietfeld M, Linker RA. Sodium in the microenvironment regulates immune responses and tissue homeostasis. Nat Rev Immunol. 2019;19:243–254. doi: 10.1038/s41577-018-0113-4 [DOI] [PubMed] [Google Scholar]
- 4.Mozaffarian D, Fahimi S, Singh GM, Micha R, Khatibzadeh S, Engell RE, Lim S, Danaei G, Ezzati M, Powles J; Global Burden of Diseases Nutrition and Chronic Diseases Expert Group. Global sodium consumption and death from cardiovascular causes. N Engl J Med. 2014;371:624–634. doi: 10.1056/NEJMoa1304127 [DOI] [PubMed] [Google Scholar]
- 5.Suckling RJ, He FJ, Markandu ND, MacGregor GA. Dietary salt influences postprandial plasma sodium concentration and systolic blood pressure. Kidney Int. 2012;81:407–411. doi: 10.1038/ki.2011.369 [DOI] [PubMed] [Google Scholar]
- 6.Dickinson KM, Clifton PM, Burrell LM, Barrett PH, Keogh JB. Postprandial effects of a high salt meal on serum sodium, arterial stiffness, markers of nitric oxide production and markers of endothelial function. Atherosclerosis. 2014;232:211–216. doi: 10.1016/j.atherosclerosis.2013.10.032 [DOI] [PubMed] [Google Scholar]
- 7.Canaud B, Kooman J, Selby NM, Taal M, Francis S, Kopperschmidt P, Maierhofer A, Kotanko P, Titze J. Sodium and water handling during hemodialysis: new pathophysiologic insights and management approaches for improving outcomes in end-stage kidney disease. Kidney Int. 2019;95:296–309. doi: 10.1016/j.kint.2018.09.024 [DOI] [PubMed] [Google Scholar]
- 8.Jantsch J, Schatz V, Friedrich D, Schröder A, Kopp C, Siegert I, Maronna A, Wendelborn D, Linz P, Binger KJ, et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 2015;21:493–501. doi: 10.1016/j.cmet.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Müller DN, Schmieder RE, Cavallaro A, Eckardt KU, Uder M, et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61:635–640. doi: 10.1161/HYPERTENSIONAHA.111.00566 [DOI] [PubMed] [Google Scholar]
- 10.Kopp C, Linz P, Wachsmuth L, Dahlmann A, Horbach T, Schöfl C, Renz W, Santoro D, Niendorf T, Müller DN, et al. (23)Na magnetic resonance imaging of tissue sodium. Hypertension. 2012;59:167–172. doi: 10.1161/HYPERTENSIONAHA.111.183517 [DOI] [PubMed] [Google Scholar]
- 11.Hammon M, Grossmann S, Linz P, Kopp C, Dahlmann A, Garlichs C, Janka R, Cavallaro A, Luft FC, Uder M, et al. 23Na magnetic resonance imaging of the lower leg of acute heart failure patients during diuretic treatment. PLoS One. 2015;10:e0141336. doi: 10.1371/journal.pone.0141336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dahlmann A, Dörfelt K, Eicher F, Linz P, Kopp C, Mössinger I, Horn S, Büschges-Seraphin B, Wabel P, Hammon M, et al. Magnetic resonance-determined sodium removal from tissue stores in hemodialysis patients. Kidney Int. 2015;87:434–441. doi: 10.1038/ki.2014.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paling D, Solanky BS, Riemer F, Tozer DJ, Wheeler-Kingshott CA, Kapoor R, Golay X, Miller DH. Sodium accumulation is associated with disability and a progressive course in multiple sclerosis. Brain. 2013;136(pt 7):2305–2317. doi: 10.1093/brain/awt149 [DOI] [PubMed] [Google Scholar]
- 14.Karg MV, Bosch A, Kannenkeril D, Striepe K, Ott C, Schneider MP, Boemke-Zelch F, Linz P, Nagel AM, Titze J, et al. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: a randomised controlled trial. Cardiovasc Diabetol. 2018;17:5. doi: 10.1186/s12933-017-0654-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitada K, Daub S, Zhang Y, Klein JD, Nakano D, Pedchenko T, Lantier L, LaRocque LM, Marton A, Neubert P, et al. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest. 2017;127:1944–1959. doi: 10.1172/JCI88532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, Sun S, Zhang Z, Chen XQ, Qin M, et al. High salt primes a specific activation state of macrophages, M(Na). Cell Res. 2015;25:893–910. doi: 10.1038/cr.2015.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neubert P, Weichselbaum A, Reitinger C, Schatz V, Schröder A, Ferdinand JR, Simon M, Bär AL, Brochhausen C, Gerlach RG, et al. HIF1A and NFAT5 coordinate Na+-boosted antibacterial defense via enhanced autophagy and autolysosomal targeting. Autophagy. 2019;15:1899–1916. doi: 10.1080/15548627.2019.1596483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hucke S, Eschborn M, Liebmann M, Herold M, Freise N, Engbers A, Ehling P, Meuth SG, Roth J, Kuhlmann T, et al. Sodium chloride promotes pro-inflammatory macrophage polarization thereby aggravating CNS autoimmunity. J Autoimmun. 2016;67:90–101. doi: 10.1016/j.jaut.2015.11.001 [DOI] [PubMed] [Google Scholar]
- 19.Ip WK, Medzhitov R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun. 2015;6:6931. doi: 10.1038/ncomms7931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496:518–522. doi: 10.1038/nature11868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. 2017;551:585–589. doi: 10.1038/nature24628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496:513–517. doi: 10.1038/nature11984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei Y, Lu C, Chen J, Cui G, Wang L, Yu T, Yang Y, Wu W, Ding Y, Li L, et al. High salt diet stimulates gut Th17 response and exacerbates TNBS-induced colitis in mice. Oncotarget. 2017;8:70–82. doi: 10.18632/oncotarget.13783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, Deng S, Herold KC, Kuchroo VK, Kleinewietfeld M, et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest. 2015;125:4212–4222. doi: 10.1172/JCI81151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Safa K, Ohori S, Borges TJ, Uehara M, Batal I, Shimizu T, Magee CN, Belizaire R, Abdi R, Wu C, et al. Salt accelerates allograft rejection through serum- and glucocorticoid-regulated kinase-1-dependent inhibition of regulatory T cells. J Am Soc Nephrol. 2015;26:2341–2347. doi: 10.1681/ASN.2014090914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binger KJ, Gebhardt M, Heinig M, Rintisch C, Schroeder A, Neuhofer W, Hilgers K, Manzel A, Schwartz C, Kleinewietfeld M, et al. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J Clin Invest. 2015;125:4223–4238. doi: 10.1172/JCI80919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nahrendorf M. Myeloid cell contributions to cardiovascular health and disease. Nat Med. 2018;24:711–720. doi: 10.1038/s41591-018-0064-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caputa G, Castoldi A, Pearce EJ. Metabolic adaptations of tissue-resident immune cells. Nat Immunol. 2019;20:793–801. doi: 10.1038/s41590-019-0407-0 [DOI] [PubMed] [Google Scholar]
- 30.Mitchell P, Moyle J. Evidence discriminating between the chemical and the chemiosmotic mechanisms of electron transport phosphorylation. Nature. 1965;208:1205–1206. doi: 10.1038/2081205a0 [DOI] [PubMed] [Google Scholar]
- 31.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862 [DOI] [PubMed] [Google Scholar]
- 32.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory aacrophages. Cell. 2016;167:457–470.e13. doi: 10.1016/j.cell.2016.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, Pearce EJ. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. 2016;45:817–830. doi: 10.1016/j.immuni.2016.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puleston DJ, Buck MD, Klein Geltink RI, Kyle RL, Caputa G, O’Sullivan D, Cameron AM, Castoldi A, Musa Y, Kabat AM, et al. Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. 2019;30:352–363.e8. doi: 10.1016/j.cmet.2019.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Opialla T, Kempa S, Pietzke M. Towards a more reliable identification of isomeric metabolites using pattern guided retention validation. Metabolites. 2020;10:E457. doi: 10.3390/metabo10110457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuich PH, Hoffmann N, Kempa S. Maui-VIA: a user-friendly software for visual identification, alignment, correction, and quantification of gas chromatography-mass spectrometry data. Front Bioeng Biotechnol. 2014;2:84. doi: 10.3389/fbioe.2014.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neubert P, Homann A, Wendelborn D, Bär AL, Krampert L, Trum M, Schröder A, Ebner S, Weichselbaum A, Schatz V, et al. NCX1 represents an ionic Na+ sensing mechanism in macrophages. PLoS Biol. 2020;18:e3000722. doi: 10.1371/journal.pbio.3000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Teijlingen Bakker N, Pearce EJ. Cell-intrinsic metabolic regulation of mononuclear phagocyte activation: findings from the tip of the iceberg. Immunol Rev. 2020;295:54–67. doi: 10.1111/imr.12848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005 [DOI] [PubMed] [Google Scholar]
- 40.Sun J, Sun J, Song B, Zhang L, Shao Q, Liu Y, Yuan D, Zhang Y, Qu X. Fucoidan inhibits CCL22 production through NF-κB pathway in M2 macrophages: a potential therapeutic strategy for cancer. Sci Rep. 2016;6:35855. doi: 10.1038/srep35855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pietzke M, Zasada C, Mudrich S, Kempa S. Decoding the dynamics of cellular metabolism and the action of 3-bromopyruvate and 2-deoxyglucose using pulsed stable isotope-resolved metabolomics. Cancer Metab. 2014;2:9. doi: 10.1186/2049-3002-2-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanonaka K, Motegi K, Arino T, Marunouchi T, Takagi N, Takeo S. Possible pathway of Na(+) flux into mitochondria in ischemic heart. Biol Pharm Bull. 2012;35:1661–1668. doi: 10.1248/bpb.b12-00010 [DOI] [PubMed] [Google Scholar]
- 44.Hernansanz-Agustín P, Choya-Foces C, Carregal-Romero S, Ramos E, Oliva T, Villa-Piña T, Moreno L, Izquierdo-Álvarez A, Cabrera-García JD, Cortés A, et al. Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature. 2020;586:287–291. doi: 10.1038/s41586-020-2551-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanin DE, Matsushita M, Klein Geltink RI, Grzes KM, van Teijlingen Bakker N, Corrado M, Kabat AM, Buck MD, Qiu J, Lawless SJ, et al. Mitochondrial membrane potential regulates nuclear gene expression in macrophages exposed to prostaglandin E2. Immunity. 2018;49:1021–1033.e6. doi: 10.1016/j.immuni.2018.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gustafsson AB. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr Cardiol. 2011;32:267–274. doi: 10.1007/s00246-010-9876-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. 2018;122:1460–1478. doi: 10.1161/CIRCRESAHA.118.310082 [DOI] [PubMed] [Google Scholar]
- 48.Tian R, Colucci WS, Arany Z, Bachschmid MM, Ballinger SW, Boudina S, Bruce JE, Busija DW, Dikalov S, Dorn GW, II, et al. Unlocking the secrets of mitochondria in the cardiovascular system: path to a cure in heart failure—a report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation. 2019;140:1205–1216. doi: 10.1161/CIRCULATIONAHA.119.040551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium-glucose cotransporter 2 inhibitors: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75:422–434. doi: 10.1016/j.jacc.2019.11.031 [DOI] [PubMed] [Google Scholar]
- 50.Baartscheer A, Schumacher CA, Wüst RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia. 2017;60:568–573. doi: 10.1007/s00125-016-4134-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.