

Abstract
Prion diseases are transmissible neurodegenerative disorders that affect mammals, including humans. The central molecular event is the conversion of cellular prion glycoprotein, PrPC, into a plethora of assemblies, PrPSc, associated with disease. Distinct phenotypes of disease led to the concept of prion strains, which are associated with distinct PrPSc structures. However, the degree to which intra- and inter-strain PrPSc heterogeneity contributes to disease pathogenesis remains unclear. Addressing this question requires the precise isolation and characterization of all PrPSc subpopulations from the prion-infected brains. Until now, this has been challenging. We used asymmetric-flow field-flow fractionation (AF4) to isolate all PrPSc subpopulations from brains of hamsters infected with three prion strains: Hyper (HY) and 263K, which produce almost identical phenotypes, and Drowsy (DY), a strain with a distinct presentation. In-line dynamic and multi-angle light scattering (DLS/MALS) data provided accurate measurements of particle sizes and estimation of the shape and number of PrPSc particles. We found that each strain had a continuum of PrPSc assemblies, with strong correlation between PrPSc quaternary structure and phenotype. HY and 263K were enriched with large, protease-resistant PrPSc aggregates, whereas DY consisted primarily of smaller, more protease-sensitive aggregates. For all strains, a transition from protease-sensitive to protease-resistant PrPSc took place at a hydrodynamic radius (Rh) of 15 nm and was accompanied by a change in glycosylation and seeding activity. Our results show that the combination of AF4 with in-line MALS/DLS is a powerful tool for analyzing PrPSc subpopulations and demonstrate that while PrPSc quaternary structure is a major contributor to PrPSc structural heterogeneity, a fundamental change, likely in secondary/tertiary structure, prevents PrPSc particles from maintaining proteinase K resistance below an Rh of 15 nm, regardless of strain. This results in two biochemically distinctive subpopulations, the proportion, seeding activity, and stability of which correlate with prion strain phenotype.
Author summary
Prion diseases are neurodegenerative diseases that include bovine spongiform encephalopathy (BSE or mad cow disease) in cattle, chronic wasting disease (CWD) in cervids and Creutzfeldt-Jakob disease (CJD) in humans. These diseases are caused by self-propagated misfolding and aggregation of the naturally occurring prion protein. Variations in the structure of prion aggregates are associated with distinct disease phenotypes, but how this prion structural heterogeneity translates into clinical presentation has been difficult to determine, largely because it is technically difficult to isolate and characterize the full range of prion structures from prion-infected brain. Here, we overcame this challenge by using a versatile fractionation technique, one that is strikingly unexplored in neurodegenerative research, and present the most detailed description, to date, of strain-specific prion subpopulations. We found that prion quaternary structure was a major contributor to structural heterogeneity. We also discovered that all prion strains studied underwent a significant structural change resulting in two distinctive subpopulations whose proportions correlated with the strain phenotype. Our work provides new insights into the molecular basis of prion strain variation and is a proof of concept that can be applied to other protein misfolding neurodegenerative disorders.
Introduction
Transmissible spongiform encephalopathies (TSEs), also known as prion diseases, comprise a group of lethal neurodegenerative disorders that affect several mammalian species including humans [1,2]. The central molecular event in TSEs is the misfolding and aggregation of the host-encoded prion glycoprotein, PrPC, into a wide variety of assemblies associated with disease, collectively called PrPSc [3,4]. During the course of the disease, a constellation of PrPSc particles, ranging from small, soluble, protease-sensitive (PrPsen) oligomers, to large, less soluble, partially protease-resistant (PrPres) fibrils, progressively accumulate in the brain. Under controlled experimental transmission conditions, a specific disease phenotype, characterized by incubation period, clinical signs of disease, PrPSc glycotype, and tissue tropism, can be reproduced in hosts with the same genetic background, leading to the operational concept of prion strains [2,5]. It is well established that different prion strains arise from different conformations of PrPSc, but within a given strain, there is also a diversity of PrPSc structures that may further contribute to phenotype [6–12]. However, the molecular basis linking PrPSc strain-specific structures and disease phenotypes remains elusive, in large part because isolating such structurally and biochemically diverse PrPSc particles is a challenging task.
One approach to isolating PrPSc particles has been sedimentation velocity. This technique has generated data that supports a strain-specific size distribution of PrPSc particles and has provided evidence for an association between PrPSc quaternary structure and its infectivity [13–17]. However, the limited resolution of this technique, differences in sample preparation and experimental conditions among laboratories, and the challenges of accurately assigning size values to isolated particles, have created inconsistent descriptions of the PrPSc subpopulations contained in a prion isolate. A higher resolution technique, size exclusion chromatography (SEC), has been also used to investigate PrPSc structural heterogeneity within prion strains [18–21], but limitations related to protein-stationary phase interaction and fractionation range have restricted SEC to the study of the smallest PrPSc oligomers present in a prion isolate, excluding from the analysis a major proportion of the largest and, potentially, most biologically relevant PrPSc particles.
We have adopted asymmetric-flow field-flow fractionation (AF4) as an ideal technique for isolating and characterizing PrPSc particles. AF4 is a flow-based separation technique widely used for the isolation and characterization of nanoparticles and polymers in the pharmaceutical industry, and is increasingly being applied to the characterization of biological macromolecules, protein complexes, extracellular vesicles, and viruses [22–24]. Two perpendicular flows, forward laminar flow and crossflow, are applied to particles, which are then separated based on their hydrodynamic properties. The technique allows for high resolution and reproducibility, and by adjusting crossflow gradients during the fractionation run, a very wide range of particle sizes can be separated, from a few nanometers to several micrometers [25]. Unlike SEC, the absence of a stationary phase greatly reduces the problems of sample-stationary phase interaction, which is particularly troublesome for “sticky” particles like large PrPSc aggregates. It also allows for fractionation of complex mixtures such as brain homogenate. The use of in-line multi-angle and dynamic light scattering (MALS and DLS) detectors allows for precise measurement of the size and shape of the eluting particles.
Surprisingly, the use of AF4 technology to study neurodegenerative disease-related protein aggregates has been very limited to date [26–29]. Within the field of prion disease research, AF4-MALS-DLS was first used to determine the size of the most infectious prion particle [26]. In this seminal work, PrPSc from 263K-infected Syrian hamster brains was first highly purified and digested with proteinase K, then disrupted by detergent, sonication and freeze-thaw before being fractionated and analyzed. Recently, we developed a method for using AF4 on brain homogenates without the need for initial purification or sonication, and with in-line DLS measurements, we were able to ascertain the size distribution of PrPSc particles in the brains of RML-infected mice [29]. Now we have applied our isolation method and AF4-MALS-DLS analysis to the characterization of inter- and intra-strain differences of subpopulations of PrPSc particles in the brains of Syrian golden hamsters infected with three different prion strains. All three strains share the same primary PrP structure; two strains (Hyper and 263K) were derived from different sources but produce almost identical phenotypes and share biochemical properties, the third (Drowsy) has a different phenotype and biochemical profile [6,10,30]. Our high-resolution fractionation and analysis provides the most detailed description, to date, of PrPSc particles in the brain at terminal stage of disease. We demonstrate that quaternary structure is a major contributor to PrPSc heterogeneity, but for all strains studied, a fundamental change, likely in secondary/tertiary structure, prevents PrPSc particles from maintaining proteinase K resistance below an Rh of 15 nm. These distinct populations of PrPsen and PrPres also have strain-specific changes in glycosylation pattern and templating activity.
Results
Solubilization step results in a high yield of soluble PrP aggregates for all strains, with no change in PK-resistance patterns compared with starting material
Our goal was to isolate PrPSc aggregates, defined as all types of PrP aggregates that are specifically associated with prion infection, whether directly infectious themselves and resistant to proteinase K or not. To solubilize PrP from membranes while preserving PrPSc assemblies close to their “natural” state, we incubated strains 263K, HY or DY brain homogenate (BH) with 2% dodecyl-β-maltoside and 2% sarkosyl sequentially, as optimized by Tixador et al. [14]. Detergent-insoluble particles that could affect the quality of our AF4 fractionations were removed by a short centrifugation. We performed batch mode DLS measurements on the supernatant and pellet and found similar sized particles were isolated for each strain: the supernatant contained particles of 3–300 nm Rh compared with 40–600 nm Rh in the pellet (Fig 1A and 1B). By immunoblot, the amount of PrP in the supernatant and pellet, before PK digestion, was similar for the three strains (Fig 1C and 1D), with a supernatant:pellet proportion of 72:28. Treatment of both supernatant and pellet with proteinase-K (PK) yielded the expected PrPres banding patterns, with the unglycosylated isoform migrating at 21 kDa for HY and 263K and 19 kDa for DY [30]. The ratio PrPres:total PrP was higher for HY and 263K than for DY with values of 0.56 (±0.05), 0.59 (±0.04) and 0.38 (±0.08), respectively, for supernatant PrP and 0.58 (±0.05), 0.57 (±0.06) and 0.29 (±0.10) for the pelleted PrP. Assuming all animals have comparable levels of PrPC, this was consistent with the fact that DY PrPSc is less resistant to PK than 263K and HY PrPSc [30].
Fig 1.

Particle size distributions determined by DLS measurements of detergent-soluble (A) and pellet (B) fractions after sequential incubation of 263K (red), HY (green), and DY (blue) brain homogenates with 2% dodecyl-β-maltoside and 2% sarkosyl, followed by 10 min centrifugation at 20,000 x g. Representative immunoblotting (C) and densitometric analysis (D) of detergent-soluble (S) and pellet (P) fractions of 263K (red), HY (green) and DY (blue). The pellet was resuspended in half of the original volume; the same volumes of S and resuspended P fractions were loaded in the polyacrylamide gel. Twice the volume of PK treated samples (20 μg/mL of PK for 1h at 37°C) were loaded in the same gel for quantification. SAF83 (dilution 1:10,000) was used as primary antibody. Bars represent average ± SE of three BHs for each prion strain.
Strains with the same phenotype have the same PrPSc size distribution
Batch mode DLS measurements of the soluble starting material indicated that particles had a size distribution of 3–300 nm Rh (Fig 1A). We, therefore, optimized the AF4 crossflow gradient for separating particles of 3–300 nm Rh within a 60 min run (see Methods for details). We injected 250 μg of total soluble protein and collected 200 μL fractions each minute. As expected, all fractograms had similar UV absorbance profiles (Fig 2A), indicating a similar amount and composition of proteins in the BH of the three strains. The Rh of the eluting particles measured by in-line DLS matched that seen in batch mode DLS, 3–300 nm Rh.
Fig 2.

A) Representative fractograms of control (NBH, black dotted line), 263K (red), HY (green), and DY (blue) detergent-solubilized BHs. Solid curves: UV signal (protein content) at 280 nm; circles: Rh (average ± SD) from thirty DLS measurements taken during the one-minute collection for each AF4 fraction; rhombus: particle length, calculated by fitting MALS signal to a rod-shape model. B) Representative immunoblots for PrP content in collected AF4 fractions; fractions from each brain homogenate were loaded in two polyacrylamide gels (gel 1: fraction 2 to 26/28, gel 2: fraction 28/30 to 50/54). SAF83 (dilution 1:10,000) was used as primary antibody. C) Size distributions of prion particles depicted as relative amount of PrP (from PrP immunoblot intensity; average ± SE) contributing to each particle size (Rh (nm)). Non-infected (dotted line), 263K (red), HY (green), DY (blue). Three brains for each prion strain were analyzed and averaged. D) Electron micrographs of fractions #47 (> 70 nm Rh) from HY (left) and DY (right) AF4 fractionation. No fibrillar particles were visible on the same fraction number from NBH fractionation. Bars = 100 nm. E) Density of PrPSc particles (average ± SE) for each particle length. 263K (red), HY (green), DY (blue). The correlation between particle length and Rh is shown as a black dotted line.
To determine PrP elution profiles, fractions were immunoblotted for PrP and PrP signal intensity quantified (Fig 2B). The PrP intensity from each fraction was plotted against the average Rh of the particles present in that fraction and the area under the curve from the entire run was normalized to 1 (Fig 2C).
Because our samples were not protease-digested before fractionation, the PrP in the sample contained normal PrPC in addition to PrPSc. To determine the contribution of PrPC to the total PrP in the size distribution curves, we analyzed uninfected normal brain homogenate (NBH) under the same fractionation conditions (Fig 2B and 2C). All PrPC from age-matched uninfected brain homogenates eluted within the first 16 fractions, with the maximum PrP signal intensity corresponding to particles of ~4 nm Rh, and no signal in particles larger than 7 nm. Conversely, prion-infected samples had PrP particles ranging from 3–300 nm Rh.
Interestingly, almost identical PrP elution patterns were seen for 263K and HY, but there was a very different pattern for DY. For 263K and HY, the PrP signal intensity increased by four times from 7 to 40 nm Rh particles, and the signal intensity remained almost constant as particle size increased beyond 40 nm Rh. In contrast, the PrP distribution for DY showed a constant decrease in PrP signal from 7 to 300 nm Rh. At ~30 nm Rh, the size distribution curves from the three strains intersected, indicating that DY contains more PrPSc particles smaller than 30 nm, and 263K and HY have more PrPSc particles larger than this.
For PrPSc particles increasing in size from 60–200 nm, particle density remains constant for 263K and HY but decreases for DY
To determine how many PrPSc particles were present in each fraction (particle density), we needed information about particle length and PrP content for each fraction. Electron microscopy images of larger fractions from HY and DY indicated that both contain fibrillar material, likely prion fibrils based on appearance (Fig 2D). In addition, previous light scattering data on purified 263K particles has revealed that assemblies of 12 nm Rh and larger are transitioning to a fibrillar form [26]. Therefore, we fit our MALS data to a rod-shaped model to determine particle lengths in fractions greater than 15 nm Rh (Fig 2A and Table 1).
Table 1. Particle density calculation.
| Fraction # | Rh (nm) | Length (nm) | PrP monomers per PrPSc particle | PrP normalized intensity | Relative number of PrPSc particles | Max. possible PrPSc particle density (particles/mL) |
|---|---|---|---|---|---|---|
| 24 | 11.6 | 39.1 | 78 | 0.08316 | 0.001065 | 3.08E+09 |
| 26 | 15.5 | 57.8 | 116 | 0.102213 | 0.000884 | 2.56E+09 |
| 28 | 19.6 | 80.0 | 160 | 0.148777 | 0.000093 | 2.69E+09 |
| 30 | 22.9 | 106.6 | 213 | 0.179116 | 0.000084 | 2.43E+09 |
| 32 | 26.9 | 142.1 | 284 | 0.224708 | 0.000079 | 2.29E+09 |
| 34 | 32.9 | 175.1 | 350 | 0.348884 | 0.000996 | 2.88E+09 |
| 36 | 38.7 | 199.4 | 399 | 0.370763 | 0.000930 | 2.69E+09 |
| 38 | 46.0 | 223.5 | 447 | 0.322006 | 0.000720 | 2.09E+09 |
| 40 | 49.2 | 253.5 | 507 | 0.301998 | 0.000596 | 1.72E+09 |
| 42 | 57.6 | 361.2 | 722 | 0.290421 | 0.000402 | 1.16E+09 |
| 44 | 69.3 | 524.2 | 1048 | 0.299313 | 0.000286 | 8.26E+08 |
| 46 | 77.9 | 704.0 | 1408 | 0.309167 | 0.000220 | 6.36E+08 (*) |
| 48 | 105.6 | 1621.9 | 3244 | 0.394646 | 0.000122 | 3.52E+08 (*) |
The Rh of particles was calculated from DLS measurements. The length of the particles was calculated fitting the MALS measurements to a rod-shape model. The number of PrP monomers per PrPSc particle was estimated from the PrPSc particle length, assuming a contribution of 0.49 nm in length per PrP monomer based on the recent parallel in-register intermolecular beta sheets (PIRIBS) model for the 263K strain. The PrP normalized intensity was determined by immunoblotting using Sha31 antibody. The relative number of PrPSc particles was calculated by dividing the PrP normalized intensity by number of PrP monomers per PrPSc particle. The maximum possible PrPSc particle density was calculated with ASTRA software (see methods) for fractions 46 and 48, and extrapolated to the rest of the fractions based on the relative number of PrPSc particles. (*) these values of particle density were calculated from MALS measurements directly, fitting the data to a rod-shape model, and assuming a rod radius of 4.25 nm. This calculation was performed for all samples; the specific values obtained for sample HY-E are shown in the table.
Once particle length was determined, we could use MALS data to directly calculate particle density (see Methods for details). However, because our starting material was brain homogenate, particles other than PrPSc were present in the fractions. To overcome this issue, we analyzed only fractions 46–50, with particles of Rh >70nm and highly enriched for PrPSc; these fractions had the highest ratio of PrP:total protein as determined by PrP immunoblotting intensity and UV absorption at 280 nm. In addition, the light scattering intensity and the corresponding particle density was much lower for NBH than for infected BHs in this region of the fractogram, suggesting that the main contribution to light scattering signal in fractions 46–50 was from large PrPSc particles (S1 Fig). The PrPSc particle densities for particles of Rh ~ 80–90 nm were 4.51x108 (± 8.54x107), 6.43x108 (±1.43x108), and 2.0x108 (± 1.61x107) particles/mL for 263K, HY, and DY, respectively (Fig 2E and Table 1).
We also wanted to ascertain PrPSc particle densities for smaller fractions. Having determined the particle lengths in each fraction, we then estimated the number of PrP monomers that would fit into fibrils of a given length, based on a parallel in-register intermolecular beta sheets (PIRIBS) architecture, with one PrP monomer present for every 0.49 nm in fibril height [31]. For particles of Rh 15 nm, this equaled particles of length ~58 nm, comprised of 116 PrP monomers (see Table 1). Although lateral bundling of PrPSc fibrils might also occur, its effect on mass per unit length calculations was not considered here.
Using the immunoblot data, we divided the PrP immunoblotting intensity from each fraction by the number of PrP monomers comprising the PrPSc fibrils at that fraction. These values provided information about the relative amounts of PrPSc particles in each fraction. Then we used the ratio of calculated particle density:PrP immunoblot intensity from fractions 46–50 to back-calculate the particle densities for the smaller fractions (Table 1).
PrPSc size distribution plots indicated that HY and 263K had almost a constant PrPSc particle density, 2.0x109 particles/mL, in the range of 60–200 nm length (from ~15 to 40 nm Rh according to DLS) (Fig 2E). Above 200 nm in length, the particle density decreased. DY PrPSc showed a more pronounced and sustained decrease in the number of particles as length increased from ~50 to 800 nm.
Glycosylation profile changes with the size of prion particle in a strain-specific manner
The ratio of di-, mono-, and un-glycosylated isoforms of PrP is one criterion for strain typing; however, 263K, HY and DY share the same PrP glycosylation pattern when unfractionated BH is analyzed by immunoblotting [6,32]. Our analysis of unfractionated BH showed the same di:mono:un-glycosylated ratio of 38:36:26 for total PrP (Figs 1A and 3A) and 55:35:10 for PrPres for all three strains. Given the differences in glycosylation ratios between total PrP and PrPres, we questioned whether changes in glycosylation pattern might occur in different subpopulations of PrPSc aggregates and, if so, whether these changes differed among strains. Indeed, immunoblot analysis of AF4 fractionations revealed strain-specific variation of PrP glycosylation patterns with the particle size.
Fig 3. Percentage of PrP glycosylation isoforms among AF4 fractions for 263K (red), HY (green) and DY (blue).

A) Percentage of di-, mono-, and un-glycosylated PrP present in detergent-solubilized BHs as calculated from immunoblots of three BHs for each strain (representative immunoblot depicted in Fig 1C). B) Percentage of di- (circles), mono- (triangles), and un-glycosylated (rhombus) PrP isoforms present in each AF4 fraction as calculated from NBH (empty symbols), 263K (red), HY (green), and DY (blue) immunoblots of three BHs for each strain (representative immunoblot shown in Fig 2B). The Rh (nm) of particles contained in fractions 16, 26, 36/38, and 52/54 are indicated in brackets. C) Percentage (average ± SE) of di- (top) and mono-glycosylated (bottom) PrP isoforms in the range of fractions where the glycoform ratio remains constant. Three BHs were analyzed for each prion strain. p-value *** ≤ 0.001.
The contribution of PrPC to the glycoform ratio in the first 16 fractions (particles smaller than 7 nm Rh) was determined by analyzing the NBH fractionation immunoblot (Fig 2B). More than 80% of PrPC eluted in fractions 6–10, where the glycoform ratio was 46:25:29 (Fig 3B). Between fractions 11 to 16, the percentage of di-glycosylated isoform increased to 75:10:15, as expected, given that extra glycan groups increase the Rh of PrPC. No appreciable PrP signal was detected after fraction 20, meaning that the contribution of PrPC to glycoform ratio calculations was limited to particles smaller than 7 nm Rh.
Given that the host PrPC is the same in all prion strains studied (Syrian golden hamster), we predicted that the glycoform ratios of PrP within fractions 6–10 would be similar across all strains. However, whereas HY and DY had almost the same glycoform ratios as NBH in these fractions, 263K had a higher proportion of di-glycosylated and less un-glycosylated isoforms (Fig 2B). This may indicate that there was less PrPC in these fractions, as suggested by the PrP size distribution analysis (Fig 2C), and/or a greater amount of small, highly di-glycosylated PrPSc oligomers contributing to the PrP signal, that shifted the glycoform ratio of this strain in these fractions.
For particles larger than 7nm Rh, 263K and HY had very similar glycosylation patterns. In particles of size range 7–15 nm Rh, ratios were 65:25:10 and 60:30:10 respectively; for particles 15–45 nm Rh, the ratio gradually shifted to 55:35:10 and 51:36:13 respectively, and then remained almost constant in particles of 45–300 nm Rh (Fig 3B and 3C). Both the reductions in di-glycosylated and increases in mono-glycosylated isoforms were statistically significant for 263K and HY (Fig 3C).
Interestingly, DY had a distinct glycoform ratio which remained relatively constant at 45:38:17 for all particles larger than 7 nm. There was a trend towards a reduction in di-glycosylated particles with increasing size, but these changes were not statistically significant (Fig 3C).
HY is comprised of more stable PrPSc particles than 263K
PrPSc structural stability is another feature used for prion strain typing. Incubation of brain homogenate with increasing concentrations of denaturant has previously demonstrated that 263K and HY are structurally more stable than DY [10,12,33,34]. Additionally, differences in the exposure of N-terminal regions in a narrow range of denaturant concentrations revealed a higher stability of HY over 263K at tertiary and secondary structure [10]. Instead of measuring the structural stability of the whole population of PrPSc particles present in a BH, we explored the dissociating effect of SDS micelles over the different sub-populations of PrPSc particles. Using the same fractionation conditions as before, we increased the concentration of sodium chloride from 20 to 150 mM in the AF4 running buffer, promoting SDS micellar formation (S2 Fig). Under SDS micellar conditions, all prion strains underwent a change in size distribution of PrPSc particles (Figs 4A, 4C and S3). As expected, the proportion of particles with Rh ~ 4 nm, corresponding to monomeric (and/or dimeric) PrP, significantly increased in all cases. For DY, the amount of monomerization was the largest, increasing from 10% to 50% of the total population. In contrast, HY and 263K particles of Rh ~ 4 nm increased from 10% to only 25% of the population, indicating fewer particles were monomerized. Interestingly, AF4 fractionation also revealed subtle but statistical differences between 263K and HY. SDS micelles increased the population of particles with Rh 4–10 nm more for 263K than for HY as measured by subtracting the cumulative curves from AF4 fractionation in the presence and absence of SDS micelles (Fig 4D). These results indicate a rank stability of PrPSc quaternary structure of HY > 263K >>> DY.
Fig 4.

Cumulative PrP signal as a function of particle size for 263K (A), HY (B), and DY (C) fractionated in the absence (light symbols) or in the presence (dark symbols) of SDS micelles. Circles, triangles, and rhombus represent data obtained from three different BHs (indicated by the letters A-I). Vertical and horizontal dotted lines intersect at 4 nm Rh and 50% of the cumulated PrP signal, respectively. D) Subtraction of the cumulative size distribution curves in the presence and absence of SDS micelles for 263K (red) and HY (green). Error bars represent SE; p-value * ≤ 0.05.
PrPres size distribution is independent of the strain phenotype
To investigate the association between PrP size and protease resistance, we PK-treated the AF4 fractions depicted in Fig 2B and quantified the amount of PrPres by immunoblotting (Fig 5A). We used Sha31, an antibody that recognizes the epitope 145–152 of the hamster PrP, which is within the PK-resistant core of PrPSc. The loss of epitope detection is interpreted as at least partial digestion of this epitope and the associated core. We therefore define PrPres as those particles whose Sha31 epitope remains stable under conditions of PK digestion. As expected, the AF4 fractions containing the smallest particles showed a lower PrP signal after PK digestion [13,15,18]. Surprisingly, despite the differences in size distribution of total PrP between HY, 263K and DY, immunoblotting analysis showed a very similar pattern of the PK resistant ~20 kDa fragment and its mono- and di-glycosylated isoforms, with a signal increase starting at fractions 26–28 in the three strains (Fig 5B). We plotted PrPres size distribution by correlating the Rh measurements of AF4 fractionation, before PK digestion, with the immunoblotting intensity of the PK-treated fractions. PrPres signal appeared rather abruptly at ~15 nm Rh, and the intensity of PrPres signal reached a maximum at ~40–50 nm Rh, and was sustained until >200 nm Rh (Fig 5C and 5D). This result strongly suggests that PrPSc particles cannot maintain PK-resistance below 15 nm Rh, regardless of strain.
Fig 5.

A) Representative immunoblot for PrPres from 263K, HY, and DY AF4 fractions; fractions from each brain homogenate sample were treated with PK and loaded in two polyacrylamide gels (gel 1: fraction 4 to 24, gel 2: fraction 26 to 52). B) Quantification of PrPres signal from 263K (red), HY (green), and DY (blue) AF4 fractions; (average ± SE). C) Size distributions of PrPres particles from 263K (red), HY (green) and DY (blue). Circles, triangles, and rhombus represent data from the three BH analyzed for each strain. Each set of data was fitted to a sigmoidal curve and the average of the curves is shown as a dashed line for each strain. D) Average size of the smallest PrP particle with increased PrPres signal. Error bars represent SE.
Within each strain, PrPres particles have similar stability regardless of size
Next, we analyzed the structural stability of PrPres particles, defined here as the presence of epitope 145–152 under PK digestion conditions, as a function of size by incubating fractions larger than 25 nm Rh with increasing concentrations of SDS followed by digestion with PK. As expected, immunoblotting analysis showed that PrPres particles were more stable for 263K than for DY (Fig 6). Under our experimental conditions, 263K PrPres intensities could be fitted to a sigmoidal curve, with 50% signal reached at 0.09–0.11% SDS. Conversely, DY PrPres signal was better fitted to a single exponential decay model with its 50% intensity occurring at 0.05–0.08% SDS (Fig 6). Regardless of inter-strains differences, the structural stability of PrPres particles within a given strain remained constant despite increasing size, at least in the range from 25 to 65 nm (Fig 6C). These results suggest that after acquiring PK resistance, no major conformational changes take place in PrP structure, at least around this epitope, other than the addition of more PrP units.
Fig 6. Conformational stability of 263K and DY PrPSc particles ranging from 25 to 65 nm Rh.

A) Representative immunoblots of 263K (top) and DY (bottom) fractions incubated with increasing concentrations of SDS and digested with PK. 3F4 (dilution 1:3,000) was used as primary antibody. B) Densitometric analysis of 263K (top) and DY (bottom) of remaining PrPres after incubation of AF4 fractions with increasing concentrations of SDS. 263K data was fitted to a sigmoidal function whereas DY data was fitted to a single decay hyperbolic function. Samples from three BH for each strain were analyzed. C) SDS concentration at which PrPres reaches half of the maximum intensity as a function of particle size. Data represents average of 3 replicates and error bars represent SE.
PrPSc particle seeding activity varies with particle size in a strain-specific manner
It has been previously demonstrated that HY has a higher templating activity than DY in vitro and in vivo [12,35,36]. Here we used RT-QuIC assay to compare the templating activity of the AF4 fractions containing PrP particles of 10, 20, 40 and 70 nm Rh from 263K, HY and DY (Fig 7). For 263K and HY, the aggregation of recombinant hamster-prion protein (SHaPrP) had a shorter lag phase when seeded with fractions containing larger particles (40 and 70nm Rh) than when seeded with the same volume of fractions containing smaller particles (10 and 20nm Rh) (Fig 7A, 7B and 7D). Interestingly, all tested DY fractions had the same seeding activity, with lag phases comparable to the slower HY and 263K fractions (Fig 7C and 7D). We then evaluated the ability of these fractions to convert PrPC into PrPres in CAD5-PrP−/− (HaPrP) cells, a murine catecholaminergic cell line lacking endogenous mouse PrP expression and expressing SHaPrP [37]. After seven passages of CAD5-PrP−/− (HaPrP) cells post-exposure to particles of 8, 10, 30 and 70 nm Rh from HY, the highest PrPres signal was found in cells treated with 30 and 70 nm Rh particles (Fig 7E). Interestingly, we also detected PrPres in cells treated with the 8 nm Rh particles, which are PrPsen particles as assessed by Sha31 antibody. While there are no reports of successful infection of CAD5-PrP−/− (HaPrP) cells with DY, surprisingly, we were able to detect PrPres signal for cells treated with 30 and 70nm Rh particles from DY, although with much less intensity than those from HY. The lack of infectivity of PrPsen DY particles, despite their intrinsic seeding activity, could be influenced by their lower structural stability, and consequent higher clearance, than the HY particles of the same size. To more directly compare the seeding activity of particles of the same size among prion strains, we seeded the RT-QuIC reaction with the same amount of total PrP monomeric units, as measured by immunoblotting. For particles smaller than 20 nm Rh, HY and 263K showed higher seeding activity than DY, whereas for particles 40 nm Rh and larger, DY particles were more efficient (Fig 7F). Of note, there was a statistically significant difference in lag phase between HY and 263K for particles bigger than 40 nm. For all these reactions, the amount of PrP added as seed was based on monomeric PrP content, not the number of PrPSc particles. Thus, a direct correlation between seeding activity and size of PrPSc was evident in the case of DY, with a shortening of lag phase as Rh increases, but for HY and 236K, this correlation was harder to establish, since both templating activity and number of PrPSc particles decreased with the increment of particle size.
Fig 7. Templating activity of 263K (red), HY (green), and DY (blue) fractions assessed by RT-QuIC assay and cell infectivity assay.

The kinetics of aggregation of recombinant SHaPrP unseeded (black dotted line) or seeded with particles of 10 (circles), 20 (squares), 40 (triangles) and 70 (rhombus) nm Rh from 263K (A), HY (B), and DY (C) are depicted. All aggregation reactions were seeded with the same volume of AF4 fractions. D) Lag phases calculated from the kinetics depicted in A, B, and C. E) Immunoblot for PrPres accumulated in CAD5-PrP−/− (HaPrP) at passage #7 after incubation with AF4 fractions containing particles of 8, 10, 30 and 70 nm Rh. Brain homogenate and non-infected cells (n.i.) were used as positive and negative controls. F) Lag phases for the aggregation reactions of recombinant SHaPrP seeded with particles of 10, 20, 40 and 70 nm Rh, using the same amount of PrP mass. Data represents average of 3 replicates and error bars represent SE; p-value * ≤ 0.05, ** ≤ 0.01, and *** ≤ 0.001.
Discussion
Prion strains consist of a spectrum of conformationally distinct PrPSc particles that encode the information for a specific disease phenotype. However, the level of intra- and inter-strain heterogeneity of these PrPSc particles, as well as the biological relevance of this heterogeneity, is poorly understood. Using an AF4-DLS-MALS configuration, we isolated and precisely determined the size distribution of PrPSc particles in the brains of Syrian golden hamsters infected with 263K, HY or DY strains at end stage of disease. In addition, we provided a detailed description of changes in glycoform profiles, resistance to protease, structural stability, and replication activity as a function of PrPSc quaternary structure. Our data allow us to propose a model that explains PrPSc heterogeneity.
PrPSc quaternary structure is associated with disease phenotype
Analysis of unfractionated PrPSc from HY and 263K brain homogenate previously demonstrated that they have very similar secondary structures, structural stabilities, replication activities, PK digestion profiles, and glycosylation patterns [6,12,38,39]. Here we show that these two prion strains with almost identical phenotype also have almost identical size distributions of PrPSc particles, further supporting the association between PrPSc quaternary structure and strain phenotype. Conversely, DY, a strain with different clinical signs, incubation period, brain titre, and brain lesion profile, has a substantially different PrPSc size distribution, with a larger population of small PrPsen particles and smaller population of large PrPres particles than HY and 263K. The PrPsen:PrPres ratio was previously correlated with the incubation period in hamster-adapted prion strains, in which “fast” strains like 263K and HY have smaller PrPsen:PrPres ratio than “slow” strains like DY [10,40]. Our results not only confirm this association between PrPsen:PrPres ratio and incubation period, but also provide a detailed description of the size distribution and biochemical features of the PrPSc particles comprising each of these two populations at end-stage of disease.
The combination of particle density, structural stability, and replication activity of PrPSc particles contributes to prion strain incubation period
In addition to the PrPsen:PrPres ratio, PrPSc chemical stability has been cited as an explanation for variable incubation periods. When unfractionated brain homogenate was treated with increasing concentrations of SDS or GdnHCl, an inverse correlation between PrPSc chemical stability and incubation period was found for hamster-adapted prion strains [12,33,34]. Interestingly, the opposite situation was described for mouse-adapted prion strains, in which accumulation of less stable PrPSc particles was found in strains with shorter incubation periods [41–43]. Two different hypotheses have been proposed to explain these converse correlations: a) the more stable hamster-PrPSc particles have a lower rate of clearance in the brain, thus more time to replicate and to exert their neurotoxic effects, shortening the incubation period; b) the less stable mouse-PrPSc particles have a higher rate of fragmentation which generates more replicative nuclei, causing accelerated PrPSc propagation and shortening the incubation period [10,12,36,42–44]. Our study shows that both structural stability and templating activity change with the size of PrPSc particles and do so in a strain-specific manner. Since the particle density of PrPSc subpopulations is also strain-specific, we propose that the incubation period of a prion strain is a consequence of the combination of the stability, the templating activity, and particle density of each PrPSc subpopulation accumulated in the brain. For example, highly replicating PrPSc particles, if only present in low amounts, will not exert much effect on the overall replication activity of the strain. This is the case for DY. We found that PrPres particles from DY have higher replication activities than PrPres from HY and 263K, possibly because their lower stability creates more replication nuclei; however, the total amount of these DY PrPres particles is much lower than the amount of the less replicative HY and 263K PrPres particles. Furthermore, the predominant population in DY is PrPsen, which has a lower replication activity than the PrPsen from HY and 263K. In addition, the lower stability of DY PrPSc particles, which favours replication by creating new seeds, could also reduce replication capacity by promoting clearance. Thus, the combination of more PrPsen particles with low replication efficiency and structural stability and fewer, less stable PrPres particles with higher replication efficiency, leads to an overall combined effect that DY is less efficient at replicating, resulting in a longer incubation period.
Protease resistance develops at a hydrodynamic radius of 15 nm, regardless of strain
Surprisingly, despite the marked differences in size distribution of total PrP (PrPsen plus PrPres) and in the ratio of PrPsen/PrPres between the “fast” and “slow” strains studied here, the size distributions of PrPres are almost identical. In all cases, we observed a transition from PrPsen to PrPres starting at Rh ~15nm. This result strongly suggests a strain-independent conformational change in PrPSc structure once particles reach 15 nm Rh. Alternately, if the smallest particles with partial resistance to PK digestion simply came from the fragmentation of larger PrPres particles in the brain or during sample preparation, the 15 nm Rh PrPres particles might not reflect the point at which conformational change occurs, but rather the smallest size at which a PK-resistant conformation can be maintained. Interestingly, we recently described the evolution of PrPSc particles in the brain of RML-infected mice over the course of disease, observing that PrPres particles were also always 15 nm Rh or larger [29].
To estimate how many PrP units would comprise a particle of 15 nm Rh, we made two assumptions, one assuming a fibrillar shape [26], and one assuming a parallel in-register intermolecular beta-sheet (PIRIBS) architectures [31]. This, combined with our MALS measurements, allowed us to estimate that fibrillar PrP particles of Rh ~15nm will have a length of ~58 nm and consist of 116 PrP monomers giving a final mass of 3944 kDa, if we use an average Mw = 34 kDa per monomer (based on the glycoform stoichiometry shown for 263K and HY, Fig 3). This size estimate is in line with other studies; sucrose gradient centrifugation and SEC studies of 263K and mouse-adapted scrapie identified PK-resistant particles starting at a Mw higher than 2000 kDa [13,18]. We further validated our assumptions by comparing our Mw calculation of larger particles to those reported by Silveira et al., on AF4-fractionated 263K. Silveira et al. found that 263K PrPres particles of Rh ~37 nm are fibrillar particles with a calculated MW of 7770 (± 4270) kDa (by MALS) and corresponding to 362 (± 198) PrP units considering a PrP molecules averaging 21.5 kDa for these PK treated particles [26], comparable to our 39 nm Rh particles with calculated 399 PrP monomers (Table 1).
We also did these calculations based on a four-rung β-solenoid structure containing two protofilaments, with two PrP monomers present for every 1.92 nm in fibril height, and a rod radius of 5.5 nm [45–48]. S1 table depicts the results for these calculations.
AF4 fractionation reveals new strain differences
Although previous studies have not detected differences in glycosylation patterns between 263K, HY and DY [30,32], our data show that the transition from PrPsen to PrPres correlates with changes in glycosylation pattern and the degree of this change is strain-specific. Thus, we observed a more pronounced variation in glycotype stoichiometry for 263K and HY than for DY, which shows a lower and almost constant percentage of di-glycosylated isoform for all PrPSc particles. Interestingly, lower levels of the di-glycosylated isoform have been associated with the accumulation of smaller and less stable PrPSc particles in rodent prion strains [42,49,50]. Recent studies have demonstrated the relevance of PrPSc glycotype stoichiometry in prion replication and faithful transmission of strain information [49,50]. Our data show that this stoichiometry either has an influence on, or is influenced by, the size of the PrPSc assemblies.
AF4 fractionation also revealed subtle differences between the almost identical prion strains HY and 263K [33,38]. Using antibodies against the N-terminal region, Safar et al. reported a higher epitope exposure for 263K than for HY, when crude brain homogenates were incubated with 2 to 3M GdnHCl [10]. Here we show that 263K PrPSc particles dissociate in SDS at a higher rate than HY particles, demonstrating a difference in stability of PrPSc assemblies between these two strains. In addition, RT-QuIC assays for the fractionated PrPSc populations revealed a higher replication activity for HY particles with Rh > 40nm.
A model for PrPSc heterogeneity
Collectively, our data suggest that the main contributor to PrPSc intra-strain heterogeneity is PrPSc quaternary structure. PrPSc particles grow in size with quasi-equivalent biochemical properties. Only one major change in the core structure (secondary/tertiary structure) takes place, and this change manifests as a switch from protease-sensitive to protease-resistant species, accompanied by a slight change in glycosylation stoichiometry, at a strain-independent size of 15 nm Rh (Fig 8). Once PrPSc becomes partially resistant to PK digestion, its conformational stability remains almost constant up to at least Rh ~70 nm (or ~500 nm fibril length), suggesting that no other major conformational changes take place in PrPSc core structure. This results in a continuum of quaternary structures, with the amount of PrPSc particles comprising each size subpopulation, along with their specific stability and templating activity, defining the incubation period, and potentially the clinical phenotype, of a particular prion strain. Thus, we could divide PrPSc particles into different sized subpopulations and represent incubation period (IP) as follows:
where k is a PrPSc subpopulation. The structural stability of PrPSc particles will influence incubation period through 1) rate of clearance, and 2) rate of templating nuclei generation by fragmentation.
Fig 8. Proposed model of PrPSc structural heterogeneity.
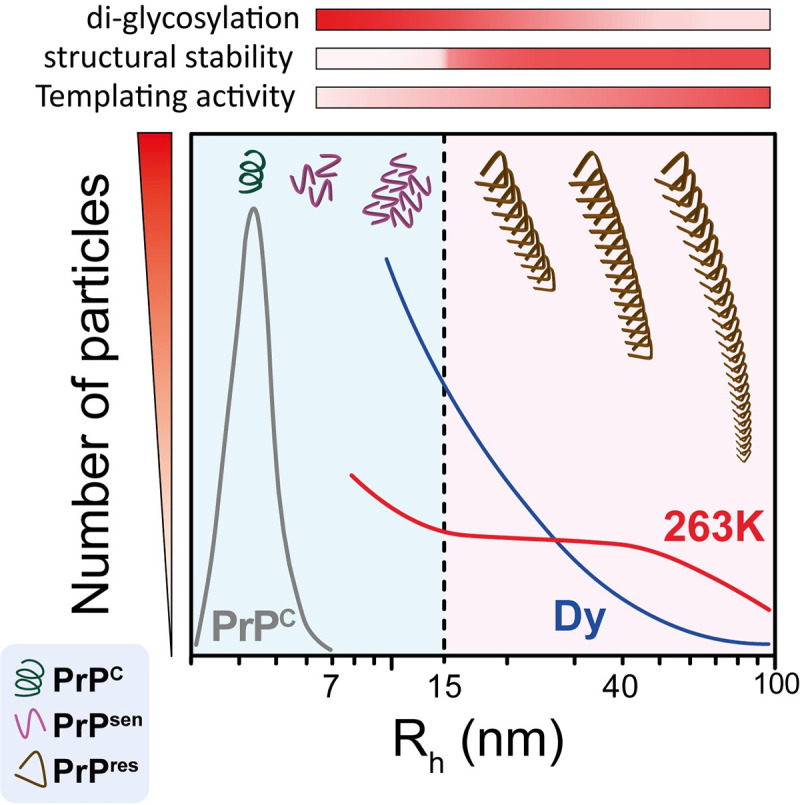
A continuum of PrPSc quaternary structures accumulate in the brain of prion-infected animals. Whereas the size of PrPSc particles is the main contributor to PrPSc heterogeneity, a major change in the core structure occurs when 15 nm Rh is reached, generating two distinctive subpopulations that define the properties of the prion strain. At this size, there is a substantial increase in resistance to proteolysis and a reduction in the incorporation of di-glycosylated PrP isoforms into the PrPSc particles. Once the mature PrPres particle is formed, the structural stability and glycosylation patterns remain constant despite further addition of PrP monomeric units. The PrPSc templating activity is also affected by the size of the particle, increasing as the particle size increases, as seen in the case of DY.
The above considerations regarding PrPSc particle sizes assume that the isolated PrPSc particles contain only prion protein, but it is likely that that other proteins and macromolecules could be interacting with PrPSc. If the degree of interaction varies with strain, this could affect the particle size distribution. Additionally, although we intentionally chose sample solubilization conditions that minimize the dissociation of PrP aggregates, the necessary presence of detergents in the solubilization and AF4 running buffers could still affect the actual size distribution of native PrP particles present in the prion infected brains, especially in the case of DY. Finally, although we divided PrPSc particles into PrPsen and PrPres populations, this was based on Sha31 immunoblot detection, which is specifically probing for the resistance of epitope 145–152, so we cannot comment on the existence of assemblies with smaller resistant cores more C-terminal to this epitope.
Conclusion
Our results show that PrPSc quaternary structure is a significant contributor towards PrPSc structural heterogeneity, resulting in biochemically distinctive PrPSc subpopulations, and the proportion of these subpopulations correlates with prion strain phenotype. Our AF4-MALS approach to separating and analyzing prion aggregates from different prion strains has implications for other protein misfolding-related neurodegenerative disorders where conformational strains have been proposed [51–57]. The structural heterogeneity of α-synuclein, amyloid β, and tau aggregates could form the molecular basis for the broad spectrum of phenotypes seen in synucleinopathies, Alzheimer’s disease, and other tauopathies. By applying our technique to these conditions, we may not only promote an understanding of strain-phenotype relationships across neurodegenerative diseases but help direct therapies towards the most relevant aggregate conformations.
Methods
Ethics statement
All animal studies described herein were performed in accordance with Canadian Council on Animal Care (CCAC) guidelines, under protocol AUP914 approved by the animal care use committee for Health Sciences Laboratory Animal Services at the University of Alberta.
Hamster-adapted prion strains / brain tissues
Brain tissues were taken from clinically affected Syrian golden hamsters (Envigo) inoculated with brain homogenates (BH) of hamster-adapted strains of different origin: transmissible mink encephalopathy-derived Hyper (HY) and Drowsy (DY), and Scrapie-derived 263K. Three 263K-infected brains were obtained from hamsters inoculated with 10% 263K-stock BH and euthanized upon presenting with overt clinical signs at 74 days post infection (dpi). Seven HY-infected brains were generated by inoculation of 10% HY-stock BH, resulting in advanced clinical disease at 70 dpi. The DY samples included two brain homogenate pools; pool 1 consisted of three brains from hamsters inoculated with a 10−5 BH dilution of a DY-stock that were euthanized with clinical signs at 240 dpi. The second drowsy pool was composed of seven DY-infected brains from hamsters inoculated with a 10−5 dilution of the pool 1 and collected after >200 dpi. Two individual DY-infected brains from hamsters euthanized at 168 and 170 dpi were generated by inoculation of a 10−2 dilution pool 2 into naïve hamsters.
Brain homogenization and sample blinding
Individual brains were weighed and homogenized in ultra-pure water (Sigma) with ceramic beads in an Omni Bead Ruptor to make 10% brain homogenates (w/v). The HY and DY pools were initially homogenized in ultrapure water using a dounce glass homogenizer. The resulting pool homogenate was further passaged through needles of different diameters, 17- 21G. All samples were aliquoted in sets of three for each strain (individual or pool homogenate) and assigned a sample number for blinded analysis. Aliquots were kept at -80°C until further analysis.
Batch mode dynamic light scattering (DLS)
DLS measurements of solubilized brain homogenate and pellets resuspended in solubilization buffer (50 mM HEPES pH 7.4; 300 mM NaCl; 10 mM EDTA; 4% (w/v) dodecyl-β-D-maltoside (Sigma)) were performed with a Malvern Zetasizer-Nano S. A 633 nm wavelength HeNe laser was used to detect backscattered light at a fixed angle of 173°. Measurements were performed at 20°C and the solution viscosity and refractive index of water was assumed for calculation purposes. The data was collected without attenuation and a minimum number of 10 consecutive runs of 10 seconds each was averaged to obtain the autocorrelation function. Particle size was calculated by the manufacturer’s software through the Stokes-Einstein equation assuming spherical shapes of the particles.
Asymmetric-flow field-flow fractionation (AF4)
Brain homogenates (10% w/v) were solubilized by adding an equal volume of solubilization buffer (50 mM HEPES pH 7.4; 300 mM NaCl; 10 mM EDTA; 4% (w/v) dodecyl-β-D-maltoside (Sigma)) and incubated for 45 min on ice. Sarkosyl (N-lauryl sarcosine; Fluka) was added to a final concentration of 2% and incubated on ice for 30 min. The sample was centrifuged (20,000xg, 10 min) at 4°C. The supernatant was collected and 350 μg total protein were subjected to asymmetrical-flow field-flow fractionation on an AF2000 Postnova system using 50 mM HEPES pH 7.4 (containing 50 mM sodium chloride and 0.05% sodium dodecyl sulfate (SDS) to minimize interaction of proteins with the channel membrane) as the running buffer. The channel was 26.5 cm in length and 350 μm in height, constructed with a trapezoidal spacer of maximal width 21 mm at the inlet, and lined with a 10 kDa cutoff polyethersulfone membrane at the accumulation wall. Samples were focused for 4 min and then eluted at a channel flow of 0.5 mL/min with constant cross-flow for the first 10 min, decreasing from 1.5 to 0.35 mL/min in the following 15 min, from 3.5 to 0 mL/min in the next 30 min, and run with no cross-flow for the last 10 min. A slot pump was run at 0.3 mL/min to concentrate the samples before they passed through the detectors. Fractions of 0.2 mL were collected. Multi-angle light scattering (MALS) and dynamic light scattering (DLS) were collected simultaneously in the in-line DAWN HELEOS II detector (Wyatt Technology), operating at a wavelength of 662 nm. Data analysis was performed with ASTRA 6.1.7.17 software (Wyatt Technology).
Hydrodynamic radius measurements from in-line DLS
DLS data were collected every 2 seconds at 140.1° scattering angle for determination of hydrodynamic radius (Rh) of the AF4 eluting particles. Fitting the data to an autocorrelation function was performed using the cumulants model. The injection of 350 μg of protein in each AF4 run ensures sufficient sample to scatter at least three times more light than the solvent, providing good signal/noise ratios for all size ranges.
Calculation of particle length using in-line MALS
Static light scattering data were collected at 16 detector angles simultaneously every 2 seconds across the fractogram. For particles with sizes above the angular variation detection limit (Rg ~10 nm), we used the MALS data to determine particle length assuming a rod-shaped particle. This assumption is supported by previous AF4-MALS-DLS and electron microscopy analysis of highly purified 263K strain PrPSc particles where it was shown that PrPSc particles became ellipsoid-spherical at Rh = 12.4 nm, and larger particles had a more fibrillary structure [26]. We also detected fibrillar structures in our larger fractions where PrP was enriched (Fig 2D). The excess Rayleigh ratio, R(θ), as a function of scattering angle, was fit to the Rayleigh-Gans approximation for an infinitely thin rod using the ASTRA software, in which the form factor, P(θ), obeys the following relationship [58]:
where u = [(π nο/λο) L sin(θ/2)], L is the rod length and is assumed to be much greater than the rod diameter, nο is the refractive index of the solvent at the incident radiation (vacuum) wavelength, and λο is the incident radiation (vacuum) wavelength.
Number of PrP monomers per particle
Knowing particle size allowed us to calculate the number of PrP monomers that would “fit” into fibrils of different lengths. Based on the recently published model for 263K PrPSc fibrils [31], we assumed that each monomer contributes with a height of 0.49 nm, giving the following calculation:
Calculation of PrPSc particle density
Based on the number of monomers present in each sized particle, we could use our immunoblot data of PrP intensities (representing relative amount of total PrP monomers in each fraction) to further calculate the relative number of PrPSc particles present in each fraction. The absolute number of PrPSc particles present in each fraction could not be directly measured because there were proteins other than PrPSc particles present; these other proteins contributed to the MALS signal that would normally be used to calculate particle density. However, we were able to determine the maximum number of PrPSc particles/mL that could be present, by analyzing MALS data from fractions 46–50. These fractions have much less protein, as measured by UV signal, but have the highest level of PrP, based on immunoblot, making them the purest PrP fraction. By assuming all MALS signal came from PrPSc particles in these fractions, we calculated the theoretical maximum number of PrPSc particles/mL (PrPSc particle density) present in these fractions. To determine the theoretical maximum PrPSc particle density for the smaller fractions, we took the ratio of PrPSc particle density:relative number of PrPSc particles from fractions 46–50 and applied it to the smaller fractions (see Table 1 for details).
For particles of uniform density and volume V, the number of particles per mL in the ith slice, ni, is proportional to the zero-angle Rayleigh ratio divided by the square of the particle´s volume:
The value of R(0) was determined by fitting the light scattering intensity as a function of angle to the rod model. The refractive index of the prion particles was set to 1.587, as this is a typical average value for protein refractive index [59]. For estimation of particle volume, we used the particle lengths calculated from MALS data and assumed a rod radius of 4.25 nm [31]. From this, the ASTRA software (Wyatt Technology) was able to calculate the number of PrPSc particles per mL for each eluting data slice across the fractogram [60].
Proteinase-K digestion
For all Proteinase-K (PK) digestions, 0.5 μg/μL of protein was incubated with 20 μg/mL of PK at 37°C for 1h and 300 rpm orbital shaking. For digestion of AF4 fractions, 10 μL of each fraction were incubated with 20 μg/mL of PK in the presence of 0.5 μg/μL bovine serum albumin (BSA) as the PK substrate to account for the low mass of total protein in eluted fractions. The reaction was terminated by addition of Pefabloc (Sigma) and incubation at 4°C for 10 min, followed by boiling with SDS-sample buffer and electrophoresis on 4–12% NuPAGE Bis-Tris gels (Invitrogen).
Conformational stability assay
The conformational stability assay was performed as described previously with slight modifications [12]. Briefly, 10 and 20 μL from 263K and DY AF4 fractions were incubated for 30 min. with increasing concentration of SDS, from 0.025 to 0.635% (w/v), at 22°C and shaken at 450 rpm. The samples were then digested with 1 μg/mL of PK for 1 h at 37°C and 450 rpm orbital shaking in the presence of 15 μg of bovine serum. The reaction was terminated by the addition of 4 mM PMSF. For 263K samples, equal volumes of 2x loading buffer (125 mM Tris HCl pH 6.8, 20% glycerol, 4 mM EDTA, 6%SDS, and 10% 2-mercapto ethanol) was added, boiled for 10 min., loaded in a 4–12% NuPAGE Bis-Tris precast gels, transferred to PVDF membranes, and probed with the 3F4 antibody (dil. 1:3,000; Sigma). To improve immunoblot PrPres signal, DY samples were methanol precipitated by adding 5x volumes chilled methanol and incubated for 2h at -30°C. The samples were centrifuged at 18,200 x g for 30 min., and the pellets were dried, resuspended in 1X loading buffer, boiled for 10 min. and subjected to SDS-PAGE and immunoblotting with 3F4 (dil. 1:3,000; Sigma).
PrP signal was analyzed by Image Quant software and PrPres intensity was fitted to sigmoidal and one phase decay models with Graph Pad Prism 8.0.
Expression and Purification of recombinant hamster PrP
Syrian hamster prion protein (SHaPrP) containing residues 90–231 (which form the protease-resistant fragment in PrPSc) was cloned into the pET-15b plasmid between the XhoI and EcoRI sites as described previously [61]. Cys residues were introduced at each terminus by mutating Ser residues in the thrombin cleavage site and the prion site S232. The 19-kDa, N-terminal His-tagged SHaPrP was expressed in Escherichia coli BL21 (DE3) and purified by FPLC (GE Healthcare) using a nickel-nitriloacetic acid (Ni-NTA column). PrP was refolded on the Ni-NTA column, with native folding confirmed by circular dichroism spectroscopy, and the purity and identity of the protein verified by SDS–PAGE and immunoblotting (3F4 antibody, Millipore).
Real-time quaking-induced conversion (RT-QuIC) assay
Recombinant PrP protein in 6M guanidine hydrochloride (GdnHCl) solution was diluted in RT-QuIC buffer (20 mM sodium phosphate pH 7.4; 130 mM NaCl; 10 mM EDTA; 0.002% SDS) to a final protein concentration of 0.2 mg/mL (and residual 0.2 M GdnHCl). Reactions were seeded with AF4 fractions containing the same amount of total PrP (based on immunoblotting intensity) in a final volume of 180 μL/well. The aggregation reactions were carried out in 96-well plates (white plate, clear bottom; Costar 3610) sealed with thermal adhesive film (08-408-240; Fisherbrand). The samples were incubated in the presence of 10 mM thioflavin T (ThT) at 42°C with cycles of 1 min shaking (700 rpm double orbital) and 1 min rest. ThT fluorescence measurements (450+/210 nm excitation and 480+/210 nm emission; bottom read) were collected every 60 minutes. There were three technical replicates per experiment.
CAD5 cell infection assay
A murine catecholaminergic cell line lacking endogenous mouse PrP expression and stably transfected with hamster PrP, CAD5-PrP−/− (HaPrP) cells, were generously provided by Dr. Joel Watts of Tanz Centre for Research in Neurodegenerative Diseases, University of Toronto [37]. CAD5-PrP−/− (HaPrP) were grown in Opti-MEM medium (Thermo Fisher Scientific) containing 10% fetal bovine serum and 0.2% penicillin-streptomycin at 37°C with 5% CO2. Two milliliters of cells suspension were combined with 120 μL of AF4 fraction and incubated in a 6-well plate for two days. Then, the media was aspirated and the cells were mechanically passaged seven times every 2–3 days and harvested for immunoblotting analysis. Cells were homogenized in 50 μL ice cold RIPA buffer. Total protein concentration was determined by BCA assay. To detect PrPres, 20 μg of protein (in 10 μL) were treated with 40 μg/mL Proteinase K (Invitrogen) at 37°C for 1 h. Then 2X loading buffer (187.5 mM Tris HCl pH 6.8, 15% glycerol, 15% SDS, 9 mM EDTA, 8 M urea, 8% βME) was added, samples were boiled for 10 min, and 15 μL were subjected to immunoblotting analysis using Sha31 (1:20,000 dilution) as primary antibody.
Negative stain electron microscopy
Carbon-coated 200 mesh copper grids (Electron Microscopy Science, USA) were glow charged for 30 seconds using a Pelco Easy Glow 100 x glow discharge unit (Ted Pella Inc, USA). Microliter amounts (2–3 μl) of the purified fractions were adsorbed on the grids for up to 10 min by sitting drop method. The grids were washed three times (3 × 50 μL) with filtered ammonium acetate (0.1 M and 0.01 M), pH 7.4 and stained with filtered 2% (w/v) uranyl acetate (Electron Microscopy Science, USA) or filtered 2% (w/v) phosphotungstate (Sigma-Aldrich). Excess stain was removed using filter paper and the grids were air-dried. The samples were viewed with a Tecnai G20 transmission electron microscope (FEI Eindhoven, NL) using an acceleration voltage of 200 kV. Electron micrographs were recorded on an Eagle 4 k × 4 k CCD camera (FEI).
Immunoblotting
Samples were prepared in Laemmli loading buffer containing SDS and 2-mercaptoethanol, boiled for 10 min, electrophoresed on 4–12% NuPAGE Bis-Tris precast gels using an Invitrogen system and transferred to polyvinyl difluoride (PVDF; Millipore) membranes (wet transfer). Blots were then incubated with primary 1:30,000 dilution of Sha31 antibodies (Spibio) in TBS-0.5% Tween 20 at 4°C overnight. 1:5,000 dilution of Sha31 was used for AF4 fractions. AP-tagged anti-mouse IgG (Promega) in TBS-0.1% Tween was used as secondary antibody. Blots were developed using AttoPhos substrate (Promega) and detected on an ImageQuant LAS 4000 (GE Healthcare). To compare distribution curves from different immunoblots, PrP intensities from each fraction were first plotted against the average Rh for that fraction, then the area under the curve for the entire run was calculated and normalized to 100% for each strain.
Statistical considerations
One-way analysis of variance (ANOVA) was used to identify group-wise differences and post-hoc Tukey’s test was used to identify pairwise differences (p < 0.05 considered significant). All analyses were performed using Graphpad Prism (v5).
Supporting information
Particle density (A) and UV signal at 280nm (B) for NBH (black), 263K (red), HY (green), and DY (blue) BHs fractionated by AF4. In fractions 46–50, higher particle density and UV signal is evident for prion-infected samples when compared with NBH.
(TIF)
SDS micelles present in the running buffer are evident in the DLS measurements, where background values are ~2–3 nm Rh (red dots).
(TIF)
Fifty mM HEPES pH 7.4 containing 150 mM sodium chloride and 0.05% SDS was used as AF4 running buffer. Three brains for 263K (red, A-C), HY (green, D-F), and DY (blue, G-I) were analyzed.
(TIF)
The Rh of particles was calculated from DLS measurements. The length of the particles was calculated fitting the MALS measurements to a rod-shape model. The number of PrP monomers per PrPSc particle was estimated from the PrPSc particle length, assuming a contribution of 1.92 nm in length per PrP monomer and a PrPSc fibril formed by two protofilaments. The PrP normalized intensity was determined by immunoblotting using Sha31 antibody. The relative number of PrPSc particles was calculated by dividing the PrP normalized intensity by number of PrP monomers per PrPSc particle. The maximum possible PrPSc particle density was calculated with ASTRA software (see methods) for fractions 46 and 48, and extrapolated to the rest of the fractions based on the relative number of PrPSc particles. (*) these values of particle density were calculated from MALS measurements directly, fitting the data to a rod-shape model, and assuming a rod radius of 5.5 nm. This calculation was performed for all samples; the specific values obtained for brain E are shown in the table.
(DOCX)
Acknowledgments
We thank Dr. Michael Woodside and Craig Garen for providing the recombinant prion protein SHaPrP substrate used in the RT-QuIC experiments, Dr. Joel Watts for generously providing the CAD5-PrP−/− (HaPrP) cells and Wyatt Technology for personal assistance in the analysis of DLS and MALS data.
Abbreviations
- AF4
asymmetric-flow field-flow fractionation
- BH
brain homogenate
- DLS
dynamic light scattering
- DY
Drowsy prion strain
- HY
Hyper prion strain
- MALS
multiangle light scattering
- NBH
normal brain homogenate
- PK
endoproteinase K
- PrP
prion protein
- PrPC
cellular form of prion protein
- PrPres
protease-resistant form of prion protein
- PrPSc
disease-associated form of prion protein
- PrPsen
protease-sensitive form of prion protein
- SHaPrP
recombinant Syrian hamster prion protein
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This research was supported in part by Genome Canada in support of the Systems Biology and Molecular Ecology of Chronic Wasting Disease project (DM), the Alberta Prion Research Institute in collaboration with the Alberta Livestock and Meat Agency (Exploration IV, 201600016/2016A003R) (VS), the CJD Foundation (LC, VS), and the Alberta Synergies in Alzheimer’s and Related Disorders (SynAD) program which is funded by the Alzheimer Society of Alberta and Northwest Territories through their Hope for Tomorrow program and the University Hospital Foundation (VS, LC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24: 519–550. doi: 10.1146/annurev.neuro.24.1.519 [DOI] [PubMed] [Google Scholar]
- 2.Weissmann C. The state of the prion. Nat Rev Microbiol. 2004;2: 861–871. doi: 10.1038/nrmicro1025 [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216: 136–144. doi: 10.1126/science.6801762 [DOI] [PubMed] [Google Scholar]
- 4.Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218: 1309–1311. doi: 10.1126/science.6815801 [DOI] [PubMed] [Google Scholar]
- 5.Bartz JC. Prion Strain Diversity. Cold Spring Harb Perspect Med. 2016;6. doi: 10.1101/cshperspect.a024349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66: 2096–2101. doi: 10.1128/JVI.66.4.2096-2101.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68: 7859–7868. doi: 10.1128/JVI.68.12.7859-7868.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A. 1994;91: 2839–2842. doi: 10.1073/pnas.91.7.2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274: 2079–2082. doi: 10.1126/science.274.5295.2079 [DOI] [PubMed] [Google Scholar]
- 10.Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4: 1157–1165. doi: 10.1038/2654 [DOI] [PubMed] [Google Scholar]
- 11.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428: 323–328. doi: 10.1038/nature02392 [DOI] [PubMed] [Google Scholar]
- 12.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 2011;7: e1001317. doi: 10.1371/journal.ppat.1001317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, et al. Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry. 2006;45: 15710–15717. doi: 10.1021/bi0615442 [DOI] [PubMed] [Google Scholar]
- 14.Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, et al. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 2010;6: e1000859. doi: 10.1371/journal.ppat.1000859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim C, Haldiman T, Surewicz K, Cohen Y, Chen W, Blevins J, et al. Small protease sensitive oligomers of PrPSc in distinct human prions determine conversion rate of PrP(C). PLoS Pathog. 2012;8: e1002835. doi: 10.1371/journal.ppat.1002835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laferrière F, Tixador P, Moudjou M, Chapuis J, Sibille P, Herzog L, et al. Quaternary structure of pathological prion protein as a determining factor of strain-specific prion replication dynamics. PLoS Pathog. 2013;9: e1003702. doi: 10.1371/journal.ppat.1003702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saverioni D, Notari S, Capellari S, Poggiolini I, Giese A, Kretzschmar HA, et al. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J Biol Chem. 2013;288: 27972–27985. doi: 10.1074/jbc.M113.477547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzaban S, Friedlander G, Schonberger O, Horonchik L, Yedidia Y, Shaked G, et al. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002;41: 12868–12875. doi: 10.1021/bi025958g [DOI] [PubMed] [Google Scholar]
- 19.Sasaki K, Minaki H, Iwaki T. Development of oligomeric prion-protein aggregates in a mouse model of prion disease. J Pathol. 2009;219: 123–130. doi: 10.1002/path.2576 [DOI] [PubMed] [Google Scholar]
- 20.Igel-Egalon A, Moudjou M, Martin D, Busley A, Knäpple T, Herzog L, et al. Reversible unfolding of infectious prion assemblies reveals the existence of an oligomeric elementary brick. PLoS Pathog. 2017;13(9): e1006557. doi: 10.1371/journal.ppat.1006557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foliaki ST, Lewis V, Islam AMT, Ellett LJ, Senesi M, Finkelstein DI, et al. Early existence and biochemical evolution characterise acutely synaptotoxic PrPSc. PLoS Pathog. 2019;15: e1007712. doi: 10.1371/journal.ppat.1007712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giddings JC. A New Separation Concept Based on a Coupling of Concentration and Flow Nonuniformities. Separation Science. 1966;1: 123–125. doi: 10.1080/01496396608049439 [DOI] [Google Scholar]
- 23.Fraunhofer W, Winter G. The use of asymmetrical flow field-flow fractionation in pharmaceutics and biopharmaceutics. Eur J Pharm Biopharm. 2004;58: 369–383. doi: 10.1016/j.ejpb.2004.03.034 [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Freitas D, Kim HS, Fabijanic K, Li Z, Chen H, et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat Cell Biol. 2018;20: 332–343. doi: 10.1038/s41556-018-0040-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eskelin K, Poranen MM, Oksanen HM. Asymmetrical Flow Field-Flow Fractionation on Virus and Virus-Like Particle Applications. Microorganisms. 2019;7. doi: 10.3390/microorganisms7110555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature. 2005;437: 257–261. doi: 10.1038/nature03989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rambaldi DC, Zattoni A, Reschiglian P, Colombo R, De Lorenzi E. In vitro amyloid Abeta(1–42) peptide aggregation monitoring by asymmetrical flow field-flow fractionation with multi-angle light scattering detection. Anal Bioanal Chem. 2009;394: 2145–2149. doi: 10.1007/s00216-009-2899-1 [DOI] [PubMed] [Google Scholar]
- 28.Marassi V, Beretti F, Roda B, Alessandrini A, Facci P, Maraldi T, et al. A new approach for the separation, characterization and testing of potential prionoid protein aggregates through hollow-fiber flow field-flow fractionation and multi-angle light scattering. Anal Chim Acta. 2019;1087: 121–130. doi: 10.1016/j.aca.2019.08.003 [DOI] [PubMed] [Google Scholar]
- 29.Eskandari-Sedighi G, Cortez LM, Yang J, Daude N, Shmeit K, Sim V, et al. Quaternary Structure Changes for PrPSc Predate PrPC Downregulation and Neuronal Death During Progression of Experimental Scrapie Disease. Mol Neurobiol. 2020. doi: 10.1007/s12035-020-02112-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol. 1992;73 (Pt 2): 329–334. doi: 10.1099/0022-1317-73-2-329 [DOI] [PubMed] [Google Scholar]
- 31.Kraus A, Hoyt F, Schwartz CL, Hansen B, Hughson AG, Artikis E, et al. Structure of an infectious mammalian prion. bioRxiv. 2021: 2021.02.14.431014. doi: 10.1101/2021.02.14.431014 [DOI] [PubMed] [Google Scholar]
- 32.Scott MR, Groth D, Tatzelt J, Torchia M, Tremblay P, DeArmond SJ, et al. Propagation of prion strains through specific conformers of the prion protein. J Virol. 1997;71: 9032–9044. doi: 10.1128/JVI.71.12.9032-9044.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peretz D, Scott MR, Groth D, Williamson RA, Burton DR, Cohen FE, et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001;10: 854–863. doi: 10.1110/ps.39201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergara J, Burton DR, et al. A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron. 2002;34: 921–932. doi: 10.1016/s0896-6273(02)00726-2 [DOI] [PubMed] [Google Scholar]
- 35.Mulcahy ER, Bessen RA. Strain-specific kinetics of prion protein formation in vitro and in vivo. J Biol Chem. 2004;279: 1643–1649. doi: 10.1074/jbc.M307844200 [DOI] [PubMed] [Google Scholar]
- 36.Shikiya RA, Langenfeld KA, Eckland TE, Trinh J, Holec SAM, Mathiason CK, et al. PrPSc formation and clearance as determinants of prion tropism. PLoS Pathog. 2017;13: e1006298. doi: 10.1371/journal.ppat.1006298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bourkas MEC, Arshad H, Al-Azzawi ZAM, Halgas O, Shikiya RA, Mehrabian M, et al. Engineering a murine cell line for the stable propagation of hamster prions. The Journal of biological chemistry. 2019;294: 4911–4923. doi: 10.1074/jbc.RA118.007135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem. 1998;273: 32230–32235. doi: 10.1074/jbc.273.48.32230 [DOI] [PubMed] [Google Scholar]
- 39.Makarava N, Savtchenko R, Baskakov IV. Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J Biol Chem. 2013;288: 33–41. doi: 10.1074/jbc.M112.419531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sajnani G, Silva CJ, Ramos A, Pastrana MA, Onisko BC, Erickson ML, et al. PK-sensitive PrP is infectious and shares basic structural features with PK-resistant PrP. PLoS Pathog. 2012;8: e1002547. doi: 10.1371/journal.ppat.1002547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Legname G, Nguyen HB, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006;103: 19105–19110. doi: 10.1073/pnas.0608970103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thackray AM, Hopkins L, Klein MA, Bujdoso R. Mouse-adapted ovine scrapie prion strains are characterized by different conformers of PrPSc. J Virol. 2007;81: 12119–12127. doi: 10.1128/JVI.01434-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, et al. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci U S A. 2009;106: 20417–20422. doi: 10.1073/pnas.0910350106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sang JC, Meisl G, Thackray AM, Hong L, Ponjavic A, Knowles TPJ, et al. Direct Observation of Murine Prion Protein Replication in Vitro. J Am Chem Soc. 2018;140: 14789–14798. doi: 10.1021/jacs.8b08311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amenitsch H, Benetti F, Ramos A, Legname G, Requena JR. SAXS structural study of PrP(Sc) reveals ~11 nm diameter of basic double intertwined fibers. Prion. 2013;7: 496–500. doi: 10.4161/pri.27190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vázquez-Fernández E, Vos MR, Afanasyev P, Cebey L, Sevillano AM, Vidal E, et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016;12: e1005835. doi: 10.1371/journal.ppat.1005835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terry C, Wenborn A, Gros N, Sells J, Joiner S, Hosszu LLP, et al. Ex vivo mammalian prions are formed of paired double helical prion protein fibrils. Open Biol. 2016;6. doi: 10.1098/rsob.160035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spagnolli G, Rigoli M, Orioli S, Sevillano AM, Faccioli P, Wille H, et al. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019;15: e1007864. doi: 10.1371/journal.ppat.1007864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katorcha E, Makarava N, Savtchenko R, Baskakov IV. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci Rep. 2015;5: 16912. doi: 10.1038/srep16912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Makarava N, Chang JC, Molesworth K, Baskakov IV. Posttranslational modifications define course of prion strain adaptation and disease phenotype. J Clin Invest. 2020;130: 4382–4395. doi: 10.1172/JCI138677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lau A, So RWL, Lau HHC, Sang JC, Ruiz-Riquelme A, Fleck SC, et al. α-Synuclein strains target distinct brain regions and cell types. Nat Neurosci. 2020;23: 21–31. doi: 10.1038/s41593-019-0541-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiang W, Yau W, Lu J, Collinge J, Tycko R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature. 2017;541: 217–221. doi: 10.1038/nature20814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015;129: 469–491. doi: 10.1007/s00401-014-1380-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Narasimhan S, Guo JL, Changolkar L, Stieber A, McBride JD, Silva LV, et al. Pathological Tau Strains from Human Brains Recapitulate the Diversity of Tauopathies in Nontransgenic Mouse Brain. J Neurosci. 2017;37: 11406–11423. doi: 10.1523/JNEUROSCI.1230-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154: 103–117. doi: 10.1016/j.cell.2013.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4: 2575. doi: 10.1038/ncomms3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522: 340–344. doi: 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- 58.Wyatt PJ. Measurement of special nanoparticle structures by light scattering. Anal Chem. 2014;86: 7171–7183. doi: 10.1021/ac500185w [DOI] [PubMed] [Google Scholar]
- 59.Young G, Hundt N, Cole D, Fineberg A, Andrecka J, Tyler A, et al. Quantitative mass imaging of single biological macromolecules. Science. 2018;360: 423–427. doi: 10.1126/science.aar5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wyatt PJ. Submicrometer Particle Sizing by Multiangle Light Scattering following Fractionation. Journal of Colloid and Interface Science. 1998;197: 9–20. doi: 10.1006/jcis.1997.5215 [DOI] [PubMed] [Google Scholar]
- 61.Gupta AN, Neupane K, Rezajooei N, Cortez LM, Sim VL, Woodside MT. Pharmacological chaperone reshapes the energy landscape for folding and aggregation of the prion protein. Nat Commun. 2016;7: 12058. doi: 10.1038/ncomms12058 [DOI] [PMC free article] [PubMed] [Google Scholar]