

Loss of UCHL1 activates an energy-dependent mitophagy via pyruvate kinase, which relieves Parkinson’s disease symptoms.
Abstract
The role of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1; also called PARK5) in the pathogenesis of Parkinson’s disease (PD) has been controversial. Here, we find that the loss of UCHL1 destabilizes pyruvate kinase (PKM) and mitigates the PD-related phenotypes induced by PTEN-induced kinase 1 (PINK1) or Parkin loss-of-function mutations in Drosophila and mammalian cells. In UCHL1 knockout cells, cellular pyruvate production and ATP levels are diminished, and the activity of AMP–activated protein kinase (AMPK) is highly induced. Consequently, the activated AMPK promotes the mitophagy mediated by Unc-51–like kinase 1 (ULK1) and FUN14 domain–containing 1 (FUNDC1), which underlies the effects of UCHL1 deficiency in rescuing PD-related defects. Furthermore, we identify tripartite motif–containing 63 (TRIM63) as a previously unknown E3 ligase of PKM and demonstrate its antagonistic interaction with UCHL1 to regulate PD-related pathologies. These results suggest that UCHL1 is an integrative factor for connecting glycolysis and PD pathology.
INTRODUCTION
Parkinson’s disease (PD) is one of the most prevalent neurodegenerative disorders. The movement disorders of patients with PD result from loss of dopaminergic (DA) neurons in the substantia nigra, a region of the midbrain (1). To maintain energetic and functional demands, DA neurons are known as one of the cell types that require high-energy supplies and thus perform glucose metabolism at a high level and produce excessive reactive oxygen species (ROS) as by-products (2, 3). As a result, DA neurons are sensitive to oxidative stress, and the mitochondrial damages induced by ROS can be a potential cause for the death of DA neurons in patients with PD. Consistently, inhibition of the electron transport chain (ETC) in mitochondria by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 1,1′-dimethyl-4,4′-bipyridinium (paraquat) causes degeneration of DA neurons in animal models and induces symptoms related to PD (4–7). Therefore, understanding the molecular connection between glucose metabolism and mitochondrial homeostasis has been imperative to uncover the molecular pathogenesis of PD.
Oxidative stress and calcium accumulation are the main factors for inducing mitochondrial damages. The damaged mitochondria should be removed by mitochondria-specific autophagy, mitophagy. Among the PD genes identified from genetic and genomic analyses of patients with PD, PTEN-induced kinase 1 (PINK1; also called PARK6) comprises a central signaling axis together with the E3 ligase Parkin (also called PARK2), and both of them play an important role in controlling mitophagy (8–12). Upon mitochondrial damages, PINK1 phosphorylates Parkin, which induces translocation of Parkin from the cytosol to the damaged mitochondria (13). Ubiquitination of various mitochondrial proteins by Parkin triggers the recruitment of mitophagy adaptor proteins and mitophagosomes (14, 15). Other PD-associated genes, such as PARK7 (DJ-1) (16), PARK8 (LRRK2) (17), PARK13 (HTRA2/Omi) (18), and PARK15 (FBXO7) (19), have been also reported to be involved in this PINK1-Parkin pathway.
Since protein ubiquitination plays a crucial role in the PINK1-Parkin pathway, ubiquitin C-terminal hydrolase L1 (UCHL1; also called PARK5) has also been regarded as an important PD-associated gene (20, 21). However, the exact role and molecular targets of UCHL1 have not been found yet. Belonging to the UCH family of deubiquitinating enzymes (DUBs), UCHL1 hydrolyzes a glycine peptide bond at the C terminus of ubiquitin and is highly expressed in the brain, accounting for 1 to 2% of total brain proteins (22, 23). Unexpectedly, one German family suffering from familial PD was identified carrying I93M mutation of UCHL1, which provides the first evidence to categorize UCHL1 as a PD-susceptible gene (21). After the initial finding, the E7A substitution of UCHL1 was associated with motor neuron dysfunctions similar to PD (24). On the contrary, several statistical studies proposed reduced incidence of PD by the S18Y polymorphism in certain populations (25–29). Despite these epidemiological findings, whether the mutations in UCHL1 are directly related to PD is still highly controversial.
In the present study, we find that the loss of UCHL1 regulates glucose metabolism and rescues the defects related to PD. Our metabolic analysis identify that specific glycolytic metabolites are decreased and an energy-dependent mitophagy pathway is induced by inhibition of UCHL1. Therefore, we propose that UCHL1 is an integrative regulator of glucose metabolism, mitochondrial homeostasis, and PD pathogenesis.
RESULTS
Loss of UCHL1 rescues the PD phenotypes induced by PINK1 and Parkin deficiency
Mitophagic dysfunction is one of the main causes of PD, and the PINK1-Parkin pathway plays a central role in controlling the mitophagy induced by mitochondrial damages. To identify a novel regulatory mechanism for the mitophagy related to PD, we conducted a small-scale genetic screen using Drosophila.
In our first screen, we genetically crossed UAS–RNA interference (RNAi) lines for 15 different fly genes orthologous to PD-associated human genes (table S1) (30) with PINK1 knockout (KO) flies (PINK1B9) (9) using mef2-GAL4 driver, which specifically knocked down PD genes in the muscle of PINK1 mutants. Through this screen, we examined whether the impaired mitochondrial morphology caused by PINK1 deficiency was rescued by the RNAi lines for PD genes (Fig. 1A and fig. S1A). In the secondary screen, we observed the mitochondrial morphology in the muscle after crossing Parkin null mutants (park1) (31) with the RNAi lines that showed positive outcomes in the first screen (Fig. 1A and fig. S1B). As a result, we isolated two RNAi lines that completely rescued the defects in mitochondrial morphology of both PINK1 and Parkin null flies. The isolated lines turned out to be the RNAi lines for Drosophila UCH (cg4265), the orthologous gene for UCHL1 in humans (Fig. 1A and fig. S2A).
Fig. 1. Loss of UCHL1 rescues the PD-related phenotypes of PINK1 and Parkin mutants in Drosophila.
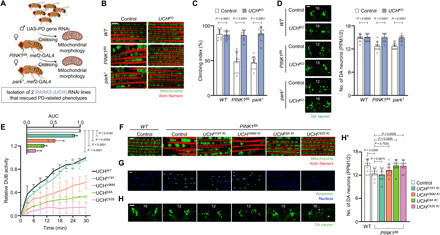
(A) Genetic scheme to screen new suppressive genes for the swollen mitochondrial phenotypes of PINK1 (PINK1B9) and Parkin (park1) null mutants. (B) Confocal fluorescence images of the adult flight muscles. Green indicates mitochondria, and red indicates actin filament. Scale bar, 5 μm. Percentage of the flies having swollen mitochondria was quantified in fig. S3C. (C) Measurement of the climbing ability in the adult flies. n = 10. (D) Confocal immunofluorescence images and numbers of DA neurons in the PPM1/2 regions of adult fly brains. n = 10. Green indicates DA neuron. Scale bar, 20 μm. The number in panels indicates the number of DA neurons in each image. (E) Measurement of the DUB activities of UCH mutant proteins. Linear graphs show relative DUB activities of the mutants normalized by the activity of UCH WT at 30 min. Bar graphs show area under the curve (AUC) of the linear graphs. n = 3. (F) Confocal fluorescence images of the adult flight muscles. Green denotes mitochondria, and red denotes actin filament. Scale bar, 5 μm. Percentage of the flies having swollen mitochondria was quantified in fig. S5A. (G) Confocal fluorescence images for terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assays of the adult flight muscles. Green denotes apoptosis, and blue denotes nucleus. Scale bar, 5 μm. Percentage of the flies showing apoptotic responses was quantified in fig. S5B. (H) Confocal immunofluorescence images (H) and numbers (H′) of DA neurons in the PPM1/2 regions of adult fly brains. n = 10. Green indicates DA neuron. Scale bar, 20 μm. The number in panels indicates the number of DA neurons in each image. Two-way analysis of variance (ANOVA) with Sidak’s multiple comparison test was used (C and D), and one-way ANOVA with Dunnett’s multiple comparison test was used (H′). Data were presented as means + SD.
To firmly establish these RNAi knockdown results, we next produced a UCH null mutant fly line (UCHKO) by introducing an early-stop frame shift mutation using the CRISPR-Cas9 system (fig. S2B) (32). UCH null flies were homozygous viable and did not show notable PD-related phenotypes (fig. S3, A to E). However, when we generated PINK1B9 or park1 flies carrying the homozygous UCHKO mutation, we found that UCH KO completely rescued the PD-related phenotypes such as crushed thoraces and abnormal wing postures observed in the null mutant flies of PINK1 and Parkin (fig. S3, A to B′) (9, 31). Other phenotypes of the PINK1- or Parkin-deficient mutants including swollen mitochondria (Fig. 1B and fig. S3C) and increased apoptotic responses in the muscle (fig. S3D) impaired climbing ability (Fig. 1C), and reduced number of DA neurons in the PPM1/2 and PPL1 regions of Drosophila adult brain (Fig. 1D and fig. S3E) were completely rescued by UCH null mutation. These data demonstrate that the loss of UCHL1 rescues the PD-related pathogenesis in PINK1 or Parkin KO flies.
Deubiquitinase activity of UCH inversely correlates with the suppression of PD phenotypes
Meanwhile, several substitutional mutations in UCHL1 have been reported to be related to neurodegeneration, especially in PD. The I93M mutation of UCHL1 was linked to PD from one German family (21), and the E7A substitution was associated with motor neuron dysfunctions similar to PD (24). On the contrary, several statistical studies proposed reduced incidence of PD by the S18Y polymorphism (20, 27, 33–35). Despite these findings, whether UCHL1 is directly related to PD is still highly controversial (26, 36). Therefore, we sought to find the molecular connections between UCHL1 mutations and PD using Drosophila and mammalian models.
Since UCHL1 serves as a DUB, we determined whether the fly ortholog, UCH, also preserves its DUB activity. We analyzed in vitro DUB activities of UCH recombinant proteins bearing H19Y, V96M, E8A, and C93S (a catalytic dead form) mutations respectively identical to human mutations, S18Y, I93M, E7A, and C90S (fig. S2A). The DUB activities of UCH H19Y, V96M, E8A, and C93S mutant proteins were measured to be approximately 100, 50, 25, and 10%, respectively, compared to that of UCH wild type (WT) (Fig. 1E). Consistently, we obtained similar results from human UCHL1 mutant proteins on the DUB activity (fig. S4A) (24, 37).
To study that the DUB activity of UCH is related to PD pathogenesis, we generated UCH knock-in (KI) flies bearing E8A, H19Y, V96M, and C93S mutations using the CRISPR-Cas9 system (fig. S2C) (38, 39). We examined various phenotypes related to PD, including swollen mitochondrial morphology, increased apoptotic responses in thoraces, loss of DA neurons, and decreased climbing ability, in these UCH KI flies, compared to WT flies. Notably, there were no significant defects or differences in the phenotypes among the flies (fig. S4, B to D).
To further investigate the relevance of the DUB activity of UCH in the PD pathogenesis induced by PINK1 and Parkin mutations, we integrated each of these UCH KI alleles into PINK1 or Parkin single KO background flies and observed changes in the PD-related phenotypes. UCHE8A and UCHC93S KI with highly decreased DUB activities (Fig. 1E) markedly alleviated the swollen mitochondrial morphology and the elevated apoptotic signals of PINK1B9 (Fig. 1, F and G, and fig. S5, A and B) and park1 flies (fig. S6, A to B′). In addition, UCHV96M KI flies, which have ~50% DUB activity of UCH WT (Fig. 1E), partially rescued the phenotypes of PINK1 (Fig. 1, F and G, and fig. S5, A and B) and Parkin null flies (fig. S6, A to B′). However, UCHH19Y KI flies, which have a similar UCH DUB activity to that of WT flies (Fig. 1E), did not rescue the defective phenotypes of PINK1B9 (Fig. 1, F and G, and fig. S5, A and B) and park1 flies (fig. S6, A to B′). Consistent with the results shown above, the reduced climbing ability and the loss of DA neurons of PINK1B9 (Fig. 1, H and H′, and fig. S5, C and D) and park1 flies (fig. S6, C to E′) were rescued by UCHE8A and UCHC93S KI, but not by UCHH19Y and UCHV96M KI, mutations. Together, we conclude that the loss of UCH DUB activity protects the PD-related pathogenesis in the flies lacking PINK1 or Parkin.
Mitophagy is elevated in UCHL1 KO cells
Having shown that the abrogation of UCH suppresses the PD phenotypes induced by PINK1 and Parkin deficiency, we then investigated the mechanism of this in mammalian systems. In particular, as PINK1 and Parkin are closely linked to mitophagy (40), we generated UCHL1 KO human embryonic kidney (HEK) 293 cell lines using the CRISPR-Cas9 system (fig. S7, A and B) (41) and measured the amounts of outer [mitofusin-1 (MFN1) and translocase of outer mitochondrial membrane 20 (TOM20)] and inner [translocase of inner mitochondrial membrane 23 (TIM23) and cytochrome C oxidase subunit IV (COXIV)] mitochondrial membrane proteins to detect mitophagy (42). When we treated UCHL1 WT or KO HEK293 cells with carbonyl cyanide m-chlorophenyl hydrazone (CCCP), the amount of mitochondrial proteins was more quickly and strongly reduced in UCHL1 KO cells compared to WT cells (Fig. 2A). To visualize mitophagy, we also used a mitophagy reporter dye (Mtphagy Dye) (43), which generates red fluorescence when the pH in mitochondria decreases as they colocalize with lysosomes. When we induced mitophagy by treating CCCP, the number of red fluorescence dots per cell in UCHL1 KO HEK293 cells doubled compared to WT cells (Fig. 2B). We also observed a similarly elevated level of mitophagy from UCHL1 KO cells treated with another mitophagy inducer, the mix of antimycin A and oligomycin (fig. S7C). These results showing that mitochondrial proteins were reduced more quickly in UCHL1 KO HEK293 cells by two different mitophagy inducers were also repeated in UCHL1 KO SH-SY5Y cells derived from DA human neuroblastoma (fig. S7, D and E).
Fig. 2. Loss of UCHL1 enhances mitophagy.
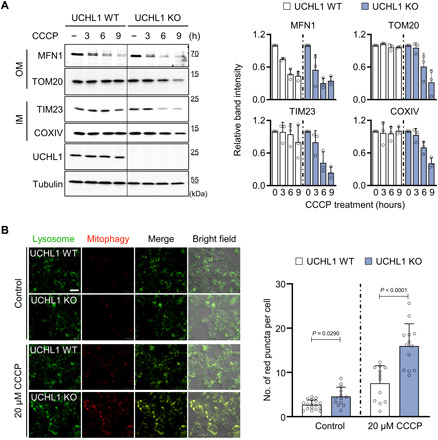
(A) Left: Immunoblot analysis of mitochondrial outer membrane (OM; MFN1 and TOM20) and inner membrane (IM; TIM23 and COXIV) proteins upon 20 μM CCCP treatment. Right: Relative quantification of the immunoblot band intensity of indicated mitochondrial proteins. n = 3. (B) Left: Confocal fluorescence images of Mtphagy Dye in HEK293 UCHL1 WT and KO cells upon 20 μM CCCP treatment. Green indicates lysosome, and red indicates mitophagy. Right: Number of Mtphagy Dye red dots per cell in HEK293 UCHL1 WT and KO cells upon 20 μM CCCP treatment. UCHL1 WT (n = 17) and KO (n = 12) without CCCP treatment (−). UCHL1 WT (n = 13) and KO (n = 13) with 20 μM CCCP treatment. Scale bar, 20 μm. Two-way ANOVA with Sidak’s multiple comparison test was used (B). All data were presented as means + SD.
Next, we asked whether the changes in the DUB activity of UCHL1 mutants can affect mitophagy. We expressed UCHL1 WT, E7A, S18Y, C90S, and I93M in UCHL1 KO cells and observed mitochondrial protein levels in the transfected cells. We found that the expression of UCHL1 E7A or C90S in UCHL1 KO cells induced mitophagy at a level similar to untransfected UCHL1 KO cells upon CCCP treatment (fig. S7F). However, the amount of mitochondrial proteins in UCHL1 KO cells expressing UCHL1 WT, S18Y, or I93M was not reduced as much as that of UCHL1 KO cells expressing UCHL1 E7A or C90S upon CCCP treatment (fig. S7F). Together with the aforementioned data, we conclude that the loss of UCHL1 DUB activity facilitates mitophagy.
Loss of UCHL1 promotes mitophagy via FUN14 domain–containing 1
We then performed another small-scale genetic screen in Drosophila against previously known mitophagy receptors to identify a downstream target of UCHL1. Mitophagy receptors orchestrate mitochondria-specific autophagy upon mitochondrial damages by recruiting LC3 proteins. Among mitochondrial proteins with LC3-interacting region motif (44), four proteins [Zonda (Zda) (45, 46), Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (Bnip3) (47), sequestosome 1 (p62) (48), and FUN14 domain–containing 1 (FUNDC1) (49)] are conserved between Drosophila and human. We drove the expression of UAS-RNAi against these genes using heat-shock (hs)–GAL4 driver in PINK1 and UCH double KO flies and observed mitochondrial morphology in the flight muscles. Although Zda, Bnip3, or p62 knockdown did not significantly change the mitochondrial morphology of PINK1 and UCH double null flies, FUNDC1 knockdown generated swollen mitochondria morphologies in the double null flies (Fig. 3A). In addition, FUNDC1 knockdown worsened other PD-related phenotypes, including impaired climbing abilities, increased apoptosis, and degeneration of DA neurons, in PINK1 and UCH double KO flies, similar to those in PINK1 KO flies (Fig. 3, B and C, and fig. S8, B to C′). All of these results observed in PINK1B9 background flies were similarly reproduced in park1 background flies (Fig. 3, B and C, and fig. S8, A to C′).
Fig. 3. Mitophagy induced by UCHL1 deficiency is mediated by FUNDC1.
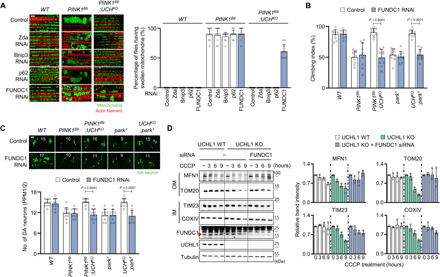
(A) Left: Confocal fluorescence images of the adult flight muscles. Right: Percentage of the flies having swollen mitochondria. n = 10. Green denotes mitochondria, and red denotes actin filament. Scale bar, 5 μm. (B) Measurement of the climbing ability in the adult flies. n = 10. (C) Confocal immunofluorescence images and numbers of DA neurons in the PPM1/2 regions of adult fly brains. n = 10. Green denotes DA neuron. Scale bar, 20 μm. The number in panels indicates the number of DA neurons in each image. (D) Left: Immunoblot analysis of mitochondrial outer (MFN1 and TOM20) and inner (TIM23 and COXIV) membrane proteins upon 20 μM CCCP and FUNDC1 small interfering RNA (siRNA) treatment. Right: Relative quantification of the immunoblot band intensity of indicated mitochondrial proteins. n = 3. Two-way ANOVA with Tukey’s multiple comparison test was used (B and C). All data were presented as means + SD.
We therefore examined whether UCHL1 interacts with FUNDC1 in mammalian cells as in Drosophila. Notably, the increased mitophagy upon CCCP treatment in UCHL1 KO cells was blocked by FUNDC1 knockdown (Fig. 3D). On the basis of these results, we conclude that FUNDC1 mediates mitophagy downstream of UCHL1.
FUNDC1-mediated mitophagy is elevated by adenosine 5′-monophosphate–activated protein kinase and Unc-51–like kinase 1 in UCHL1 KO cells and Drosophila
In previous reports, activated adenosine 5′-monophosphate (AMP)–activated protein kinase (AMPK) phosphorylates Unc-51–like kinase 1 (ULK1) at S555 (44, 50), which induces mitophagy via activation of FUNDC1 (51). To examine whether UCHL1 is involved in this mechanism, we determined the activation status of endogenous ULK1 and AMPK in UCHL1 WT and KO cells. Increased phosphorylation of AMPK at T172 and ULK1 at S555 was observed in UCHL1 KO cells compared to UCHL1 WT cells (Fig. 4A and fig. S9A). Upon treatment of compound C, a potent AMPK inhibitor, the phosphorylation of AMPK and ULK1 was suppressed in UCHL1 KO cells comparable to that in WT cells (Fig. 4A and fig. S9A), indicative of the AMPK-mediated phosphorylation of ULK1 in the KO cells. Therefore, to examine whether UCHL1 regulates mitophagy through AMPK and ULK1, we knocked down AMPK or ULK1 in UCHL1 KO cells using small interfering RNA (siRNA) transfection. As a result, the increased mitophagy in UCHL1 KO cells was blocked when siRNA of AMPK (Fig. 4B and fig. S9B) or ULK1 was transfected (Fig. 4C and fig. S9C), suggesting that the UCHL1-mediated mitophagy is induced by activation of AMPK and ULK1 in mammals.
Fig. 4. FUNDC1-mediated mitophagy in UCHL1 KO cells and flies is induced by AMPK and ULK1.
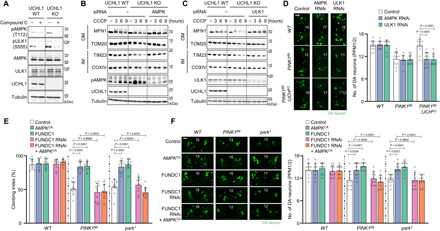
(A) Immunoblot analysis of AMPK and ULK1 phosphorylation in HEK293 UCHL1 WT and KO cells without (−) or with (+) 10 μM compound C treatment. The band intensity of AMPK and ULK1 phosphorylation was quantified in fig. S9A. (B) Immunoblot analysis of mitochondrial outer (MFN1 and TOM20) and inner (TIM23 and COXIV) membrane proteins upon 20 μM CCCP and AMPKα siRNA treatment. The band intensity of the four mitochondrial proteins (MFN1, TOM20, TIM23, and COXIV) was quantified in fig. S9B. (C) Immunoblot analysis of mitochondrial outer (MFN1 and TOM20) and inner (TIM23 and COXIV) membrane proteins upon 20 μM CCCP and ULK1 siRNA treatment. The band intensity of the four mitochondrial proteins (MFN1, TOM20, TIM23, and COXIV) was quantified in fig. S9C. (D) Confocal immunofluorescence images (left) and numbers (right) of DA neurons in the PPM1/2 regions of adult fly brains. n = 10. Green indicates DA neuron. Scale bar, 20 μm. The number in panels indicates the number of DA neurons in each image. (E) Measurement of the climbing ability in the adult flies. n = 10. (F) Confocal immunofluorescence images (left) and numbers (right) of DA neurons in the PPM1/2 regions of adult fly brains. n = 10. Green indicates DA neuron. Scale bar, 20 μm. The number in panels indicates the number of DA neurons in each image. Two-way ANOVA with Sidak’s multiple comparison test was used (D to F). All data were presented as means + SD.
To confirm these observations in Drosophila, we knocked down AMPK or ULK1 in the double KO flies lacking PINK1 and UCH or Parkin and UCH. Because of developmental lethality caused by hs-GAL4 and mef2-GAL4 drivers, we used tyrosine hydroxylase (th)–GAL4 driver to knock down AMPK or ULK1 in the DA neurons of PINK1 and UCH or Parkin and UCH double KO flies. The number of DA neurons in the PPM1/2 and PPL1 regions was significantly reduced in PINK1 and Parkin null flies, and this DA neuron phenotype was almost completely rescued by UCH KO (Fig. 4D and fig. S9, D and E). However, the concurrent knockdown of AMPK or ULK1 blocked this rescuing effect of UCH KO (Fig. 4D and fig. S9, D and E), supporting the evolutionarily conserved roles of AMPK and ULK1 downstream of UCHL1 in Drosophila.
Furthermore, we confirmed the interaction between AMPK and FUNDC1 in PINK1 or Parkin null flies. Similar to previous studies, we identified that swollen mitochondria and increased apoptosis in the thoraces of PINK1B9 or park1 flies were mitigated by overexpression of constitutively active AMPK (AMPKCA) (52, 53) or FUNDC1 under hs-GAL4 driver (fig. S10, A to B′). In addition, the decreased climbing abilities and the loss of DA neurons of PINK1 or Parkin null mutants were rescued by AMPKCA or FUNDC1 overexpression (Fig. 4, E and F, and fig. S10, C and C′). The rescued phenotypes of PINK1B9 or park1 flies by expressing AMPKCA were reversed by concurrently expressing FUNDC1 RNAi (Fig. 4, E and F, and fig. S10, A to C′), suggesting that AMPK rescues the PD-related phenotypes via FUNDC1 in Drosophila.
UCHL1 stabilizes pyruvate kinase leading to regulation of the energy-dependent mitophagy
AMPK is a master regulator of cellular energy homeostasis responding to AMP/adenosine 5′-triphosphate (ATP) levels. This led us to hypothesize that the loss of UCHL1 could have affected energy levels in the cell to enhance mitophagy. To address this possibility, we measured the endogenous ATP levels in UCHL1 KO cells in comparison to WT cells and found lower ATP levels in UCHL1 KO cells (Fig. 5A). We hence supposed that UCHL1 is related to ATP production processes such as glycolysis, oxidative phosphorylation (OXPHOS), and citric acid cycle. To examine which of the three processes is related to UCHL1, we measured ATP levels from UCHL1 WT and KO cells treated with 2-deoxy-d-glucose (2-DG), rotenone, or oligomycin. Rotenone and oligomycin are inhibitors of ETC in the OXPHOS system (54), and 2-DG inhibits glycolysis as a glucose analog (55). We found that ATP levels were halved in UCHL1 KO cells compared to UCHL1 WT cells, and their ATP levels were proportionally further reduced when treated with rotenone or oligomycin (Fig. 5A). However, upon 2-DG treatment, we unexpectedly could not observe differences in ATP levels between UCHL1 WT and KO cells (Fig. 5A). Furthermore, we validated relative ATP levels in fruit flies treated with these drugs or expressing RNAi affecting glycolysis and OXPHOS system. Similar to the cell data, UCH KO flies displayed decreased ATP levels compared to control flies, and their ATP levels remained lower than WT flies when rotenone or oligomycin was fed. However, UCH KO flies showed similar levels of ATP compared to control flies when 2-DG was fed (fig. S11A). Consistent with these pharmacologic data, the ATP levels of UCH KO flies with glucose transporter 1 (GLUT1) knockdown were comparable to those of the GLUT1 knockdown flies in WT background. However, flies with knockdown of NADH (reduced form of nicotinamide adenine dinucleotide) dehydrogenase ubiquinone iron-sulfur protein 3 (NDUFS3) and ATP synthase subunit β (ATPsynβ), which are the components of the first and the last OXPHOS complexes, respectively, showed further reduced ATP levels in UCH KO background (fig. S11B). These pharmacologic and genetic data from mammalian cells and fruit flies consistently support that UCHL1 targets glycolysis.
Fig. 5. PKM is a direct target of UCHL1 to control mitophagy.
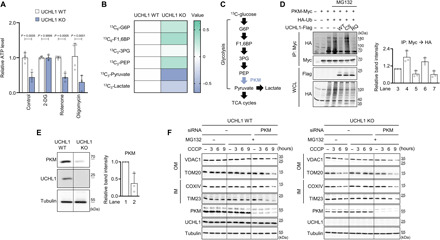
(A) Measurement of ATP levels in HEK293 UCHL1 WT or KO cells treated with 25 mM 2-DG, 1 μM rotenone, or 10 μM oligomycin. n = 4. (B) Measurement of glycolytic metabolites in HEK293 UCHL1 WT and KO cells after feeding 13C-glucose. The data were shown as a heatmap based on liquid chromatography–mass spectrometry metabolomics results. The row displays metabolites, and the column represents samples. Metabolites decreased are displayed in blue, while metabolites increased are displayed in green. The brightness of each color corresponds to the magnitude of the difference when compared with average value. n = 3. (C) Scheme of the glycolysis pathway. PKM converts phosphoenolpyruvate (PEP) to pyruvate. (D) Left: Immunoblot analysis of PKM ubiquitination in HEK293 cells coexpressing UCHL1 WT, C90S (CS), or R178Q (RQ). The cells were cotransfected with the plasmids carrying Myc-tagged PKM, hemagglutinin (HA)–tagged ubiquitin (Ub), and Flag-tagged UCHL1 WT, C90S, or R178Q and then treated with 20 μM MG132 for 4 hours before cell lysis. IP, immunoprecipitation; WCL, whole-cell lysate. Right: Relative quantification of the anti-HA immunoblot band intensity of the immunoprecipitated proteins by Myc antibody. n = 3. (E) Left: Immunoblot analysis of endogenous PKM in UCHL1 WT and KO HEK293 cells. Right: Relative quantification of the immunoblot band intensity of PKM. n = 3. (F) Immunoblot analysis of mitochondrial outer (VDAC1 and TOM20) and inner (COXIV, and TIM23) membrane proteins upon 20 μM CCCP, 20 μM MG132, and PKM siRNA treatment in UCHL1 WT or KO HEK293 cells. The band intensity of the four mitochondrial proteins (VDAC1, TOM20, TIM23, and COXIV) was quantified in fig. S11C. Two-way ANOVA with Sidak’s multiple comparison test was used (A). All data were presented as means + SD.
To pinpoint the target of UCHL1, we traced intermediate metabolites from each glycolytic step by culturing cells with 13C-labeled glucose and compared the amount of glycolytic metabolites in UCHL1 WT and KO cells. Notably, most of the metabolites before the pyruvate conversion step were increased, whereas the amount of pyruvate was highly decreased in UCHL1 KO cells compared to UCHL1 WT cells (Fig. 5B). Since pyruvate is converted from phosphoenolpyruvate (PEP) by pyruvate kinase (PKM) (Fig. 5C) (56), we suspected that PKM could be regulated by UCHL1. Therefore, we performed an experiment measuring the ubiquitination status of PKM in HEK293 cells while overexpressing UCHL1 WT, C90S (a catalytic dead form), or R178Q (a hyperactive form) (57) under the treatment with MG132 to inhibit proteasomal degradation (58). We observed that the expression of UCHL1 WT or R178Q decreased the ubiquitination of PKM proteins, whereas the expression of UCHL1 C90S mutant did not (Fig. 5D). To further examine whether UCHL1 deficiency would decrease PKM stability, we compared the amount of endogenous PKM proteins between UCHL1 WT and KO cells. Notably, the amount of endogenous PKM proteins was highly decreased in UCHL1 KO cells (Fig. 5E) compared to WT HEK293 cells, suggesting that UCHL1 stabilizes PKM through its DUB activity to regulate cellular ATP levels. In addition, we examined whether the expression level of PKM could regulate the UCHL1-dependent mitophagy. As UCHL1 stabilized PKM through its DUB activity, we used MG132 for blocking proteasomal degradation of the ubiquitinated PKM. The elevated mitophagy in UCHL1 KO cells was blocked by MG132 treatment (Fig. 5F, right, and fig. S11C, right). However, mitophagy was induced again when treating MG132 and PKM siRNA together to UCHL1 KO cells (Fig. 5F, right, and fig. S11C, right), confirming that the amount of PKM protein is critical to induce UCHL1-dependent mitophagy. In UCHL1 WT cells, the level of mitophagy was not altered by MG132 treatment alone since UCHL1 deubiquitinated PKM (Fig. 5F, left, and fig. S11C, left). However, similar to UCHL1 KO cells, mitophagy was elevated by cotreatment of MG132 and PKM siRNA in UCHL1 WT cells (Fig. 5F, left, and fig. S11C, left).
Furthermore, we examined the phosphorylation of AMPK and ULK1 in UCHL1 WT and KO cells expressing PKM. The elevated phosphorylation of AMPK and ULK1 in UCHL1 KO cells decreased with PKM overexpression to the phosphorylation levels, comparable with those in UCHL1 WT cells overexpressing PKM (fig. S11D). In addition, we found that the increased mitophagy in UCHL1 KO cells was blocked by overexpressing PKM proteins (fig. S11E). All of these results indicate that UCHL1 targets PKM proteins to regulate mitophagy via AMPK and ULK1 signaling.
Tripartite motif–containing 63 ubiquitinates PKM proteins and plays an antagonistic role of UCHL1
To identify an E3 ligase playing an antagonistic role of UCHL1, we used databases from the Biological General Repository for Interaction Datasets (BioGRID) (59). According to BioGRID results, PKM interacts with 311 proteins. Among them, seven E3 ligases [Parkin; mouse double minute 2 homolog; tripartite motif–containing 63 (TRIM63); HECT and RLD domain–containing E3 ubiquitin ligase family member 1; HECT, UBA, and WWE domain–containing E3 ubiquitin protein ligase 1; ligand of numb-protein X 1; and STIP1 homology and U-box–containing protein 1] are linked to PKM. Therefore, we examined which of the seven E3 ligases could ubiquitinate PKM. We identified that the exogenous expression of TRIM63 elevated the ubiquitin levels of PKM proteins (Fig. 6A). As PKM was ubiquitinated by TRIM63 and stabilized by UCHL1 in a protein ubiquitination–dependent manner, we tested whether the TRIM63-mediated ubiquitination of PKM proteins could be reduced by UCHL1. As a result, the ubiquitination of PKM proteins by TRIM63 was decreased by expressing UCHL1 WT, but not by expressing UCHL1 C90S (Fig. 6B). In addition, the amount of PKM proteins was elevated by TRIM63 knockdown in both UCHL1 WT and UCHL1 KO cell lines (Fig. 6C). However, the decreased PKM proteins in UCHL1 KO cells were recovered as much as those in UCHL1 WT cells under TRIM63 siRNA treatment (Fig. 6C). We also found that the enhanced mitophagy in UCHL1 KO cells was normalized by treating TRIM63 siRNA (Fig. 6D). These results demonstrate that TRIM63 and UCHL1 function antagonistically to control PKM ubiquitination and protein stability.
Fig. 6. TRIM63 E3 ligase and UCHL1 antagonistically regulates PKM ubiquitination and protein stability.
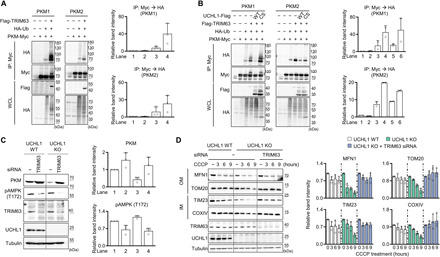
(A) Left: Immunoblot analysis of PKM ubiquitination in HEK293 cells coexpressing TRIM63. The cells were cotransfected with the plasmids carrying Myc-tagged PKM, HA-tagged ubiquitin, and Flag-tagged TRIM63. Right: Relative quantification of the anti-HA immunoblot band intensity of the immunoprecipitated proteins by Myc antibody. n = 3. (B) Left: Immunoblot analysis of PKM ubiquitination in HEK293 cells coexpressing both TRIM63 and UCHL1 WT or C90S. The cells were cotransfected with the plasmids carrying Myc-tagged PKM, HA-tagged ubiquitin, Flag-tagged TRIM63, and Flag-tagged UCHL1 WT or C90S. Right: Relative quantification of the anti-HA immunoblot band intensity of the immunoprecipitated proteins by Myc antibody. n = 2. (C) Left: Immunoblot analysis of endogenous PKM and AMPK phosphorylation in UCHL1 WT and KO HEK293 cells with TRIM63 siRNA expression. Right: The band intensity of PKM and AMPK phosphorylation. n = 3. (D) Left: Immunoblot analysis of mitochondrial outer (MFN1 and TOM20) and inner (TIM23 and COXIV) membrane proteins upon 20 μM CCCP and TRIM63 siRNA treatment. Right: Relative quantification of the immunoblot band intensity of indicated mitochondrial proteins. n = 3.
Loss of UCHL1 rescues PD phenotypes through stabilizing PKM
To further study the interaction between UCHL1 and PKM or TRIM63 in vivo, we knocked down Drosophila PKM (encoded by cg7070), the ortholog of human PKM, by expressing PKM RNAi or Drosophila TRIM63 (TRIM9, encoded by cg31721) in PINK1 or Parkin null flies under the hs-GAL4 driver. Consistent with the mammalian data, PKM knockdown or TRIM9 overexpression ameliorated the PD-related phenotypes of PINK1B9 and park1 flies, including decreased climbing abilities, loss of DA neurons, swollen mitochondria, and increased apoptosis (Fig. 7, A and B, and fig. S12, A to C), similar to the results of UCH KO. We further confirmed their genetic interaction using transgenic flies overexpressing PKM and knocking down TRIM9 by hs-GAL4 driver in PINK1 and UCH or Parkin and UCH double null mutants. The rescued PD-related phenotypes of PINK1B9 or park1 by UCH KO were reversed by the concurrent overexpression of PKM or TRIM9 knockdown [climbing ability (Fig. 7C and fig. S14E), mitochondrial morphology (figs. S13A and S14, A and A′), apoptosis in the muscles (figs. S13B and S14, B and B′), and loss of DA neurons (Fig. 7D and figs. S13C and S14, C and D′)]. Collectively, these data confirm that the loss of UCHL1 destabilizes PKM and subsequently activates mitophagy, which alleviates the PD-related pathogenesis induced by PINK1 or Parkin mutations in Drosophila.
Fig. 7. Loss of UCHL1 blocks PD-related phenotypes in a PKM-dependent manner.
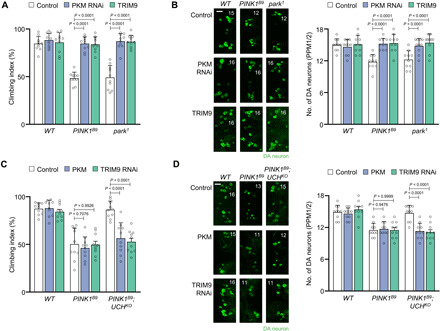
(A) Measurement of the climbing ability in the adult flies. n = 10. (B) Confocal immunofluorescence images (left) and numbers (right) of DA neurons in the PPM1/2 regions of adult fly brains. n = 10. Green denotes DA neuron. Scale bar, 20 μm. The number in panels indicates the number of DA neurons in each image. (C) Measurement of the climbing ability in the adult flies. n = 10. (D) Confocal immunofluorescence images (left) and numbers (right) of DA neurons in the PPM1/2 regions of adult fly brains. n = 10. Green denotes DA neuron. Scale bar, 20 μm. The number in panels indicates the number of DA neurons in each image. Two-way ANOVA with Sidak’s multiple comparison test was used (A to D). All data were presented as means + SD.
Last, we examined the interaction between PKM and the AMPK-ULK1-FUNDC1 signaling axis in fruit flies. We found that the swollen mitochondria in the muscle of PINK1 or Parkin mutant flies were rescued by PKM knockdown. However, the rescuing effect of PKM knockdown was blocked by the additional knockdown of FUNDC1 (fig. S15A). In addition, we observed that the DA neuronal death of PINK1 or Parkin KO flies in the PPM1/2 and PPL1 regions of the fly brain was rescued by PKM RNAi expression, and the concurrent expression of FUNDC1 RNAi blocked this rescuing effect of PKM knockdown (fig. S15, B and C). We then examined the genetic interaction between AMPK/ULK1 and PKM in the DA neurons in PINK1 and Parkin mutants. Expectedly, the anti–cell death effects of PKM knockdown in the DA neurons of PINK1B9 and park1 mutants were blocked by the concurrent expression of AMPK or ULK1 RNAi (fig. S16, A to B′). Furthermore, we identified the interaction between PKM and TRIM9 in Drosophila. TRIM9 overexpression rescued the swollen mitochondria phenotype of PINK1 and Parkin mutants, and these rescuing effects of TRIM9 overexpression were almost completely blocked by the concurrent expression of PKM (fig. S16C). Together, we conclude that PKM is antagonistically regulated by UCHL1 and TRIM63, and its glycolytic activity controls AMPK, ULK1, FUNDC1, and mitophagy in both mammalian cells and fruit flies.
DISCUSSION
In this study, we have identified a critical role of UCHL1 in regulating both glycolysis and energy-dependent mitophagy pathway. UCHL1 mutations have been known to induce PD, which defines UCHL1 as a PD-susceptible gene. However, our study revealed that UCHL1 mutations with loss of function rather alleviated PD pathogenesis by promoting mitophagy via sequential activation of AMPK, ULK1, and FUNDC1. In addition, the loss of UCHL1 inhibited the last step of glycolysis by reducing the protein stability of PKM and consequently rescued the defects related to PD. Furthermore, these rescuing effects are regulated by TRIM63, a previously unknown E3 ligase for PKM. These findings led us to propose an integrative mechanism of UCHL1 in between glycolysis and PD pathogenesis.
There were previous reports that PKM or pyruvate is associated with PD pathogenesis. In glioblastoma cell lines, the interaction between PKM and Parkin increases under glucose starvation, and Parkin ubiquitinates PKM to decrease its activity (60). These and our results suggest that PKM might be one of the key factors for controlling PD pathogenesis. However, we here found that the PD-related phenotypes in Parkin KO were completely alleviated by the loss of UCHL1, although the ubiquitination of PKM by Parkin was absent. That is, the deubiquitination site of PKM by UCHL1 is distinct from the ubiquitination site of PKM by Parkin, implying that a novel E3 ligase, not Parkin, plays an antagonistic role of UCHL1. Accordingly, we found TRIM63 as the E3 ligase and proved that controlling TRIM63 could regulate PD pathologies. Furthermore, in the blood of some patients with PD, increased pyruvate concentration and decreased pyruvate dehydrogenase E1 subunit β expression were observed (61). The mitochondrial pyruvate carrier (MPC) is involved in transport of pyruvate across the inner membrane of mitochondria. Pharmacological inhibition of MPC was recently found to be neuroprotective in multiple neurotoxin-based and genetic models of neurodegeneration, which are relevant to PD. Specifically, chemical inhibition of MPC in MPTP-treated mice and cultured human midbrain dopamine neurons increases survival of DA neurons in substantia nigra and augments striatal dopamine production (62). Together, elevating PKM activity or increasing pyruvate level correlates PD pathologies. Therefore, controlling the cellular PKM activity or pyruvate concentration could be a potential target to treat PD pathogenesis.
On the basis of our data that inhibition of UCHL1 rescues the symptoms related to PD, pharmacological inhibition of UCHL1 DUB activity would be a potential therapeutic strategy for managing PD. 5-Chloro-1-[(2,5-dichlorophenyl) methyl]-1H-indole-2,3-dione (LDN-57444) has been reported to be a potent UCHL1 inhibitor (63), and we have examined its inhibitory effect through our DUB assays. As a result, LDN-57444 could inhibit the activity of both human UCHL1 and Drosophila UCH (fig. S17A). We observed that the LDN-57444 treatment induced mitophagy in PINK1 or Parkin KO mouse embryonic fibroblast (MEF) cell lines (fig. S17B). Moreover, the drug treatment activated AMPK signaling in HEK293 or SH-SY5Y cell lines (fig. S17C). Therefore, we sought to examine whether the administration of LDN-57444 ameliorates any PD-related phenotypes in our Drosophila models. However, as LDN-57444 was very unstable under experimental conditions and the toxicity of dimethyl sulfoxide that dissolves LDN-57444 was fatal to fruit flies, the immediate usage of the drug was inappropriate for further experiments. We are currently screening specific inhibitors for UCHL1 with better physicochemical properties and less toxicity in both flies and mammalian cells.
The PINK1-Parkin pathway checks mitochondrial damages and distinguishes damaged mitochondria by labeling them with ubiquitins for initiating mitophagy. However, we here found that the UCHL1-controlled mitophagy is triggered by suppression of PKM and consequent inhibition of glycolysis. These results implicate that mitophagy can be commenced by nonmitochondrial factors and the UCHL1-controlled mitophagy may not distinguish between damaged and undamaged mitochondria. Therefore, we suggest that the mitophagy induced by UCHL1 deficiency plays a house-keeping role in mitochondrial homeostasis depending on cytosolic energy status.
In conclusion, we have identified that UCHL1 plays a critical role in regulating mitophagy independent of the PINK1-Parkin pathway and PKM is the responsible target of UCHL1 in controlling mitophagy and PD pathogenesis. On the basis of these data, we propose that UCHL1 is a possible target for the treatment of PD and that metabolic activities and nutritional factors are critical measures in regulating PD pathogenesis, in addition to previously known genetic and environmental factors.
MATERIALS AND METHODS
Plasmid constructs and chemical reagents
WT UCHL1 (NM_004181) was cloned into pcDNA3.1 zeo (+) C-terminal Myc-tagged vector or pCMV 14 C-terminal Flag-tagged vector. UCHL1 mutants (E7A, S18Y, C90S, I93M, and R178Q) were generated using a site-directed point mutagenesis method. N-terminal hemagglutinin (HA)–tagged human ubiquitin was cloned into pRK5 vector. The C-terminal Myc-tagged human PKM (PKM1, NM_002654.5; PKM2, AB528306.1) was cloned into pcDNA3.1 zeo (+) vector. The N-terminal Flag-tagged human TRIM63 (NM_032588.3) was cloned into pCMV10 vector. Cells were treated with CCCP (Calbiochem), rotenone (Sigma-Aldrich), oligomycin (Sigma-Aldrich), 2-DG (Sigma-Aldrich), antimycin A (Sigma-Aldrich), compound C (Sigma-Aldrich), or carbobenzoxy-Leu-Leu-leucinal (MG132, Calbiochem).
Cell culture and transfection
HEK293, HEK293T, and SH-SY5Y cells were used. HEK293 and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Welgene, Korea) supplemented with 10% fetal bovine serum (Invitrogen) at 37°C in a humidified atmosphere composed of 5% CO2. SH-SY5Y cells were cultured in DMEM (Welgene, Korea) supplemented with 20% fetal bovine serum (Gibco) at 37°C in a humidified atmosphere composed of 5% CO2. HEK293T cells were transfected using polyethylenimine reagent (Sigma-Aldrich). HEK293 cells were transfected using Lipofectamine 3000 (Invitrogen) as instructed by the manufacturer. For siRNA transfection, we used RNAiMAX (Invitrogen).
Generation of UCHL1 KO HEK293 and SH-SY5Y cell lines
The CRISPR genome editing technique was used for the generation of UCHL1 KO cells. To generate UCHL1 KO HEK293 cells, the guide RNA sequence (GTGGCGCTTCGTGGACGTGC) was cloned into the PX459 vector (#62988, Addgene). We generated UCHL1 KO cells using the previously reported method (41). The plasmid was transfected into HEK293 cells. Forty-eight hours after transfection, transfected cells were selected by puromycin (5 μg/ml) for 3 days, and then single colonies were transferred onto 96-well plates with one colony in each well. The UCHL1 KO clones were screened by immunoblot analysis with rabbit anti-UCHL1 antibody (Cell Signaling Technology).
To generate UCHL1 KO SH-SY5Y cells, the guide RNA sequence (TCCGCAGGTGCTGTCCCGGC) was cloned into pLenti-CRISPRv2 vector. SH-SY5Y cells were seeded with about 70% cell density at 100-mm plate. After 24 hours, cells were transfected with pLenti-CRISPRv2, pmDC-gag/pol, pRSV-rev, and pmDK-VSVg using Lipofectamine LTX (Invitrogen). After 3 days, cell media were filtered using 0.22-μm filter (Satorius Stedim Biotech) for collecting lentiviral particles and stored at −80°C. SH-SY5Y cells were cultured at six-well plate and were added with 500 μl of filtered lentiviral particles for transduction. Seventy-two hours after transduction, cells were selected by puromycin (5 μg/ml) for 3 days, and then single colonies were transferred onto 96-well plates with one colony in each well.
Antibodies
For immunoblot analysis, the following antibodies were used: mouse anti-MFN1 (Abcam), rabbit anti-TOM20 (Cell Signaling Technology), mouse anti-TIM23 (BD Biosciences), rabbit anti-COXIV (Cell Signaling Technology), rabbit anti-UCHL1 (Cell Signaling Technology), mouse anti-tubulin (Developmental Studies Hybridoma Bank), rabbit anti–phospho-AMPKα (T172, Cell Signaling Technology), rabbit anti–phoshpo-ULK1 (S555, Cell Signaling Technology), mouse anti-AMPK (Abcam), rabbit anti-ULK1 (Cell Signaling Technology), rabbit anti-FUNDC1 (Novus Biologicals), mouse anti-VDAC1 (Abcam), mouse anti-NDUFS3 (Abcam), rabbit anti-Flag (Cell Signaling Technology), rabbit anti-HA (Cell Signaling Technology), mouse anti-Myc (MBL, Japan), mouse anti-TRIM63 (Santa Cruz Biotechnology), and rabbit anti-PKM antibody (Cell Signaling Technology). Peroxidase-conjugated secondary antibodies were purchased from the Jackson laboratory.
Immunoprecipitation and immunoblotting
For immunoprecipitation, cells were lysed using a lysis buffer A [20 mM tris (pH 7.5), 100 mM NaCl, 1 mM EDTA, 2 mM EGTA, 50 mM β-glycerophosphate, 50 mM NaF, 1 mM sodium vanadate, 2 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), leupeptin (10 μg/ml), pepstatin A (1 μg/ml), and 1% Triton X-100] and were subjected to immunoprecipitation and immunoblotting according to standard procedures. Cell lysates were centrifuged at 13,000 rpm for 20 min at 4°C and were incubated overnight after the addition of primary antibodies. Lysates were then incubated with protein A/G agarose beads for 2 hours at 4°C, washed four times in detergent-free lysis buffer A, and eluted with 2× Laemmli buffer at 95°C. Immunoblot analysis on mitochondrial proteins was performed using radioimmunoprecipitation assay buffer [50 mM tris (pH 8.0), 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40, 0.1% SDS, 2 mM DTT, 1 mM PMSF, leupeptin (10 μg/ml), and pepstatin A (1 μg/ml)]. Total protein was quantified using the BCA Protein Assay Kit (Pierce). Lysates were subjected to SDS–polyacrylamide gel electrophoresis analysis, followed by immunoblotting according to standard procedures. The blots were developed and viewed under LAS-4000 (Fujifilm).
Fly stocks
The Drosophila lines used in the experiments were mef2-GAL4 (27390, Bloomington Drosophila Stock Center), hs-GAL4 (2077, Bloomington Drosophila Stock Center), th-GAL4 (8848, Bloomington Drosophila Stock Center), PINK1B9 (34749, Bloomington Drosophila Stock Center), park1 (34747, Bloomington Drosophila Stock Center), UAS-Zda RNAi (5482R-2, Japanese National Institute of Genetics), UAS-Bnip3 RNAi (5059R-1, Japanese National Institute of Genetics), UAS-p62 RNAi (10360R-1, Japanese National Institute of Genetics), UAS-FUNDC1 RNAi (5676R-1, Japanese National Institute of Genetics), UAS-AMPK RNAi (3051R-1, Japanese National Institute of Genetics), UAS-ULK1 RNAi (10967R-1, Japanese National Institute of Genetics), UAS-AMPKCA (previously described) (52), UAS-FUNDC1 (F004027, FlyORF), UAS-PKM RNAi (7070R-2, Japanese National Institute of Genetics), UAS-TRIM9 RNAi (31721R-2, Japanese National Institute of Genetics), UAS-TRIM9 (received from B. Ye) (64), UAS-GLUT1 RNAi (28645, Bloomington Drosophila Stock Center), UAS-NDUFS3 RNAi (12079R-1, Japanese National Institute of Genetics), and UAS-ATPsynβ (8189R-1, Japanese National Institute of Genetics). Drosophiila PKM cDNA (UFO11109) was obtained from Drosophila Genomics Resource Center (Indiana University), and UAS-PKM-HA was generated by microinjecting pUAST-PKM-HA into w1118 embryos. The UAS-PD gene RNAi lines used in Fig. 1A were described in table S1. All flies were grown in standard cornmeal-yeast-agar medium at 25°C.
Generation of UCH KO and KI flies
For generation of UCH KO flies, we used CAS-0001 fly from Japanese National Institute of Genetics. After selecting a Cas9 target site as far forward as possible in the UCH first exon, we made single-guide RNA (sgRNA) using in vitro transcription as previously described (32). As both UCH gene and nos-Cas9 are on the second chromosome, we crossed CAS-0001 with w1118 and collected embryos. After injecting sgRNA into the embryos, the adult male flies were crossed with bc/cyo. To exclude the case that DNA break had occurred on the nos-Cas9 chromosome, we sorted out the flies that did not carry nos-Cas9 by polymerase chain reaction (PCR). We then amplified the target loci by PCR and analyzed the sequence.
For generation of UCH KI flies, we injected three different kinds of plasmids into fly embryos: Cas9 expression vector (pHsp70-Cas9; #45945, Addgene), sgRNA expression vectors, and donor plasmids for homologous recombination (38). To construct sgRNA expression vectors, 24–base pair (bp) oligonucleotides with 20-bp sgRNA sequence and 4-bp Bbs1 restriction enzyme site were synthesized and inserted into pU6-Bbs1-chiRNA vector (#45946, Addgene). For preparing donor plasmids, we extracted fly genomic DNA and amplified the homology arms that have a length of about 2 kb and a protospacer adjacent motif (PAM) sequence at the center by PCR. After cloning the homology arms into pBS vector, we made each point mutation by site-directed mutagenesis. In addition, as the homology arms need the sequences that sgRNA could recognize, sgRNA sequences in the homology arms were modified by a site-directed mutagenesis to avoid changing the UCH protein sequence. Last, all the three completed vectors were injected into fly embryos to generate various UCH KI flies. The KI flies were identified by PCR and subsequent DNA sequencing.
Measurement of ATP levels
We used the ATP Bioluminescence Assay Kit HS II (Roche) and analyzed ATP levels according to the manufacturer’s instruction. To measure ATP levels in cells, HEK293 WT or KO cells were cultured in 96 wells for 24 hours, and we treated rotenone (Sigma-Aldrich), oligomycin (Sigma-Aldrich), and 2-DG (Sigma-Aldrich) for 12 hours before measuring ATP levels. Samples were incubated with cell lysis reagents for 5 min at room temperature and were added with luciferase reagents. The luminescence was measured by Tecan Plate Reader Infinite 200. Three independent experiments were performed for data quantification. To measure ATP levels in flies, five third instar larvae were collected and ground in lysis reagents. After lysis, luciferase reagents were added to the supernatants, and the luminescence was measured by Tecan Plate Reader Infinite 200. Feeding flies with 2-DG (65), rotenone (66), and oligomycin (67) was performed as previously described.
Detecting mitophagy in cells
Mitophagy was measured using a mitophagy detection kit (Dojindo Molecular Technologies, Korea) according to the manufacturer’s instruction. HEK293 UCHL1 WT or KO cells were seeded on 10-mm cover slips in 12-well plates and cultured at 37°C overnight. HEK293 cells were treated with 20 μM CCCP for 12 hours and measured mitophagy as suggested by the manufacturer’s instruction (43). The cells were observed by LSM 710 confocal microscope. Three independent experiments were performed for quantification, and the red fluorescence dots per cell were counted using ImageJ.
Glucose isotope tracing for metabolomics
For metabolite extraction from cells, 200 μl of ice-cold mixture of methanol, acetonitrile, and distilled water (5:3:2) were mixed with cell pellet (cell number = 5 × 106), and 400 μl of ice-cold mixture of methanol and acetonitrile (5:3:2) was added to the 100 μl of culture media. This mixture was vortexed for 20 s and frozen in liquid nitrogen for 2.5 min. It was then thawed at 25°C for 1.5 min. This freeze-and-thaw cycle was repeated twice, and the cell homogenate was centrifuged at 15,000g for 20 min at 4°C. The supernatant was concentrated by vacuum evaporation in VS-802 Speed-Vac (VISION SCIENTIFIC, Korea). The dried samples were reconstituted in 25 μl of chilled mixture of acetonitrile/water (1:1) for mass spectrometry analysis. 13C-labeled metabolites derived from the U-13C glucose tracer were measured by Q Exactive focus mass spectrometer (Thermo Fisher Scientific, Germany). The liquid chromatography separation was performed with a ZIC-pHILIC column (150 × 2.1 mm, 5 μm; Merck KGaA, Germany) on ACQUITY UPLC system (Waters). The column temperature was set at 35 °C with a flow rate of 0.2 ml/min, and the analytes were eluted with a linear gradient [mobile phase A: acetonitrile; mobile phase B: 10 mM ammonium carbonate in water (pH 9.0)] as follows: 80% A in 0 to 4.5 min, 80 to 5% in 4.5 to 13.5 min, 5% in 13.5 to 19.5 min, 5 to 80% 19.5 to 22.5 min, and 80% 22.5 to 26.5 min. Before injection, samples were kept at 4°C in an autosampler, and the injection volume was 5 μl. For the mass detection, Thermo Orbitrap was operated in full scan of negative ion mode with mass range from 64 to 830 mass/charge ratio (m/z), and the instrument parameters were as follows: 4.2 kV of spray voltage; capillary temperature at 270°C; and sheath gas, auxiliary gas, and sweep gas at flow rates of 46, 12, and 6 arbitrary units, respectively. Metabolites were identified by accurate mass measurement (accuracy of <6 parts per million) and confirmed by comparing the m/z and retention time with those of the standard compounds (table S2). The peak area of metabolites and those isotopologues was measured using Xcalibur software (Thermo Fisher Scientific).
For sample preparation, UCHL1 WT and KO HEK293 cells were cultured in 60-mm plate with DMEM media (Gibco) for 48 hours. Cells were washed twice with Dulbecco’s phosphate-buffered saline (DPBS; Gibco), and media were changed with 25 mM U-13C-glucose (Cambridge Isotope Laboratories) in DMEM without glucose (Gibco) supplemented with 10% dialyzed fetal bovine serum (Gibco). Cells were harvested after 12-hour incubation in U-13C-glucose media. Cells were trypsinized and centrifuged at 5000 rpm for 3 min at 4°C. Cell pellets were washed twice with DPBS, were frozen in liquid nitrogen, and stored at −80°C before metabolite extraction.
Quantification of wing and thorax abnormality in Drosophila
To quantify the abnormality of wing and thorax, the percentage of 3-day-old male flies showing crushed thorax and downturned or upturned wing of 10 flies was measured. Ten independent experiments were performed for quantification (68).
Mitochondria staining and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling assay in Drosophila
Three-day-old male flies were fixed with 4% paraformaldehyde. After fixation, the thoraces of flies were collected and stained. For mitochondria staining, streptavidin was used to observe mitochondria (1:200; Alexa Fluor 488 streptavidin, Invitrogen), and phalloidin was used to observe actin filament (1:200; phalloidin-tetramethylrhodamine B isothiocyanate, Merck). For terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay, an in situ cell death detection kit (Roche) was used to visualize apoptosis, and Hoechst was used to stain nucleus (1:200; Hoechst 33258, Invitrogen).
To quantify the percentage of flies having mitochondrial abnormalities, a fly with a mitochondrion having a length more than 5 μm and the width more than 3 μm was defined as the fly having abnormal mitochondria. To quantify the percentage of flies showing apoptosis, a fly having more than 10 TUNEL dots in any of six dorsal longitudinal muscles of the indirect flight muscles was defined as the fly showing apoptosis (68). Ten 3-day-old male flies were counted to quantify these percentages. Ten independent experiments were performed for data quantification.
Climbing assay in Drosophila
Three-day-old male flies were transferred in groups of 10- to 18-cm-long vials and were incubated for 5 min at room temperature for recovery period. All the flies were moved to the bottom of the vial by gently tapping, the percentage of flies arriving over 15 cm within 12 s was measured. The assay was repeated 10 times per one group of the flies, and 10 groups of each genotype were performed for data quantification.
DA neuron staining in Drosophila
Thirty-day-old male flies were fixed with 4% paraformaldehyde, and the brains were dissected. DA neurons were stained with anti-TH mouse antibody (1:200; ImmunoStar). For counting the number of DA neurons, the brains were observed by LSM 710 confocal microscope (Carl Zeiss) via Z-stack analysis. DA neurons from 10 flies of each genotype were measured for data quantification.
Generation of recombinant UCHL1 proteins
Escherichia coli BL21 (DE3) strain was used for transformation, cloning, and protein production. Human UCHL1, Drosophila UCH, and their mutants (E7A, S18Y, C90S, and I93M in human UCHL1/ E8A, H19Y, C93S, and V96M in Drosophila UCH) were cloned into pGEX 4T-1 vector. Twenty milliliters of overnight precultures were used to inoculate 500 ml of LB supplemented with ampicillin. Cultures were incubated at 37°C with 180 rpm shaking. When the optical density at 600 nm reached 0.6, protein expression was induced by adding 0.1 mM isopropyl-β-d-thiogalactopyranoside for about 1 to 2 hours. Cells were harvested by centrifugation at 12,000 rpm for 30 min at 4°C. Cell pellets were resuspended in a lysis buffer [25 mM tris-HCl (pH 7.5), 150 mM NaCl, and 1 mM EDTA] supplemented with protease inhibitors [1 mM PMSF, leupeptin (10 μg/ml), and pepstatin A (1 μg/ml)] and 0.5% Triton X-100. Cells were disrupted by sonication, and the lysate was clarified by centrifugation at 10,000 rpm for 10 min at 4°C. Glutathione S-transferase (GST)–tagged proteins in supernatant were purified by binding to GST beads (GE Healthcare) for 2 hours at 4°C. GST beads were washed three to four times with a lysis buffer without Triton X-100. GST-tagged proteins were eluted with an elution buffer [10 mM reduced glutathione, 50 mM tris-HCl, and 5% glycerol (pH 8.0)].
DUB activity assay
The DUB activity of each UCHL1 or UCH mutant protein was measured using a DUB activity assay kit (K485, BioVision). UCHL1 or UCH protein (1 mg/ml) was used, and DUB substrate [ubiquitin-7-amido-4-methylcoumarin (UB-AMC)] was diluted into DUB assay buffer (1:44). To initiate reaction, 5 μl of UCHL1 or UCH protein and 5 μl of DUB substrate were mixed. Using the Tecan Plate Reader Infinite 200, the fluorescence emitted from DUB reaction and the AMC standard were measured every 3 min for 30 min (24).
Statistical analysis
A blind manner was used in all experiments and analysis. Image areas were selected randomly during observing samples. For computing P values, one-way analysis of variance (ANOVA; Dunnett’s multiple comparison test and Tukey’s multiple comparison test), two-way ANOVA (Sidak’s multiple comparison test and Tukey’s multiple comparison test), and multiple t tests were used. All the tests were examined via GraphPad Prism v.8 for the statistics.
Acknowledgments
We would like to thank FlyBase, Bloomington Drosophila Stock Center, Vienna Drosophila Resource Center, Japanese National Institute of Genetics, FlyORF, and Drosophila Genomics Resource Center for fly stocks and related information. We also thank B. Ye for TRIM63-related fly lines. Funding: This study was supported by National Research Foundation of Korea grants funded by the Korean government (MSIT) (NRF-2019R1A2C2005810 and NRF-2020R1A5A1018081 to J.C. and NRF-2018R1A3B1052328 to S.P.) and the grant HI17C0328 to J.C. from the Korea Health Technology Research and Development Project, through the Korea Health Industry Development Institute, funded by the Ministry of Health and Welfare. S.J.H., D.L., and J.C. were supported by BK21 Plus Program from the Ministry of Education, Republic of Korea. S.J.H. and W.J.X. were supported by National Research Foundation of Korea (NRF-2021R1I1A1A01042946 and NRF-2020R1I1A1A01072189, respectively). Author contributions: S.J.H., D.L., and J.C. designed this research, and E.C. and S.C. initiated the research. S.J.H., D.L., W.J.X., S.P., and J.C. performed the experiments and analyzed the data. S.J.H., D.L., S.C., S.M., and J.C. wrote the manuscript, and all authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabg4574/DC1
REFERENCES AND NOTES
- 1.Rabey J. M., Hefti F., Neuromelanin synthesis in rat and human substantia nigra. J. Neural. Transm. Park. Dis. Dement. Sect. 2, 1–14 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Bolam J. P., Pissadaki E. K., Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 27, 1478–1483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Surmeier D. J., Obeso J. A., Halliday G. M., Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukushima T., Gao T., Tawara T., Hojo N., Isobe A., Yamane Y., Inhibitory effect of nicotinamide to paraquat toxicity and the reaction site on complex I. Arch. Toxicol. 71, 633–637 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Vila M., Przedborski S., Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 365–375 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Richardson J. R., Quan Y., Sherer T. B., Greenamyre J. T., Miller G. W., Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol. Sci. 88, 193–201 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Callio J., Oury T. D., Chu C. T., Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J. Biol. Chem. 280, 18536–18542 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greene J. C., Whitworth A. J., Kuo I., Andrews L. A., Feany M. B., Pallanck L. J., Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 4078–4083 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J. M., Chung J., Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M., Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Poole A. C., Thomas R. E., Andrews L. A., McBride H. M., Whitworth A. J., Pallanck L. J., The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 105, 1638–1643 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J. W., Yang L., Beal M. F., Vogel H., Lu B., Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U.S.A. 103, 10793–10798 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Springer W., Kahle P. J., Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 7, 266–278 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Jin S. M., Youle R. J., PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harper J. W., Ordureau A., Heo J. M., Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Joselin A. P., Hewitt S. J., Callaghan S. M., Kim R. H., Chung Y. H., Mak T. W., Shen J., Slack R. S., Park D. S., ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 21, 4888–4903 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Smith W. W., Pei Z., Jiang H., Moore D. J., Liang Y., West A. B., Dawson V. L., Dawson T. M., Ross C. A., Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 18676–18681 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plun-Favreau H., Klupsch K., Moisoi N., Gandhi S., Kjaer S., Frith D., Harvey K., Deas E., Harvey R. J., McDonald N., Wood N. W., Martins L. M., Downward J., The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 9, 1243–1252 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Burchell V. S., Nelson D. E., Sanchez-Martinez A., Delgado-Camprubi M., Ivatt R. M., Pogson J. H., Randle S. J., Wray S., Lewis P. A., Houlden H., Abramov A. Y., Hardy J., Wood N. W., Whitworth A. J., Laman H., Plun-Favreau H., The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 16, 1257–1265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J., Zhao C. Y., Si Y. M., Liu Z. L., Chen B., Yu L., ACT and UCH-L1 polymorphisms in Parkinson’s disease and age of onset. Mov. Disord. 17, 767–771 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Leroy E., Boyer R., Auburger G., Leube B., Ulm G., Mezey E., Harta G., Brownstein M. J., Jonnalagada S., Chernova T., Dehejia A., Lavedan C., Gasser T., Steinbach P. J., Wilkinson K. D., Polymeropoulos M. H., The ubiquitin pathway in Parkinson’s disease. Nature 395, 451–452 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Wilkinson K. D., Lee K. M., Deshpande S., Duerksen-Hughes P., Boss J. M., Pohl J., The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 246, 670–673 (1989). [DOI] [PubMed] [Google Scholar]
- 23.Larsen C. N., Krantz B. A., Wilkinson K. D., Substrate specificity of deubiquitinating enzymes: Ubiquitin C-terminal hydrolases. Biochemistry 37, 3358–3368 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Bilguvar K., Tyagi N. K., Ozkara C., Tuysuz B., Bakircioglu M., Choi M., Delil S., Caglayan A. O., Baranoski J. F., Erturk O., Yalcinkaya C., Karacorlu M., Dincer A., Johnson M. H., Mane S., Chandra S. S., Louvi A., Boggon T. J., Lifton R. P., Horwich A. L., Gunel M., Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 3489–3494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lincoln S., Vaughan J., Wood N., Baker M., Adamson J., Gwinn-Hardy K., Lynch T., Hardy J., Farrer M., Low frequency of pathogenic mutations in the ubiquitin carboxy-terminal hydrolase gene in familial Parkinson’s disease. Neuroreport 10, 427–429 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Maraganore D. M., Lesnick T. G., Elbaz A., Chartier-Harlin M. C., Gasser T., Krüger R., Hattori N., Mellick G. D., Quattrone A., Satoh J. I., Toda T., Wang J., Ioannidis J. P. A., de Andrade M., Rocca W. A.; the UCHL1 Global Genetics Consortium , UCHL1 is a Parkinson’s disease susceptibility gene. Ann. Neurol. 55, 512–521 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Carmine Belin A., Westerlund M., Bergman O., Nissbrandt H., Lind C., Sydow O., Galter D., S18Y in ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) associated with decreased risk of Parkinson’s disease in Sweden. Parkinsonism Relat. Disord. 13, 295–298 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Kyratzi E., Pavlaki M., Stefanis L., The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells. Hum. Mol. Genet. 17, 2160–2171 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Xilouri M., Kyratzi E., Pitychoutis P. M., Papadopoulou-Daifoti Z., Perier C., Vila M., Maniati M., Ulusoy A., Kirik D., Park D. S., Wada K., Stefanis L., Selective neuroprotective effects of the S18Y polymorphic variant of UCH-L1 in the dopaminergic system. Hum. Mol. Genet. 21, 874–889 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Blauwendraat C., Nalls M. A., Singleton A. B., The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cha G. H., Kim S., Park J., Lee E., Kim M., Lee S. B., Kim J. M., Chung J., Cho K. S., Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc. Natl. Acad. Sci. U.S.A. 102, 10345–10350 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bassett A., Liu J. L., CRISPR/Cas9 mediated genome engineering in Drosophila. Methods 69, 128–136 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Maraganore D. M., Farrer M. J., Hardy J. A., Lincoln S. J., McDonnell S. K., Rocca W. A., Case-control study of the ubiquitin carboxy-terminal hydrolase L1 gene in Parkinson’s disease. Neurology 53, 1858–1860 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Satoh J., Kuroda Y., A polymorphic variation of serine to tyrosine at codon 18 in the ubiquitin C-terminal hydrolase-L1 gene is associated with a reduced risk of sporadic Parkinson’s disease in a Japanese population. J. Neurol. Sci. 189, 113–117 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Elbaz A., Levecque C., Clavel J., Vidal J. S., Richard F., Corrèze J. R., Delemotte B., Amouyel P., Alpérovitch A., Chartier-Harlin M. C., Tzourio C., S18Y polymorphism in the UCH-L1 gene and Parkinson’s disease: Evidence for an age-dependent relationship. Mov. Disord. 18, 130–137 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Healy D. G., Abou-Sleiman P. M., Casas J. P., Ahmadi K. R., Lynch T., Gandhi S., Muqit M. M. K., Foltynie T., Barker R., Bhatia K. P., Quinn N. P., Lees A. J., Gibson J. M., Holton J. L., Revesz T., Goldstein D. B., Wood N. W., UCHL-1 is not a Parkinson’s disease susceptibility gene. Ann. Neurol. 59, 627–633 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Liu Y., Fallon L., Lashuel H. A., Liu Z., Lansbury P. T. Jr., The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 111, 209–218 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Gratz S. J., Ukken F. P., Rubinstein C. D., Thiede G., Donohue L. K., Cummings A. M., O’Connor-Giles K. M., Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics 196, 961–971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gratz S. J., Harrison M. M., Wildonger J., O'Connor-Giles K. M., Precise genome editing of Drosophila with CRISPR RNA-guided Cas9. Methods Mol. Biol. 1311, 335–348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pickrell A. M., Youle R. J., The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazarou M., Sliter D. A., Kane L. A., Sarraf S. A., Wang C., Burman J. L., Sideris D. P., Fogel A. I., Youle R. J., The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwashita H., Torii S., Nagahora N., Ishiyama M., Shioji K., Sasamoto K., Shimizu S., Okuma K., Live cell imaging of mitochondrial autophagy with a novel fluorescent small molecule. ACS Chem. Biol. 12, 2546–2551 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Laker R. C., Drake J. C., Wilson R. J., Lira V. A., Lewellen B. M., Ryall K. A., Fisher C. C., Zhang M., Saucerman J. J., Goodyear L. J., Kundu M., Yan Z., Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8, 548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim G. G., Lim K. L., Parkin-independent mitophagy-FKBP8 takes the stage. EMBO Rep. 18, 864–865 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhujabal Z., Birgisdottir Å. B., Sjøttem E., Brenne H. B., Øvervatn A., Habisov S., Kirkin V., Lamark T., Johansen T., FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 18, 947–961 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanna R. A., Quinsay M. N., Orogo A. M., Giang K., Rikka S., Gustafsson Å. B., Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T., p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Liu L., Feng D., Chen G., Chen M., Zheng Q., Song P., Ma Q., Zhu C., Wang R., Qi W., Huang L., Xue P., Li B., Wang X., Jin H., Wang J., Yang F., Liu P., Zhu Y., Sui S., Chen Q., Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Tian W., Li W., Chen Y., Yan Z., Huang X., Zhuang H., Zhong W., Chen Y., Wu W., Lin C., Chen H., Hou X., Zhang L., Sui S., Zhao B., Hu Z., Li L., Feng D., Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 589, 1847–1854 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Wu W., Tian W., Hu Z., Chen G., Huang L., Li W., Zhang X., Xue P., Zhou C., Liu L., Zhu Y., Zhang X., Li L., Zhang L., Sui S., Zhao B., Feng D., ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15, 566–575 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee J. H., Koh H., Kim M., Kim Y., Lee S. Y., Karess R. E., Lee S. H., Shong M., Kim J. M., Kim J., Chung J., Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 447, 1017–1020 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Ng C. H., Guan M. S. H., Koh C., Ouyang X., Yu F., Tan E. K., O'Neill S. P., Zhang X., Chung J., Lim K. L., AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J. Neurosci. 32, 14311–14317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jastroch M., Divakaruni A. S., Mookerjee S., Treberg J. R., Brand M. D., Mitochondrial proton and electron leaks. Essays Biochem. 47, 53–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wick A. N., Drury D. R., Nakada H. I., Wolfe J. B., Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 224, 963–969 (1957). [PubMed] [Google Scholar]
- 56.Romano A. H., Conway T., Evolution of carbohydrate metabolic pathways. Res. Microbiol. 147, 448–455 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Rydning S. L., Backe P. H., Sousa M. M. L., Iqbal Z., Øye A. M., Sheng Y., Yang M., Lin X., Slupphaug G., Nordenmark T. H., Vigeland M. D., Bjørås M., Tallaksen C. M., Selmer K. K., Novel UCHL1 mutations reveal new insights into ubiquitin processing. Hum. Mol. Genet. 26, 1217–1218 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Hayashi M., Saito Y., Kawashima S., Calpain activation is essential for membrane fusion of erythrocytes in the presence of exogenous Ca2+. Biochem. Biophys. Res. Commun. 182, 939–946 (1992). [DOI] [PubMed] [Google Scholar]
- 59.Oughtred R., Stark C., Breitkreutz B. J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R., Zhang F., Dolma S., Willems A., Coulombe-Huntington J., Chatr-aryamontri A., Dolinski K., Tyers M., The BioGRID interaction database: 2019 update. Nucleic Acids Res. 47, D529–D541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu K., Li F., Han H., Chen Y., Mao Z., Luo J., Zhao Y., Zheng B., Gu W., Zhao W., Parkin regulates the activity of pyruvate kinase M2. J. Biol. Chem. 291, 10307–10317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmed S. S., Santosh W., Kumar S., Christlet H. T., Metabolic profiling of Parkinson’s disease: Evidence of biomarker from gene expression analysis and rapid neural network detection. J. Biomed. Sci. 16, 63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghosh A., Tyson T., George S., Hildebrandt E. N., Steiner J. A., Madaj Z., Schulz E., Machiela E., McDonald W. G., Galvis M. L. E., Kordower J. H., Van Raamsdonk J. M., Colca J. R., Brundin P., Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci. Transl. Med. 8, 368ra174 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Liu Y., Lashuel H. A., Choi S., Xing X., Case A., Ni J., Yeh L. A., Cuny G. D., Stein R. L., Lansbury P. T. Jr., Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol. 10, 837–846 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Yang L., Li R., Kaneko T., Takle K., Morikawa R. K., Essex L., Wang X., Zhou J., Emoto K., Xiang Y., Ye B., Trim9 regulates activity-dependent fine-scale topography in Drosophila. Curr. Biol. 24, 1024–1030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown E. B., Shah K. D., Faville R., Kottler B., Keene A. C., Drosophila insulin-like peptide 2 mediates dietary regulation of sleep intensity. PLOS Genet. 16, e1008270 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doktór B., Damulewicz M., Pyza E., Overexpression of mitochondrial ligases reverses rotenone-induced effects in a Drosophila model of Parkinson’s disease. Front. Neurosci. 13, 94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bahadorani S., Hur J. H., Lo T. Jr., Vu K., Walker D. W., Perturbation of mitochondrial complex V alters the response to dietary restriction in Drosophila. Aging Cell 9, 100–103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ham S. J., Lee D., Yoo H., Jun K., Shin H., Chung J., Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 117, 4281–4291 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabg4574/DC1