

This study provides new insights into how the soil microbiome of urban greenspaces differs from surrounding natural ecosystems.
Abstract
The structure and function of the soil microbiome of urban greenspaces remain largely undetermined. We conducted a global field survey in urban greenspaces and neighboring natural ecosystems across 56 cities from six continents, and found that urban soils are important hotspots for soil bacterial, protist and functional gene diversity, but support highly homogenized microbial communities worldwide. Urban greenspaces had a greater proportion of fast-growing bacteria, algae, amoebae, and fungal pathogens, but a lower proportion of ectomycorrhizal fungi than natural ecosystems. These urban ecosystems also showed higher proportions of genes associated with human pathogens, greenhouse gas emissions, faster nutrient cycling, and more intense abiotic stress than natural environments. City affluence, management practices, and climate were fundamental drivers of urban soil communities. Our work paves the way toward a more comprehensive global-scale perspective on urban greenspaces, which is integral to managing the health of these ecosystems and the well-being of human populations.
INTRODUCTION
Urban greenspaces, such as parks and residential gardens, are critically important for promoting mental and physical well-being and for reducing morbidity and mortality (1). According to the United Nations, 68% of the global population will live in cities by 2050, increasing the environmental and social stresses for the billions of humans living in urban areas (2). Urban greenspaces make up most of the open spaces available for recreational activities in urban areas such as sport and social engagement and play important roles in curbing pollution, reducing noise, and lowering air temperatures (1–3). Urban greenspaces also provide habitat for a myriad of plants and animals including a wide range of bird species (e.g., pigeons), terrestrial and arboreal mammals (e.g., squirrels), and diverse above- and below-ground invertebrates (3, 4). Similarly, soils in urban greenspaces are also home to a diverse community of microbes, including archaea, bacteria, fungi, and protists (4–6). This soil biodiversity plays important roles in maintaining ecosystem services such as soil fertility, plant health, productivity, and waste decomposition (7). Moreover, human exposure to soil microbes has been shown to be beneficial to human health by promoting effective immunoregulation functions and reducing allergies (8). However, some soil microbes (e.g., Mycobacterium, Listeria, and Fusarium spp.) can also have substantial negative consequences for the sustainability of urban greenspaces and for animal, human, and plant health. For instance, some soil microbial taxa harbor antibiotic resistance genes and could potentially influence the health of people and animals by reducing our ability to fight human diseases (9, 10). Unfortunately, unlike with urban birds, plants, and mammals, we have a very limited understanding of the identity and function of the soil organisms inhabiting our urban parks and gardens. Previous microbial studies in urban greenspaces have focused on specific locations (e.g., New York’s Central Park) (4, 5), specific groups of soil organisms (e.g., mycorrhizal fungi) (6), or specific microbial functions (e.g., denitrification) (11). However, we currently lack a global perspective on the soil microbiome of urban greenspaces and how these soil microbiomes compare to those found in nearby natural ecosystems. A comprehensive evaluation must consider multiple cities with contrasting populations, vegetation, and climates and include a wide range of functional and taxonomic groups of soil organisms. Improving our understanding of soil organisms associated with urban parks is a critical step toward building a better understanding of the role of these fundamental organisms in controlling the functioning and health of urban environments and toward managing the sustainable development of urban greenspaces.
Here, we report results of the first global field assessment of the soil microbiome in urban greenspaces, aiming to (i) compare the diversity and community composition of microbial taxonomic and functional groups of urban greenspaces with those of nearby “natural” ecosystems; (ii) identify the soil microbial taxa (bacteria, archaea, protists, and fungi) that are consistently found to be residents of urban greenspaces across the globe; (iii) evaluate the socioeconomic and environmental factors linked to the structure of the soil microbiome of urban greenspaces; and (iv) assess the microbial functional attributes characterizing soils in urban greenspaces including genes associated with pathogenesis, greenhouse gas emissions, nutrient cycling, and abiotic stress.
RESULTS AND DISCUSSION
To achieve these goals, we carried out a field survey across 112 sites from 17 countries spanning six continents (Fig. 1A, table S1, and fig. S1). We used an experimental design that included paired urban and nearby natural sites located across 56 globally distributed locations (statistical “blocks”; Fig. 1A, table S1, and fig. S1). In each location (e.g., Cape Town, South Africa; fig. S1), we established a 30 m by 30 m plot in a representative urban greenspace (e.g., an urban forest or lawn in a city park; fig. S1) and a nearby, relatively undisturbed natural ecosystem resembling the original vegetation representative of these locations (e.g., a natural grassland; fig. S1). Urban greenspaces were located mostly in public parks and large residential gardens and comprised a mixture of open areas with lawns, scattered trees, patches of shrubs, and associated flowerbeds. We refer to these ecosystems as urban greenspaces hereafter (1). Natural areas near these cities were unmanaged or seminatural forests, shrublands, and grasslands, many of them relict ecosystems. Urban greenspaces and natural ecosystems strongly differ in vegetation structure and supported lower levels of plant species richness (fig. S2 and table S2). Agricultural lands were not included in this study. Our database comprises a wide range of urban areas including relatively small (population < 50,000; e.g., Alice Springs, Australia), medium (population, 50,000 to 1 million; e.g., Ljubljana, Slovenia), and large cities (population > 5 million; e.g., Beijing, China; table S1), and spanned a large range in vegetation types (forests, grasslands, and shrublands) and climatic regions (arid, temperate, tropical, and cold ecosystems; table S1). Our sampling was explicitly designed to account for the spatial heterogeneity in our plots (fig. S1; Materials and Methods). At each plot, we collected three composite soil samples (to 7.5 cm in depth) along with information on the dominant vegetation type (e.g., forest), vegetation structure in three 30-m transects (plant species richness, plant cover, and proportion of locations with ectomycorrhizal dominant plant species; table S2 and fig. S1) and management practices (irrigation, fertilization, and mowing). A total of 336 composite topsoil samples were analyzed for this study as detailed in Materials and Methods.
Fig. 1. The diversity and structure of the soil microbiome in urban greenspaces across the globe.
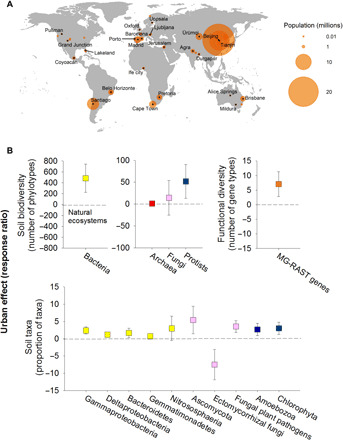
(A) The locations of the 56 surveyed cities included in this study along with their respective human populations. The names of some of the largest cities are included in this figure (see table S1 for details on all cities). (B) The changes [site-level response ratios, mean ± 95% confidence interval (CI)] in microbial diversity and the proportional abundances of significant taxa, comparing urban greenspaces to nearby natural ecosystems (n = 56 response ratios). Points above the dashed line indicate that the diversity or relative abundances of those taxa are higher in urban greenspaces compared to the corresponding natural areas near each city. Complementary figures showing results for other groups of organisms can be found in figs. S7 to S10. Phylotypes, ASVs; MG-RAST, metagenomics rapid annotation using subsystem technology.
To achieve our first aim, we characterized the biodiversity [number of amplicon sequence variants (ASVs); taxa that share 100% sequence similarity in the targeted gene region] and community composition (proportional abundances of ASVs, phyla/class, and functional groups) of bacteria, archaea, protists, and fungi in the soil samples from 56 paired cities and natural vegetated areas using ribosomal RNA (rRNA) gene amplicon sequencing (Materials and Methods). Diversity within each group was calculated at an equivalent sequencing depth across all samples using plot-level ASVs based on three soil composite samples per plot (Materials and Methods). We compared the diversity and proportion of soil organisms using two statistical approaches that explicitly considered our sampling using a blocked design (paired urban and natural ecosystems): (i) response ratios (i.e., changes in the values of microbial attributes from natural to cities; see Materials and Methods for correlations with other choices of response ratios) and (ii) nested permutational multivariate analysis of variance (PERMANOVA) comparing urban to natural ecosystems (56 paired locations; see Materials and Methods for details).
Our analyses show that urban greenspaces harbor soil microbiomes that are distinct from adjacent natural ecosystems (Figs. 1 to 8 and figs. S3 to S10). Urban greenspaces harbored communities of soil protists and bacteria that were, on average, 12 and 17%, respectively, more diverse than the adjacent natural ecosystems, with no significant differences in the richness of fungi and archaea (Fig. 1B and fig. S3). However, our analyses also revealed that, at the global scale, urban greenspaces tended to host more homogeneous microbial communities across cities than those found across natural ecosystems (Betadisper P < 0.001; Fig. 2, A and B, and figs. S4 to S6). Specifically, microbial communities were between 25% (protists) and 101% (fungi) more homogeneous in urban greenspaces than in nearby natural ecosystems (+39% for bacteria and +83% for archaea; Fig. 2B). In other words, our analyses show a greater similarity in the community composition of archaea, bacteria, fungi, and protists across the 56 globally distributed urban greenspaces than across the corresponding natural ecosystems (Fig. 2B). This result was maintained across contrasting geographic, climatic, and vegetation contexts (fig. S6). We further show that patterns in soil community similarity for different soil organisms (archaea, bacteria, fungi, and protists) are highly correlated across communities, both across cities and across natural ecosystems (Fig. 3). This finding suggests that the homogenization effects of urban greenspaces and the potential environmental drivers of these effects are shared across distinct groups of soil organisms. Human-induced land-use changes such as the conversion of forests to pasture have been reported to result in the biotic homogenization of soil microbial communities (12). Moreover, the importance of land-use intensification (e.g., livestock grazing) in generating cross-site multitrophic homogenization has been previously reported at larger spatial scales (13). Likewise, homogenization of bacterial communities has also been reported in dust samples collected from urban areas compared with more rural areas across the United States (14). Our results, emphasizing a global convergence (reduced geographic variation) in the soil microbiome of urban greenspaces, are consistent with the effects of urbanization on bird (15), plant, and invertebrate communities (16). Together, our findings provide novel evidence that urban greenspaces are important hot spots for the local (alpha) diversity of some microbial groups such as bacteria and protists worldwide. However, we also show that the geographic variation in microbial community composition is typically lower across cities worldwide than across natural ecosystems, suggesting a global homogenization in the soil microbiome of urban greenspaces.
Fig. 8. Selected relationships between socioeconomic, management, and environmental drivers and the proportional abundances or richness of selected functional genes across urban greenspaces (A to E) (n = 56 urban greenspaces).
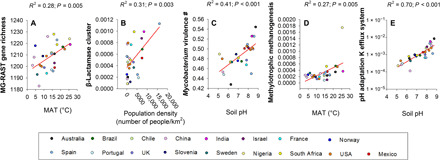
Fig. 2. Global homogenization in the soil microbiome of urban greenspaces.
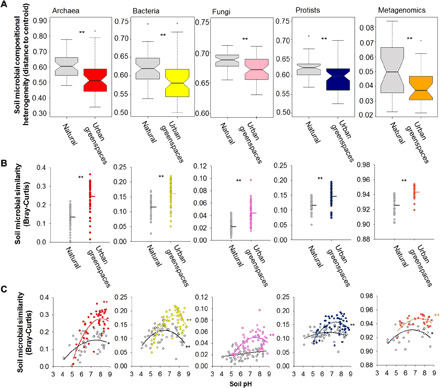
(A) The soil community composition heterogeneity of soil microorganisms in natural ecosystems and urban greenspaces. Boxes include median and 25th/75th percentile of the distances to the group centroid derived from betadisper (vegan R package). Asterisks indicate significant differences in compositional heterogeneity based on permutation test for homogeneity of multivariate dispersions (Materials and Methods). (B) Information on the within-group (urban or natural) community similarity for archaea, bacteria, fungi, protists, and metagenomics. Dots represent the average similarity (Bray-Curtis) in the soil community composition of each natural/urban site compared with the rest of natural/urban sites, respectively. In these panels, higher community similarity values indicate that the soils harbor communities that are more similar in composition, compared with the rest of the sites, across the 56 surveyed locations. The solid lines show mean values (n = 112 urban greenspaces and natural ecosystems). Asterisks indicate significant differences in nested PERMANOVA analyses using a block design as described in the Materials and Methods. (C) The relationship between soil pH and within-group (urban or natural) community similarity for archaea, bacteria, fungi, and protists. **P < 0.01; *P < 0.05.
Fig. 3. Relationships between the within-group (urban or natural) community similarity of archaea, bacteria, fungi, and protists.
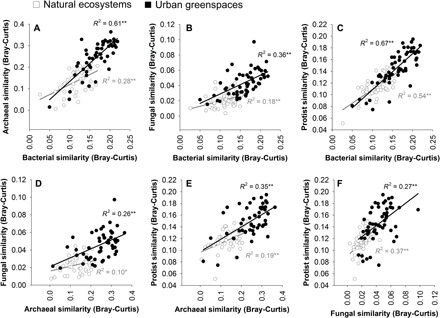
(A to F) Dots represent the average similarity in community composition of each natural/urban site compared with the rest of natural/urban sites, respectively. In these panels, higher community similarity values indicate that the soils harbor communities that are more similar in composition, compared with the rest of the sites, across the 56 surveyed locations (n = 56 urban greenspaces or natural ecosystems). **P < 0.01; *P < 0.05.
Soils from urban greenspaces harbored communities of archaea, bacteria, fungi, and protists distinct from those found in natural ecosystems, with urban greenspaces consistently supporting a significantly greater proportion of Gammaproteobacteria, Deltaproteobacteria, Bacteroidetes, Gemmatimonadetes, Ascomycota, Chlorophyta, and Amoebozoa (Fig. 1D and figs. S7 to S10). These phyla/classes include multiple fast-growing organisms (e.g., members from Gammaproteobacteria and Bacteroidetes) that could take advantage of the often fertilized and irrigated conditions found in urban ecosystems (Fig. 1D). The Amoebozoa group, for example, includes a wide variety of species that feed on bacteria and could more likely thrive in urban systems because of irrigation practices more often providing the water films needed to sustain their activities (Fig. 1D). Urban greenspaces also supported a higher proportion of Chlorophyta, important photosynthetic organisms, which often colonize bare soils in urban greenspaces (Fig. 1D) (17). Our results further indicate that the soils in urban greenspaces have a lower proportion of ectomycorrhizal and ericoid mycorrhizal fungi (but not arbuscular mycorrhizal fungi) compared with those from adjacent natural areas (Fig. 1D and figs. S7 to S10). These results are consistent in plots dominated by both ectomycorrhizal (EcM) and no-EcM plants (table S2 and fig. S11). The negative impact of urban environments on ectomycorrhizal fungi has been previously reported, at a local scale, in Southern Finland (6). Our findings further indicate that urban greenspaces support a higher proportion of fungal parasites and plant pathogens that are often economically important pests (Fig. 1D and figs. S8 and S10) (18).
Identifying the members of the soil microbiome that commonly reside in urban greenspaces is fundamental for the appropriate management of these habitats, as revealed by studies on macro-organisms (19). Here, we identified the indicative members of the soil microbiome of urban greenspaces across the globe (urban greenspaces taxa hereafter). We focused on those microbial phylotypes (ASVs) that (i) were relatively ubiquitous (>25% of all cities), (ii) were classified as “species indicators” of greenspaces using the algorithm of the “indicspecies” R package (see Materials and Methods), and (iii) showed statistically significantly higher proportions in urban rather than natural environments (see Materials and Methods for a more detailed description of these analyses). On the basis of these three criteria, we identified a total of 539 phylotypes (i.e., 488 bacteria, 47 protists, 2 archaea, and 2 fungi from a total of 142 genera) characterizing the soil microbiome of urban greenspaces (Fig. 4A and table S3). These patterns were consistent across geographical regions, climates, and vegetation types (Fig. 4B). The urban greenspace taxa included important fungal and oomycete plant pathogens such as Fusarium intricans, Pythium rostratifingens, and Pythium uncinulatum, fungal decomposers such as Mortierella elongata, archaeal nitrifiers such as Nitrososphaera sp., bacteria such as Streptomyces and Pseudomonas spp., and multiple species of bacteria-feeding amoebae (table S3 for sequences). Our results suggest that, similar to what has been found for birds (e.g., pigeons) and plants (e.g., roses), many comparable microbial species thrive in urban ecosystems across the globe.
Fig. 4. Microbial phylotypes comprising the urban greenspace soil microbiome.
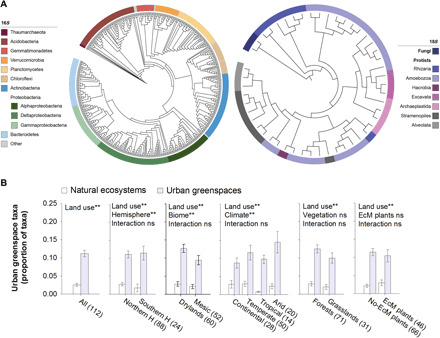
(A) Two phylogenetic trees showing the diversity of prokaryotic and eukaryotic organisms identified as being characteristic of urban greenspaces. See table S3 for the complete list of taxa and their representative sequences. (B) The proportion (mean ± SE) of the microbial phylotypes comprising the urban greenspace microbiome across continents, climates, and vegetation types (mean ± SE; n in brackets). ns, not significant; **P < 0.01. P values are based on nested PERMANOVA analyses using a block design as described in the Materials and Methods. EcM, ectomycorrhizal.
To improve the sustainable management of urban greenspaces, we need to understand how environmental and socioeconomic changes will affect important structural and functional attributes of the soil microbiome. Here, we identified the most important socioeconomic factors [in terms of per capita gross domestic product (GDP) and population density], vegetation structure (plant diversity, plant cover, and presence of ectomycorrhizal dominant plants), park management practices (irrigation, fertilization, and mowing), and environmental factors (climate, solar radiation, total plant cover, and soil properties) associated with the soil microbiome of urban greenspaces (n = 56 urban greenspaces). Socioeconomic and climatic factors such as GDP per capita and high temperatures could influence the soil microbiome of urban greenspaces by increasing environmental stress associated with disturbance, pollution, and climatic stress. As expected, we found that soil properties and climate significantly influenced the soil microbiome across cities. For instance, we found a well-established association between soil pH and bacterial richness (20), a positive correlation between the proportion of Cercozoa and mean annual precipitation (21), and a positive relationship between bacterial and protist richness (Figs. 5 and 6 and figs. S12 to S14) (22). Likewise, soil pH was also the most important factor associated with the changes in the soil community similarity across cities, which mostly supported curvilinear relationships (Fig. 2C). Plant diversity was negatively related to bacterial and archaeal community similarity, as well as to the proportion of urban greenspace taxa (Fig. 6).
Fig. 5. Socioeconomic, management, and environmental drivers of the soil microbiome in urban greenspaces.
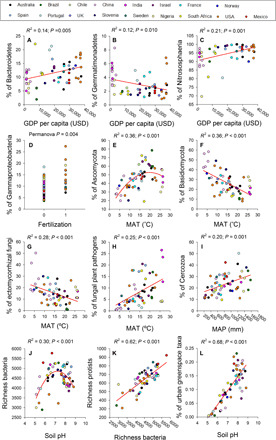
(A to L) Selected relationships between the proportional abundances or diversity of soil organisms and site characteristics across urban areas (n = 56 urban greenspaces). USD, U.S. dollars; MAT, mean annual temperature; MAP, mean annual precipitation.
Fig. 6. Spearman correlation analyses aiming to identify the most important socioeconomic factors, management practices, and environmental drivers of the taxonomic and functional properties of the soil microbiome of urban greenspaces (n = 56 urban greenspaces).
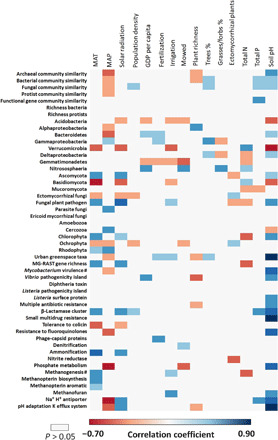
Statistically nonsignificant correlations (P > 0.05) are shown in white. Total P and N = Soil total P and N.
Our findings show that economic metrics, park management practices, and climate are important factors associated with the richness and community composition of the soil microbiome in urban greenspaces (figs. S12 and S13). Our results indicate that warmer and more irrigated cities supported a greater proportion of fungal plant pathogens (Figs. 5 and 6 and figs. S12 and S13) and revealed that warmer and more densely populated cities also had a lower proportion of symbiotic ectomycorrhizal fungi—a pattern that could also be influenced by a reduction in ectomycorrhizal hosts at lower latitudes (23). Warming and disturbance have been shown to regulate the distribution of fungal pathogens and mycorrhizal organisms in natural terrestrial environments worldwide (24). In addition, more affluent cities were characterized by soils with a greater proportion of Nitrososphaeria (often involved in nitrification) and potentially rapid-growing Gammaproteobacteria (e.g., Pseudomonas sp.) (see other examples in Fig. 5 and figs. S12 and S13). These patterns are likely the result of fertilizer applications to greenspace soils, given that similar taxonomic shifts have been observed in grasslands worldwide receiving elevated nutrient inputs (25). Thus, our findings suggest that changes in temperature, as well as differences in important socioeconomic and management factors, are largely associated with the soil microbiome of our greenspaces.
To gain a deeper understanding of the functional attributes of the urban greenspace soil microbiome, we obtained shotgun metagenomic data from a subset of 54 soil samples (one of the soil composite samples from a selection of 27 cities and their corresponding natural ecosystems). This selection covered a wide range of cities from contrasting climates and populations and 17 countries from both hemispheres (table S1). Consistent with our results for bacterial richness, soils from urban greenspaces typically had greater functional gene richness than adjacent natural terrestrial ecosystems (Fig. 1C and fig. S15). We found further evidence of global homogenization of the soil functional microbiome of urban greenspaces (Fig. 2), with functional gene profiles from urban greenspaces being more homogeneous across cities worldwide than the variability among natural ecosystems (Fig. 2, B and C, and fig. S15)—a pattern likely associated with the higher soil pH in urban greenspaces compared with natural ecosystems (Fig. 2 and figs. S12 and S14).
The microbial communities in urban greenspaces had distinct functional gene profiles (Figs. 1C and 7, A to C, and figs. S4 to S6 and S15). We further investigated well-known functional genes associated with pathogenesis, greenhouse gas emissions, nutrient cycling, and abiotic stress (Fig. 7) and found that urban greenspaces had a higher proportion of plant and human pathogens (26–29), greenhouse gas emission, and nitrogen and phosphorus cycle genes. For instance, soils from urban greenspaces had a higher proportion of genes associated with Mycobacterium virulence. This was especially important in the most alkaline soils [Figs. 6 to 8 and (15)] and is in agreement with our observation that one of the most abundant species in the soil microbiome of urban greenspaces is a member of the genus Mycobacterium (table S3). Although most soil mycobacteria are nonpathogenic (28), others are known to be important pathogens of humans and animals and can cause important respiratory infections (28, 29). Moreover, compared with natural ecosystems, urban greenspaces had a greater proportion of genes associated with Listeria and diphtheria toxins, Vibrio pathogenesis islands, and key antibiotic resistance genes (e.g., β-lactamases in Streptococcus, which includes penicillin) (Fig. 7A and fig. S15) (26, 27), which could all potentially influence human health. The proportion of genes coding for Vibrio pathogenesis islands was higher in more affluent cities and that of genes coding for β-lactamases in Streptococcus in warmer, more densely populated cities (Fig. 4C and fig. S12). We also found higher proportions of viral genes in urban greenspaces than in natural ecosystems, particularly in fertilized greenspaces (Fig. 7A and fig. S15). We note that gene annotations are approximate, and we do not know whether these particular genes detected may affect human health outcomes. Therefore, extrapolating and linking the occurrence of particular soil microbial genes to human health needs to be further investigated in the future to better understand how those microbes found in urban soils may affect human health.
Fig. 7. The functional attributes of the soil microbiome in urban greenspaces across the globe.
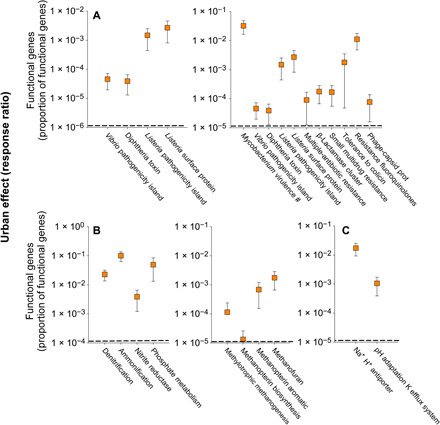
(A to C) The changes (site-level response ratios) in the proportion of selected functional genes associated with human diseases and antibiotic resistance (A), nutrient cycles (B), and abiotic stress (C) from natural to urban ecosystems (mean ± 95% CI; n = 56 response ratios). Points above the dashed line indicate a positive response ratio, with the selected gene category or soil property being relatively more abundant in urban greenspace soils compared with the corresponding “natural” sites.
Our results also indicate that soils from urban greenspaces included a higher proportion of bacterial genes associated with N and P cycling, likely associated with the fact that nitrogen and P are important fertilizers in urban greenspaces (from atmospheric deposition and direct fertilizer application). We also found a greater proportion of genes associated with archaeal methylotrophic methanogenesis (30) and denitrification processes, especially in irrigated and mowed greenspaces (Figs. 5 to 7B and fig. S15). This is important because it suggests that urban greenspaces could potentially be important sources of greenhouse gas emissions (methane and nitrous oxide) to the atmosphere (11). However, direct measurements of these soil processes are required to determine how these patterns in gene abundances might relate to actual process rates in urban greenspaces. Last, we found a higher proportion of gene copies associated with bacterial tolerance to alkaline or saline conditions (Figs. 5 to 7C and fig. S15), which are typical of many urban greenspaces (fig. S14). Together, these findings suggest that the soil microbiome of urban greenspaces supports a wide variety of potentially pathogenic organisms and microbes associated with important soil biogeochemical conditions and processes. The potential consequences of these relationships justify more detailed future investigations to better understand the functioning of urban soils and their contributions to environmental and human health.
In summary, we found that urban greenspaces are important hot spots of local soil microbial taxonomic and functional diversity but also support a global homogenization in the structure and function of the soil microbiome. More specifically, we show that urban soils across the globe harbor more similar microbiomes than would be expected from comparable analyses of soils from adjacent natural ecosystems. Our analyses indicate that soils from urban greenspaces are characterized by higher proportions of fast-growing bacteria, algae, nitrifiers, and important plant pathogens, which were particularly dominant in the warmer, more affluent, and more intensively managed greenspaces. Last, our results indicate that the urban greenspace microbiome harbors a greater proportion of genes associated with greenhouse gas emissions (denitrification and methanogenesis), as well as elevated proportions of genes associated with human pathogens and antibiotic resistance (e.g., β-lactamases), which may potentially have important implications for human health. Together, our study represents the first global assessment of the structure and functional attributes of the soil microbiome of urban greenspaces worldwide.
MATERIALS AND METHODS
Study sites
A total of 112 ecosystems across 17 countries and six continents (table S1 and Fig. 1A) were included in this study. Our survey targeted well-established urban parks and large residential gardens. We also targeted relatively undisturbed natural ecosystems including seminatural forests, grasslands, and shrublands close to cities or relict forests maintaining their original vegetation and embedded within urban spaces. These natural ecosystems were selected to represent the most common ecosystem type in each location in the absence of urbanization. Natural ecosystems were, on average, 22.8 ± 3.3 km apart from urban greenspaces. Mean annual precipitation and temperature in the selected cities ranged from 210 to 1577 mm and 3.1° to 26.4°C, respectively. In each location, we surveyed a 30 m by 30 m plot using three parallel transects of equal length, spaced 15 m down the part. We also collected information on the vegetation structure of each location based on the three 30-m transects. This information includes plant species richness, plant cover, and proportion of locations with ectomycorrhizal dominant plant species (table S2).
Soils were collected from 56 paired urban greenspaces and nearby natural ecosystems (Fig. 1A, table S1, and fig. S1) between 2017 and 2019. Samples were over the three 30-m transects (fig. S1). To account for spatial heterogeneity in our plots, three composite soil samples (from five soil cores, top 7.5-cm depth) were collected under the most common environments (vegetation and open areas between plant canopies covered by bare soils and nonvascular plants) found at each plot (fig. S1). A total of 336 composite soil samples were collected for this study. After field collection, each composite soil sample was divided into two subsamples; one subsample was immediately frozen at −20°C for molecular analyses, while the other subsample was air-dried for chemical analyses. Soil pH had a similar range for natural (4.1 to 8.6) and urban greenspaces (5.1 to 8.8), with urban greenspaces having slightly more alkaline soils (fig. S14).
Soil biodiversity sequencing
Amplicon sequencing
Soil DNA was extracted from each of the 336 composite soil samples using the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The diversity of soil archaea, bacteria, and protists was measured via amplicon sequencing using the Illumina MiSeq platform (Illumina Inc., CA, USA). DNA was shipped to the University of Colorado Boulder where all samples were processed using the same standardized protocol. A total of 336 soil composite samples (three composite samples per plot; fig. S1) were sequenced aiming to characterize the soil biodiversity of urban greenspaces. To characterize the richness (number of phylotypes) and community composition (proportion of phylotypes; relative abundance; percentage) of archaea, bacteria, and fungi, a portion of the prokaryotic 16S and eukaryotic 18S rRNA genes were sequenced using the 515F/806R and Euk1391f/EukBr primer sets (4, 22), respectively. Bioinformatic processing was performed using DADA2 (31). Phylotypes (i.e., ASVs) were identified at the 100% identity level. The ASV abundance tables were rarefied at 5000 (bacteria via 16S rRNA gene), 100 (archaea via 16S rRNA gene), and 1000 (protists via 18S rRNA gene) sequences per sample, respectively, to ensure even sampling depth within each belowground group of organisms. Protists are defined as all eukaryotic taxa, except fungi, invertebrates (Metazoa), and vascular plants (Streptophyta). The richness and community composition of fungi were determined via full-length 18S ITS (internal transcribed spacer) amplicon sequencing using the primers ITS9mun/ITS4ngsUni and the PacBio Sequel II platform in the University of Tartu (32). Bioinformatic processing was performed as explained above (ASVs at 100% similarity). The fungi ASV abundance table was rarefied at 1000 sequences per sample. See fig. S16 for rarefaction curves. Richness units are number of phylotypes (ASVs). Proportion of taxa units are percentages.
Rarefaction cross-validation
Before conducting further analyses, we ensured that our choice of rarefaction level, taken to maximize the number of samples in our study, was not obscuring our results. Thus, using the samples with the highest sequence per sample yield, we tested for the impact of different levels of rarefaction on soil biodiversity. We found highly statistically significant correlations between the diversities and community compositions of soil archaea (rarefied at 100 versus 500 sequences per sample), bacteria (rarefied at 5000 versus 12,500 sequences per sample), fungi (rarefied at 1000 versus 5000 sequences per sample), and protists (rarefied at 1000 versus 5000 sequences per sample), for a subset of samples wherein high numbers of sequences were available. These analyses provide evidence that our choice of rarefaction level did not affect our results or conclusions (fig. S17).
Plot-level estimations of soil biodiversity
Before data and statistical analyses, within-plot information on all soil (e.g., pH) and microbial variables (rarefied ASV tables), derived from three composite soil samples per plot, were averaged to obtain plot-level estimates (33). This allowed us to relate the spatial heterogeneity in our plots (fig. S1) to our metrics of soil biodiversity (number of ASVs) and community composition (proportion of taxa). This approach allowed us to obtain plot-level estimates of the proportion and number of phylotypes at the 112 studied sites (56 urban and 56 natural paired ecosystems) for bacteria, fungi, and protists based on 336 composite soil samples from five soil cores each. The only exception was archaea, for which we only obtained high-resolution information for 92 plots including 39 urban/natural paired ecosystems. Using these averaged plot-level ASV abundance tables, we calculated the richness (number of ASVs) of the most prevalent prokaryotic and eukaryotic organisms in our soil samples.
Microbial functional diversity
A composite sample per plot was sequenced for the entire metagenome in 27 paired urban/natural ecosystems (54 samples). More than 500 ng of DNA per soil sample was isolated for shotgun metagenomic (34, 35) sequencing using the DNeasy PowerSoil DNA Isolation Kit (Qiagen Inc., USA) according to the manufacturer’s protocol. Sequencing was performed using an Illumina HiSeq (Illumina Inc., USA) at Majorbio in Shanghai, China. Raw reads {PE150 [150–base pair (bp) paired-end reads]} were trimmed to remove low-quality reads as follows. First, the SeqPrep software (https://github.com/jstjohn/SeqPrep) was used to remove the adapter sequences. Second, the library sickle (https://github.com/najoshi/sickle) was used to trim the reads from the 5′ end to the 3′ end using a sliding window (size, 50 bp; 1-bp step). If the mean quality of bases inside a window dropped below 20, then the remainder of the read below the quality threshold was trimmed. Quality-trimmed reads that were shorter than 50 bp or containing N (ambiguous bases) were discarded.
The original sequences of the 54 samples were annotated using Subsystem Technology [MG-RAST (metagenomics rapid annotation using subsystem technology); www.mg-rast.org)] (36) to perform quality control and automated annotation as well as produce taxonomic and functional assignments. MG-RAST generates taxonomic assignments based on the SEED subsystem database by DIAMOND software (version 0.9.32) by best-hit classification with a maximum E value of 1 × 10−5, a minimum identity of 60%, and a minimum alignment length of 25 amino acids for proteins and functional categories. The resulting table was parsed at SEED subsystem level 3 by software SUPER-FOCUS. The proportion of 1276 groups of functional genes was determined from these analyses. This information was used to analyze patterns in the community composition and similarity of functional genes. Moreover, we investigated particular groups of genes that are known to be associated with pathogenesis, greenhouse gas emissions, nutrient cycling, and abiotic stress (Fig. 7). The richness of functional genes was determined from a rarefied gene table including 5.8 million annotated reads per sample.
Statistical analyses
Comparing the soil microbiome of urban greenspaces and natural ecosystems
We compared the richness (number of ASVs) and community composition (proportion of ASVs, phyla/classes, or functional genes and guilds) of soil organisms using two complementary statistical approaches aiming to explicitly consider our sampling using a block design (paired urban and natural ecosystems):
1) Response ratios. We calculated the changes in the values in the proportion and richness of soil microbial taxonomic and functional variables from natural to cities as follows: Change variable A = Value variable A urban − Value variable A natural. This approach yielded similar results to other response ratios such as percentage changes: (Value variable A urban − Value variable A natural) × 100/Value variable A natural and lnRR: ln (Value variable A urban + 1) − ln (Value variable A natural + 1). In these formulas, “variable A” represents any variable included in this study (e.g., richness of bacteria).Thus, on average, the response ratio used in the main analyses in this paper was highly correlated with that calculated from % changes (average r = 0.84; P < 0.001) and lnRR (average r = 0.99; P < 0.001) across all microbial variables included in this study. The response ratio used in our paper has the advantage that it is easy to interpret (changes in the proportion of taxa or richness) compared with other indexes such as lnRR that implies stronger data transformation (37).
2) Nested PERMANOVA analyses (38) using a block design (to account for our paired natural/urban ecosystem design) testing for differences in the values associated with the richness and proportions of microbial variables in urban greenspaces versus natural ecosystems. We used the function “adonis” in the R package “Vegan” (39) and the term “strata” (block) to conduct these analyses. This approach is expected to yield similar results to the response ratio approach and provided complementary statistical support to our study.
This study is designed to capture the global variability of soil microbiomes in natural and urban greenspaces. In this respect, we have 56 natural and urban replicates globally distributed. We would like to clarify that within-city replication would be considered pseudo-replication in this design. While within-plot replication is fundamental when comparing two factors at a local scale or in an experiment, including within-plot replicates could result in important pseudo-replication issues when evaluating the relationship between environmental factors (e.g., climate) and the diversity and proportions of taxa and functional genes across locations.
Soil community homogenization analyses
We tested for a homogenization effect of urban greenspaces cf. natural ecosystems on the soil microbiome (archaea, bacteria, fungi, protists, and metagenomics). To such an end, we used the betadisper R function (39) to calculate the dispersion within each group (urban greenspaces and natural ecosystems) based on a Bray-Curtis dissimilarity matrix. Subsequently, the permutetest function (39) was used to compare dispersions between urban greenspaces and natural ecosystems.
To further explore these results, we determined the average within-group (urban or natural) community similarity of each plot using a Bray-Curtis similarity matrix. Locations with higher community similarity values indicate that the soils harbor communities that are more similar in composition across natural or urban ecosystems. These analyses were done for archaea, bacteria, fungi, protists, and metagenomics. Significant differences in community similarity were further tested using nested PERMANOVA analyses with a block design (site) as described above.
Identifying the soil microbiome of urban greenspaces
To assess the soil microbiome associated with urban greenspaces (urban greenspaces taxa), we followed the three steps. First, using the information from the plot-level ASV abundance table, we kept those microbial phylotypes (bacterial, fungal, and protist ASVs) that were present in at least 25% of the studied cities (40). Second, we then used the function “multipatt” in the R package indicspecies (41) to identify those microbial phylotypes that were significant indicators of city greenspaces compared with natural ecosystems. In particular, this function studies the association between species patterns and combinations of groups of sites and identifies what species are most likely to be indicators of a given group of sites (here, urban greenspaces). Last, we conducted nested PERMANOVA tests, as explained above, to ensure that the proportion of the selected phylotypes was significantly higher in urban than natural ecosystems. We always used a P < 0.01 in these analyses to ensure that we retained only those microbial phylotypes strongly associated with the urban park-associated soil microbiome. We conducted these analyses independently for archaea, bacteria, fungi, and protists. The proportion of “urban greenspaces” taxa (i.e., urban cluster; Fig. 3 and table S3) was calculated as the average of all taxa selected as archaea, bacteria, fungi, or protist phylotypes that were consistently common in urban greenspaces. Before conducting these analyses, the proportion of microbial taxa was previously standardized (0 to 1) (42). We also calculated the difference in the proportion of selected urban greenspaces taxa from natural to urban pairs using the response ratio explained above. We did this calculation using the average standardized relative abundance of all urban greenspaces taxa. It should be noted that this calculation yielded the exact same result as when first obtaining the individual response ratios for the proportion of each urban phylotype and then averaging them out (r = 1.00; P < 0.001).
Environmental drivers of the soil microbiome of urban greenspaces
We first used random forest modeling to identify the major factors associated with the diversity, proportion, and community similarity of soil organisms in urban greenspaces (n = 56). The importance of each variable is calculated as the percentage of increase in the mean square error (MSE) from multiple regression trees. Put simply, when an important environmental factor is missing from the model, the MSE increases, indicating that this factor is an important predictor of a given response variable. We then used Spearman correlations to test for the link between soil biodiversity and the proportion of the urban park core microbiome with multiple socioeconomic and environmental factors in 56 urban greenspaces. We also used Spearman correlations to evaluate the socioeconomic and environmental drivers associated with functional microbial diversity and the proportion of functional genes. Spearman rank correlations are a nonparametric approach that does not require normality of data or homogeneity of variances and measures the strength and direction of the association between two ranked variables. In addition, unlike Pearson correlations, Spearman rank correlations can be used to associate two variables regardless of whether they are ordinal, interval, or ratio.
Soil radiation and climatic information were extracted from the WorldClim database (www.worldclim.org/data/index.html). Soil pH was measured with a pH meter, in a 1:2.5 mass:volume soil and water suspension. Total nitrogen (N) in the soil was analyzed using an elemental analyzer (C/N Flash EA 112 Series-Leco Truspec). Total soil P was determined, after nitric-perchloric acid digestion, using an inductively coupled plasma optical emission spectrometer (ICAP 6500 DUO; Thermo-Scientific, Waltham, MA, USA). Population density was determined using the latest available city censuses using official national statistical sources. We used population density rather than the total population because it is a more complete index of the human pressure on urban environments. Population density was positively correlated to total population (Spearman ρ = 0.33; P = 0.010). Moreover, we collected information about mowing, irrigation, and fertilization treatments in the urban greenspaces. GDP per capita, which provides information on the monetary value of final goods and services in the regions for the cities surveyed, was extracted from that dataset in (43), to provide information on the economic activity for each location. Plant structure data (plant diversity and cover) were determined in the three 30-m transects within each plot (fig. S1) (44). The ectomycorrhizal preferences of the dominant plant species in each plot were determined in the field using (table S2) (45).
Acknowledgments
We would like to thank C. Walsh and R. Ochoa-Hueso for advice on bioinformatics and statistical analyses. We also thank M. Martin for revising the English of the manuscript. In addition, we thank J. Owojori for connecting us with our sampling collaborator in Nigeria, A. R. Bamigboye. Funding: M.D.-B. and this project were supported by a 2019 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation (URBANFUN) and by the BES grant agreement no. LRB17\1019 (MUSGONET). M.D.-B. is also supported by a Ramón y Cajal grant from the Spanish Ministry of Science and Innovation (RYC2018-025483-I). N.F. was supported by grants from the U.S. National Science Foundation (DEB1556090 and DEB1542653). L.T. acknowledges support from Norway-Baltic collaboration grant EMP442 and Estonian Science Foundation grant PRG632. B.K.S. acknowledges a research award by the Humboldt Foundation and funding from the Australian Research Council (DP190103714). F.A. is supported by ANID FONDECYT 11180538 and 1170995. S.A. is funded by ANID FONDECYT 1170995 and ANID ANILLO ACT192027. F.B. and J.L.M. acknowledge support from the Spanish Ministry and FEDER funds for the project AGL2017-85755-R, the i-LINK+ 2018 (LINKA20069) from CSIC, as well as funds from “Fundación Séneca” from Murcia Province (19896/GERM/15). C.P. acknowledges support from the Spanish State Plan for Scientific and Technical Research and Innovation (2013–2016), award reference AGL201675762-R (AEI/FEDER, UE). M.B. acknowledges support from a Juan de la Cierva Formación grant from the Spanish Ministry of Economy and Competitiveness (FJCI-2018-036520-I). T.P.M. would like to acknowledge contributions from the National Research Foundation of South Africa and cities involved in the South African survey. Slovenian coauthors were supported by the research project J4-1766 “Methodology approaches in genome-based diversity and ecological plasticity study of truffles from their natural distribution areas” and the Research Program in Forest Biology, Ecology, and Technology (P4-0107) of the Slovenian Research Agency. J.D. and A. Rey acknowledge support from the FCT (IF/00950/2014 and SFRH/BDP/108913/2015, respectively), as well as from the MCTES, FSE, UE, and the CFE (UIDB/04004/2020) research unit financed by FCT/MCTES through national funds (PIDDAC). J.P.V. acknowledges financial support from SERB (Science and Engineering Research Board) (EEQ/2017/000775) India. J.-Z.H. and H.-W.H. are financially supported by Australian Research Council (DP170101628). The BBVA Foundation accepts no responsibility for the opinions, statements, and contents included in the project and/or the results thereof, which are entirely the responsibility of the authors. Author contributions: M.D.-B. developed the original idea of the analyses presented in the manuscript and coordinated all field and laboratory operations. Field data were collected by M.D.-B., D.J.E., Y.-R.L., A.R.B., J.L.B.-P., J.G.I., T.P.M., C.S., P.T., E.Z., J.P.V., L.W., J.W., T.G., M.B., G.F.P.-B., T.U.N., A.L.T., X.-Q.Z., J.D., A. Rodriguez,, X.Z., F.A., S.A., C.P., and A. Rey. Laboratory analyses were done by M.D.-B., N.F., H.-W.H., J.-Z.H., F.B., J.L.M., and L.T. Bioinformatic analyses were done by N.F., B.S., J.-T.W., B.K.S., and C.C.-D. Statistical analyses were done by M.D.-B. The manuscript was written by M.D.-B. and edited by N.F. and D.J.E., with contributions from all coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The data associated with this study can be found in the following: https://figshare.com/s/529c98609cd88e78e1a3; DOI: 10.6084/m9.figshare.12930986.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabg5809/DC1
REFERENCES AND NOTES
- 1.World Health Organization, Urban Green spaces: A Brief for Action (2017). [Google Scholar]
- 2.United Nations Department of Economic and Social Affairs Population Division, World Urbanization Prospects: The 2018 Revision (2018). [Google Scholar]
- 3.E. Gómez-Baggethun, Å. Gren, D. N. Barton, J. Langemeyer, T. McPhearson, P. O’Farrell, E. Andersson, Z. Hamstead, P. Kremer, Urban ecosystem services, in Urbanization, Biodiversity and Ecosystem Services: Challenges and Opportunities, T. Elmqvist, M. Fragkias, J. Goodness, B. Güneralp, P. J. Marcotullio, R. I. McDonald, S. Parnell, M. Schewenius, M. Sendstad, K. C. Seto, C. Wilkinson, Eds. (Springer, 2013). [Google Scholar]
- 4.Ramirez K. S., Leff J. W., Barberán A., Bates S. T., Betley J., Crowther T. W., Kelly E. F., Oldfield E. E., Shaw E. A., Steenbock C., Bradford M. A., Wall D. H., Fierer N., Biogeographic patterns in below-ground diversity in New York City’s Central Park are similar to those observed globally. Proc. R. Soc. B 281, 20141988 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charlop-Powers Z., Pregitzer C. C., Lemetre C., Ternei M. A., Maniko J., Hover B. M., Calle P. Y., McGuire K. L., Garbarino J., Forgione H. M., Charlop-Powers S., Brady S. F., Urban park soil microbiomes are a rich reservoir of natural product biosynthetic diversity. Proc. Natl. Acad. Sci. U.S.A. 113, 14811–14816 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hui N., Liu X., Kotze D. J., Jumpponen A., Francini G., Setälä H., Ectomycorrhizal fungal communities in urban parks are similar to those in natural forests but shaped by vegetation and park age. Appl. Environ. Microbiol. 83, e01797-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagg C., Bender S. F., Widmer F., van der Heijden M. G. A., Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. U.S.A. 111, 5266–5270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rook G. A., Brunetm L. R., Microbes, immunoregulation, and the gut. Gut 54, 317–320 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forsberg K. J., Reyes A., Wang B., Selleck E. M., Sommer M. O. A., Dantas G., The shared antibiotic resistome of soil bacteria and human pathogens. Science 337, 1107–1111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez J. L., Antibiotics and antibiotic resistance genes in natural environments. Science 321, 365–367 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Raciti S. M., Burgin A. J., Groffman P. M., Lewis D. N., Fahey T. J., Denitrification in suburban lawn soils. J. Environ. Qual. 40, 1932–1940 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Rodrigues J. L. M., Pellizari V. H., Mueller R., Baek K., Jesus E. C., Paula F. S., Mirza B., Hamaoui G. S., Tsai S. M., Feigl B., Tiedje J. M., Bohannan B. J. M., Nusslein K., Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 110, 988–993 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gossner M. M., Lewinsohn T. M., Kahl T., Grassein F., Boch S., Prati D., Birkhofer K., Renner S. C., Sikorski J., Wubet T., Arndt H., Baumgartner V., Blaser S., Blüthgen N., Börschig C., Buscot F., Diekötter T., Jorge L. R., Jung K., Keyel A. C., Klein A. M., Klemmer S., Krauss J., Lange M., Müller J., Overmann J., Pašalić E., Penone C., Perović D. J., Purschke O., Schall P., Socher S. A., Sonnemann I., Tschapka M., Tscharntke T., Türke M., Venter P. C., Weiner C. N., Werner M., Wolters V., Wurst S., Westphal C., Fischer M., Weisser W. W., Allan E., Land-use intensification causes multitrophic homogenization of grassland communities. Nature 540, 266–269 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Barberán A., Ladau J., Leff J. W., Pollard K. S., Menninger H. L., Dunn R. R., Fierer N., Continental-scale distributions of dust-associated bacteria and fungi. Proc. Natl. Acad. Sci. U.S.A. 112, 5756–5761 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murthy A. C., Fristoe T. S., Burger J. R., Homogenizing effects of cities on North American winter bird diversity. Ecosphere 7, e01216 (2016). [Google Scholar]
- 16.McKinney M. L., Effects of urbanization on species richness: A review of plants and animals. Urban Ecosyst. 11, 161–176 (2008). [Google Scholar]
- 17.B. A. Whitton, Diversity, ecology and taxonomy of the cyanobacteria, in Photosynthetic Prokaryotes, N. H. Mann, N. G. Carr, Eds. (Plenum Press, 1992). [Google Scholar]
- 18.Barford E., Crop pests advancing with global warming. Nature 10.1038/nature.2013.13644 , (2013). [Google Scholar]
- 19.Beninde J., Veith M., Hochkirch A., Biodiversity in cities needs space: A meta-analysis of factors determining intra-urban biodiversity variation. Ecol. Lett. 18, 581–592 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Fierer N., Jackson R. B., The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 103, 626–631 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliverio A. M., Geisen S., Delgado-Baquerizo M., Maestre F. T., Turner B. L., Fierer N., The global-scale distributions of soil protists and their contributions to belowground systems. Sci. Adv. 4, eaax8787 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delgado-Baquerizo M., Bardgett R. D., Vitousek P. M., Maestre F. T., Williams M. A., Eldridge D. J., Lambers H., Neuhauser S., Gallardo A., García-Velázquez L., Sala O. E., Abades S. R., Alfaro F. D., Berhe A. A., Bowker M. A., Currier C. M., Cutler N. A., Hart S. C., Hayes P. E., Hseu Z. Y., Kirchmair M., Peña-Ramírez V. M., Pérez C. A., Reed S. C., Santos F., Siebe C., Sullivan B. W., Weber-Grullon L., Fierer N., Changes in belowground biodiversity during ecosystem development. Proc. Natl. Acad. Sci. U.S.A. 116, 6891–6896 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steidinger B. S., Crowther T. W., Liang J., Van Nuland M. E., Werner G. D. A., Reich P. B., Nabuurs G. J., De-Miguel S., Zhou M., Picard N., Herault B., Zhao X., Zhang C., Routh D., Peay K. G.; GFBI Consortium , Climatic controls of decomposition drive the global biogeography of forest-tree symbioses. Nature 569, 404–408 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Delgado-Baquerizo M., Guerra C. A., Cano-Díaz C., Egidi E., Wang J. T., Eisenhauer N., Singh B. K., Maestre F. T., The proportion of soil-borne pathogens increases with warming at the global scale. Nat. Clim. Chang. 10, 550–554 (2020). [Google Scholar]
- 25.Leff J. W., Jones S. E., Prober S. M., Barberán A., Borer E. T., Firn J. L., Harpole W. S., Hobbie S. E., Hofmockel K. S., Knops J. M. H., McCulley R. L., la Pierre K., Risch A. C., Seabloom E. W., Schütz M., Steenbock C., Stevens C. J., Fierer N., Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc. Natl. Acad. Sci. U.S.A. 112, 10967–10972 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bahram M., Hildebrand F., Forslund S. K., Anderson J. L., Soudzilovskaia N. A., Bodegom P. M., Bengtsson-Palme J., Anslan S., Coelho L. P., Harend H., Huerta-Cepas J., Medema M. H., Maltz M. R., Mundra S., Olsson P. A., Pent M., Põlme S., Sunagawa S., Ryberg M., Tedersoo L., Bork P., Structure and function of the global topsoil microbiome. Nature 560, 233–237 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Bhatia R., Antimicrobial Resistance in developing Asian countries: Burgeoning challenge to global health security demanding innovative approaches. Glob. Biosecur. 1, 50–54 (2019). [Google Scholar]
- 28.Walsh C., Gebert M. J., Delgado-Baquerizo M., Maestre F. T., Fierer N., A global survey of mycobacterial diversity in soil. Appl. Environ. Microbiol. 85, e01180-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gebert M. J., Delgado-Baquerizo M., Oliverio A. M., Webster T. M., Nichols L. M., Honda J. R., Chan E. D., Adjemian J., Dunn R. R., Fierer N., Ecological analyses of mycobacteria in showerhead biofilms and their relevance to human health. mBio 9, e01614-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanwonterghem I., Evans P. N., Parks D. H., Jensen P. D., Woodcroft B. J., Hugenholtz P., Tyson G. W., Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 1, 16170 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P., DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 7, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tedersoo L., Anslan S., Bahram M., Drenkhan R., Pritsch K., Buegger F., Padari A., Hagh-Doust N., Mikryukov V., Gohar D., Amiri R., Hiiesalu I., Lutter R., Rosenvald R., Rähn E., Adamson K., Drenkhan T., Tullus H., Jürimaa K., Sibul I., Otsing E., Põlme S., Metslaid M., Loit K., Agan A., Puusepp R., Varik I., Kõljalg U., Abarenkov K., Regional-scale in-depth analysis of soil fungal diversity reveals strong pH and plant species effects in Northern Europe. Front. Microbiol. 11, 1953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maestre F. T., Delgado-Baquerizo M., Jeffries T. C., Eldridge D. J., Ochoa V., Gozalo B., Quero J. L., García-Gómez M., Gallardo A., Ulrich W., Bowker M. A., Arredondo T., Barraza-Zepeda C., Bran D., Florentino A., Gaitán J., Gutiérrez J. R., Huber-Sannwald E., Jankju M., Mau R. L., Miriti M., Naseri K., Ospina A., Stavi I., Wang D., Woods N. N., Yuan X., Zaady E., Singh B. K., Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. U.S.A. 112, 15684–15689 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fierer N., Leff J. W., Adams B. J., Nielsen U. N., Bates S. T., Lauber C. L., Owens S., Gilbert J. A., Wall D. H., Caporaso J. G., Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. U.S.A. 109, 21390–21395 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fierer F., Ladau J., Clemente J. C., Leff J. W., Owens S. M., Pollard K. S., Knight R., Gilbert J. A., McCulley R. L., Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342, 621–624 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Overbeek R., Olson R., Pusch G. D., Olsen G. J., Davis J. J., Disz T., Edwards R. A., Gerdes S., Parrello B., Shukla M., Vonstein V., Wattam A. R., Xia F., Stevens R., The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.J. Koricheva, J. Gurevitch, K. Mengersen, Handbook of Meta-analysis in Ecology and Evolution (Princeton Univ. Press, 2013). [Google Scholar]
- 38.Anderson M. J., A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001). [Google Scholar]
- 39.J. Oksanen, F. Guillaume Blanchet, M. Friendly, R. Kindt, P. Legendre, D. M. Glinn, P. R. Minchin, R. B. O’Hara, G. L. Simpson, P. Solymos, M. Henry H. Stevens, E. Szoecs, H. Wagner, vegan: Community Ecology Package, R package (2019).
- 40.Delgado-Baquerizo M., Reich P. B., Trivedi C., Eldridge D. J., Abades S., Alfaro F. D., Bastida F., Berhe A. A., Cutler N. A., Gallardo A., García-Velázquez L., Hart S. C., Hayes P. E., He J. Z., Hseu Z. Y., Hu H. W., Kirchmair M., Neuhauser S., Pérez C. A., Reed S. C., Santos F., Sullivan B. W., Trivedi P., Wang J. T., Weber-Grullon L., Williams M. A., Singh B. K., Multiple elements of soil biodiversity drive ecosystem functions across biomes. Nat. Ecol. Evol. 4, 210–220 (2020). [DOI] [PubMed] [Google Scholar]
- 41.M. deCaceres, F. Jansen, N. Dell, Indicspecies: Relationship between Species and Groups of Sites, R package (2020).
- 42.Wang L., Delgado-Baquerizo M., Wang D., Isbell F., Liu J., Feng C., Liu J., Zhong Z., Zhu H., Yuan X., Chang Q., Liu C., Diversifying livestock promotes multidiversity and multifunctionality in managed grasslands. Proc. Natl. Acad. Sci. U.S.A. 116, 6187–6192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kummu M., Taka M., Guillaume J. H. A., Gridded global datasets for Gross Domestic Product and Human Development Index over 1990-2015. Sci. Data 5, 180004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maestre F. T., Quero J. L., Gotelli N. J., Escudero A., Ochoa V., Delgado-Baquerizo M., Garcia-Gomez M., Bowker M. A., Soliveres S., Escolar C., Garcia-Palacios P., Berdugo M., Valencia E., Gozalo B., Gallardo A., Aguilera L., Arredondo T., Blones J., Boeken B., Bran D., Conceicao A. A., Cabrera O., Chaieb M., Derak M., Eldridge D. J., Espinosa C. I., Florentino A., Gaitan J., Gatica M. G., Ghiloufi W., Gomez-Gonzalez S., Gutierrez J. R., Hernandez R. M., Huang X., Huber-Sannwald E., Jankju M., Miriti M., Monerris J., Mau R. L., Morici E., Naseri K., Ospina A., Polo V., Prina A., Pucheta E., Ramirez-Collantes D. A., Romao R., Tighe M., Torres-Diaz C., Val J., Veiga J. P., Wang D., Zaady E., Plant species richness and ecosystem multifunctionality in global drylands. Science 335, 214–218 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soudzilovskaia N. A., Vaessen S., Barcelo M., He J., Rahimlou S., Abarenkov K., Brundrett M. C., Gomes S. I. F., Merckx V., Tedersoo L., FungalRoot: Global online database of plant mycorrhizal associations. New Phytol. 227, 955–966 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabg5809/DC1