

Fluid shear sensing via TRPM7 limits tumor cell intravasation by activating RhoA/myosin and calmodulin/IQGAP1/Cdc42 pathways.
Abstract
Tumor cell intravasation preferentially occurs in regions of low fluid shear because high shear is detrimental to tumor cells. Here, we describe a molecular mechanism by which cells avoid high shear during intravasation. The transition from migration to intravasation was modeled using a microfluidic device where cells migrating inside longitudinal tissue-like microchannels encounter an orthogonal channel in which fluid flow induces physiological shear stresses. This approach was complemented with intravital microscopy, patch-clamp, and signal transduction imaging techniques. Fluid shear–induced activation of the transient receptor potential melastatin 7 (TRPM7) channel promotes extracellular calcium influx, which then activates RhoA/myosin-II and calmodulin/IQGAP1/Cdc42 pathways to coordinate reversal of migration direction, thereby avoiding shear stress. Cells displaying higher shear sensitivity due to higher TRPM7 activity levels intravasate less efficiently and establish less invasive metastatic lesions. This study provides a mechanistic interpretation for the role of shear stress and its sensor, TRPM7, in tumor cell intravasation.
INTRODUCTION
Cell migration and intravasation play a critical role in the dissemination of cancerous cells from a primary tumor to secondary sites in the body. Cells in vivo migrate within three-dimensional (3D) extracellular matrices and through 3D longitudinal channel-like tracks occurring naturally in diverse anatomical features, such as between the connective tissue and the basement membrane of nerve or muscle or epithelium, in and along blood vessels, and in bone cavities, or generated by leader cancer cells or cancer-associated stromal cells (1, 2). These tissue microtracks (1) have widths varying from 3 to 30 μm and lengths up to 600 μm.
For a migrating cell to intravasate (i.e., to enter the circulation), it must cross the endothelial cell layer and resist the exposure to shear stress from the circulating fluid that is considered to be detrimental to the tumor cell (3). Although disruption of endothelial cell-cell junctions is considered to be a key mediator of intravasation (3), human tumor cells were found to intravasate in a zebrafish model only in locations where the vasculature was being remodeled but not at intact vessels (4). Along these lines, tumor-associated blood vessels are leaky because of the disruption of normal vascular function by paracrine signaling (such as secretion of tumor necrosis factor–α), proteolytic degradation, induction of remodeling/angiogenesis, and hypoxia (3). In mouse models of mammary carcinoma, tumor-associated leaky vasculature frequently displayed intercellular holes between endothelial cells ranging from 2 to 5 μm in diameter (5). In vitro studies also reveal that breast cancer cells crossing the endothelium create disruptions of ≥20 μm in width (6).
Shear stress, defined as the force per unit area (dyne/cm2) caused by the movement of adjacent layers of fluid with different velocities, is a physiologically relevant physical cue, which may affect the process of intravasation. Tumor cell intravasation typically occurs in the vicinity of the tumor where angiogenesis-induced capillary sprouts grow (3) and the shear stress is relatively low (0.2 to 6 dyne/cm2) (7). It is widely believed that intravasation preferentially occurs in regions of decreased fluid flow because low shear stress facilitates tumor cell survival in the circulation (3). We here provide a molecular interpretation based on the ability of normal (noncancerous) or tumor cells to sense and respond to shear stress during their transition from migration to intravasation. We identified transient receptor potential melastatin 7 (TRPM7) as the key fluid shear sensor and demonstrate that cells displaying higher sensitivity to fluid shear due to higher TRPM7 activity levels intravasate less efficiently in vitro and in vivo and establish less invasive metastatic lesions. We further elucidate the signaling pathway downstream of TRPM7 sensing.
RESULTS
Fibroblasts reverse migration direction upon sensing shear flow at their leading edge that results in calcium influx
Cells are subjected to fluid shear stress during their intravasation into the circulatory system. To determine how cells respond to fluid shear, we modeled the transition from migration to intravasation using a microfluidic device where cells moving inside longitudinal, collagen I–coated microchannels encounter an orthogonal 2D-like channel in which fluid flow is controlled to generate prescribed shear stress levels ranging from 0 dyne/cm2 (static conditions) to 0.5 and 5 dyne/cm2 (Fig. 1A). Primary human dermal fibroblast cells, GM05565 (Coriell Institute), spontaneously migrated from the 2D seeding region through microchannels (Fig. 1B). Eighty percent of these cells exited into the far 2D region in the absence of fluid flow (static conditions) (Fig. 1C and movie S1). In marked contrast, a substantial percentage (>40 to 60%) of primary dermal fibroblasts, which reached the microchannel end and sensed fluid flow (~0.5 dyne/cm2) in the apposing 2D-like area, reversed migration direction and remained in the microchannels (Fig. 1, B and C, and movie S1). This shear stress–induced direction change was independent of the microchannel width (Fig. 1C) and observed in channels as narrow as 6 or 3 μm. Because the dermal fibroblast cells infrequently entered these smallest microchannels due to their size, we focused on 10-μm-wide microchannels, which impose physical confinement and are similar in size to openings observed in the endothelial layer during disruption by tumor cells (5, 6). In addition to dermal fibroblasts, newborn human foreskin fibroblasts (NuFFs) and Chinese hamster ovary (CHO) cells were also sensitive to shear stress and reversed direction at the end of microchannels under fluid flow but not static conditions (Fig. 1, D and E). To decouple the effect of shear stress from shear rate on fibroblast reversal, we held the shear rate constant (62.5 s−1) but increased the shear stress from 0.5 to 5 dyne/cm2 by increasing the viscosity of the medium by 10-fold (fig. S1A). The extent of fibroblast cell reversal increased with increasing shear stress at a fixed shear rate (fig. S1B), suggesting that cells sense and respond to the applied force and hence shear stress.
Fig. 1. Fibroblasts reverse migration direction upon sensing fluid shear at their leading edge, which promotes rapid entry of extracellular calcium.
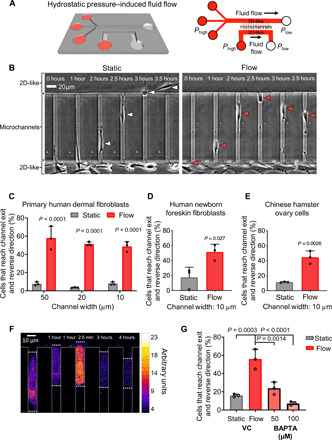
(A) Perspective view and schematic of the microfluidic device consisting of an array of parallel microchannels arranged in a ladder-like configuration and sandwiched orthogonally between two large 2D-like channels (W = 400 μm, H = 30 μm) where fluid flow is controlled. Cell migration was tracked in collagen I–coated microchannels of prescribed width (W = 10, 20, or 50 μm), height (H = 10 μm), and length (L = 200 μm). (B) In the absence of flow (static conditions), most of primary dermal fibroblasts migrate from the 2D-like seeding area into microchannels and exit to the other side into the apposing 2D-like area (white arrowheads). In contrast, 40 to 60% of these cells reverse migration direction when they reach the microchannel end and sense fluid flow in the apposing 2D-like area (red arrowheads). (C) Percentage of primary human dermal fibroblasts, (D) human newborn foreskin fibroblasts, or (E) CHO migrating cells that reach the end of the microchannels and then reverse their migration direction to remain in the microchannels under static or flow (0.5 dyne/cm2) conditions in the 2D-like regions. (F) Use of Fluo-4 Direct to visualize intracellular calcium in a migrating dermal fibroblast cell as it reaches the end of a microchannel where fluid flow is present. Scale bar, 10 μm. (G) Percentage of BAPTA- or vehicle control (VC)–treated cells that reverse migration direction in the presence of shear flow (0.5 dyne/cm2). Data represent the means ± SD from three independent experiments. Statistical comparisons were made between static and flow using two-way analysis of variance (ANOVA) followed by Sidak’s multiple comparisons test (C) or Student’s t test (D and E) or one-way ANOVA followed by Tukey’s post hoc test (G). See also fig. S1.
Calcium (Ca2+) signaling contributes to persistent cell migration, chemotaxis, and responses to physical cues (8, 9). To elucidate the role of Ca2+ signaling in shear-induced direction change, migrating fibroblasts were loaded with the calcium indicator dye Fluo-4 Direct (9). Cells that reached the microchannel end and protruded into the apposing 2D-like area where shear flow (0.5 dyne/cm2) prevailed exhibited a rapid and sharp increase in Fluo-4 Direct fluorescence, indicative of increased intracellular calcium concentration (Fig. 1F); these cells switched migration direction and subsequently restored their basal intracellular calcium levels (Fig. 1F). Entry of extracellular calcium into the cell is required for shear-mediated direction change, as chelating extracellular calcium with 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) caused migrating fibroblasts to exit microchannels in a dose-dependent manner (Fig. 1G). Together, these data suggest that while normal fibroblasts can migrate inside confining microtracks, they actively sense shear flow and avoid intravasation via a Ca2+-dependent pathway.
TRPM7-mediated calcium influx triggers the reversal of fibroblast migration direction upon fluid shear sensing
Because of the relatively low throughput of visualizing the intracellular Ca2+ levels during cell exit from the microchannels, we examined large cell populations in the 2D seeding area in the presence or absence of fluid shear. Cells loaded with Fluo-4 Direct were imaged for 1 min under static (no flow) conditions and then subjected to hydrostatic pressure–driven flow. Cell exposure to shear stress (0.5 dyne/cm2) caused a sharp spike in intracellular Ca2+ concentration (Fig. 2, A and B) similar to that observed when confined cells protruded into the apposing 2D-like area where fluid flow prevailed.
Fig. 2. TRPM7 is the key fluid shear sensor, which mediates the reversal of fibroblast migration direction.
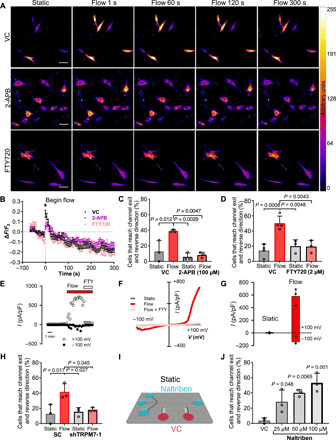
(A and B) Fluo-4 Direct fluorescence in vehicle control, 2-aminoethoxydiphenyl borate (2-APB)–, or fingolimod hydrochloride (FTY720)–treated dermal fibroblasts in a 2D-like channel imaged every 6 s for 1 min under static conditions followed by exposure to shear flow. Scale bars, 50 μm. (B) Quantification of Fluo-4 Direct fluorescence (ΔF/F0) from (A), where ΔF = F − F0, F = fluorescence at a given time point, and F0 = average fluorescence intensity under static conditions. Data represent means ± SEM for n > 25 cells pooled from three independent experiments. *P < 0.05, two-way ANOVA followed by Tukey’s post hoc test. Percentage of cells that reverse migration direction following treatment with (C) 2-APB (100 μM), (D) FTY720 (2 μM) or vehicle control. (E) Time course of whole-cell TRPM7-like cationic currents recorded at +100 and −100 mV in wild-type primary fibroblasts exposed to flow condition (10 μl/s) followed by exposure to 10 μM FTY720. (F) Current-voltage relationships of whole-cell cationic currents recorded under static, flow, and flow + FTY conditions. (G) Mean current densities measured under static and flow conditions in wild-type primary fibroblasts (n = 6). P = 0.02 paired t test between static versus flow. (H) Percentage of scramble control (SC) or TRPM7-knockdown cells that reverse migration direction under static or flow conditions. (I) Schematic of microfluidic device after replacement of the medium in the channel opposite to the seeding channel with medium containing naltriben or vehicle control. (J) Percentage of fibroblasts that reverse migration direction under static conditions at the end of the microchannels in response to naltriben or vehicle control as shown in (I). Data represent means ± SD from n ≥ three independent experiments. P values calculated one-way ANOVA followed by Tukey’s post hoc test unless otherwise stated in the legend. See also figs. S2 and S3.
Mechanosensitive ion channels can be activated by physical cues, including shear stress, to enable the uptake of extracellular Ca2+ into the cell (8). We tested the involvement of different mechanosensitive ion channels in the generation of shear stress–induced Ca2+ influx by using general and specific inhibitors. 2-Aminoethoxydiphenyl borate (2-APB) inhibits the influx of extracellular Ca2+ via blocking several TRP channels, including TRPC1, TRPC6, and TRPM7, and impedes store-operated Ca2+ release by inhibiting the inositol triphosphate receptor (9). 2-APB (100 μM) significantly dampened the Ca2+ response of primary dermal fibroblasts to shear stress in 2D (Fig. 2, A and B). TRPM7 has been implicated in the steering of cell migration in response to external hydraulic pressure (9). Inhibiting TRPM7 with fingolimod hydrochloride (FTY720) (9) also markedly reduced the shear-mediated increase in intracellular Ca2+ concentration (Fig. 2, A and B). Consistently, most of the migrating fibroblasts treated with 2-APB (Fig. 2C) or the TRPM7 inhibitor FTY720 (Fig. 2D) exited from the microchannels in the presence of shear stress into the apposing 2D-like area, suggesting that the mechanosensitive TRPM7 ion channel may serve as a shear stress sensor for these cells. The presence of TRPM7 channels in human dermal fibroblasts and their activation by shear stress were further confirmed by whole-cell patch-clamp recordings. Time course of shear stress–induced cationic current activation in a human dermal fibroblast and its inhibition by the use of the TRPM7 inhibitor FTY720 are shown in Fig. 2E. The corresponding current/voltage curves obtained at different times are shown in Fig. 2F. In the absence of perfusion (static conditions), fibroblasts exhibited low basal currents that increased upon opening of flow directed toward the cell attached to the recording pipette. Currents activated by shear stress presented the typical TRPM7 outward rectification and were blocked following FTY720 addition to the perfusion solution. Mean normalized currents obtained from human dermal fibroblasts at −100 mV and +100 mV under static and flow conditions are shown in Fig. 2G.
To assess the involvement of other potential shear stress sensitive channels, such as TRP vanilloid 4 (TRPV4) (10) and Piezo1 (11), in regulating the exit of fibroblasts under fluid flow conditions, we used a panel of selective pharmacological inhibitors. While the presence of TRPV4 has already been established for human dermal fibroblasts (12), the activity of Piezo channels in these cells, to the best of our knowledge, has not been reported. Therefore, we tested the presence of Piezo1 channels in human dermal fibroblasts recorded under cell-attached patch-clamp recording conditions (fig. S2A). Under this recording configuration, the cell membrane inside the pipette tip is stretched by negative suction pulses applied with a high-speed pressure clamp. Typical Piezo1-like currents were recorded (fig. S2A) that increased with increasing pressure pulses (fig. S2B). Inhibition of Piezo1/2 and TRPC1/C6 with GsMTx-4 (13, 14) or TRPV4 with GSK2193874 (15) failed to alter the migration pattern of fibroblasts in the presence or absence of flow in the apposing 2D-like channel (fig. S3, A and B). In marked contrast, the general TRP and mechanosensitive ion channel inhibitor ruthenium red enabled fibroblasts to exit into shear flow (fig. S3C), as also observed with 2-APB.
To confirm the ion channel(s) responsible for extracellular Ca2+ uptake in the presence of shear flow, and given that the 2-APB inhibitor targets the TRPC1, TRPC6, and TRPM7 channels, we designed short hairpin RNAs (shRNAs) to specifically knock down each of these channels. Knockdown was confirmed by quantitative polymerase chain reaction (qPCR) (fig. S3D) and verified by Western blot for TRPM7 (fig. S3E). TRPC1 or TRPC6 silencing failed to affect the shear-induced direction change of fibroblasts (fig. S3, F and G). On the other hand, TRPM7 knockdown, but not scramble control, resulted in cells exiting the microchannels at a similar rate in the absence or presence of shear flow, confirming that TRPM7 confers shear sensitivity in normal fibroblasts (Fig. 2H). To further establish whether TRPM7 activation alone is sufficient to trigger reversal of fibroblast migration, we filled the seeding channel and microchannels with control media and then added media containing the TRPM7 activator naltriben (16), to the 2D-like channel on the far side of the microchannels (Fig. 2I). Consequently, migrating cells that reach the end of the microchannels will experience TRPM7 activation under static conditions induced by the presence of naltriben. Naltriben caused fibroblasts to reverse their migration direction in a dose-dependent manner under static (no flow) conditions (Fig. 2J), thereby validating TRPM7 as a key upstream sensor of shear flow–mediated cell reversal.
Fluid shear activates myosin-II contractility, which is required for the reversal of fibroblast migration direction
To elucidate the downstream mechanism underlying the reversal of migration direction of fibroblasts upon sensing shear flow, we examined their actin cytoskeleton dynamics using Lifeact–green fluorescent protein (GFP)–expressing cells. Fibroblasts (>90%) migrated inside the microchannels with a mesenchymal (protrusion-based) morphology, which is indicative of an actin polymerization–driven migration mechanism (Fig. 3, A and B). About 50% of cells that reversed their migration direction after sensing shear flow switched from a protrusive to a bleb-based migration mode (Fig. 3, A and B), which was characterized by intense actin localization at the cell poles and the formation of sphere-like membrane bulges, termed blebs (17, 18). While most (>90%) of the fibroblasts that exited the microchannels into the apposing 2D-like channels under static conditions spread fully and exhibited a mesenchymal phenotype, only half of the exiting fibroblasts when flow was present were able to spread, and the rest remained round or adopted a bleb-based morphology (Fig. 3C). The bleb-based and rounded morphologies induced by shear flow in 2D and inside microchannels suggest increased cellular actomyosin contractility (17–21). Furthermore, physical cues, such as shear stress, can increase cellular contractility via mechanosensitive Ca2+ signaling (9, 20). Along these lines, fibroblasts in 2D that were exposed to shear stress for 5 min displayed increased phosphorylation (Ser19) of myosin light chain 2 (pMLC) compared with cells maintained under static conditions (Fig. 3, D and E). This shear-induced activation of myosin-II contractility was dependent on TRPM7, as treatment with FTY720 abolished the increase of pMLC (Fig. 3, D and E). The functional role of actomyosin contractility in the reversal of migration direction of fibroblasts upon sensing shear flow was demonstrated by the use of blebbistatin, which suppressed the shear-induced directional change of fibroblasts in microchannels (Fig. 3F).
Fig. 3. Fluid shear activates myosin-II contractility, which is required for reversal of fibroblast migration direction.
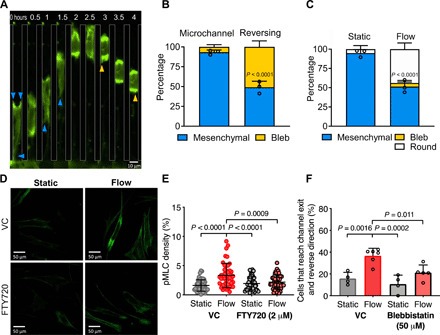
(A) Representative images of a migrating fibroblast expressing Lifeact-GFP imaged by epifluorescence at ×20 magnification. Shear flow is present perpendicular to the microchannel exits. Blue arrowheads mark cell protrusions. Yellow arrowheads mark cell blebs. (B) Migration phenotype of Lifeact-GFP–expressing fibroblasts in microchannels with flow present at the exits. Left bar, phenotype analysis of all cells migrating in the microchannel before reaching the end. Right bar, phenotype analysis of cells that have reached the end of the microchannel and reversed their migration direction. (C) Percentage of Lifeact-GFP–expressing fibroblasts exhibiting a protrusive, blebbing, or round morphology after exiting the microchannel into a 2D-like area under static (left) or flow (right) conditions. P value was calculated by two-way ANOVA followed by Sidak’s multiple comparisons test. Data represent means ± SD from three independent experiments. Data points represent percentage values of protrusive cells from individual experiments. (D) Representative confocal images of cells in 2D under static conditions or exposed to shear flow for 5 min and then fixed and immunostained for pMLC. (E) Quantification of pMLC fluorescence density. Data points represent values for individual cells pooled from three independent experiments. (F) Percentage of cells that reverse migration direction when treated with blebbistatin (50 μM) or a vehicle control. Data represent means ± SD from ≥3 independent experiments. Data points represent the percentage value from individual experiments. P values in (E) and (F) were calculated by one-way ANOVA followed by Tukey’s multiple comparisons test.
RhoA/myosin-II and IQGAP1-Cdc42 act in concert to mediate the reversal of fibroblast migration direction
The small guanosine triphosphatase (GTPase) protein RhoA is activated by diverse physical cues and has been extensively studied because of its regulation of cellular contractility and migration (18, 22). Using confocal fluorescence lifetime imaging microscopy (FLIM) coupled with a Förster resonance energy transfer (FRET)–based RhoA activity biosensor (17, 18), we demonstrate that shear stress activated RhoA, as evidenced by the decreased fluorescence lifetimes of fibroblasts subjected to shear flow relative to static conditions (Fig. 4, A and B). Consistent with RhoA activation by shear stress, inhibition of Rho-associated protein kinase (ROCK) by Y27632 abolished the shear-induced direction change of fibroblasts (Fig. 4C). To determine whether RhoA activation alone was sufficient to reverse the migration direction of the fibroblasts, we used the optoGEF-RhoA CIBN-CAAX system, which controls the subcellular activation of RhoA with high spatiotemporal accuracy by targeting the Rho Guanine Nucleotide Exchange Factor 11 (ARHGEF11) activator to the plasma membrane in response to blue light (23). Light stimulation of the leading edge of fibroblasts migrating inside microchannels caused rapid localization of ARHGEF11 at the cell anterior followed by a reversal of migration direction (Fig. 4, D to F). In comparison, stimulation of RhoA activity at the trailing edge of migrating cells maintained forward migration (Fig. 4F). To further establish that RhoA activation at the leading edge specifically played a key role in reversing cell migration direction, we used the optoGEF-RhoA CIBN-Mito system to locally deactivate RhoA by targeting ARHGEF11 to the mitochondria (23). Down-regulation of RhoA activity at either the cell anterior or posterior did not alter cell migration direction (fig. S4, A and B) indicating that while up-regulation of RhoA activity at the leading edge was sufficient to reverse cell migration direction, its activity was dispensable for maintaining migration direction over short periods of time (~30 min). Together, these results suggest that RhoA is activated at the leading edge of migrating cells when they encounter shear stress at the end of microchannels, causing them to reverse their migration direction. Because RhoA activation mediates fluid shear–induced cell reversal (Fig. 4, A to C) and given the negative cross-talk between RhoA and Rac1 (24, 25), we predicted that Rac1 inhibition would exacerbate the extent of shear-induced cell reversal and confirmed it via the use of the selective inhibitor NSC 23766 (fig. S4C).
Fig. 4. RhoA and Cdc42 act in concert to mediate the reversal of migration direction.
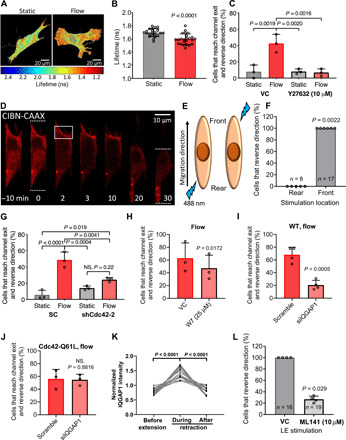
(A) Representative pixelwise heatmaps of RhoA FRET fluorescence lifetimes (ns) in fibroblasts under static or flow conditions. (B) Quantification of FLIM-FRET lifetimes. Data points represent the average fluorescent lifetime over a whole cell, pooled from three independent experiments. (C) Percentage of cells that reverse migration direction following treatment with Y27632 (10 μM) or vehicle control. (D) Fibroblasts expressing OptoGEF-RhoA and CAAX-CIBN-GFP. Dotted lines indicate the cell’s leading and trailing edges during confined migration at t = 0 and 30 min. For 2 min, the cell moves upward and is then stimulated with blue light in the region enclosed by the box. OptoGEF-RhoA enrichment is observed at the plasma membrane in this region, and the cell reverses its migration direction. (E) OptoGEF-RhoA–expressing fibroblasts migrating in microchannels were stimulated with blue light at the front or rear. (F) Percentage of cells that reversed their migration direction after optogenetic stimulation at the front or rear. Data points represent percentage of cells from an individual experiment. n = number of cells assayed. Percentage of (G) scramble control or Cdc42-KD cells, (H) vehicle control or W7-treated cells, (I) scramble control or siIQGAP1 cells, and (J) and scramble control or siIQGAP1 cells transduced with the constitutively active Cdc42-Q61L that reverse migration direction. NS, not significant; WT, wild type. (K) GFP-IQGAP1 intensity at cell edge during extension-retraction events (n = 18 cells from five independent experiments). (L) Percentage of OptoGEF-RhoA–expressing fibroblasts treated with ML141 (10 μM) or vehicle control that reverse their migration direction after stimulation with blue light at the leading edge (LE). Data represent means ± SD from three independent experiments (C and G to J). Statistical comparisons were made using Student’s t test (B and H to J) or one-way (C and G) or two-way ANOVA (K) followed by Tukey’s post hoc test and the Mann-Whitney U test (L and F). See also figs. S4 and S5.
Persistent cell migration is characterized by distinct protein activity at the cell leading and trailing edges, and disruption of cell front/rear polarity alters the migration direction (8, 22). We hypothesized that when migrating cells reached the end of the microchannel and experienced force from shear flow, actomyosin contractility was activated at the leading edge of the cell, thereby disrupting the front/rear polarity. Fibroblasts were then forced to reestablish polarity, leading most of the cells to reverse their migration direction in accordance with the locally activated myosin-II. Notably, some fibroblasts recovered their initial polarity and managed to exit the microchannel into the apposing 2D space. Cdc42 is a major cell polarity protein normally active at the leading edge of migrating cells (22), which may be involved in the reestablishment of front/rear polarity and reversal of migration direction of fibroblasts after experiencing shear stress. Inhibition of Cdc42 with the drug ML141 (fig. S5A) or by knockdown with either of two shRNA sequences (Fig. 4G and fig. S5, B and C) markedly suppressed the percentage of fibroblasts that reversed migration direction upon encountering shear flow. This finding suggests that reestablishment of cell polarity signaling downstream of Cdc42 was required for fibroblasts to respond to the shear stimulus. We wished to further evaluate how the two Rho family GTPases, RhoA and Cdc42, work together to facilitate cell reversal. The IQGAP scaffolding protein family has been shown to facilitate cross-talk across diverse regulatory pathways. While expression of IQGAP2 is primarily limited to liver and IQGAP3 is limited to brain, testis, lung, and spleen, IQGAP1 is ubiquitously expressed. We verified the higher abundance of IQGAP1 in comparison to IQGAP2 or IQGAP3 in fibroblasts using qPCR (fig. S5D). IQGAP1 has been implicated in Ca2+-dependent Cdc42 signaling via calmodulin (26). Prior studies also revealed the enrichment of IQGAP1 at retracting edges together with calmodulin (27). We thus hypothesized that the elevated intracellular Ca2+ in response to the shear stress leads to Cdc42-mediated cellular repolarization via the action of calmodulin and IQGAP1. Inhibition of calmodulin activity via W7 led to a reduction in the percentage of fibroblasts that reversed their direction in response to shear flow (Fig. 4H). Moreover, IQGAP1 knockdown in wild-type fibroblasts via small interfering RNA (siRNA; fig. S5E) caused a marked decrease of cell reversal when cells experienced shear flow (Fig. 4I). Intriguingly, expression of the constitutively active Cdc42-Q61L (28) rescued the reversal phenotype of IQGAP1-knockdown cells to levels of scramble controls (Fig. 4J), thereby placing Cdc42 downstream of IQGAP1. Time-lapse imaging of GFP-IQGAP1–transfected cells on 2D revealed an increase in IQGAP1 accumulation at the retracting edges of fibroblasts (Fig. 4K and fig. S5F).
While inhibition of RhoA/myosin-II–mediated contractility and Cdc42 independently reduce cell reversal (Figs. 3F and 4, C and G), we wished to determine whether activity of both RhoA and Cdc42 are essential for successful cell reversal. To this end, we inhibited Cdc42 using ML141 in optoGEF-RhoA CIBN-CAAX–expressing cells and then stimulated the leading edge of migrating fibroblasts with blue light. Cdc42 inhibition markedly reduced the percentage of fibroblasts that reversed their migration direction after RhoA activation (Fig. 4L), revealing that both RhoA and Cdc42 activities are required for cell reversal. Cumulatively, these data illustrate that while up-regulation of RhoA activity is sufficient to mediate shear stress–responsive fibroblast reversal, a functional IQGAP1-Cdc42 signaling pathway is critical for such reversal, and acts in concert with RhoA/myosin-II.
Fibrosarcoma cells have markedly reduced TRPM7 activity and capacity to respond to fluid shear
During cancer metastasis, migrating tumor cells encounter diverse physical stimuli stemming from the architecture of the extracellular matrix, local tissue stiffness and compression, and shear stresses in the circulatory system. On the basis of fibroblasts’ aversion to exiting microchannels into shear flow, we hypothesized that cancer cells arising from this cell type, such as fibrosarcoma, must evade this mechanism to successfully intravasate into the tumor-associated circulatory system, where shear stress would otherwise stimulate Ca2+ influx, thereby triggering the reversal of cell migration direction and preventing their entry into the circulation. In contrast to normal fibroblasts, migrating HT-1080 fibrosarcoma cells consistently exited the microchannels under both static conditions and into shear stress regimes similar to those of tumor microvasculature (0.5 dyne/cm2) (Fig. 5A). Increasing the shear stress level to 5 dyne/cm2, by increasing the shear rate from 62.5 to 625 s−1, caused ~50% of HT-1080 fibrosarcoma cells to reverse their migration direction at the end of the microchannels, which corresponds roughly to the proportion of fibroblasts observed at 0.5 dyne/cm2. These data suggest that fibrosarcoma cells have a 10-fold higher shear stress threshold for avoiding intravasation. Along these lines, when migrating fibroblasts encountered a shear stress of 5 dyne/cm2, an even higher proportion reversed their migration direction compared with that at 0.5 dyne/cm2 (Fig. 5B).
Fig. 5. HT-1080 fibrosarcoma cells display reduced TRPM7 activity, fluid shear sensitivity, and RhoA activity.
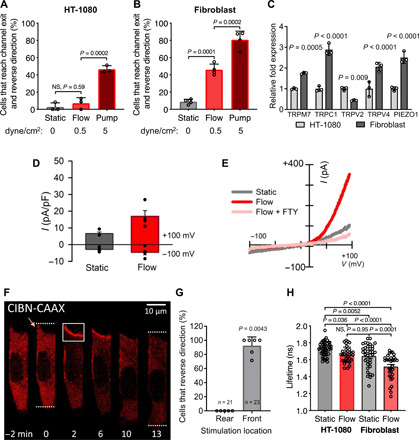
(A) Percentage of HT-1080 fibrosarcoma or (B) primary dermal fibroblasts that reverse migration direction at the end of the microchannels under static or prescribed shear flow conditions. (C) Relative mRNA expression of HT-1080 and fibroblast cells assessed by qPCR. Data represent means ± SD from ≥3 independent experiments (A to C). (D) Current-voltage relationships of whole-cell cationic currents recorded from HT-1080 fibrosarcoma cells under static and flow conditions. (E) Mean current densities measured under static, flow, and flow + FTY conditions in human wild-type primary fibroblasts (n = 4). P = 0.03 paired t test static versus flow. (F) HT-1080 cells expressing OptoGEF-RhoA and CAAX-CIBN-GFP. Dotted lines indicate the cell’s leading and trailing edges during confined migration at t = 0 or 13 min. Yellow arrow indicates a leading edge protrusion at t = 0. For 2 min, the cell moves upward and is then stimulated with blue light in the region indicated by the white box. OptoGEF-RhoA enrichment is observed at the plasma membrane in this region, and the cell reverses its migration direction. (G) Percentage of HT-1080 cells that reversed their migration direction after optogenetic stimulation at the front or rear. Data points represent percentage of cells from an individual experiment. n = number of cells assayed. (H) Comparison of RhoA FLIM for HT-1080 and fibroblasts in 2D under static or flow conditions. HT-1080 cells have reduced RhoA activity under static conditions compared with fibroblasts. Flow increases HT-1080 RhoA activity to similar levels to fibroblasts under static conditions but not to the level of fibroblasts in flow. Statistical comparisons were made using one-way ANOVA followed by Tukey’s post hoc test (A to C and H) or the Mann-Whitney U test (G).
In light of the role of TRPM7 in the reversal of fibroblast migration direction, we hypothesized that the reduced sensitivity of HT-1080 fibrosarcoma cells to fluid shear may be due to altered expression and/or activity of this mechanosensitive ion channel. qPCR revealed that the HT-1080 cells have significantly lower expression levels of TRPM7, TRPC1, TRPV4, and Piezo1 but elevated expression of TRPV2 compared with normal fibroblasts (Fig. 5C). As TRPM7 is required for shear-induced direction change in fibroblasts, it is likely that its down-regulation is responsible for the HT-1080 cells’ ability to exit the microchannels into shear flow. Compared to fibroblasts, HT-1080 cells exhibited reduced TRPM7 current in the presence of shear flow, as evidenced by whole-cell patch-clamp recordings (Fig. 5, D and E). Current-voltage relationships of HT-1080 cells show low basal cationic currents that were activated by fluid perfusion under vehicle control conditions but not in the presence of a TRPM7 inhibitor (Fig. 5, D and E).
As RhoA activation at the leading edge was sufficient to induce fibroblast reversal, we used the optoGEF-RhoA CIBN-CAAX system to test whether this pathway could also trigger the same response in migrating HT-1080 cells. Leading edge stimulation with blue light caused ARHGEF11 localization to the HT-1080 cell anterior followed by a switch in migration direction (Fig. 5, F and G). On the other hand, RhoA activation at the cell rear failed to alter the migration direction of HT-1080 cells (Fig. 5G). Because RhoA activation at the leading edge was sufficient to cause migrating HT-1080 cells and fibroblasts to reverse direction, we questioned whether these cells exhibit altered RhoA activation in response to shear stress, which may also explain the difference in shear-induced cell reversal between these two populations. Using FLIM-FRET, we measured the relative levels of RhoA activity in both cell types under static and flow (0.5 dyne/cm2) conditions. Shear stress up-regulated RhoA activity in HT-1080 cells albeit to markedly lower levels than fibroblasts (Fig. 5H). Normal fibroblasts relative to HT-1080 cells also displayed significantly elevated RhoA activity levels under static (no flow) conditions, which were comparable to those of shear-activated HT-1080 cells (Fig. 5H). Together, HT-1080 cell migration direction can be controlled by RhoA activation; while shear stress activates RhoA in these cells, their baseline RhoA activity is considerably lower than that of fibroblasts, which ultimately contributes to the reduced shear sensitivity of HT-1080 cells.
HT-1080 fibrosarcoma cells gain acute shear sensitivity upon overexpressing TRPM7 and display reduced invasion out of the primary tumor and intravasation
To test whether the lower TRPM7 expression levels displayed by HT-1080 cells relative to fibroblasts underlie their reduced sensitivity to shear stress, we examined the impact of TRPM7–yellow fluorescent protein (YFP) overexpression in HT-1080 fibrosarcoma cell function in vitro and in vivo. HT-1080 cells overexpressing TRPM7-YFP presented higher basal (static condition) and flow-activated currents sensitive to FTY720 than mock (YFP) transfected cells (Fig. 6A). Moreover, a markedly increased percentage of TRPM7-YFP–overexpressing cells as compared with their YFP-tagged controls reversed migration direction upon experiencing shear flow in the presence of 50 μM naltriben (Fig. 6B). In the absence of naltriben, cells also displayed enhanced shear sensitivity although they failed to reverse back into the channels (fig. S6A).
Fig. 6. HT-1080 fibrosarcoma cells gain acute shear sensitivity upon overexpressing TRPM7 and display reduced invasion out of the primary tumor and intravasation.
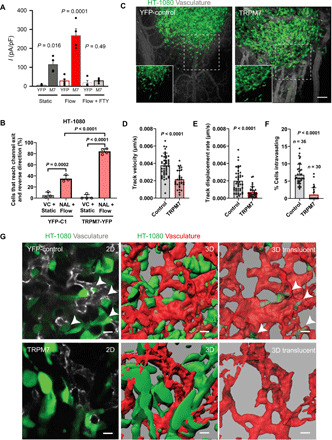
(A) Mean current densities measured under static and flow conditions in the presence or absence of FTY in HT-1080 cells transfected with YFP or mouse TRPM7-YFP. (B) Percentage of HT-1080 cells transfected with either TRPM7-YFP or YFP-C1 that reverse migration direction under static or shear flow conditions. Cells experiencing shear flow were treated with 50 μM naltriben (NAL) while those under static conditions with vehicle control. Data represent means ± SD from ≥3 independent experiments with >40 total cells analyzed per condition. Statistical comparison was performed using one-way ANOVA followed by Tukey’s post hoc test. (C) Representative images showing primary tumors formed by control (YFP-C1) or TRPM7-overexpressing (TRPM7-YFP) HT-1080 cells. Images are maximum intensity projections of 3D reconstructions. Insets show YFP signal alone from the areas marked by dashed squares. Quantification of average cancer cell (D) track velocity and (E) displacement rate at the invasive fronts of tumors formed by control or TRPM7-overexpressing HT-1080 cells. (F) Quantification of cancer cell intravasation rate for tumors formed by control or TRPM7-overexpressing HT-1080 cells. (G) Representative images from the primary tumors formed by control or TRPM7-overexpressing HT-1080 cells showing single optical sections (left), 3D reconstructions (middle), and 3D translucent rendering of vasculature with intravasated cancer cells inside the vascular lumen (right). White arrowheads point to intravasating HT-1080 cells at vascular wall breaches. In (D) to (F), data are means + SD with statistical comparison made using unpaired t test (D and E) or Mann-Whitney test (F). Scale bars, 100 μm (C) or 20 μm (G). See also fig. S6.
To generalize our data showing a key role for TRPM7 in shear stress sensing and reversal of migration direction, we examined the human MDA-MB-231 adenocarcinoma cells. Akin to HT-1080 fibrosarcoma cells, MDA-MB-231 cells displayed a similar trend of increased reversal in response to increasing shear stress levels, which was abolished by TRPM7 knockout (fig. S6B).
To test the role of TRPM7 in the context of intravasation and formation of metastatic lesions in vivo, we used the chicken chorioallantoic membrane (CAM) ex ovo model of human cancer metastasis (29, 30). In this unique model, fluorescently labeled human HT-1080 cells were implanted into the CAM tissue or injected directly into the CAM vasculature, thereby allowing for real-time high-resolution imaging of cancer cell intravasation and invasion at the tumor front or of metastatic lesions, respectively (29, 30). YFP-labeled control or TRPM7-YFP–overexpressing HT-1080 cells readily formed tumors when implanted into the CAM tissue (Fig. 6C). Notably, time-lapse analysis of cancer cell invasion at the tumor fronts reveals that YFP-control HT-1080 cells rapidly invaded out the main tumor mass maintaining a high degree of directionality, while TRPM7-YFP–overexpressing HT-1080 cells were significantly slower and less directional as manifested by their decreased cell track velocity and displacement rate, respectively (Fig. 6, D and E, and movie S2). Cancer cell intravasation requires directional and persistent formation of invadopodia that is necessary for vascular wall breaching (31). We hypothesized that because TRPM7 overexpression inhibited directional cancer cell invasion and mediates aversion to environments in which fluid flow generates shear stress, it will block cancer cell intravasation as well. High-magnification imaging analysis of tumors formed by control and TRPM7-overexpressing cells shows that while control cells readily interacted with tumor vasculature displaying multiple cases of vascular wall breaching, TRPM7-overexpressing cells failed to penetrate into blood vessels (Fig. 6, F and G).
The ability to form overt, invasive metastatic lesions is a bottleneck step of metastatic cascade. We thus investigated whether targeting TRPM7 function inhibits this step of metastasis as well. To this end, we intravascularly injected YFP-control or TRPM7-YFP–overexpressing HT-1080 cells and imaged metastatic lesions 5 days after injection. Control cells were found to rapidly invade at metastatic sites displaying the typical “scattered” invasive phenotype. In contrast, TRPM7-YFP–overexpressing cells formed less invasive metastatic lesions where smaller, rounded HT-1080 cells were clustering together within the CAM tissue (Fig. 7, A to C). Intravital time-lapse analysis of control and TRPM7-overexpressing metastatic lesions reveals that similar to what was observed at the invasive tumor fronts, TRPM7 overexpression led to the decreased cancer cell track velocity (Fig. 7D and movie S3). Together, our data show that TRPM7 overexpression inhibits HT-1080 cancer cell invasion both at the levels of tumor front expansion and intravasation and metastatic lesion formation.
Fig. 7. TRPM7 overexpression in HT-1080 fibrosarcoma cells inhibits invasive metastatic lesion formation.
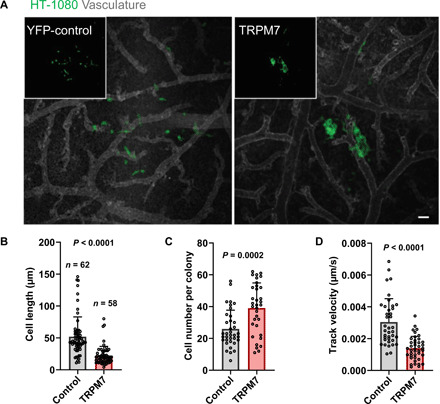
(A) Representative images showing metastatic lesions formed by control (YFP-C1) or TRPM7-overexpressing (TRPM7-YFP) HT-1080 cells. Images are maximum intensity projections of 3D reconstructions. Insets show YFP signal alone. Quantification of average (B) cancer cell length, (C) cancer cell number, and (D) cancer cell track velocity within the metastatic lesions formed by control or TRPM7-overexpressing HT-1080 cells. Data are means + SD with statistical comparison made using unpaired t test. Scale bar, 50 μm.
DISCUSSION
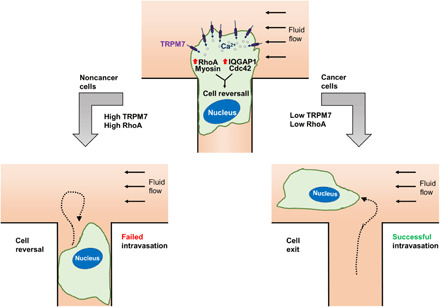
Shear stress is a physiologically relevant physical cue that regulates diverse processes in health and disease, including tumor cell intravasation. It is believed that intravasation typically occurs in regions of low fluid shear because high shear promotes tumor cell killing (3). We here provide a molecular interpretation based on the fluid shear sensors present in different fibroblast and fibroblast-derived cancer cells. Cancer cells displaying lower sensitivity to shear stress intravasate efficiently relative to normal controls. We found TRPM7 to be the key shear stress sensor. The enhanced sensitivity to shear stress of normal fibroblasts relative to HT-1080 fibrosarcoma cells is attributed to their higher TRPM7 expression and activity levels. We further propose a model (Fig. 8) where shear stress activates TRPM7 through deformation of the plasma membrane and triggers extracellular Ca2+ influx, which up-regulates RhoA/myosin-II contractility and concurrently disrupts cell front-rear polarity via a calmodulin/IQGAP1/Cdc42 pathway, thereby ultimately mediating the reversal of migration direction.
Fig. 8. Schematic summarizing the cascade of signaling events following cell exposure to fluid shear.
While sophisticated in vitro models take into account the interactions between tumor cells and endothelial cells during the process of intravasation (32, 33), a simpler, yet relevant, model system is needed to delineate how cells sense and respond to fluid shear during intravasation in the absence of any confounding factors that arise from the introduction of endothelial cells. This is further corroborated by studies showing that tumor cells intravasate in locations where the vasculature was being remodeled but not at intact vessels (4). Tumor-associated leaky vasculature (34) frequently displayed intercellular holes between endothelial cells ranging from 2 to 5 μm in diameter (5), whereas breast cancer cells crossing the endothelium in vitro create disruptions of ≥20 μm in width (6). These openings observed in the endothelial layer in vitro and in vivo are similar in size to those in our microfluidic device.
Cells sense and respond to different types of mechanical cues, such as fluid shear, hydraulic pressure, and compression, through various mechanisms, including mechanosensitive ion channels, integrins, the glycocalyx, and the nucleus (9, 18, 20, 35, 36). Ion channel activation by shear stress/fluid flow has been widely studied, particularly in endothelial cells (37). However, the actual mechanism by which ion channels are gated in response to shear stress has yet to be delineated, and as such, future work is needed to fill this gap in our knowledge. On the basis of what is known about the activation of mechanosensitive ion channels, we speculate that fluid shear could activate TRPM7 via different mechanisms, such as shear-dependent cell membrane deformation and bending, changes in membrane tension, or direct alteration of TRPM7 channel conformation itself. TRPM7 was recently reported to be the key sensor of hydraulic pressure in MDA-MB-231 breast cancer cells, which mediates extracellular Ca2+ uptake and supports a thicker cortical actomyosin meshwork (9). Calcium signaling can activate RhoA-mediated contractility by protein kinase C–α activation of p115RhoGEF (38). Once activated, RhoA increases the level of pMLC through ROCK and by directly inhibiting myosin light chain phosphatase (39). In line with these observations, chelation of extracellular calcium with BAPTA or inhibition of ROCK activity or myosin-II function blocked the shear-induced directional change of fibroblasts, thereby promoting their intravasation in the presence of shear flow. Although fibroblasts typically exhibit a mesenchymal phenotype, their exposure to fluid shear triggers RhoA activation, which supports a bleb-based phenotype (17, 18, 40). Along these lines, half of the fibroblasts, which reversed migration direction to avert exposure to fluid flow, displayed a blebbing phenotype. In accord with our findings, in vitro and in vivo studies of intravasation have detected the formation of blebs as cells protrude into a vessel, and bleb formation is often accompanied with failure to intravasate (4, 6).
Cell migration is regulated by the coordinated action of the local activity of Rho GTPases Cdc42 and Rac1 at the cell leading edge and RhoA at the trailing edge (22, 25, 41). In concert with this model, optogenetic stimulation of RhoA activity at the leading, but not trailing, edge of a migrating cell was sufficient to induce the reversal of migration direction for both normal fibroblasts and HT-1080 fibrosarcoma cells. This directional change requires not only ROCK and myosin-II activity, which are downstream of RhoA activation, but also Cdc42. This is further substantiated by data showing that Cdc42 inhibition prevents the directional change of fibroblast migration induced by optogenetic RhoA activation at the cell anterior. Optogenetic activation of Cdc42 on any edge of the cell leads to persistent migration in that direction in various cell types, such as mouse RAW 264.7 macrophage-like cells, mouse embryonic fibroblasts, and HeLa human cervical cancer cells (41–43). Localized activation of Cdc42 on one edge of a cell leads to myosin-II accumulation at the opposite (trailing) edge (42), and this myosin-II activity is necessary for persistent directional migration even during sustained Cdc2 activation at the leading edge (43). These observations are consistent with our data showing that a sudden increase in RhoA activity at the cell leading edge disrupts its established front/rear polarity. RhoA and Cdc42 have several mechanisms for cross-talk (44). We hypothesize that RhoA/myosin-II and Cdc42 maintain distinct zones of activity in migrating fibroblasts and that shear or optogenetic activation of RhoA disrupts established Cdc42 zones at the cell leading edge, often leading to Cdc42 redistribution to the former trailing edge, where its activity causes a reversal of migration direction.
Shear stress has been reported to induce entry of extracellular Ca2+ through TRPM7 in fibroblasts (8). This mechanical force evokes localized Ca2+ flickers at the leading edge in the presence of a global Ca2+ gradient in the opposite direction with increased Ca2+ concentrations polarized to the cell rear (8, 38). Although highly localized Ca2+ flickers were not observed in our experiments, it is possible that the increased total intracellular Ca2+ in response to shear flow disrupted the front-to-back Ca2+ gradient, thereby preventing sustained forward migration.
TRPM7 has been implicated in promoting breast cancer migration and metastasis by inhibition of myosin-II contractility (45). These authors reported that TRPM7 knockdown conferred a “contractile” phenotype, suppressed migration velocity and wound closure, and metastasis (45). These findings are in distinct contrast to (i) the “slender” shape observed in prior studies (8, 40) and in this work, (ii) our previous data showing that TRPM7 promotes actomyosin contractility (9), and (iii) the slower track velocity and displacement at the front of in vivo tumors of HT-1080 cells overexpressing TRPM7. It is also noteworthy that Kaplan-Meier analysis of human patients shows that patients with osteosarcoma, breast, gastric, and liver cancer expressing high levels of TRPM7 had improved survival (fig. S7, A to D). These results are in accordance with our findings showing that high TRPM7 expression interferes with fibrosarcoma cell intravasation, which may help explain the better prognosis of patients with osteosarcoma.
The reversing phenotype observed in both fibroblasts and HT-1080 fibrosarcoma cells is modulated by the activity of TRPM7. Although the influx of extracellular Ca2+ triggered by TRPM7 activation appears sufficient to cause most HT-1080 fibrosarcoma cells to reverse direction, probably occurring at specific microdomains generated in the proximity of TRPM7, similar to what has been shown for TRPV4-dependent modulation of calmodulin signaling (46), we cannot fully discard that another mechanotransduction pathway plays a cooperative role with Ca2+ signaling to regulate intravasation. For instance, RhoA can be activated in a Ca2+-independent manner via force transduction through integrins (47) or the glycocalyx (35). In line with this scenario, optogenetic activation of RhoA is sufficient to trigger the fibrosarcoma cells to reverse their migration direction and avert exposure to fluid shear. Moreover, as the fibrosarcoma cells migrate significantly faster than their normal counterparts tested here, they have reduced time to transduce mechanical signals before exiting the microchannels, which may allow them to exit these channels before responding to an inhibitory cue. Shear aversion may also be overcome by collective cell migration, where clusters of cells migrating together prevent leader cells exposed to shear from reversing migration direction. Although fibroblasts are absent from the bloodstream in healthy individuals, cancer-associated fibroblasts have been identified circulating in the bloodstream of most patients with prostate and breast cancer with metastatic disease (48, 49), often in clusters with tumor cells. This suggests both that fibroblasts can survive for some time in the circulatory system and that heterotypic collective migration of cancer and fibroblast cells may contribute to intravasation.
While IQGAP1 has been identified as a prometastatic factor (50), its role in shear sensitivity of normal fibroblasts has not been previously explored. IQGAP1 displays uniform distribution in healthy colon and liver tissues but is deregulated in cancerous lesions (51). IQGAP1 can bind to multiple proteins using its >100 binding partners (26) and thus may have potentially divergent effects on different stages of the metastatic cascade. By using a simplified intravasation model and using constitutively active Cdc42, we demonstrated that IQGAP1 acts upstream of Cdc42 and blocks normal fibroblast intravasation. Our proposed model (Fig. 8) of calmodulin/IQGAP1/Cdc42 in shear-induced cell reversal of migration direction highlights yet another role of IQGAP1 and its scaffolding function in integrating diverse signaling pathways.
Recent work points to mechanosensitive ion channels as important elements for cells to probe their physical microenvironment (9, 36, 52), conferring cells advantages to migrate under un/restricted environments, to invade and proliferate. We now report on how normal fibroblasts use the TRPM7 mechanosensitive cation channel to detect and avoid shear stress, thereby protecting them from entering the blood stream. We propose that cancer cells repress this mechanism, by reducing TRPM7 activity and/or its downstream signaling, to intravasate.
MATERIALS AND METHODS
Cells and cell culture
Human dermal fibroblasts (GM05565) were purchased from the Coriell Institute (Camden). NuFFs were a gift from S. Gerecht (Johns Hopkins). CHO and human MDA-MB-231 adenocarcinoma cells were purchased from the American Type Culture Collection. HT-1080 cells were a gift from D. Wirtz (Johns Hopkins). Cells were maintained in tissue culture flasks at 37°C and 5% CO2. GM05565, CHO, MDA-MB-231, and HT-1080 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin (Gibco). NuFFs were maintained in DMEM with high glucose and without sodium pyruvate (Gibco) supplemented with 10% (v/v) FBS. Cells were passaged using 0.05% trypsin-EDTA (Gibco).
Fabrication of microfluidic device
The polydimethylsiloxane-based microfluidic device consisted of an array of parallel microchannels of prescribed length (L = 200 μm) and height (H = 10 μm) and different widths (W = 3, 6, 10, 20, and 50 μm). These microchannels were orthogonally sandwiched by 2D-like seeding and media channels with a width of 400 μm and a height of 30 μm. The devices were created using standard multilayer photolithography and replica molding as described previously (9, 17, 18, 53, 54). Before cell migration experiments, the microfluidic devices were coated with a solution of rat tail collagen I (20 μg/ml) (Gibco) for 1 hour at 37°C.
Cell seeding, treatment with pharmacological agents, and imaging
Cells were passed and resuspended in media to a concentration of 1 × 106 to 2 × 106 cells/ml. Twenty microliters of cell suspension was added to the cell inlet and incubated for 5 min at 37°C and 5% CO2. Cell suspension was then moved from the cell inlet to the cell outlet and incubated for another 5 min to ensure even seeding of the microchannels. Cells were incubated for 1 to 2 hours at 37°C and 5% CO2 until they entered the microchannels. After the cells entered the microchannels, media in the device was changed to generate the proper hydrostatic pressure gradient as described below and, in select experiments, included a pharmacological agent or vehicle control. Directly after the media change, cell behavior was captured through a 10×/0.30 numerical aperture Ph1 objective lens in 10-min intervals for 14 hours by a Digital Sight Qi1Mc camera mounted on an Eclipse Ti-E Inverted microscope (Nikon) equipped with a stage top incubator (Tokai Hit Co., Shizuoka, Japan) maintained at 37°C with 5% CO2 and 100% humidity. In select experiments, cells were treated with the following pharmacological agents and corresponding vehicle controls (purchased from Sigma-Aldrich unless otherwise stated): 2-APB (Calbiochem; 100 μM), FTY720 (2 μM), naltriben (25 to 100 μM), GSK2193874 (15 μM), BAPTA (50 or 100 μM), ruthenium red (10 μM), blebbistatin (50 μM), Y27632 (10 μM), ML141 (Santa Cruz Biotechnology; 10 μM), ionomycin (1 to 10 μM), W7 hydrochloride (Tocris; 25 μM), and NSC 23766 (Tocris; 50 μM).
Generation of shear flow via hydrostatic pressure gradient
Cells were seeded in the microfluidic devices. Once firmly adhered to the collagen I–coated seeding channel, 50 μl of media was placed in each of the six wells of the microfluidic device, such that the hydrostatic pressure in the device was equal everywhere and the medium was at rest (static). At this point, the devices were incubated at 37°C and 5% CO2 for 1 to 2 hours, until cells had entered and migrated to the center of most of the microchannels. Then, the medium was aspirated from all wells, and 167 μl of fresh media were placed in the inlet wells to the seeding and media channels, such that a hydrostatic gradient was generated across these larger channels, while pressure across the perpendicular microchannels was balanced. For static conditions, the medium was also placed in the outlet wells so that hydrostatic pressure was balanced everywhere.
The flow of aqueous fluids in microfluidic devices can be modeled by the Navier-Stokes equation for Newtonian fluids with no body forces (55). For laminar fluid flow, the fluid flow rate resulting from the pressure gradient can be modeled in the channels as (56)
| (1) |
where μ is the fluid viscosity (approximated by the viscosity of water), L is the length of the channel, Q is the volumetric flow rate, W is the channel width, and H is its height. While the exact solution for flow in rectangular ducts requires expansion of a Taylor series, a good approximation of the wall shear stress (τw) on the lower surface in front of the microchannel exits (where migrating cells contact fluid flow) exists on the basis of the low aspect ratio (α, defined height over width) of our microfluidic channels (0.075), the assumptions of steady-state and low Reynolds number (Re << 1) (57)
| (2) |
where m and n are empirically derived constants with m = 1.7 + 0.5α −1.4, where α is the aspect ratio as defined above and n = 2 for α < 1/3. Using these equations, we verified that the Re based on the area-averaged flow velocity is ~0.03 (Re << 1).
Fluid flow from the inlet well to the outlet well reduces the pressure gradient over time. Equating the change in fluid volume in the inlet well rate over time to the fluid flow rate
| (3) |
where a is the cross-sectional area of the inlet/outlet wells and h is the difference in height between these wells. Rearrangement and application of integration limits leads to
| (4) |
that can be solved for the final form
| (5) |
Equation 5 can be used to determine the change in pressure gradient as a function of time. Updating of the pressure gradient driving the flow rate in Eqs. 1 and 2 reveals how hydrostatically driven shear stress in our system changes as a function of time. From the solution of time-dependent shear stress (fig. S8A), we verified that the stress levels are maintained between 75 and 50% of the initial levels within the first 3.5 and 8.5 hours. Most cell reversal or exit events were observed during this time window.
Generation of shear flow using syringe pump
For fluid access, one inlet and one outlet well were punched using a 1-mm-diameter biopsy punch for both the large cell seeding and media channels. Microfluidic devices were assembled and coated with collagen I. Cells were seeded and allowed to enter the microchannels as described above. Microbore tubing (Cole-Parmer) was connected to the outlet wells, which drained to separate waste collection vessels to allow verification of equal flow through each channel (fig. S8B). To generate shear stress, a syringe pump (Harvard Apparatus) was fit with a 10-ml luer-lock syringe (BD) full of media. The syringe was connected to a blunt-tip needle fit into the microbore tubing. The tubing passed through a splitter (Cole-Parmer), and the outlets of the splitter were connected to microbore tubing entering the two microfluidic device channel inlets. The pump volumetric flow rate was set to control the channel shear stress based on Eq. 2, and the volumetric flow rate was verified by measuring the volume in the waste collection vessels at the end of the experiment. Because of the limitation of the media volume in the syringe, these experiments were stopped after 4 hours, which is consistent with when most cells exit the channels or when reversal events were observed in other experiments.
In experiments aimed to decouple effects of shear stress from shear rate, we held the shear rate constant (62.5 s−1) but increased the shear stress from 0.5 to 5 dyne/cm2 by increasing the viscosity of the medium from 0.8 to 8 cP via addition of 500-kDa dextran (Spectrum Chemical). As a control, equimolar amount of 6-kDa dextran (Alfa Aesar) was added to the basal medium at 0.8 cP to keep the molarity constant in media of different viscosities. Viscosity was measured using Cannon-Fenske capillary viscometers. The flow of 8-cP media was maintained by use of a syringe pump while hydrostatic pressure was used to drive flow in the devices with 0.8-cP media. Shear rate was calculated by taking the ratio of shear stress to viscosity.
Fluo-4 Direct
Fluo-4 Direct working solution was prepared 2× according to the manufacturer’s instructions (Sigma-Aldrich). Working solution was diluted 1× with cell media, and cells already seeded in microfluidic devices were treated with this 1× solution for 1 hour at 37°C and 5% CO2. Next, Fluo-4 Direct solution was removed and replaced with 167-μl regular media in every well followed by incubation for 1 hour to allow the cells to come to rest after any physical stimulation associated with the media change. Microfluidic devices were then placed on an A1 confocal microscope (Nikon) equipped with a stage top incubator and cage (Tokai Hit Co.) maintained at 37°C with 5% CO2 and 100% humidity. Cells were imaged in 6-s intervals for 5 min under static (no flow) conditions. Flow was induced by aspirating the media from the two outlet wells, and then imaging was resumed. For drug experiments, the drug or vehicle control was introduced during the 1-hour incubation period before imaging. Fluorescence intensity of the collected images was analyzed using ImageJ (National Institutes of Health). Fluorescence intensity of individual cells was quantified by outlining the cell at each time point using polygonal regions of interest.
Electrophysiological recording
Endogenous stretch-activated piezo-like cationic currents in fibroblasts were recorded in the cell-attached configuration. Borosilicate glass patch pipettes, with a tip resistance of 1 to 1.5 megohm, were filled with a solution containing 130 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM TEA-Cl, and 10 mM Hepes (adjusted to pH 7.3 with NaOH). The bath solutions contained 140 mM KCl, 1 mM MgCl2, 10 mM glucose, and 10 mM Hepes (adjusted to pH 7.3 with tris). This extracellular potassium was used to zero the membrane potential, allowing better control of the voltage (−80 mV) across the membrane patch using an EPC10-USB patch-clamp amplifier (HEKA Elektronik, Germany). Stimulation of the Piezo1 channel was performed using a high-speed pressure clamp (HSPC-1) from ALA Scientific Instruments. Sequential negative pressure pulses of 250 ms from −10 to −70 mmHg (Δ−10 mmHg) were applied every 10 s. Currents were recorded at 10 kHz and low-pass–filtered at 0.5 kHz. Whole-cell TRPM7-like cationic currents were recorded from fibroblasts and HT-1080 fibrosarcoma cells under static (no flow) or shear stress conditions (perfusion at 10 μl/s). Cells were held at 0 mV, and ramps from −100 to +100 mV (400 ms) were applied at a frequency of 0.2 Hz. Ramp data were acquired at 10 kHz and low-pass–filtered at 1 kHz. Experiments were performed at room temperature (22° to 26°C). Whole-cell currents were measured using pipettes (1.6 to 2 megohm) filled with a solution containing 140 mM CsCl, 10 mM Hepes, 1 mM EGTA, 4 mM Na2ATP, and 0.3 mM Na3GTP (pH 7.2 to 7.3 and 295 to 300 mosmol/liter). The external solution contained 110 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 10 mM Hepes, and 5 mM glucose (pH 7.3 to 7.4 and 305 to 310 mosmol/liter).
siRNA and plasmid transfection
For siRNA knockdown, scramble (sc-37007) and IQGAP1 (sc-35700) siRNA was purchased from Santa Cruz Biotechnology. Cells were transiently transfected with siRNA using the Lipofectamine RNAiMax Kit (Invitrogen) by following the manufacturer’s protocol.
The pcDNA3.1 plasmid expressing mouse TRPM7 tagged with YFP was a gift from T. Gudderman (Ludwig-Maximilians Universitat, Munich, Germany), and control YFP (pEYFP-C1) was purchased from Clontech. The GFP-IQGAP1 plasmid was a gift from J. Schober (Southern Illinois University Edwardsville) (27). Plasmid pcDNA3-EGFP-Cdc42(Q61L) was a gift from K. Hahn and purchased from Addgene (plasmid no. 12600). For transient transfections, 60 to 80% confluent HT-1080 cells or fibroblasts were transfected using a Lipofectamine 3000 reagent following the manufacturer’s recommendations.
shRNA design and cloning and Lifeact imaging
Silencing RNA sequences to relevant genes were retrieved from the Gene Perturbation Platform (Broad Institute). The targeting sequences are as follows: Scramble control (GCACTACCAGAGCTAACTCAGATAGTACT); human TRPC1 sequence-1 (TTCTCGTGAATTGGAAGTTAT); human TRPC1 sequence-2 (GCCCACCTGTAAGAAGATAAT); human TRPC6 sequence-1 (GTCCACTTGAAGCCATATTAT); human TRPC6 sequence-2 (CGCTCCACAAGCCTATCTATA); human TRPM7 sequence-1 (GTCTTGCCATGAAATACTC); human TRPM7 sequence-2 (CAAGAGATGATAAGCTGTTTA); human Cdc42 sequence-1 (CGGAATATGTACCGACTGTTT); human Cdc42 sequence-2 (CCTGATATCCTACACAACAACC).
Sense and antisense silencing sequences were added on either end of a short hairpin loop, and complete forward and reverse sequences were ordered as custom DNA oligomers (Life Technologies). To generate shRNA lentiviral plasmids, we subcloned the targeting oligomer sequences or nontargeting scramble control into the pLVTHM lentiviral plasmid (a gift from D. Trono, plasmid no. 12247) using Mlu I and Cla I as restriction sites. Cdc42 targeting sequences and scramble control sequences were subcloned into the pLKO.1 lentiviral plasmid (a gift from R. Weinberg, plasmid no. 8453) using Age I and Eco RI as restriction sites. Sequence integrity and orientation was verified by Sanger Sequencing (JHU Genetic Resources Core Facility).
The pLVTHM, pLKO.1, pLenti.PGK.Lifeact-GFP.W (gifts from R. Lansford, plasmid no. 51010), RHOA2G FRET biosensor (a gift from O. Pertz, plasmid nos. 40176 and 40179), psPAX2 (a gift from D. Trono, plasmid no. 12260), and pMD2.G (a gift from D. Trono, plasmid no. 12259) plasmids were purchased from Addgene.
To produce lentivirus, 293T/17 cells were cotransfected with psPAX2, pMD2.G, and the appropriate lentiviral plasmid. Lentivirus was collected after 48 hours after transfection, filtered through 0.45-μm filters (Thermo Fisher Scientific) and concentrated by centrifugation (50,000g for 2 hours at 4°C). Cells were transduced for 24 hours with media containing lentiviral particles. Cells transduced with pLKO.1 were selected with puromycin (0.5 μg/ml; Gibco) 48 hours after transduction and maintained with this concentration. Migration experiments with cells transduced with pLVTHM were also imaged using 488-nm light, and only cells expressing GFP (encoded on pLVTHM) were analyzed.
Quantitative polymerase chain reaction
Standard qPCR techniques were applied as described previously (58) using the following primers: PIEZO1: F-(5′-TTCCTGCTGTACCAGTACCT-3′) and R-(5′-AGGTACAGCCACTTGATGAG-3′); TRPC1: F-(5′-GATGTGCTTGGGAGAAATGC-3′) and R-(5′-AATGACAGGTGCAACATCCA-3′); TRPC6: F-(5′-AGACAATGGCGGTCAAGTTC-3′) and R-(5′-TCCCAGAAAAATGGTGAAGG-3′); TRPM7: F-(5′-GGAGATGCCCTCAAAGAACA-3′) and R-(5′-TGCTCAGGGGGTTCAATAAG-3′); TRPV2: F-(5′-CAAACCGATTTGACCGAGAT-3′) and R-(5′-GTTCAGCACAGCCTTCATCA-3′); TRPV4: F-(5′- CCCGTGAGAACACCAAGTTT-3′) and R-(5′- GTGTCCTCATCCGTCACCTC-3′); IQGAP1: F-(5′-TTCTATGCAGCTTTCTCGGG-3′) and R-(5′-CTGTCGAACTAAGTATCCACGG-3′); IQGAP2: F-(5′-GCTAGGGGAAATCGGCGAG-3′) and R-(5′-TGCAGAGAGCCTTTCATCGT-3′); IQGAP3: F-(5′-GTTCGGCAGAAGTTTGCTGAGC-3′) and R-(5′-CACTCCAGGTAAATCTTCCG CTG-3′).
Lifeact and IQGAP1 imaging
Fibroblasts expressing Lifeact-GFP were seeded in microfluidic devices and imaged through a 20×/0.4 numerical aperture Ph1 lens using an Eclipse Ti-E Inverted microscope (Nikon) equipped with an Intensilight C-HGFIE arc lamp (Nikon) and the appropriate excitation and emission filters (Nikon). Fibroblasts expressing GFP-IQGAP1 were seeded in microfluidic devices and imaged using an A1 confocal microscope (Nikon) with a 60× oil objective and fluorescein isothiocyanate filters. Temperature (37°C) and CO2 (5%) were maintained using a controlled humidity stage top incubator and cage (Tokai Hit).
Fluorescence images were analyzed using ImageJ (National Institutes of Health). For each cell, a kymograph was drawn at the cell edge during an extension-retraction event, and GFP-IQGAP1 intensities were measured before, during, and after retraction (fig. S5F). Three sets of these extension-retraction events were considered per cell, and measured intensity values were plotted after normalization of the raw intensity to the average intensity value from each event.
Immunofluorescence and actin staining
Cells were fixed with 4% formaldehyde solution (Thermo Fisher Scientific), permeabilized with 1% Triton X-100 (Sigma-Aldrich), blocked against nonspecific adhesion with 2% bovine serum albumin (Sigma-Aldrich) and 1% goat serum (Thermo Fisher Scientific), immunostained for target proteins, and then imaged on an A1 confocal microscope (Nikon). The primary antibody, anti-pMLC (Ser19, 3671, Cell Signaling Technology), was administered at the manufacturer’s recommended concentration. Quantification of pMLC density was performed as described in (59). For actin labeling, cells were stained with rhodamine phalloidin (Thermo Fisher Scientific) at the manufacturer’s recommended concentration.
Fluorescence lifetime imaging microscopy
For measurements of RhoA activity, cells stably transduced with pLentiRHOA2G were imaged using a Zeiss LSM 780 microscope fitted to a PicoQuant FLIM system consisting of the PicoHarp 300 time-correlated single-photon counting (TCSPC) module, Sepia II laser control module, and two hybrid PMA-04 detectors. A PeCon environment chamber was used to maintain the cells at 37°C and 5% CO2 during imaging.
The FLIM data were acquired with an Apochromat 40×/1.1W Corr 27 lens (Zeiss), using a 440-nm diode excitation laser pulsed at 32 MHz and reflected to the sample through an 80/20 mirror (Zeiss). Emission light below 488 nm was collected at the PicoQuant PMA-04 hybrid detector by a dichroic mirror after it passed through a transparent plate and optic fiber. Images were acquired at a single scan of 1024 × 1024 pixels with a pinhole size between 3- and 1.5-μm z-sections (to limit the emission photon count rate below 10% of the laser excitation rate). TCSPC data were acquired in 8-ps time bins within 31.25-ns time window.
The FLIM data were processed with SymPhoTime 64 (PicoQuant) software by using a custom-written script for the calculation of the internal response function from 100 data points with no smoothing. A constant threshold was applied to eliminate fluorescent signal from outside the cell area, and the data were binned to obtain 600 to 1000 photons per binned pixel. Then, a three-exponential reconvolution was used to fit the fluorescence decays into every binned pixel. As the RhoA FRET sensor was mostly excluded from the nucleus resulting in an inconsistent and weak photon output, the nuclear pixels were manually excluded by drawing a free region of interest around the nucleus during the final segmentation. Next, using the software, we calculated the amplitude-weighted fluorescence lifetime average of the total cell (17, 18).
Optogenetics
The cryptochrome 2 (CRY2)–CIBN light-gated dimerizer system was used to control the subcellular localization of RhoA with high spatiotemporal accuracy (23). This system relies on the fusion of the catalytic (DHPH) domain of the RhoA-GEF, ARHGEF11, to CRY2-mCherry and its GFP-labeled dimerization partner, CIBN, engineered to bind to the plasma or mitochondrial membrane. Light-induced localization of optoGEF-RhoA to the plasma membrane via the CAAX anchor (CAAX-CIBN-GFP) promotes cortical RhoA activity, while targeting of optoGEF-RhoA to the mitochondria via mito-CIBN-GFP reduces cortical RhoA activity (23). ARHGEF11(DHPH)-CRY2-mCherry, CAAX-CIBN-GFP, and mito-CIBN-GFP were gifts from X. Trepat (Institute for Bioengineering of Catalonia). Plasmids were encapsulated in lentivirus as described above. Cells were stably transduced with ARHGEF11(DHPH)-CRY2-mCherry and then with either mito-CIBN-GFP or CAAX-CIBN-GFP. Cells were seeded in microfluidic devices and incubated under static conditions at 37°C and 5% CO2 until they migrated into W = 10 μm by H = 10 μm microchannels. The microfluidic devices were transferred to a stage top incubation system (Tokai Hit Co., Shizuoka, Japan) mounted on an A1 confocal microscope (Nikon). Cell migration was monitored by imaging the mCherry channel in real time to identify the leading and trailing edges. A rectangular area was marked at the cell leading or trailing edge and stimulated with 488-nm light at 1% laser power for 1 s. Stimulations were repeated 20× at 10-s intervals to reach consistent localization of ARHGEF11 to the membrane or mitochondria. mCherry images were recorded after each stimulation to monitor the localization. Following stimulation, cell migration and ARHGEF11 were monitored through further imaging of mCherry.
Western blotting
Western blots were performed as previously described (17, 18, 58) using NuPage 4 to 12% bis-tris gels and the following antibodies (at the manufacturer’s recommended concentrations): anti-TRPM7 (mouse, clone S74-25, Thermo Fisher Scientific MA5-27620), anti-Cdc42 (rabbit, clone 11A11, Cell Signaling Technology 2466), and anti-IQGAP1 (mouse, clone 9, Santa Cruz Biotechnology, sc-376021) with glyceraldehyde-3-phosphate dehydrogenase (rabbit, clone 14C10, Cell Signaling Technology 2118) or β-actin (mouse, clone C4, BD Biosciences 612656) as a loading control. Secondary antibodies (used at 1:1000 dilution): anti-mouse immunoglobulin G (IgG) horseradish peroxidase (HRP)–linked antibody (Cell Signaling Technologies) and anti-rabbit IgG HRP-linked antibody (Cell Signaling Technologies).
CRISPR-Cas9–mediated knockout of TRPM7
Cells with stable TRPM7 knockout were previously used and verified via immunoblotting in (9).
Ex ovo chick embryo cancer xenograft model
Fertilized White Leghorn chicken eggs were obtained from the University of Alberta Poultry Research Centre and maintained in a humidified incubator at 38°C. After 4 days of incubation, embryos were removed from their shells using a Dremel tool with a cutting wheel and maintained under shell-less conditions, in a covered dish in a humidified air incubator at 38°C and 60% humidity as previously described (29, 30). All procedures were performed according to the University of Alberta guidelines.
For the primary tumor or primary tumor invasive front imaging, day 10 chicken embryos were injected with 1 × 105 HT-1080 cells (YFP-control or TRPM7-YFP–overexpressing) in phosphate-buffered saline directly in between CAM ectoderm and endoderm layers. Sterilized, rounded (22 mm) coverslips were applied on top of the tumor 24 hours after tumor cell application, and tumors or invasive tumor fronts were imaged 5 days after tumor cell inoculation.
For metastatic colony imaging, day 10 chicken embryos were intravenously injected with 2.5 × 104 HT-1080 cells (control or TRPM7-overexpressing). Sterilized, rounded (22 mm) coverslips were applied on top of the CAM-containing metastatic colonies 24 hours after cancer cell injection. Metastatic colonies were allowed to grow for 4 days, and individual, nonoverlapping metastatic colonies were selected for imaging.
Image acquisition and analysis
Real-time imaging of cancer cell invasion was performed by acquiring 4D image series of single cancer cells within the CAM tissue. A 50- to 300-μm image stack was acquired every 20 min in 2- to 5-μm step size increments for 6 to 12 hours. A Nikon A1R upright microscope (Nikon) fitted with a temperature-regulated enclosure and a range of Nikon microscope objectives [10×, 25× (WI) and 63× (oil)] was used for multicolor imaging of cancer cells and chicken embryo CAM vasculature. Image drift was corrected using the ImageJ Stack_Reg plugin (Biomedical Imaging Group, https://sites.imagej.net/BIG-EPFL/plugins/) before tracking analysis. Time 0 was defined as the time of the first image capture. At least 30 individual cells were tracked for control cells and TRPM7 overexpression mutant using a built-in Volocity Object tracking module. Track velocity was calculated as average speed of the track. Track displacement rate (productivity) was calculated using the built-in Volocity module as total track displacement (straight-line distance from the first track position to the last) divided by track time. At least three independent animals were used for each experiment. For quantification of intravasating cells per field, 63× z-stack images were acquired (1-μm step) for at least five independent tumors, and individual optical sections were scored for vascular wall–breaching cancer cells. An individual cell was considered intravasating if it was protruding at least 5 μm into the vascular lumen. For 3D reconstructions of vasculature and intravasated tumor cells, the 63× z-stacks were imported into an Imaris imaging analysis suite. First, a 3D reconstruction of both the vasculature and tumor cells was built (isosurfaces). Second, the tumor cells that are outside of the vascular lumen were “masked” and removed from the 3D rendering. The isosurface that represents the vascular wall was rendered “translucent” to allow the visualization of intravasated tumor cells. Results were plotted as intravasating cell percentage. For quantification of the cancer cell number per colony or cancer cell length, 25× z-stack images were acquired (2- to 5-μm step). Cancer cells were manually counted using Nikon Elements software, whereas cancer cell length was measured using the software’s built-in length measurement module. All experimental data were plotted and analyzed for statistical significance using GraphPad Prism 8.
Kaplan-Meier survival analysis
Analysis for osteosarcoma was performed by using R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). Analysis for breast, ovarian, and gastric cancer was done by using Kaplan-Meier plotter (https://kmplot.com) (60). Auto scan mode was used in both tools to choose the cutoff between high- and low-expression cohorts.
Statistical analysis
Data represent the means ± SD of ≥3 independent experiments, and data points denote the percentage of cell reversal from each experiment in which ≥30 cells were analyzed per condition, unless otherwise specified. The D’Agostino-Pearson omnibus normality test was used to determine whether data are normally distributed. Datasets with Gaussian distributions were compared using Student’s t test (two-tailed), one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test, or two-way ANOVA followed by Sidak’s or Tukey’s multiple comparisons tests. For log-normal distribution, statistical comparison was made after logarithmic transformation of the data followed by unpaired Student’s t test (two-tailed). For comparing two non-Gaussian distributions, the nonparametric Mann-Whitney test was used. Statistical significance was defined as P < 0.05. Calculations were performed using GraphPad Prism 7, 8, or 9 (GraphPad Software).
Acknowledgments
The pcDNA3.1 plasmid expressing mouse TRPM7 tagged with YFP was a gift from T. Gudderman (Ludwig-Maximilians Universitat, Munich, Germany). The GFP-IQGAP1 plasmid was a gift from J. Schober (Southern Illinois University Edwardsville). ARHGEF11(DHPH)-CRY2-mCherry, CAAX-CIBN-GFP, and mito-CIBN-GFP were gifts from X. Trepat (Institute for Bioengineering of Catalonia). Supplementary materials include figs. S1 to S8 and movies S1 to S3. Funding: This line of research was supported by the National Institutes of Health through grants R01-CA183804 (K.K.), R01-CA254193 (K.K.), and R01-GM134542 (K.K.); Alberta Cancer Foundation no. 26491 (J.D.L.); the Spanish Ministry of Science, Education and Universities through grant RTI2018-099718-B-100 (M.A.V.); and an institutional “Maria de Maeztu” Programme for Units of Excellence in R&D and FEDER funds (M.A.V.). Author contributions: C.L.Y. and K.B.: conception and design, collection and assembly of in vitro data, data analysis and interpretation, and manuscript writing. K.S. and J.D.L.: collection and assembly of in vivo data, data analysis and interpretation, and manuscript writing. S.A.S. and J.-C.G. performed patch-clamp experiments and analyzed and interpreted data. S.T. and P.M. performed select in vitro experiments and analyzed data. M.A.V. and K.K.: conception and design, data analysis and interpretation, and manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabh3457/DC1
REFERENCES AND NOTES
- 1.Weigelin B., Bakker G.-J., Friedl P., Intravital third harmonic generation microscopy of collective melanoma cell invasion: Principles of interface guidance and microvesicle dynamics. Dermatol. Int. 1, 32–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolf K., Alexander S., Schacht V., Coussens L. M., von Andrian U. H., van Rheenen J., Deryugina E., Friedl P., Collagen-based cell migration models in vitro and in vivo. Semin. Cell Dev. Biol. 20, 931–941 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang S. P., Cabrera R. M., Segall J. E., Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 311, C1–C14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoletov K., Montel V., Lester R. D., Gonias S. L., Klemke R., High-resolution imaging of the dynamic tumor cell vascular interface in transparent zebrafish. Proc. Natl. Acad. Sci. U.S.A. 104, 17406–17411 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hashizume H., Baluk P., Morikawa S., McLean J. W., Thurston G., Roberge S., Jain R. K., McDonald D. M., Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 156, 1363–1380 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong A. D., Searson P. C., Mitosis-mediated intravasation in a tissue-engineered tumor-microvessel platform. Cancer Res. 77, 6453–6461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stapor P. C., Wang W., Murfee W. L., Khismatullin D. B., The distribution of fluid shear stresses in capillary sprouts. Cardiovas. Eng. Technol. 2, 124–136 (2011). [Google Scholar]
- 8.Wei C., Wang X., Chen M., Ouyang K., Song L. S., Cheng H., Calcium flickers steer cell migration. Nature 457, 901–905 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao R., Afthinos A., Zhu T., Mistriotis P., Li Y., Serra S. A., Zhang Y., Yankaskas C. L., He S., Valverde M. A., Sun S. X., Konstantopoulos K., Cell sensing and decision-making in confinement: The role of TRPM7 in a tug of war between hydraulic pressure and cross-sectional area. Sci. Adv. 5, eaaw7243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartmannsgruber V., Heyken W. T., Kacik M., Kaistha A., Grgic I., Harteneck C., Liedtke W., Hoyer J., Köhler R., Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLOS ONE 2, e827 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J., Hou B., Tumova S., Muraki K., Bruns A., Ludlow M. J., Sedo A., Hyman A. J., McKeown L., Young R. S., Yuldasheva N. Y., Majeed Y., Wilson L. A., Rode B., Bailey M. A., Kim H. R., Fu Z., Carter D. A., Bilton J., Imrie H., Ajuh P., Dear T. N., Cubbon R. M., Kearney M. T., Prasad K. R., Evans P. C., Ainscough J. F. X., Beech D. J., Piezo1 integration of vascular architecture with physiological force. Nature 515, 279–282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma S., Goswami R., Merth M., Cohen J., Lei K. Y., Zhang D. X., Rahaman S. O., TRPV4 ion channel is a novel regulator of dermal myofibroblast differentiation. Am. J. Physiol. Cell Physiol. 312, C562–C572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spassova M. A., Hewavitharana T., Xu W., Soboloff J., Gill D. L., A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. U.S.A. 103, 16586–16591 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alcaino C., Knutson K., Gottlieb P. A., Farrugia G., Beyder A., Mechanosensitive ion channel Piezo2 is inhibited by D-GsMTx4. Channels 11, 245–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung M., Bao W., Behm D. J., Brooks C. A., Bury M. J., Dowdell S. E., Eidam H. S., Fox R. M., Goodman K. B., Holt D. A., Lee D., Roethke T. J., Willette R. N., Xu X., Ye G., Thorneloe K. S., Discovery of GSK2193874: An orally active, potent, and selective blocker of transient receptor potential vanilloid 4. ACS Med. Chem. Lett. 8, 549–554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hofmann T., Schäfer S., Linseisen M., Sytik L., Gudermann T., Chubanov V., Activation of TRPM7 channels by small molecules under physiological conditions. Pflugers Arch. 466, 2177–2189 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Mistriotis P., Wisniewski E. O., Bera K., Keys J., Li Y., Tuntithavornwat S., Law R. A., Perez-Gonzalez N. A., Erdogmus E., Zhang Y., Zhao R., Sun S. X., Kalab P., Lammerding J., Konstantopoulos K., Confinement hinders motility by inducing RhoA-mediated nuclear influx, volume expansion, and blebbing. J. Cell Biol. 218, 4093–4111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wisniewski E. O., Mistriotis P., Bera K., Law R. A., Zhang J., Nikolic M., Weiger M., Tuntithavornwat S., Afthinos A., Zhao R., Kalab P., Scarcelli G., Friedl P., Konstantopoulos K., Dorsoventral polarity directs cell responses to migration track geometries. Sci. Adv. 6, eaba6505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paul C. D., Mistriotis P., Konstantopoulos K., Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 17, 131–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venturini V., Pezzano F., Catala Castro F., Hakkinen H. M., Jimenez-Delgado S., Colomer-Rosell M., Marro M., Tolosa-Ramon Q., Paz-Lopez S., Valverde M. A., Weghuber J., Loza-Alvarez P., Krieg M., Wieser S., Ruprecht V., The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science 370, eaba2644 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Petrie R. J., Yamada K. M., Multiple mechanisms of 3D migration: The origins of plasticity. Curr. Opin. Cell Biol. 42, 7–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridley A. J., Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 36, 103–112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valon L., Marín-Llauradó A., Wyatt T., Charras G., Trepat X., Optogenetic control of cellular forces and mechanotransduction. Nat. Commun. 8, 14396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guilluy C., Garcia-Mata R., Burridge K., Rho protein crosstalk: Another social network? Trends Cell Biol. 21, 718–726 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hung W. C., Chen S. H., Paul C. D., Stroka K. M., Lo Y. C., Yang J. T., Konstantopoulos K., Distinct signaling mechanisms regulate migration in unconfined versus confined spaces. J. Cell Biol. 202, 807–824 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hedman A. C., Smith J. M., Sacks D. B., The biology of IQGAP proteins: Beyond the cytoskeleton. EMBO Rep. 16, 427–446 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foroutannejad S., Rohner N., Reimer M., Kwon G., Schober J. M., A novel role for IQGAP1 protein in cell motility through cell retraction. Biochem. Biophys. Res. Commun. 448, 39–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nalbant P., Hodgson L., Kraynov V., Toutchkine A., Hahn K. M., Activation of endogenous Cdc42 visualized in living cells. Science 305, 1615–1619 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Leong H. S., Robertson A. E., Stoletov K., Leith S. J., Chin C. A., Chien A. E., Hague M. N., Ablack A., Carmine-Simmen K., McPherson V. A., Postenka C. O., Turley E. A., Courtneidge S. A., Chambers A. F., Lewis J. D., Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 8, 1558–1570 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Stoletov K., Willetts L., Paproski R. J., Bond D. J., Raha S., Jovel J., Adam B., Robertson A. E., Wong F., Woolner E., Sosnowski D. L., Bismar T. A., Wong G. K., Zijlstra A., Lewis J. D., Quantitative in vivo whole genome motility screen reveals novel therapeutic targets to block cancer metastasis. Nat. Commun. 9, 2343 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gligorijevic B., Wyckoff J., Yamaguchi H., Wang Y., Roussos E. T., Condeelis J., N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 125, 724–734 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ehsan S. M., Welch-Reardon K. M., Waterman M. L., Hughes C. C., George S. C., A three-dimensional in vitro model of tumor cell intravasation. Integr. Biol. 6, 603–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zervantonakis I. K., Hughes-Alford S. K., Charest J. L., Condeelis J. S., Gertler F. B., Kamm R. D., Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U.S.A. 109, 13515–13520 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hapach L. A., Mosier J. A., Wang W., Reinhart-King C. A., Engineered models to parse apart the metastatic cascade. NPJ Precis. Oncol. 3, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi Z. D., Tarbell J. M., Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann. Biomed. Eng. 39, 1608–1619 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H. P., Alisafaei F., Adebawale K., Chang J., Shenoy V. B., Chaudhuri O., The nuclear piston activates mechanosensitive ion channels to generate cell migration paths in confining microenvironments. Sci. Adv. 7, eabd4058 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerhold K. A., Schwartz M. A., Ion channels in endothelial responses to fluid shear stress. Physiology 31, 359–369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei C., Wang X., Zheng M., Cheng H., Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 24, 254–261 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Lessey E. C., Guilluy C., Burridge K., From mechanical force to RhoA activation. Biochemistry 51, 7420–7432 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao R., Cui S., Ge Z., Zhang Y., Bera K., Zhu L., Sun S. X., Konstantopoulos K., Hydraulic resistance induces cell phenotypic transition in confinement. Sci. Adv. 7, eabg4934 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Beco S., Vaidžiulytė K., Manzi J., Dalier F., di Federico F., Cornilleau G., Dahan M., Coppey M., Optogenetic dissection of Rac1 and Cdc42 gradient shaping. Nat. Commun. 9, 4816 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Neill P. R., Kalyanaraman V., Gautam N., Subcellular optogenetic activation of Cdc42 controls local and distal signaling to drive immune cell migration. Mol. Biol. Cell 27, 1442–1450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimmerman S. P., Asokan S. B., Kuhlman B., Bear J. E., Cells lay their own tracks - optogenetic Cdc42 activation stimulates fibronectin deposition supporting directed migration. J. Cell Sci. 130, 2971–2983 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaughan E. M., Miller A. L., Yu H.-Y., Bement W. M., Control of local Rho GTPase crosstalk by Abr. Curr. Biol. 21, 270–277 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Middelbeek J., Kuipers A. J., Henneman L., Visser D., Eidhof I., van Horssen R., Wieringa B., Canisius S. V., Zwart W., Wessels L. F., Sweep F. C., Bult P., Span P. N., van Leeuwen F. N., Jalink K., TRPM7 is required for breast tumor cell metastasis. Cancer Res. 72, 4250–4261 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Donate-Macian P., Jungfleisch J., Perez-Vilaro G., Rubio-Moscardo F., Peralvarez-Marin A., Diez J., Valverde M. A., The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat. Commun. 9, 2307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swartz M. A., Lund A. W., Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer 12, 210–219 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Jones M. L., Siddiqui J., Pienta K. J., Getzenberg R. H., Circulating fibroblast-like cells in men with metastatic prostate cancer. Prostate 73, 176–181 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ao Z., Shah S. H., Machlin L. M., Parajuli R., Miller P. C., Rawal S., Williams A. J., Cote R. J., Lippman M. E., Datar R. H., El-Ashry D., Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Res. 75, 4681–4687 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Hebert J. D., Tian C., Lamar J. M., Rickelt S., Abbruzzese G., Liu X., Hynes R. O., The scaffold protein IQGAP1 is crucial for extravasation and metastasis. Sci. Rep. 10, 2439 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rotoli D., Morales M., Maeso M. D. C., Garcia M. D. P., Gutierrez R., Valladares F., Avila J., Diaz-Flores L., Mobasheri A., Martin-Vasallo P., Alterations in IQGAP1 expression and localization in colorectal carcinoma and liver metastases following oxaliplatin-based chemotherapy. Oncol. Lett. 14, 2621–2628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pardo-Pastor C., Rubio-Moscardo F., Vogel-Gonzalez M., Serra S. A., Afthinos A., Mrkonjic S., Destaing O., Abenza J. F., Fernandez-Fernandez J. M., Trepat X., Albiges-Rizo C., Konstantopoulos K., Valverde M. A., Piezo2 channel regulates RhoA and actin cytoskeleton to promote cell mechanobiological responses. Proc. Natl. Acad. Sci. U.S.A. 115, 1925–1930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balzer E. M., Tong Z., Paul C. D., Hung W. C., Stroka K. M., Boggs A. E., Martin S. S., Konstantopoulos K., Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 26, 4045–4056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong B. S., Shah S. R., Yankaskas C. L., Bajpai V. K., Wu P. H., Chin D., Ifemembi B., ReFaey K., Schiapparelli P., Zheng X., Martin S. S., Fan C. M., Quinones-Hinojosa A., Konstantopoulos K., A microfluidic cell-migration assay for the prediction of progression-free survival and recurrence time of patients with glioblastoma. Nat. Biomed. Eng. 5, 26–40 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tong Z., Cheung L. S., Stebe K. J., Konstantopoulos K., Selectin-mediated adhesion in shear flow using micropatterned substrates: Multiple-bond interactions govern the critical length for cell binding. Integr. Biol. 4, 847–856 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Bahrami M., Yovanovich M. M., Culham J. R., Pressure drop of fully-developed, laminar flow in microchannels of arbitrary cross-section. J. Fluids Eng. 128, 1036–1044 (2006). [Google Scholar]
- 57.Young E. W., Simmons C. A., Macro- and microscale fluid flow systems for endothelial cell biology. Lab Chip 10, 143–160 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Yankaskas C. L., Thompson K. N., Paul C. D., Vitolo M. I., Mistriotis P., Mahendra A., Bajpai V. K., Shea D. J., Manto K. M., Chai A. C., Varadarajan N., Kontrogianni-Konstantopoulos A., Martin S. S., Konstantopoulos K., A microfluidic assay for the quantification of the metastatic propensity of breast cancer specimens. Nat. Biomed. Eng. 3, 452–465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hung W. C., Yang J. R., Yankaskas C. L., Wong B. S., Wu P. H., Pardo-Pastor C., Serra S. A., Chiang M. J., Gu Z., Wirtz D., Valverde M. A., Yang J. T., Zhang J., Konstantopoulos K., Confinement sensing and signal optimization via Piezo1/PKA and myosin II pathways. Cell Rep. 15, 1430–1441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagy A., Lanczky A., Menyhart O., Gyorffy B., Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 8, 9227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabh3457/DC1