

Abstract
The renin-angiotensin system (RAS) is fundamental to COVID-19 pathobiology, due to the interaction between the SARS-CoV-2 virus and the angiotensin-converting enzyme 2 (ACE2) coreceptor for cellular entry. The prevailing hypothesis is that SARS-CoV-2-ACE2 interactions lead to an imbalance of the RAS, favoring proinflammatory angiotensin II (ANG II)-related signaling at the expense of the anti-inflammatory ANG-(1–7)-mediated alternative pathway. Indeed, multiple clinical trials targeting this pathway in COVID-19 are underway. Therefore, precise measurement of circulating RAS components is critical to understand the interplay of the RAS on COVID-19 outcomes. Multiple challenges exist in measuring the RAS in COVID-19, including improper patient controls, ex vivo degradation and low concentrations of angiotensins, and unvalidated laboratory assays. Here, we conducted a prospective pilot study to enroll 33 patients with moderate and severe COVID-19 and physiologically matched COVID-19-negative controls to quantify the circulating RAS. Our enrollment strategy led to physiological matching of COVID-19-negative and COVID-19-positive moderate hypoxic respiratory failure cohorts, in contrast to the severe COVID-19 cohort, which had increased severity of illness, prolonged intensive care unit (ICU) stay, and increased mortality. Circulating ANG II and ANG-(1–7) levels were measured in the low picomolar (pM) range. We found no significant differences in circulating RAS peptides or peptidases between these three cohorts. The combined moderate and severe COVID-19-positive cohorts demonstrated a mild reduction in ACE activity compared with COVID-19-negative controls (2.2 ± 0.9 × 105 vs. 2.9 ± 0.8 × 105 RFU/mL, P = 0.03). These methods may be useful in designing larger studies to physiologically match patients and quantify the RAS in COVID-19 RAS augmenting clinical trials.
Keywords: ACE2, acute respiratory distress syndrome, angiotensin, critical care, SARS-CoV-2
INTRODUCTION
Binding of SARS-CoV-2 to the angiotensin-converting enzyme 2 (ACE2) coreceptor for viral entry has focused attention on the renin-angiotensin system (RAS) in the pathophysiology of COVID-19. The prevailing hypothesis is that SARS-CoV-2-ACE2 binding/internalization increases the ratio of angiotensin II (ANG II) to ANG-(1–7) due to loss of ACE2 activity, promoting oxidative stress, inflammation, fibrosis, and increased vascular tone (1, 2). This finding has prompted speculation on the role of the RAS as a disease modifier, and whether pharmacological manipulation of the RAS might improve outcomes in patients with COVID-19.
The RAS has tissue specificity, though the shedding of membrane-bound ACE2 by ADAM metallopeptidase domain 17 (ADAM17) leads to circulating levels of catalytically active ACE2 (3, 4). Thus, measurement of circulating angiotensins and their associated enzymes in patients with COVID-19 may give important insight into the state of the RAS in COVID-19. However, accurate assessment of circulating angiotensins is challenging, given their low endogenous concentrations (5–50 pM), rapid degradation ex vivo, and the nonspecificity of commercial ELISA assays (5–7). Another challenge in the COVID-19 literature is that many studies using “healthy controls” without physiologically matched COVID-19-negative controls may incorrectly attribute RAS activation to SARS-CoV-2 rather than those events associated with severe acute illness (8). Collectively, these issues have led to uncertainty on the state of the circulating RAS in COVID-19.
To address these issues and evaluate the feasibility of measuring circulating RAS components using validated techniques, we conducted a prospective observational pilot study at two academic medical centers. Using validated laboratory assays, we compared concentrations of RAS peptides and peptidases in patients hospitalized for COVID-19 and patients hospitalized with acute non-COVID-19 respiratory illness.
METHODS
The study was performed under a public health surveillance determination or verbal consent (Wake Forest Baptist Medical Center, IRB00066371). The study included patients ≥18 yr of age; with symptoms of acute respiratory infection, with one or more of the following symptoms: cough, fever (>37.5°C/99.5°F), shortness of breath, or sore throat; and tested for SARS-CoV-2 by PCR within the last 10 days with planned admission to the study hospital. Patients were prospectively enrolled in the following cohorts: 1) SARS-CoV-2 infection with moderate acute hypoxic respiratory failure (AHRF) receiving supplemental oxygen at 1–6 L/min (moderate COVID-19, n = 12); 2) no evidence of SARS-CoV-2 infection with moderate AHRF receiving supplemental oxygen at 1–6 L/min (moderate AHRF controls, n = 11); and 3) SARS-CoV-2 infection with severe AHRF defined by the use of humidified high-flow nasal O2, noninvasive, or invasive mechanical ventilation (severe COVID-19, n = 10 study participants). Exclusions were hospitalization >5 days, home or current use of ACE inhibitor or ARB, severe chronic liver disease (Child–Pugh ≥12), comfort care measures in place, home-assisted ventilation except continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BIPAP) for sleep-disordered breathing, underlying malignancy, or other condition with estimated life expectancy <1 mo, pregnancy, prisoner, home supplemental oxygen use, or end-stage renal disease.
Whole blood was collected into EDTA-containing tubes with protease inhibitors (peptide measurement) and a separate tube for serum (peptidase activity) (9, 10). EDTA tubes were inverted five times following collection, placed in ice, and centrifuged at 2,200 rpm at 4°C within 2 h. Serum tubes were allowed to clot in an upright position at room temperature for ∼60 min before centrifugation at 2,200 rpm for 20 min at 4°C. Following centrifugation, plasma was obtained and aliquoted into a 15 mL polypropylene tube, serum was aliquoted into microcentrifuge tubes, and all samples were stored at −80°C. Blinded, deidentified patient samples were sent to the Wake Forest School of Medicine and processed at the Wake Forest Biomarker Analytical Core or by the Chappell laboratory. Samples were processed for plasma ANG II and ANG-(1–7), as well as serum ACE, ACE2, and prolyloligopeptidase (POP) activities. In addition to ACE2, we assessed POP activity in serum as this peptidase, which can convert both ANG I and ANG II to ANG-(1–7) was recently shown to solely contribute to the circulating levels of ANG-(1–7) in mice (9); however, the contribution of POP (or ACE2) to circulating human ANG-(1–7) is currently unknown.
Angiotensin Peptides and Peptidase Activities
Stored plasma was thawed on ice, extracted on Sep-Pak C18 columns (Waters Corp., Milford, MA), and quantified in duplicates by radioimmunoassay (RIA) for ANG II (IBL America, Minneapolis, MN, sensitivity of 2.0 pg/mL; intra-assay and interassay coefficients of variation of 8% and 20%, respectively) and ANG-(1–7) (custom RIA, sensitivity of 2.5 pg/mL; intra-assay and interassay coefficients of variation of 12% and 22%, respectively) (10, 11). Serum ACE, ACE2, and POP activities were measured in duplicates by fluorescent assays using the substrates Mca-Arg-Pro-Pro-Gly-Phe-Ser-Ala-Phe-Lys-DNP, MCA-Ala-Pro-Lys-DNP and Z-Arg-Pro-AMC, respectively, all at a final concentration of 10 µM (Enzo Life Sciences, VWR, Atlanta, GA). Since these fluorescent substrates are not entirely specific, additional peptidase inhibitors were added to prevent nonspecific conversion of each substrate as described previously (7, 10, 12). Specific activities for the peptidase assays were determined by addition of either the ACE inhibitor lisinopril, the ACE2 inhibitor MLN4760, or the POP inhibitor KYP-2047, all at a final concentration of 10 μM. Serum samples were diluted with the nonionic detergent Triton X-100 to a final concentration of 1.0% and incubated for 24 h on ice to lyse the viral coat (9). For the ACE and POP assays, 10 µL of serum was added to a final volume of 200 µL, 25 mM of HEPES (pH 7.4), 125 mM NaCl, and 10 μM ZnCl2. For ACE2, 400 µL of the Triton-treated serum was incubated with 200 µL of the strong anion exchanger Q Sepharose (Sigma Aldrich, St. Louis, IL ) in 25 mM HEPES, pH 7.0 for 20 min at room temperature to bind the peptidase (13). The gel was washed twice (2 × 1.5 mL in HEPES 125 mM NaCl) to remove endogenous inhibitors and excess Triton detergent. ACE2 was eluted in 250 µL of the HEPES buffer, 1 M NaCl and the eluted fraction containing ACE2 (100 μL) was added to a final volume of 200 μL of 25 mM HEPES buffer pH 7.0 and 10 μM ZnCl2. Serum samples were read in 96-well black plates with clear bottom wells using a SpectraMax plate reader (Molecular Devices, San Jose, CA). Peptidase activities were expressed as relative fluorescent values (RFU) per milliliter of serum.
Statistical Analysis
Normally distributed means were compared by one-way analysis of variance (three groups) and by Student’s t test (two groups). Kruskal–Wallis and Wilcoxon rank-sum tests were applied for three and two groups for non-normally distributed means. Associations of surrogate ACE2 and POP enzyme activity with direct measures were evaluated by simple linear regression.
RESULTS
The COVID-19-negative and COVID-19-positive moderate AHRF cohorts were physiologically similar, with a modified sequential organ failure assessment (mSOFA) score of 1.3 ± 0.5 vs. 1.8 ± 0.7 (P = 0.5) and a fraction of inspired oxygen () of 0.3 ± 0.0 vs. 0.3 ± 0.0 (P = 0.5). The severe AHRF COVID-19-positive cohort had increased organ failures (mSOFA 4.1 ± 1.9 vs. 1.6 ± 0.7, P < 0.001), increased oxygen requirements ( of 0.6 ± 0.1 vs. 0.3 ± 0.05, P < 0.001), increased ICU length of stay (11.1 ± 8.0 days vs. 0.5 ± 1.2 days, P = 0.002), and increased mortality (30% vs. 0%, P = 0.006) compared with the moderate cohorts. See Table 1 for full demographic and outcome data.
Table 1.
Cohort demographics and outcomes
| Moderate ARF |
Severe ARF |
||
|---|---|---|---|
| COVID-19 Negative (n = 11) |
COVID-19 Positive (n = 12) | COVID-19 Positive (n = 10) | |
| Age, yr | 50.8 (19) | 44.1 (17.2) | 56.1 (18.3) |
| Male | 5 (45.5%) | 6 (50%) | 7 (70%) |
| Ethnicity | |||
| Hispanic | 0 | 6 (50%) | 4 (40%) |
| Non-Hispanic | 11 (100%) | 6 (50%) | 5 (50%) |
| Other | 0 | 0 | 1 (10%) |
| Race | |||
| Black | 1 (9.1%) | 3 (25%) | 2 (20%) |
| White | 10 (90.9%) | 2 (16.7%) | 4 (40%) |
| Other | 0 (0%) | 7 (58.3%) | 4 (40%) |
| BMI, kg/m2 | 23.7 (9.2) | 35.4 (15.5) | 30.8 (7.0) |
| ESRD | 0 | 0 | 0 |
| Home steroids | 1 (9%) | 0 | 0 |
| Home immunosuppressant | 1 (9%) | 0 (0%) | 0 (%) |
| Sx duration, days | 3.7 (3) | 6.8 (2.8) | 8.8 (3.8) |
| COVID-19 ordinal scale* | 4.1 (0.3) | 4.0 (0.0) | 3 (0.5) |
| mSOFA | 1.3 (0.5) | 1.8 (0.8) | 4.1 (1.9) |
| Vasopressors | 0 (0%) | 0 (0%) | 0 (0%) |
| (enrollment) | 0.3 (0.0) | 0.3 (0.0) | 0.6 (0.1) |
| Respiratory support | |||
| Mechanical ventilation | 0 (0%) | 0 (0%) | 1 (10%) |
| Noninvasive ventilation | 0 (0%) | 0 (0%) | 1 (10%) |
| High-flow nasal oxygen | 0 (0%) | 0 (0%) | 7 (70%) |
| Nasal cannula | 10 (90.9%) | 12 (100%) | 1 (10%) |
| Other | 1 (9.1%) | 0 (0%) | 0 (0%) |
| Remdesivir | 0 | 4 (33.3%) | 6 (60%) |
| Corticosteroid tx | 3 (27%) | 6 (50%) | 9 (90%) |
| AKI requiring dialysis | 0 | 0 | 1 |
| Mortality | 0 | 0 | 3 (30%) |
| ICU-LOS | 0.7 (1.6) | 0.3 (0.6) | 11.1 (8.0) |
| ICU-free days | 25.1 (8.4) | 27.8 (0.6) | 13.0 (11.3) |
| Ventilator-free days | 22.0 (11.3) | 28.0 (0.0) | 16.9 (14.3) |
| Hospital-free days | 18.9 (9.8) | 22.5 (2.7) | 9.2 (9.8) |
*COVID-19 ordinal scale: 1 = dead; 2 = hospitalized on mechanical ventilation or ECMO; 3 = hospitalized on noninvasive ventilation or high-flow nasal oxygen; 4 = hospitalized on supplemental oxygen; 5 = hospitalized not on supplemental oxygen; 6 = not hospitalized. AKI, acute kidney injury; ARF, acute respiratory failure; ECMO, extracorporeal membrane oxygenation; ESRD, end-stage renal disease; , fraction of inspired oxygen; ICU, intensive care unit; LOS, length of stay; mSOFA, modified sequential organ failure assessment.
Median [interquartile range (IQR)] plasma ANG-(1–7) and ANG II values were 10.8 [7.1,16.2] and 15.3 [8.0,31.1] pM, respectively, and did not differ by the three study groups (Fig. 1). Three patients (13.6%) in the COVID-19-positive cohorts had ANG-(1–7) values greater than one standard deviation above the mean (107 to 217 pM range), whereas the maximum ANG-(1–7) concentration in the COVID-19-negative cohort was 19 pM. Serum ACE, ACE2, and POP activities were also similar in the study groups (Fig. 1).
Figure 1.

Results from each participant are shown for COVID-19-negative patients with moderate AHRF, COVID-19-positive patients with moderate AHRF, and COVID-19-positive patients with severe AHRF. Bars represent the mean and SD values for plasma levels of angiotensin II (ANG II) (A), angiotensin-(1–7) [ANG-(1–7)] (B), prolyloligopeptidase (POP) (C), angiotensin-converting enzyme (ACE) (D), and angiotensin-converting enzyme 2 (ACE2) (E). AHRF, acute hypoxic respiratory failure.
We further analyzed the data by grouping the moderate and severe COVID-19-positive cohorts and comparing them with COVID-19-negative patients. Using these criteria, we also found no differences in ANG II, ANG-(1–7), ACE2, or POP in COVID-19-negative versus COVID-19-positive patients. We did find that ACE activity was reduced in the COVID-19-positive patients versus COVID-19-negative patients (2.2 ± 0.9 × 105 vs. 2.9 ± 0.8 × 105 RFU/mL, P = 0.03) (Fig. 2). The ANG-(1–7):ANG II ratios that are often reported as surrogates for ACE2 activity, did not correlate with either ACE2 or POP activities (Fig. 3).
Figure 2.

Results from each partcipant are shown for peptide levels grouped by COVID-19-negative patients with moderate AHRF and combined moderate and severe COVID-19-positive patients with AHRF. Bars represent the mean and SD values for plasma levels of angiotensin II (ANG II) (A), angiotensin-(1–7) [ANG-(1–7)] (B), ANG-(1–7):ANG II ratio (C), angiotensin-converting enzyme (ACE) (D), angiotensin-converting enzyme 2 (ACE2) (E), and prolyloligopeptidase (POP) (F). AHRF, acute hypoxic respiratory failure.
Figure 3.
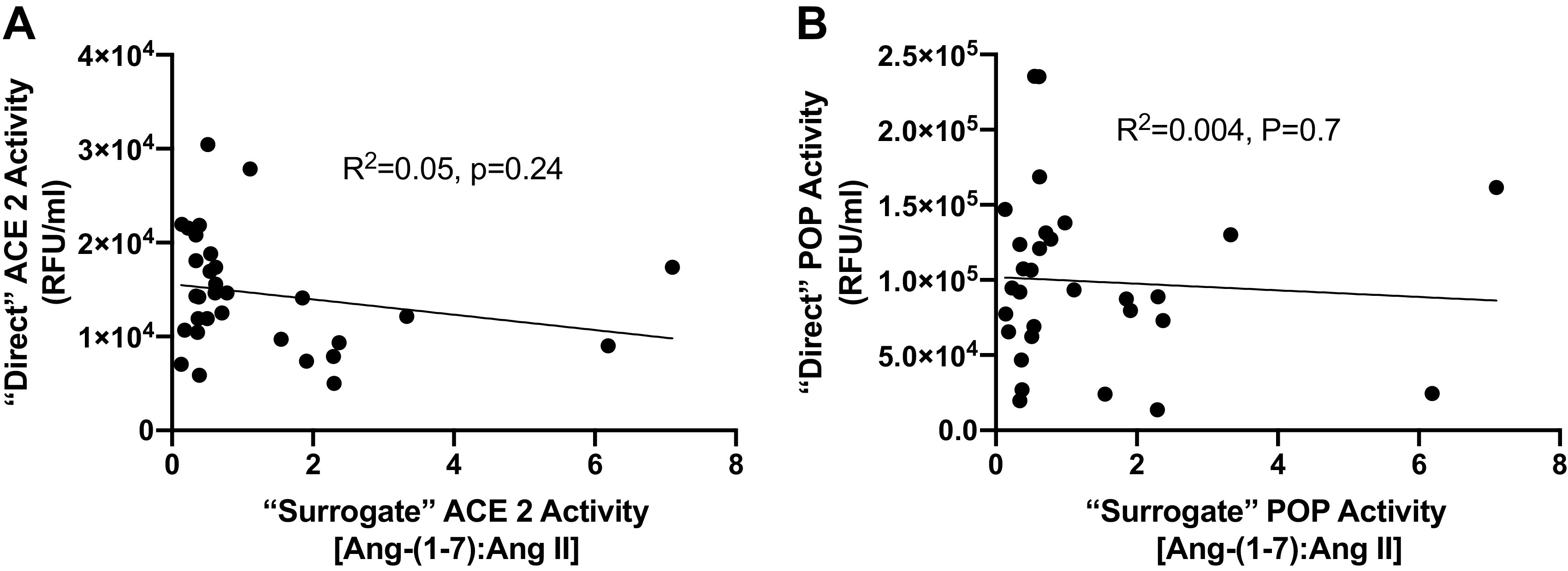
Correlation between “direct” and “surrogate” ACE2 or POP activity. Results from each participant are shown and represent the direct serum ACE2 activity (A) and POP activity (B) plotted against “surrogate” enzymatic activities using the corresponding plasma ANG-(1–7):ANG II peptide ratio. Each dot represents the paired corresponding enzymatic activity and peptide ratio for an individual patient. ACE2, angiotensin-converting enzyme 2; ANG, angiotensin; POP, prolyloligopeptidase.
DISCUSSION
In this prospective pilot study, we enrolled patients with moderate and severe COVID-19 and COVID-19-negative physiologically matched controls. We measured five components of the circulating RAS with validated measures and obtained values that are well within the physiological concentrations accepted in the field (6). This information may be useful for future studies quantifying the circulating RAS in COVID-19 and the effect of RAS augmentation on circulating angiotensins.
Our data highlight the importance of proper controls, and the importance of carefully assessing the circulating RAS. The challenges of quantifying the circulating RAS are, in part, due to the multiple peptidases contributing to the generation of ANG-(1–7) from ANG II or ANG I and the variability of assays used to quantify this pathway (7). For example, application of the ANG-(1–7):Ang II ratio as a surrogate for ACE2 activity may not accurately reflect ACE2 activity as other peptidases (e.g., renin, POP, ACE, aminopeptidases, neprilysin, and dipeptidyl aminopeptidases) contribute to the overall processing (both generation and metabolism) of ANG II and ANG-(1–7) in the circulation (9, 14). Moreover, the contribution of these or other peptidases in tissues that may actively participate in the local release of ANG-(1–7) into the circulation is unknown (15).
Other than a mild reduction in ACE activity in COVID-19-positive patients, we did not find significant differences in RAS components in our study. Interestingly, we did find that three patients in the COVID-19-positive cohorts had significant elevation of ANG-(1–7); a finding not seen in the COVID-19-negative controls. Since ACE is the primary pathway for the metabolism of ANG-(1–7) in the circulation, it is possible that the downregulation of ACE in the COVID-19-positive patients might contribute to increased ANG-(1–7) (16). However, the clinical significance of these findings are unknown and larger studies are needed to further explore this possibility.
A number of possibilities may underlie the failure to find differences in many of the circulating RAS components. First, the circulating RAS may not reflect the local tissue levels, which may be particularly altered in the lungs in COVID-19 (17). An assessment of the lung compartment in COVID-19 is more challenging, though others have shown that bronchoalveolar lavage in patients with COVID-19 is feasible (18). Second, our study is likely underpowered to reveal true differences in circulating RAS components. For example, based on the distributions of angiotensins we observed in this study, we calculated that we would have 80% power to detect differences in circulating ANG II and ANG-(1–7) between COVID-19-negative and COVID-19-positive hospitalized patients with ∼127 participants per group. In similar regard, we were underpowered to explore the relationship between other potential confounding covariates that may alter the RAS in patients with COVID-19, such as age, sex, body mass index (BMI), and corticosteroid use (19–21). For instance, dexamethasone, which is now a standard of care in severe COVID-19 has been shown to upregulate ACE and ACE2 (20, 21). Finally, our study only sampled the circulating RAS at one time point, and assessment of serial time points may shed additional insight on how the RAS changes over time in COVID-19.
Ultimately, clinical trials are needed to determine the extent that modulating the RAS in COVID-19 improves outcomes. Indeed, the measurement of circulating angiotensins in this context will provide value to better understand the influence of the RAS on patient outcomes and the response of the circulating RAS to drugs targeting this pathway in COVID-19 and beyond.
GRANTS
This work was supported by the United States Centers for Disease Control and Prevention through contract 75D30119C05670 (to D.C.F. and W.H.S).
DISCLAIMERS
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.C.F., K.W.G., J.D.C., W.H.S., and M.C.C. conceived and designed research; D.C.F., T.M.G., and M.C.C. performed experiments; D.C.F., C.L.S., and M.C.C. analyzed data; D.C.F. and M.C.C. interpreted results of experiments; D.C.F. and C.L.S. prepared figures; D.C.F., S.P.C., and W.H.S. drafted manuscript; D.C.F., K.W.G., C.L.S., S.P.C., T.M.G., J.D.C., W.H.S., and M.C.C. edited and revised manuscript; D.C.F. and M.C.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Mary LaRose, Lina Purcell, Jakea Johnson, Adrienne Baughman, and the Wake Forest Biomarker Analytical Core.
REFERENCES
- 1.South AM, Diz DI, Chappell MC. COVID-19, ACE2, and the cardiovascular consequences. Am J Physiol Heart Circ Physiol 318: H1084–H1090, 2020. doi: 10.1152/ajpheart.00217.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pathangey G, Fadadu PP, Hospodar AR, Abbas AE. Angiotensin-converting enzyme 2 and COVID-19: patients, comorbidities, and therapies. Am J Physiol Lung Cell Mol Physiol 320: L301–L330, 2021. doi: 10.1152/ajplung.00259.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai ZW, Hanchapola I, Steer DL, Smith AI. Angiotensin-converting enzyme 2 ectodomain shedding cleavage-site identification: determinants and constraints. Biochemistry 50: 5182–5194, 2011. doi: 10.1021/bi200525y. [DOI] [PubMed] [Google Scholar]
- 4.Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, Chappell MC, Wohlford-Lenane C, McCray PB, Jr.. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol 297: L84–L96, 2009. doi: 10.1152/ajplung.00071.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sparks MA, South AM, Badley AD, Baker-Smith CM, Batlle D, Bozkurt B, Cattaneo R, Crowley SD, Dell'Italia LJ, Ford AL, Griendling K, Gurley SB, Kasner SE, Murray JA, Nath KA, Pfeffer MA, Rangaswami J, Taylor WR, Garovic VD. Severe acute respiratory syndrome coronavirus 2, COVID-19, and the renin-angiotensin system: pressing needs and best research practices. Hypertension 76: 1350–1367, 2020. doi: 10.1161/HYPERTENSIONAHA.120.15948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chappell MC, Pirro NT, South AM, Gwathmey TM. Concerns on the specificity of commercial ELISAs for the measurement of angiotensin (1-7) and angiotensin II in human plasma. Hypertension 77: e29–e31, 2021. doi: 10.1161/HYPERTENSIONAHA.120.16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol 310: H137–H152, 2016. doi: 10.1152/ajpheart.00618.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, Wang Z, Li J, Li J, Feng C, Zhang Z, Wang L, Peng L, Chen L, Qin Y, Zhao D, Tan S, Yin L, Xu J, Zhou C, Jiang C, Liu L. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci 63: 364–374, 2020. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serfozo P, Wysocki J, Gulua G, Schulze A, Ye M, Liu P, Jin J, Bader M, Myöhänen T, García-Horsman JA, Batlle D. Ang II (angiotensin II) conversion to angiotensin-(1-7) in the circulation is POP (prolyloligopeptidase)-dependent and ACE2 (angiotensin-converting enzyme 2)-independent. Hypertension 75: 173–182, 2020. doi: 10.1161/HYPERTENSIONAHA.119.14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.South AM, Nixon PA, Chappell MC, Diz DI, Russell GB, Jensen ET, Shaltout HA, O’shea TM, Washburn LK. Association between preterm birth and the renin-angiotensin system in adolescence: influence of sex and obesity. J Hypertens 36: 2092–2101, 2018. doi: 10.1097/HJH.0000000000001801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brosnihan KB, Chappell MC. Measurement of angiotensin peptides: HPLC-RIA. Methods Mol Biol 1527: 81–99, 2017. doi: 10.1007/978-1-4939-6625-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goossens F, De Meester I, Vanhoof G, Scharpé S. A sensitive method for the assay of serum prolyl endopeptidase. Eur J Clin Chem Clin Biochem 30: 235–238, 1992. [PubMed] [Google Scholar]
- 13.Lew RA, Warner FJ, Hanchapola I, Yarski MA, Ramchand J, Manohar J, Burrell LM, Smith AI. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp Physiol 93: 685–693, 2008. [Erratum in Exp Physiol 100: 988, 2015]. doi: 10.1113/expphysiol.2007.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reindl-Schwaighofer R, Hodlmoser S, Eskandary F, Poglitsch M, Bonderman D, Strassl R, Aberle JH, Oberbauer R, Zoufaly A, Hecking M. ACE2 elevation in severe COVID-19. Am J Respir Crit Care Med 203: 1191–1196, 2021. doi: 10.1164/rccm.202101-0142LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chappell MC, Gomez MN, Pirro NT, Ferrario CM. Release of angiotensin-(1-7) from the rat hindlimb: influence of angiotensin-converting enzyme inhibition. Hypertension 35: 348–352, 2000. doi: 10.1161/01.hyp.35.1.348. [DOI] [PubMed] [Google Scholar]
- 16.Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 31: 362–367, 1998. doi: 10.1161/01.hyp.31.1.362. [DOI] [PubMed] [Google Scholar]
- 17.Campbell DJ. Clinical relevance of local renin angiotensin systems. Front Endocrinol (Lausanne) 5: 113, 2014. doi: 10.3389/fendo.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pickens CO, Gao CA, Cuttica M, Smith SB, Pesce L, Grant R, Kang M, Morales-Nebreda L, Bavishi AA, Arnold J, Pawlowski A, Qi C, Budinger GS, Singer BD, Wunderink RG. Bacterial superinfection pneumonia in SARS-CoV-2 respiratory failure (Preprint). medRxiv 2021. doi: 10.1101/2021.01.12.20248588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jehpsson L, Sun J, Nilsson PM, Edsfeldt A, Swärd P. Serum renin levels increase with age in boys resulting in higher renin levels in young men compared to young women, and soluble angiotensin-converting enzyme 2 correlates with renin and body mass index. Front Physiol 11: 622179, 2020. doi: 10.3389/fphys.2020.622179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fishel RS, Eisenberg S, Shai SY, Redden RA, Bernstein KE, Berk BC. Glucocorticoids induce angiotensin-converting enzyme expression in vascular smooth muscle. Hypertension 25: 343–349, 1995. doi: 10.1161/01.hyp.25.3.343. [DOI] [PubMed] [Google Scholar]
- 21.Sinha S, Cheng K, Schaffer AA, Aldape K, Schiff E, Ruppin E. In vitro and in vivo identification of clinically approved drugs that modify ACE2 expression. Mol Syst Biol 16: e9628, 2020. doi: 10.15252/msb.20209628. [DOI] [PMC free article] [PubMed] [Google Scholar]