

SUMMARY
Pathogenic Th17 cells drive inflammation in autoimmune disease, yet the molecular programming underlying Th17 cell pathogenicity remains insufficiently understood. Activation of Toll-like receptor 2 (TLR2) increases Th17 cell inflammatory potential, but little is known regarding the mechanistic outcomes of TLR2 signaling in Th17 cells. Here, we demonstrate that TLR2 is comparable to IL-23 in inducing pathogenicity and increasing the migratory capacity of Th17 cells. We perform RNA sequencing of Th17 cells stimulated though the TLR2 pathway and find differential expression of several genes linked with the Th17 genetic program as well as genes not previously associated with pathogenic Th17 cells, including Ipcef1. Enforced expression of Ipcef1 in Th17 cells abolishes the TLR2-dependent increases in migratory capacity and severely impairs the ability of Th17 cells to induce experimental autoimmune encephalomyelitis. This study establishes the importance of the TLR2 signaling pathway in inducing Th17 cell pathogenicity and driving autoimmune inflammation.
Graphical Abstract
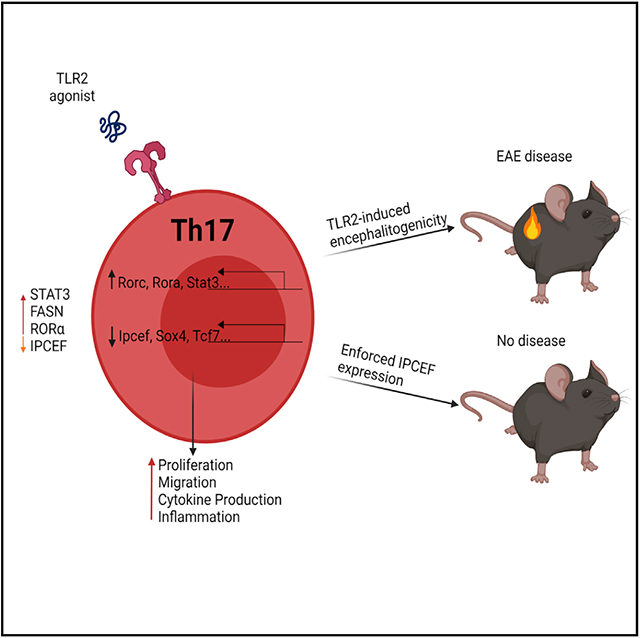
In brief
Marks et al. demonstrate an important role for TLR2 in driving inflammatory T cell function. They also reveal that IPCEF is a target for TLR2 signaling and that IPCEF expression inhibits pathogenic T cell migration in a model of autoimmune inflammation.
INTRODUCTION
Pattern recognition receptors, including Toll-like receptors (TLRs), are vital for the function of the immune system. TLRs recognize both microbial products and endogenous danger signals and are typically triggered in cells of the innate immune response to facilitate pathogen clearance and to help shape adaptive immunity (Kawai and Akira, 2007). In addition to expression on innate immune cells and other non-hematopoietic cells, TLRs are expressed and highly functional in both T and B lymphocytes (Kawasaki and Kawai, 2014; Janeway and Medzhitov, 2002). CD4+ T cells, in particular, have been shown to express various TLRs, including TLR2, TLR4, TLR1, TLR6, and TLR9 (Kabelitz, 2007; Reynolds et al., 2010). A recent study demonstrated that the TLR adaptor molecule TIRAP is induced by T cell receptor (TCR) signaling through the mechanistic target of rapamycin complex 1 (mTORC1) and is required for TLR2-induced interferon-γ (IFN-γ) production in T helper 1 (Th1) cells and CD8+ T cells. Furthermore, TLR2 signaling activates mTORC1 (Imanishi et al., 2020). TLRs have known roles in regulatory CD4+ T cells (Treg); several TLRs are expressed at higher levels compared to conventional CD4+ T cells. TLR2 activation in Tregs blocks suppressive function and increases interleukin-17 (IL-17) production, but also induces proliferation that conversely may result in increased suppression (Nawijn et al., 2013; Sutmuller et al., 2006; Dai et al., 2009; Nyirenda et al., 2011). Activation of TLR4 on Tregs also results in a loss of suppression (Nyirenda et al., 2011). Th17 cells express certain TLRs, including TLR4 and TLR2, as well as TLR1 and TLR6, which heterodimerize with TLR2 (Reynolds et al., 2010, 2012). Activation of TLR4 promotes Th17 cell proliferation, migration, and the development of experimental autoimmune encephalomyelitis (EAE), a mouse model for multiple sclerosis (MS). However, TLR4 does not appear to contribute to Th17 lineage commitment (Reynolds et al., 2012; McAleer et al., 2010; Dias et al., 2019). Importantly, activation of TLR2 directly contributes to Th17 differentiation and function (Dias et al., 2019; Reynolds et al., 2010).
Th17 cells are instrumental for several autoimmune diseases, including MS, psoriasis, and rheumatoid arthritis (Jadidi-Niaragh and Mirshafiey, 2011; Yasuda et al., 2019). Th17 cells identified in autoimmune diseases are often incredibly proliferative, produce high levels of IL-17, and frequently gain expression of the cytokines granulocyte macrophage-colony-stimulating factor (GM-CSF) and IFN-γ, as well as the transcription factor T-bet (Hirota et al., 2011; Yosef et al., 2013). These Th17 cells are deemed “pathogenic” due to their deleterious inflammatory function (Lee et al., 2012). While both IL-23 and IL-1 can contribute to the differentiation of pathogenic Th17 cells, the molecular programming underlying their pathogenicity remains insufficiently understood (Ghoreschi et al., 2010; Chung et al., 2009; McGeachy et al., 2009; Yosef et al., 2013). Furthermore, very little is known regarding the outcomes TLR2 signaling in Th17 cells.
We have previously shown that the activation of TLR2 with the synthetic agonists PAM3CSK4 (TLR2/1) or FSL-1 (TLR2/6) during Th17 differentiation results in the increased production of IL-17A and IL-17F and promotes the proliferation of Th17 cells in vitro (Reynolds et al., 2010). We demonstrated that TLR2 expression was also necessary for CD4+ T cells to induce severe EAE disease, likely through the recognition of endogenous danger signals (Reynolds et al., 2010). Importantly, TLR2 activation in human CD4+ T cells likewise promotes Th17 cell generation and IL-17 expression (Nyirenda et al., 2011; Zhao et al., 2015). While these previous studies highlight the importance of TLR2 in regulating Th17 cell differentiation and effector function, it remains unclear whether TLR2 activation is sufficient to induce pathogenicity in Th17 cells.
Th17 cells must migrate to the site of antigen for effector function; during EAE, they infiltrate the central nervous system (CNS) and direct inflammation against cognate neuroantigen. Th17 cells uniformly express chemokine receptor 6 (CCR6), the receptor for chemokine ligand 20 (CCL20) (Yamazaki et al., 2008; Kim, 2009). Previous work has demonstrated a requirement for CCR6 on Th17 cells to induce EAE or experimental arthritis (Yamazaki et al., 2008; Hirota et al., 2007; Dohlman et al., 2013). However, it remains unknown whether alternative pathogenic stimuli affects the migratory capacity of Th17 cells. In this study, we demonstrate that interaction protein for cytohesin exchange factors (IPCEF)/Pip3E inhibits TLR2-dependent Th17 cell migration. IPCEF is a relatively understudied scaffolding protein that associates with cytohesin2/ARF nucleotide-binding site opener (ARNO), allowing for Rac activation, which subsequently leads to lamellipodia formation and membrane ruffling (Venkateswarlu, 2003; White et al., 2010; Attar et al., 2012). Furthermore, silencing IPCEF expression in MDCK cells results in the loss of lamellipodia and reduces migration (White et al., 2010).
Here, we demonstrate that TLR2 activation of antigen-specific Th17 cells is sufficient to induce encephalitogenicity and, subsequently, severe EAE disease. RNA sequencing (RNA-seq) revealed 745 differentially expressed genes in TLR2-stimulated Th17 cells compared to Th17 cells polarized with transforming growth factor-β (TGF-β) and IL-6 alone. Several of these transcriptome changes have previously been associated with Th17 cell pathogenicity; however, we also have identified several factors that appear to be unique to TLR2 activation, including IPCEF. IPCEF expression in Th17 cells surprisingly diminishes the migratory capacity of pathogenic Th17 cells in vitro and furthermore inhibits the ability of TLR2-activated Th17 cells to induce EAE disease.
RESULTS
TLR2 signaling does not enhance Th17 cell differentiation in combination with IL-1β and IL-23
Our previous research, using TLR2 germline knockout mice, demonstrated that TLR2 signaling promotes Th17 cell responses (Reynolds et al., 2010). Th17 cells polarized with TGF-β and IL-6, along with the TLR2 agonists PAM3CSK4 or FSL-1, exhibited increased IL-17 production. Given that polarizing Th17 cells with TGF-β and IL-6 is not as potent for realizing inflammatory potential as compared to the IL-1β, IL-6, and IL-23 polarizing Th17 cell condition (Ghoreschi et al., 2010; McGeachy et al., 2009; Gaffen et al., 2014), we for the first time assessed the effect of TLR2 ligation on this more pathogenic condition. We will refer to Th17 cells differentiated with TGF–β and IL-6 as “Th17(β)” cells and those differentiated with IL-1β, IL-6, and IL-23 as “Th17(IL-1/IL-23)” cells.
We used TLR2f/f mice (floxed exon 2) to delete Tlr2 in the presence of Cre recombinase. Naive CD4+ T cells were stimulated with αCD3 and αCD28 under polarizing conditions for 1 day before the deletion of TLR2 by retroviral delivery of Cre. Control cells were treated with a mock vector (empty vector; RV-Mock) The expression of TLR2 in Th17(IL-1/IL-23) and Th17(β) cells was assessed with and without the retroviral delivery of Cre (Figures S1A and S1B). Surprisingly, TLR2 expression was markedly higher in Th17(IL-1/23) cells stimulated with PAM3CSK4 (Figure S1B). However, despite enhanced TLR2 expression, PAM3CSK4 stimulation did not influence IL-17 production in Th17(IL-1/IL-23) cells as we had observed for Th17(β) cells (Reynolds et al., 2010) (Figures 1A and 1B). We observed similar results when Th17(IL-1/23) cells were stimulated with FSL-1 (not shown).
Figure 1. TLR2 signaling increases the proliferation of Th17(β) cells but does not synergize with IL-1β and IL-23.
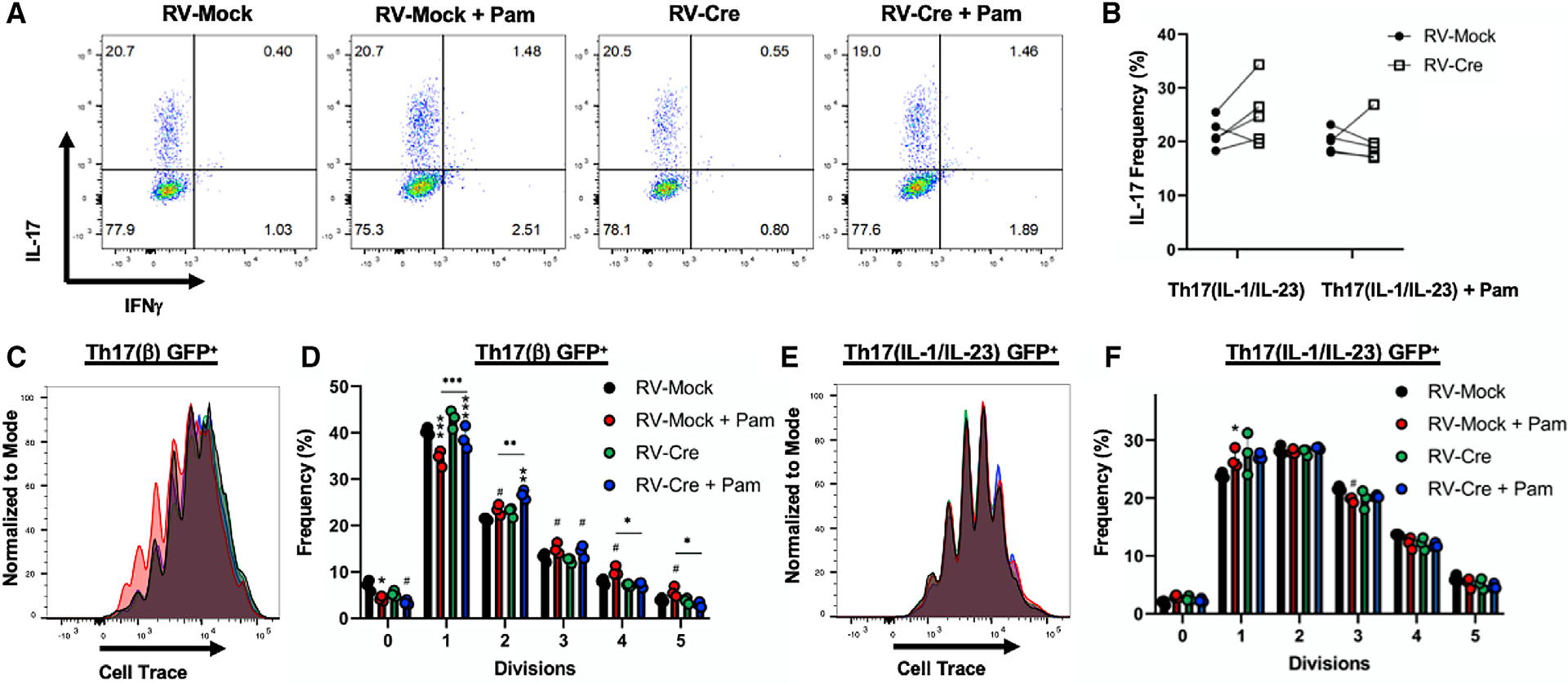
(A) Representative flow cytometry analysis (IL-17 and IFN-γ) of TLR2f/f Th17(IL-1/IL-23) cells following 4-day differentiation and transduction of control vector (RV-Mock) or Cre (RV-Cre).
(B) Compilation of data from 2 representative experiments.
(C–F) Representative proliferation histograms and pooled analysis of TLR2f/f Th17(β) (C and D) or Th17(IL-1/IL-23) (E and F) cells following 4-day differentiation. n =3 technical replicates per group. Cells were transduced with RV-Mock or RV-Cre 1 day following initial TCR activation and gated on retrovirus (GFP+) for analysis. Data are representative of 3 individual experiments and are presented as means ± SDs. Comparisons between untreated and PAM3CSK4-stimulated cells are shown directly above the bars, while comparisons between RV-Mock and RV-Cre are shown by symbols above the lines. #p < 0.05, *p < 0.01, **p < 0.001, ***p <0.0001 by 2-way ANOVA.
Sustained signaling is necessary for TLR2-dependent proliferative effects
We previously demonstrated that PAM3CSK4 stimulation drives Th17(β) cell proliferation in combination with TCR activation (Reynolds et al., 2010). Therefore, we investigated whether TLR2 deletion following TCR activation could affect proliferation by Th17(β) or Th17(IL-1/IL-23) cells in vitro. Th17(β) cells differentiated with PAM3CSK4 and receiving control retrovirus (empty vector; RV-Mock) exhibited more proliferation, illustrated by the frequency of cells dividing ≥4 times, with substantially fewer dividing 1 or 0 times. Furthermore, the deletion of TLR2 1 day after TCR activation (RV-Cre) abrogated this effect (Figures 1C and 1D), indicating that sustained TLR2 signaling is required for the increased proliferative activity of TLR2-stimulated Th17(β) cells. Still, Cre-treated cells had reductions in the 0–1 division and an increase in the 2–3 division sub-populations in comparison to RV-Mock, demonstrating that simultaneous TCR and TLR2 engagement does enhance early cell division.
We also analyzed the effect of TLR2 ligation on the more highly proliferative Th17 (IL-1/IL-23) subset. The addition of PAM3CSK4 did not further enhance proliferation (Figures 1E and 1F). Thus, TLR2 activation does not synergize with IL-1β and IL-23 in enhancing Th17(IL-1/IL-23) cell IL-17 production or proliferation as previously observed for TGF(β) cells. However, given the high expression of TLR2 observed in these cells (Figure S1B), these in vitro findings do not rule out the possibility of TLR2 signaling contributing to other mechanisms underlying IL-1β- and/or IL-23-mediated Th17 cell inflammation, especially in vivo.
TLR2 induces encephalitogenicity in Th17 cells
Our work demonstrated that TLR2 signaling in effector CD4+ T cells promotes EAE disease and encephalitogenic cytokine production (Reynolds et al., 2010). Those studies, however, were conducted using a total CD4+ T cell transfer model of EAE, which did not directly address Th17 cells. Therefore, to assess whether TLR2 activation alone is sufficient to induce Th17 pathogenicity, we performed adoptive transfer EAE using myelin oligodendrocyte glycoprotein 35–55 (MOG35–55)-specific cells expanded with peptide alone or with the addition of either IL-23 or PAM3CSK4. Expanding MOG35–55(MOG)-specific CD4+ T cells in the presence of IL-23 induces Th17 cells that cause EAE upon transfer, while expansion with MOG alone does not (McGeachy et al., 2009; Kroenke et al., 2008; El-Behi et al., 2011). MOG-specific cells were generated by immunizing IL-17-GFP mice with MOG35–55 emulsified in incomplete Freund’s adjuvant (IFA) with lipopolysaccharides (LPS). We used IFA and LPS as an adjuvant rather than complete Freund’s adjuvant (CFA) to prevent the mycobacterium present in CFA from triggering TLR2 during Th17 cell priming. Furthermore, to abrogate the effects of TLR2 on antigen-presenting cells (APCs) during expansion, we also similarly immunized TLR2−/− mice. After 7 days’ immunization, CD4+ T cells isolated from spleens and draining lymph nodes of immunized IL-17-GFP mice were combined with CD4−APCs from the TLR2−/− mice. Th17 cells were expanded for 8 days with MOG in the absence or presence of either 5 ng/mL IL-23 or 1 μg/mL PAM3CSK4. Following expansion, GFP+ cells expanded with PAM3CSK4 or IL-23 produced similar amounts of IL-17 and IFN-γ (Figure S2A). As expected, CD4+ T cells expanded with MOG alone did not proliferate to the same extent compared to the other two groups. Following expansion, we then sorted and transferred 5 × 105 GFP+ cells intravenously (i.v.) into Rag1−/− mice followed by a booster dose of MOG/IFA/LPS to induce EAE. Mice receiving GFP+ cells expanded with either PAM3CSK4 or IL-23 induced robust EAE disease, while those expanded with MOG alone did not (Figures 2A and 2B). Initially, we did not observe differences between PAM3CSK4 or IL-23 expansion; however, at the later stage, PAM3CSK4 expansion led to increases in disease incidence (Figures 2A and 2B). Thus, TLR2 activation alone is sufficient to induce Th17 cell encephalitogenicity.
Figure 2. TLR2 induces encephalitogenicity in Th17 cells.
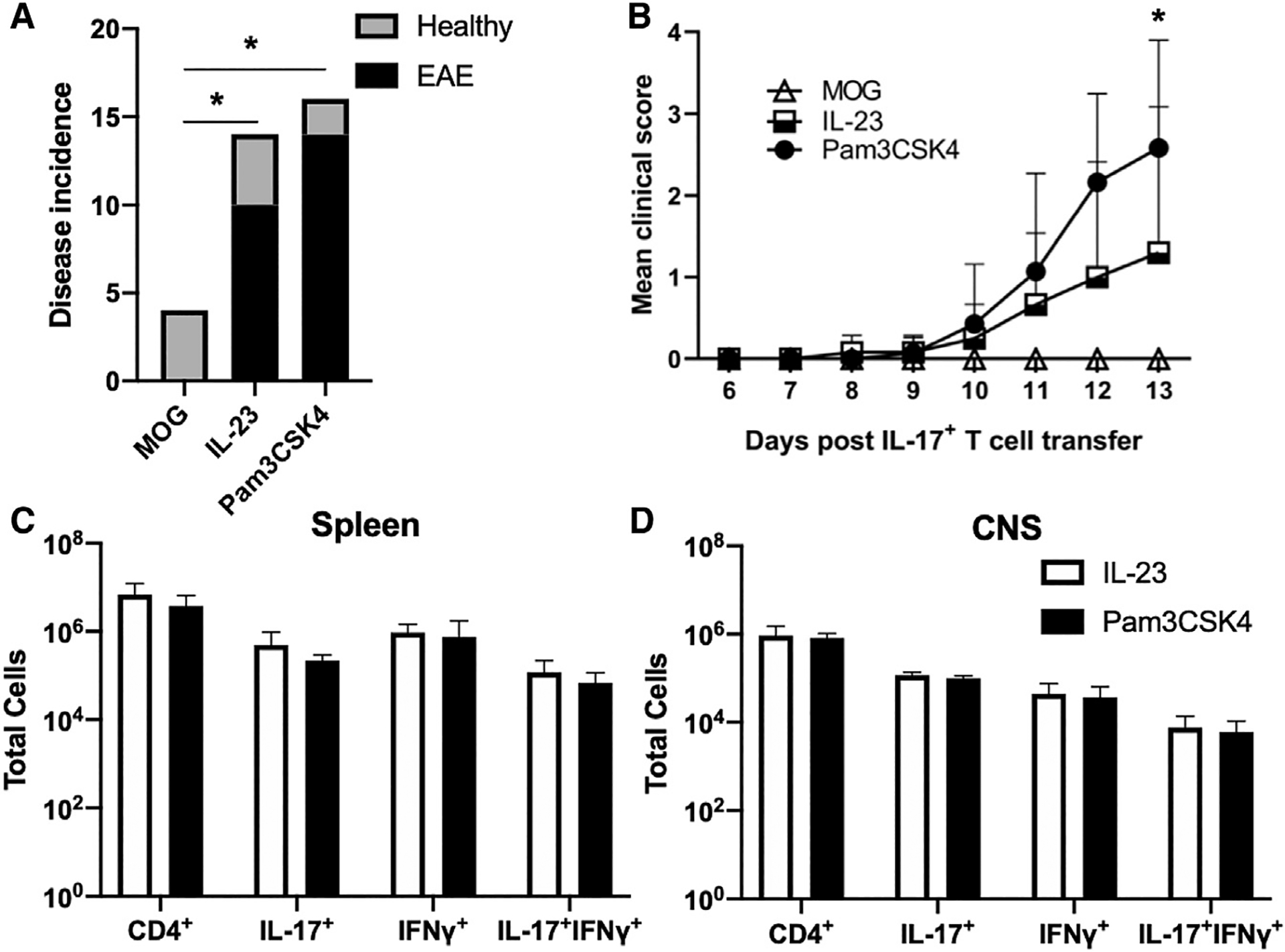
(A) Incidence of EAE in mice following transfer of IL-17+ cells expanded with MOG35–55 alone, MOG35–55 + IL-23, or MOG35–55 + PAM3CSK4. *p < 0.01 (Fisher’s exact test). n = 4–16 pooled biological replicates per group.
(B) Mean clinical scores of EAE mice were assessed until the experimental endpoint. *p < 0.01 (2-way ANOVA).
(C) Mean cell numbers of CD4+, CD11B+, CD4+IL-17+, CD4+IFN-γ+, and CD4+IL-17+IFN-γ+ isolated from CNS tissues of individual mice.
(D) Mean cell numbers of the same populations isolated from spleens of EAE mice. Data are representative of 3 independent experiments and are presented as means ± SDs.
Next, we determined the effects of TLR2 activation on Th17 cell infiltration and encephalitogenic cytokine production in the spleen and CNS tissues. We found that there were no differences in the total number of CD4+ T cells or the numbers of IL-17+, IFN-γ+, or IL-17+IFN-γ+ CD4+ T cells in the spleens of EAE mice receiving PAM3CSK4- or IL-23-treated cells (Figure 2C). Within the CNS, we observed similar numbers of infiltrating CD4+ T cells, as well as comparable IL-17 production by PAM3CSK4- or IL-23-activated CD4+ T cells (Figure 2D). Furthermore, we observed similar production of other encephalitogenic cytokines, namely IFN-γ and GM-CSF (Figures 2D and S2B). Collectively, our data indicate that TLR2 induces Th17 cell pathogenicity and helps promote Th17-driven autoimmune inflammation.
TLR2 activation alters the transcriptional program of differentiating Th17 cells
Since the activation of TLR2 is sufficient to promote Th17 cell encephalitogenicity, we investigated whether TLR2 signaling promotes a genetic program permissive for pathogenicity. We performed RNA-seq on Th17 cells at day 3 of differentiation to investigate both early and late transcriptional changes. We used biological duplicates polarized under the Th17(β) condition in the absence or presence of PAMSCSK4, while naive CD4+ T cells served as a control. We found that 390 genes were upregulated in Th17 cells treated with PAM3CSK4 compared to untreated Th17 cells. We identified several genes associated with the Th17 cell lineage, including Rora, Tnf, Il21, Il17f, and Csf2 (Figures 3A and 3B; Table S1), which verified our previous results (Reynolds et al., 2010). Fasn was also enhanced with TLR2 stimulation, further confirming our study showing this gene enhanced in TLR2-activated or IL-1β and IL-23-polarized Th17 cells (Young et al., 2017). Other notable genes include but are not limited to the Th17 pioneering factors Batf and Irf4; proliferation and survival genes Ccnd2, Cdk6, and Bcl2; and the CD8-determining lineage factor, Runx3, which stabilizes and promotes pathogenicity of IFN-γ-expressing Th17 cells (Wang et al., 2014; Ciofani et al., 2012). We also found enhanced expression of Hif1a in TLR2-activated Th17 cells, which is involved in promoting Th17 differentiation in response to the hypoxic conditions associated with rapid proliferation (Figures 1C and 1D) (Shi et al., 2011). To further validate our RNA-seq results, we performed qPCR, focusing on TLR2-enhanced genes mostly unreported by previous studies (Figure S3A). For all of the genes analyzed, we observed very similar results. Importantly, since PAM3CSK4 is a TLR1/2 ligand, we also validated this dataset with FSL-1 (TLR2/6 agonist) treatment, which demonstrated that differing paths to TLR2 activation result in comparable transcriptome changes (Figure S3A).
Figure 3. TLR2 activation alters the transcriptional program of differentiating Th17 cells.
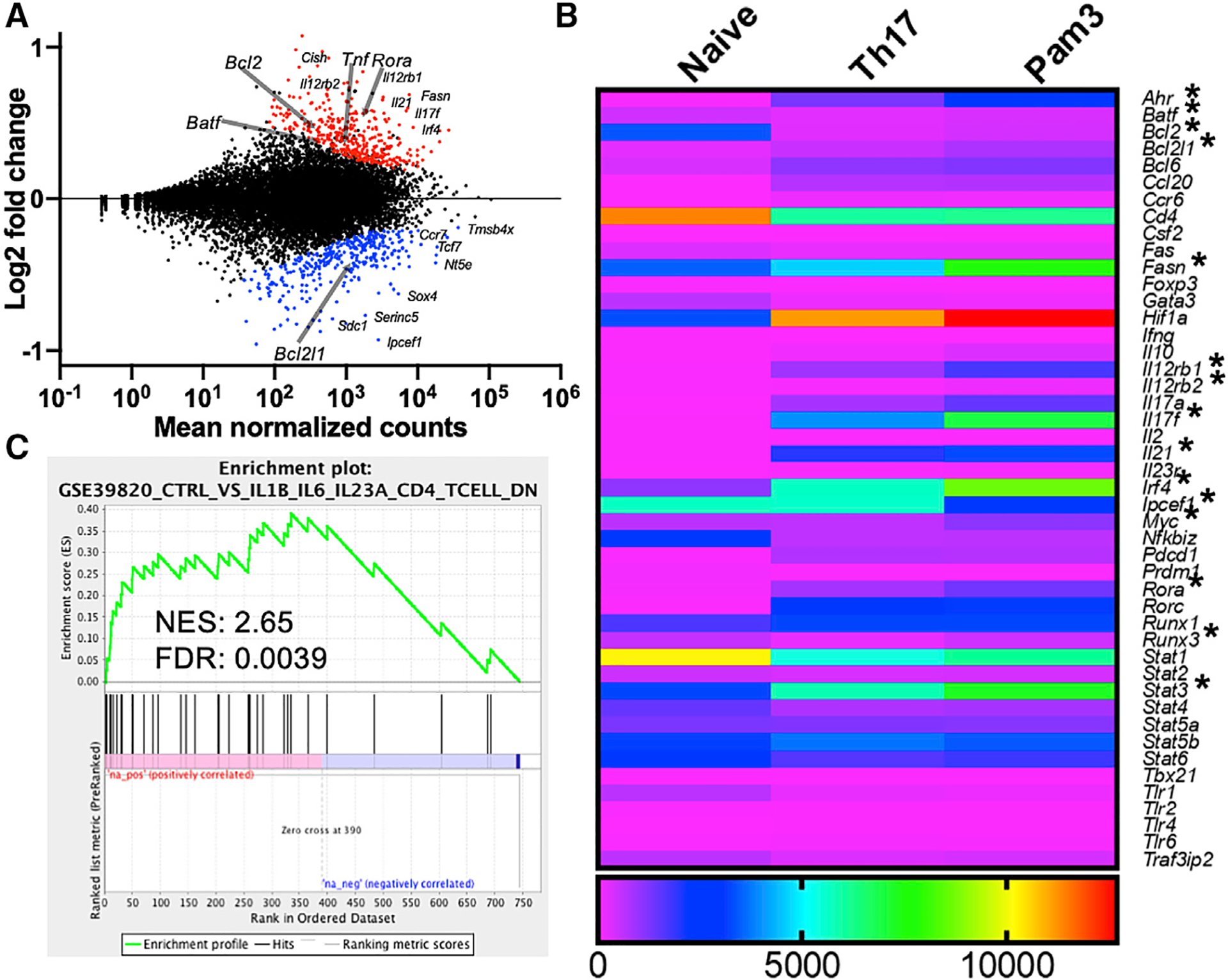
(A) Graphical representation of genes upregulated (red) or downregulated (blue) in PAM3CSK4-activated Th17(β) cells compared to non-PAM-treated Th17 cells by RNA sequencing. n = 2 biological replicates per group.
(B) Heatmap comparing expression levels of selected genes from naive CD4+, Th17(β) cells, and Th17(β) cells stimulated with PAM3CSK4. *p < 0.01 (FDR).
(C) Gene set enrichment analysis of TLR2-stimulated Th17 cells compared to Th17 cells generated with IL-1, IL-6, and IL-23. Normalized enrichment score (NES) and statistical significance (FDR) are listed.
Conversely, we found that 355 genes were downregulated in response to TLR2 activation, including genes associated with other CD4+ T cell subsets such as Gata3 and Stat1. Genes associated with immunosuppression or antiinflammatory activity, including Foxp3 and Il10, were also substantially downregulated, suggesting that TLR2 triggers pro-inflammatory effector functions at the transcriptional level (Figures 3A and 3B). Consequently, TLR2 activation in Th17(β) cells induces transcriptional changes that promote Th17 pathogenicity by both recognized and unique means.
TLR2-activated Th17 cells exhibit gene expression patterns permissive to pathogenicity
We next addressed whether TLR2 ligation causes similar programmatic changes compared to cells polarized with IL-23 and IL-1β, known contributors to pathogenicity (Ghoreschi et al., 2010). Gene set enrichment analysis (GSEA) was used to investigate transcripts differentially expressed with TLR2 activation compared to those expressed with IL-1β, IL-6, and IL-23 stimulation (Ghoreschi et al., 2010; Lee et al., 2012). Expression patterns in TLR2-activated Th17(β) cells were similar to those enriched in the Th17(IL-1/IL-23) condition (normalized enrichment score [NES] of 2.65; Figure 3C). In addition, genes enriched in our dataset exhibited a similar pattern to those enriched in Th17 cells polarized with TGF-β, IL-6, and IL-23 as opposed to only TGF-β and IL-6 (Figure S3B). Conversely, we found a negative correlation (NES of −2.77; Figure S3C) when comparing our dataset with a published set of genes bound by FoxP3 (Zheng et al., 2007), indicating that TLR2 induces inflammatory rather than suppressive outcomes in TGF-β-polarized Th17 cells.
TLR2 activation induces the downregulation of IPCEF in Th17 cells
Our RNA-seq results identified several genes that were upregulated in Th17 cells following TLR2 activation. Our discoveries include genes known to be induced in pathogenic Th17 cells (Figure 3C), as well as genes that appear to be unique to TLR2 stimulation. However, we were also interested in the genes suppressed following TLR2 activation, which led to studies on Ipcef. IPCEF is a scaffolding protein that binds the coiled-coil domain of cytohesin2/ARNO, allowing for Rac activation. Our RNA-seq results (Figures 3A and 3B) demonstrated that IPCEF was severely reduced (2-fold) following TLR2 activation. In fact, differential expression of IPCEF between untreated and TLR2-stimulated Th17 cells was the most significant of all gene expression comparisons by false discovery rate (FDR; p = 5.89E–32). Therefore, we further investigated the potential role of IPCEF in TLR2-stimulated Th17 cells and the overall function of IPCEF in pathogenic Th17 cells.
We confirmed that IPCEF expression is reduced following TLR2 activation in Th17(β) cells through both mRNA and protein analysis (Figures 4A and 4B). Importantly, IPCEF expression was also significantly lower in the more proliferative Th17(IL-1/IL-23) and Th1 subsets. We next investigated whether IPCEF expression affects Th17 cell cytokine expression, proliferation, or viability in vitro through retroviral delivery of an ectopic IPCEF expression construct (Figures S4A and S4B). We used isoform 1 since it is the canonical sequence, as opposed to the other two mouse isoforms that are generated by alternative splicing. Following the transduction of Th17 cells with RV-IPCEF or RV-Mock during differentiation, we observed no differences in IL-17 expression in Th17(β) cells or Th17(IL-1/IL-23) cells with or without the addition of PAM3CSK4 (Figures 5A and 5B). Furthermore, we found that cellular proliferation (Figures 5C and 5D) and viability (Figure 5E) remained unchanged in cells receiving either RV-IPCEF or RV-Mock.
Figure 4. IPCEF is downregulated in Th17 cells following TLR2 ligation.
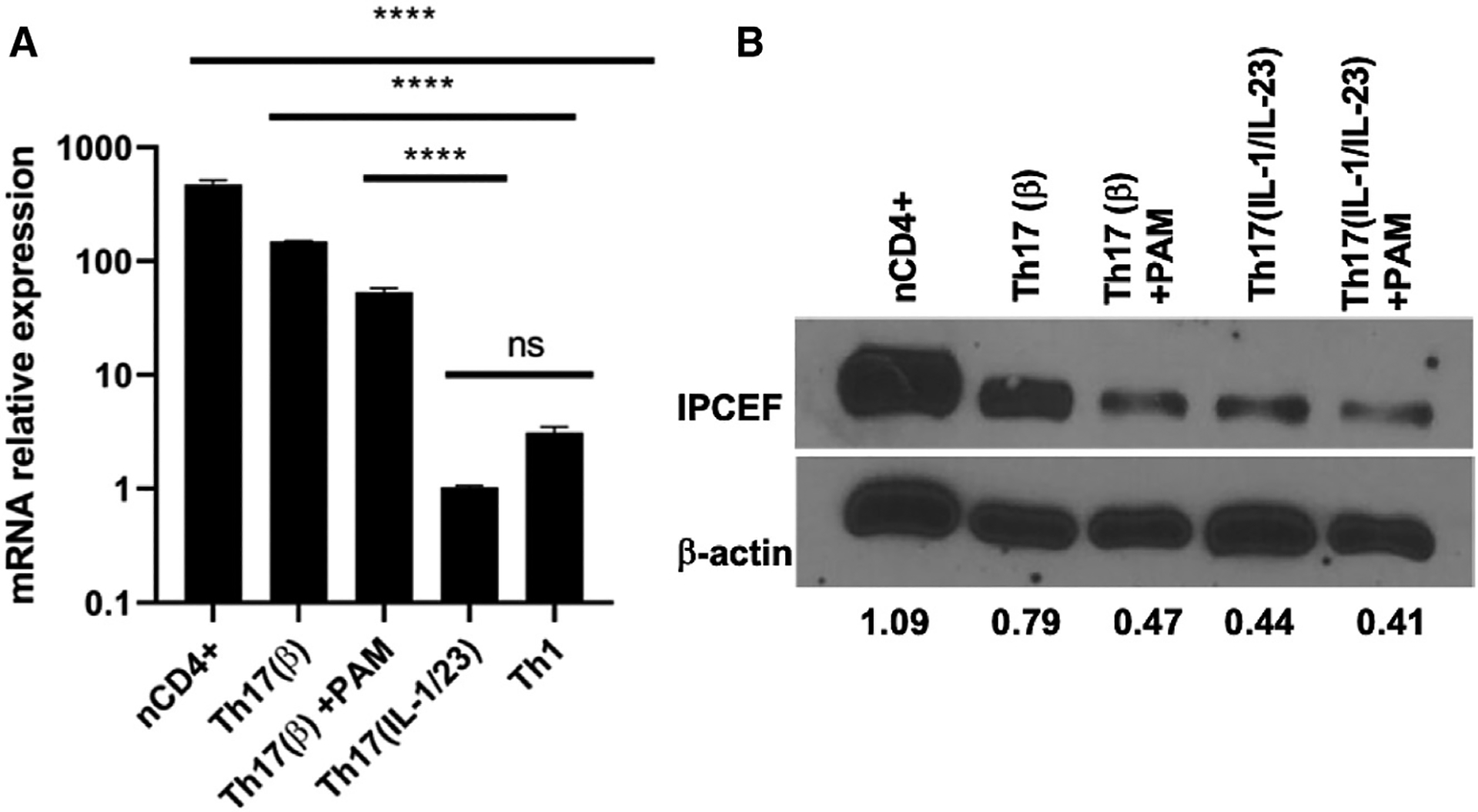
(A) mRNA expression (means ± SDs) of Ipcef1 in Th17(β) cells ± PAM3CSK4, Th17(IL-1/IL-23), and Th1 cells. n = 3 technical replicates per group. ****p < 0.0001 by 1-way ANOVA. ns, not statistically significant.
(B) Protein expression of IPCEF was examined in naive CD4+, Th17(β), and Th17(IL-1/IL-23) cells in the presence or absence of PAM3CSK4. Lower numbers represent densitometry measures of the IPCEF:actin ratio. Data are representative of 3 independent experiments.
Figure 5. IPCEF expression in Th17 cells does not affect IL-17 production, proliferation, or viability.
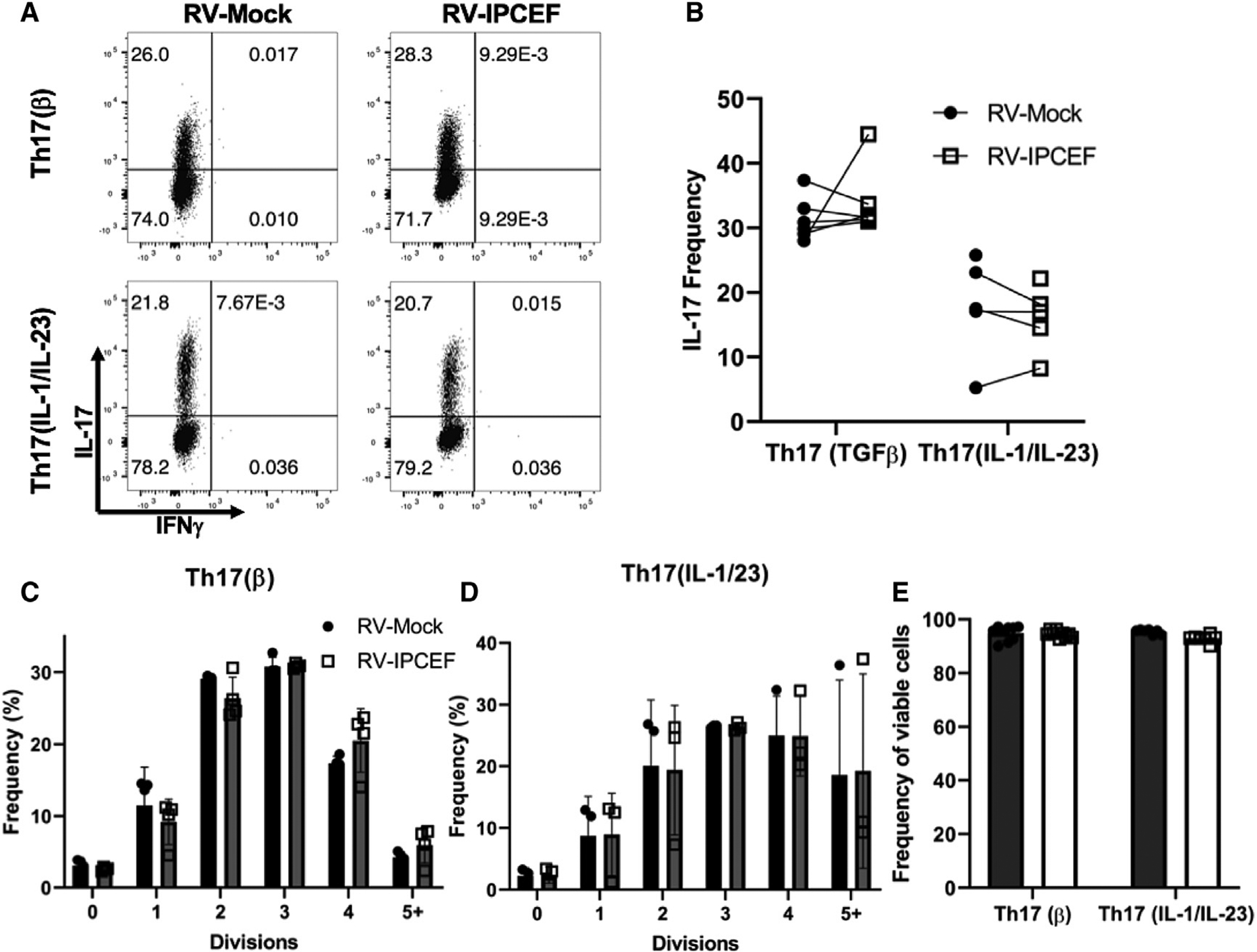
(A) Representative flow cytometry plots (IL-17 and IFN-γ) following 4-day differentiation of Th17(β) and Th17(IL-1/IL-23) cells transduced with RV-Mock or ectopic vector containing Ipcef1 (RV-IPCEF).
(B) Pooled IL-17 expression data of 3 independent experiments.
(C and D) Proliferation analysis following 4-day differentiation and transduction of Th17(β) cells (C) and Th17(IL-1/IL-23) cells (D).
(E) Frequency of viable cells was measured by viability staining of the aforementioned groups. Data are presented as means ± SDs. All data are representative of 3 independent experiments.
IPCEF impairs the migration of pathogenic Th17 cells
IPCEF is involved in cell polarization and migration of MDCK cells (White et al., 2010), leading us to hypothesize that inflammatory Th17 cells modulate IPCEF expression to promote migration. To address this question, we used a Transwell system to assess Th17 cell migration toward chemokine gradients in vitro. We investigated migration toward CCL20 because Th17 cells universally express CCR6 and are dependent on the CCR6-CCL20 interaction to infiltrate the CNS during EAE (Yamazaki et al., 2008; Reboldi et al., 2009). Th17 cells were transduced with RV-IPCEF or RV-Mock (empty vector) 1 day following polarization with either the Th17(β) or Th17(IL-1/IL-23) conditions in the absence or presence of PAM3CSK4. After a 4-day differentiation, RV+ Th17 cells were sorted, re-stimulated with αCD3, serum starved, and then plated in the upper chamber of the Transwell. After 48 h, we measured the number of cells migrating toward the 25 ng/mL CCL20 gradient loaded into the bottom chamber. Triggering TLR2 in Th17 cells substantially increased migratory capacity in both Th17(β) and Th17(IL-1/IL-23) conditions (Figure 6A). Thus, TLR2 activation promotes the migration of Th17 cells in a chemotactic manner. Surprisingly, the overexpression of IPCEF in PAM3CSK4-treated cells significantly impaired migration toward CCL20, resulting in a migratory capacity that was comparable to untreated Th17 cells. This observation was consistent in both PAM3CSK4-activated Th17(β) cells and Th17(IL-1/IL-23) polarized cells (Figure 6A). In the absence of TLR2 stimulation, however, enforced IPCEF expression did not affect the ability of Th17(β) cells to migrate, while Th17(IL-1/23) cells were only moderately impaired.
Figure 6. IPCEF impairs migration of pathogenic Th17 cells.
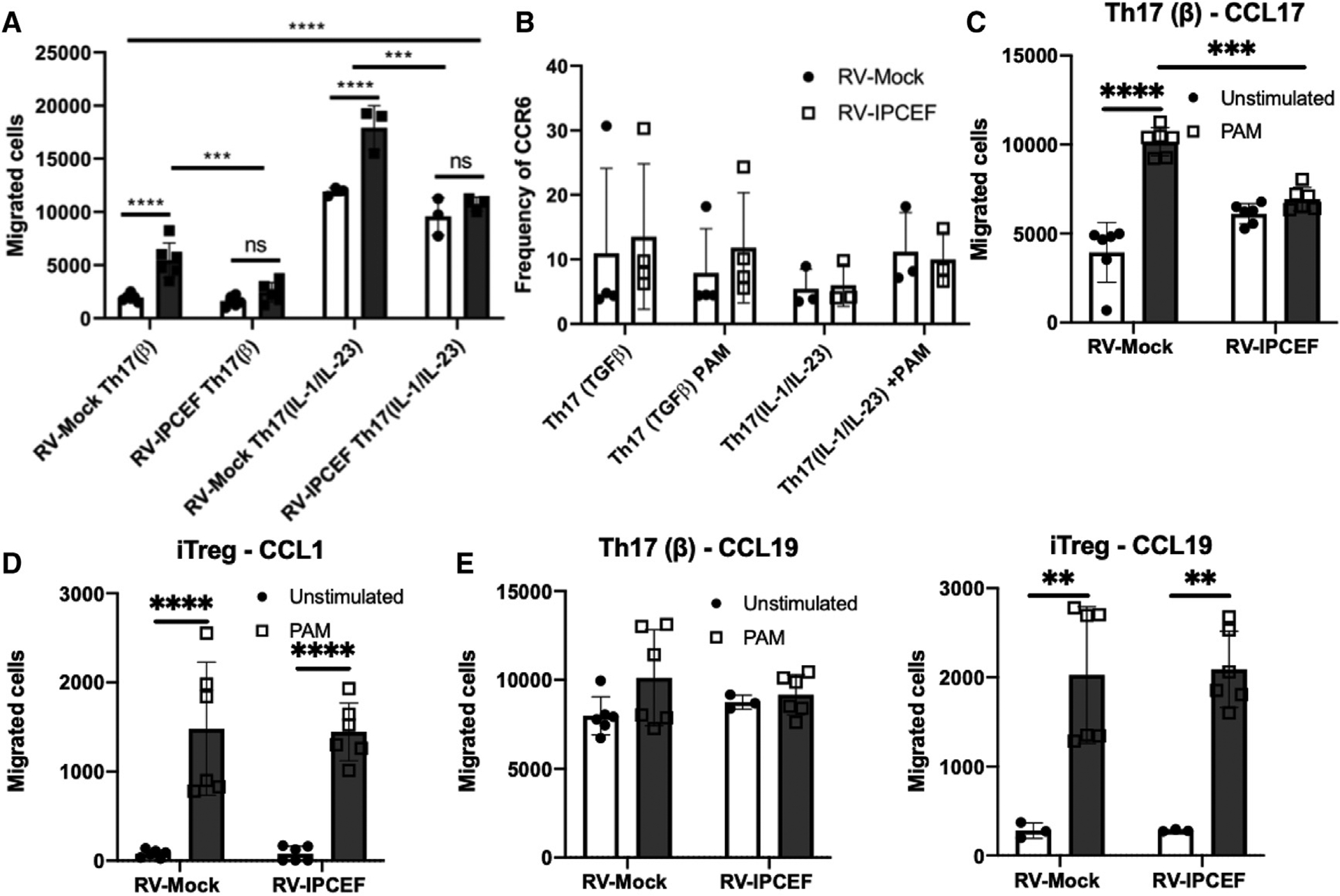
(A) Th17 cells were polarized as indicated, followed by transduction with RV-Mock or RV-IPCEF. RV+ cells were then sorted, reactivated, and plated on a Transwell for 48 h to determine the number of cells migrating toward a CCL20 gradient. n = 3 technical replicates per group.
(B) The frequency of CCR6 expression in Th17 cells, as indicated, was assessed by flow cytometry, and CCR6+ frequencies were pooled for analysis.
(C–E) Th17 or iTreg cells were transduced and plated on a Transwell, as described in (A). The number of cells migrating toward CCL17 (C), CCL1 (D), or CCL19 (E) was then assessed. **p < 0.01, ***p < 0.001, ****p < 0.0001 (2-way ANOVA). Data are representative of 3 independent experiments and are presented as means ± SDs.
Since we observed that PAM3CKS4-treated Th17 cells are more migratory toward CCL20, we investigated whether TLR2 stimulation affects CCR6 expression. Our previous work demonstrated that TLR2 activation moderately increased CCR6 expression at the mRNA level in comparison to untreated Th17 cells (Reynolds et al., 2010). We observed that this trend was actually reversed by RNA-seq (Table S1), but this comparison failed to reach statistical significance as defined by FDR. Therefore, we analyzed CCR6 surface expression by flow cytometry to obtain a definitive answer. We observed that CCR6 expression is unaffected by TLR2 activation in both Th17(β) or Th17(IL-1/IL-23) cells (Figure 6B). CCR6 expression was likewise unchanged in Th17 cells that overexpress IPCEF (Figure 6B). These data collectively indicate that TLR2-induced migration does not depend on increased CCR6 expression and may, instead, be regulated by a change in IPCEF expression.
We further investigated whether TLR2 activation or IPCEF expression affected the migration of Th17(β) cells toward other chemokines. We tested migration toward gradients of CCL17 (recognized by CCR4) or CCL19, a homeostatic lymphoid tissue signal recognized by CCR7 (Comerford et al., 2013). TLR2 activation resulted in increased migration toward CCL17, an effect that was similarly blunted with IPCEF overexpression (Figure 6C). Surprisingly, TLR2 activation or IPCEF expression did not change Th17 cell migration toward CCL19, indicating that the TLR2-IPCEF axis specifically targets inflammatory migration into tissues rather than homeostatic lymphoid organ trafficking. Similar to our observations with CCR6, TLR2 activation or IPCEF overexpression did not affect CCR4 or CCR7 expression (Figure S5A).
We also investigated TLR2 and IPCEF in other Th cell subsets. IPCEF expression is severely downregulated in Th1 cells regardless of TLR2 activation (Figures 4A and S5C) compared to naive CD4+ T cells. However, Th1 cells exhibited reduced viability upon ectopic IPCEF expression (data not shown), which prevented us from analyzing the migratory potential. Thus, we investigated these factors in the migration of induced Treg (iTreg) cells toward CCL1 (CCR8) and CCL19 (CCR7). Surprisingly, we found that iTregs were almost completely non-migratory toward either signal in vitro unless stimulated with PAM3CSK4 (Figures 6D and 6E). The overexpression of IPCEF, however, did not alter iTreg migration to gradients of either CCL1 or CCL19. Consequently, we investigated the expression of IPCEF in iTreg cells. Similar to what we observed for Th17 cells, TLR2 stimulation strongly reduces IPCEF expression in Treg cells (Figure S5B). We also found that both PAM3CSK4 treatment and IPCEF overexpression reduced CCR8, but not CCR7, expression (Figure S5B). These results suggest that these pathways act quite differently in Treg cells compared to Th17 cells, leading us to investigate relative IPCEF expression across the Th1, Th17(β), and Treg subsets. We found that iTreg cells strikingly have much more IPCEF expression compared to Th17 or Th1 cells. While TLR2 stimulation certainly reduces IPCEF in Treg cells, this expression still remains higher than that observed in unstimulated Th17 or Th1 cells (Figure S5C). Therefore, TLR2 and IPCEF dynamics in Treg cell migration are different compared to Th17 cells, warranting further study in this Th subset.
We thought it likely that altered IPCEF expression in Th17 cells affects Rac activation and thus the ability of TLR2-activated Th17 cells to migrate toward a CCL20 or CCL17 gradient, but it may not affect homeostatic migration (Cernuda-Morollón et al., 2010). To determine whether TLR2 or IPCEF overexpression alters Rac1 activation in Th17 cells, we evaluated active Rac1 following Th17 differentiation and RV-Mock or IPCEF overexpression. As expected, we found that TLR2 stimulation alone induced pull-down of more active Rac1 (Figure S6). Likewise, the overexpression of IPCEF leads to an increase in active Rac1. Interestingly, IPCEF overexpression in PAM3CSK4-stimulated Th17 cells actually decreased the pull-down of active Rac1 compared to Th17 cells overexpressing IPCEF alone. This result can somewhat be explained by our observation that TLR2-stimulated Th17 cells have less IPCEF overexpression compared to respective controls (Figure S5A). However, in the case of TLR2 stimulation, mock or IPCEF transduction resulted in the same amount of active Rac1 pull-down. Overall, these data suggest that additional factors outside of Rac1 are involved in the regulation of inflammatory Th17 cell migration. Studies are under way to address these possibilities.
IPCEF expression in Th17 cells results in a loss of CNS infiltration and a lack of EAE disease
The observed effect of IPCEF on the migration of Th17 cells in vitro led us to examine the consequence of IPCEF in pathogenic Th17 cells in vivo. To determine the importance of IPCEF along with its dependence on TLR2, we used an adoptive transfer EAE approach similar to that of Figure 3. Both WT and TLR2−/− mice were first immunized with MOG emulsified in IFA containing LPS. MOG-activated CD4+ T cells were then expanded with TLR2−/− APCs and PAM3CSK4. On the third day of expansion, we purified CD4+ T cells and transduced them with either RV-IPCEF or RV-Mock (empty) vectors, before returning the CD4+ T cells to the expansion culture. Following 8 days of expansion, we sorted and transferred the transduced CD4+ T cells into Rag1−/− mice and injected a booster dose of MOG/IFA/LPS and pertussis toxin to induce EAE. Of note, we did not observe changes to cytokine expression with ectopic IPCEF expression in CD4+ T cells before transfer (data not shown). Remarkably, we found that mice receiving the IPCEF overexpressing CD4+ T cells had significantly less incidence of disease in which only 1 of 11 mice developed EAE (Figure 7A). Subsequently, the overall disease severity was considerably reduced in these animals (Figure 7B).
Figure 7. Enforced IPCEF expression in Th17 cells reduces Th17 migration and EAE disease.
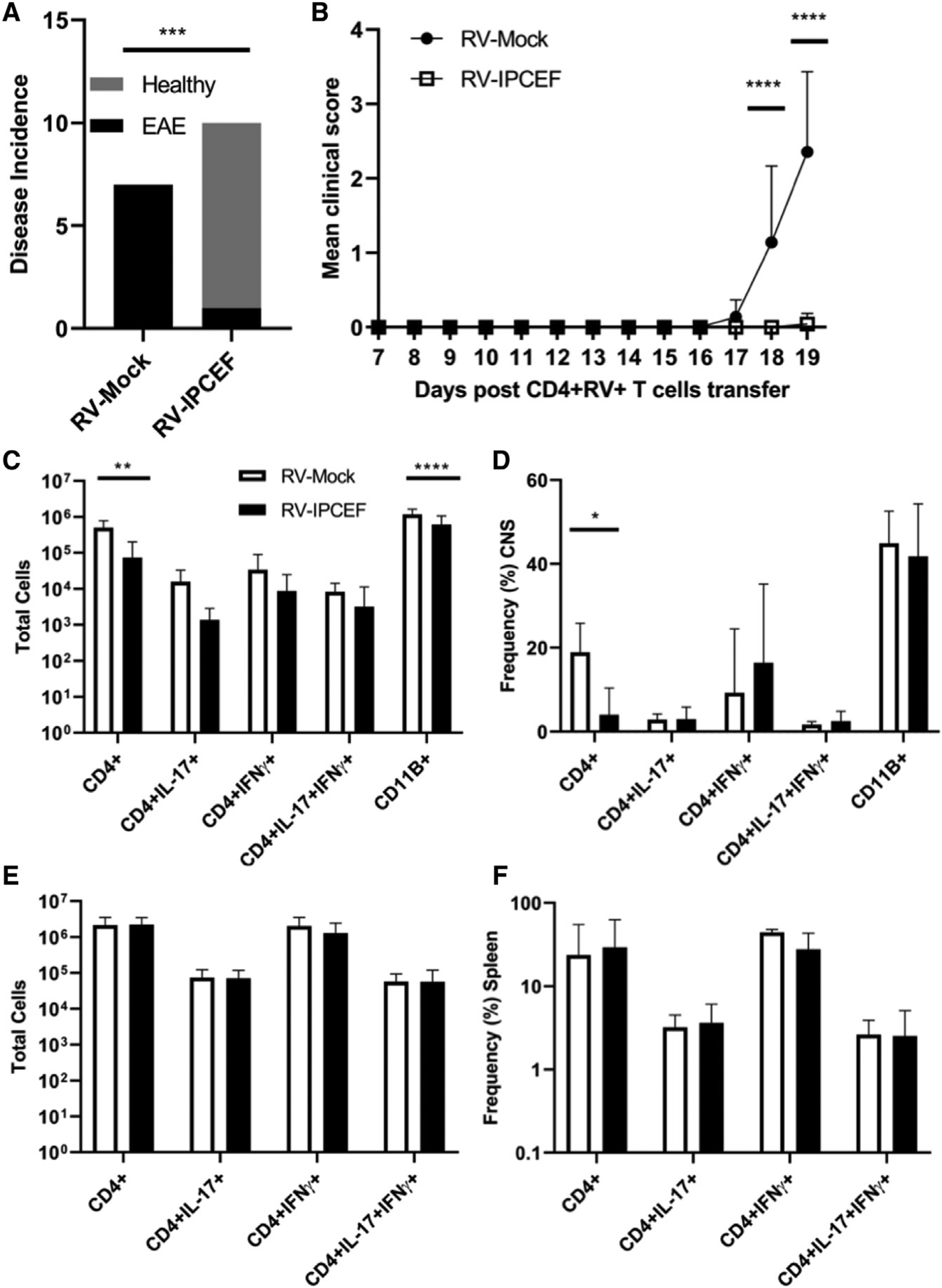
(A) EAE incidence in mice following transfer of CD4+RV+ cells expanded with PAM3CSK4 and transduced with either RV-Mock or RV-IPCEF. ***p < 0.001 (Fisher’s exact test).
(B) Mean clinical scores (+SDs) of the mice described in (A) up to the experimental endpoint. ****p < 0.0001 (2-way ANOVA).
(C and D) Mean cell numbers (C) and (D) frequencies of CD4+, CD11B+, CD4+IL-17+, CD4+IFN-γ+, and CD4+IL-17+IFN-γ+ cells isolated from the CNS as assessed by flow cytometry and pooled (means ± SDs). *p < 0.01, ****p < 0.0001 (2-way ANOVA).
(E and F) Mean (+SDs) cell numbers (E) and (F) frequencies of the same populations identified in spleens of EAE mice. Data are representative of 3 independent experiments.
Due to our observations that IPCEF is involved in suppressing TLR2-dependent migration in vitro (Figure 6A), we surmised that the infiltration of CD4+ T cells into the brains and spinal cords of EAE mice would be substantially decreased. The number of CD4+ T cells migrating into the CNS of mice receiving IPCEF-expressing cells was minimal in comparison to animals receiving mock-transduced cells (Figure 7C). Accordingly, there were substantially fewer numbers of IL-17+CD4+ and IFN-γ+CD4+ cells infiltrating into the CNS in the mice receiving the IPCEF+CD4+ T cells (Figure 7C). However, the frequency of IL-17, IFN-γ, and GM-CSF expression in the infiltrating CD4+ cells was unchanged, further supporting the notion that IPCEF does not affect encephalitogenic cytokine production (Figure 7D).
We thought it likely that the IPCEF+CD4+ T cells would remain restricted to the spleen based on normal homeostatic trafficking but have a reduced ability to migrate toward CCL20 (Figure 6). We were therefore surprised to observe that the number of CD4+ cells recovered from the spleens were similar between groups as was the expression of IL-17 and IFN-γ (Figures 7E and 7F). Thus, IPCEF-overexpressing cells appeared to remain viable due to their presence in the spleen, but they clearly had not migrated to the CNS during the course of the disease. We also assessed the capability of these splenic cells to recognize their cognate antigen by using a recall assay and observed no differences in the capacity of the splenocytes from either group of EAE mice to produce IL-17 or IFN-γ (Figures S7A and S7B).
Since IPCEF-overexpressing CD4+ cells do not migrate to the brain and spinal cord, we examined the expression of CCR6 ex vivo. We found that CCR6 expression was not changed between IPCEF-overexpressing or control CD4+ cells taken either from the spleen or from the CNS (data not shown). This corroborated what we observed in vitro: IPCEF expression does not alter CCR6 expression, but rather affects the migratory capability of Th17 cells through a different manner. Future experiments are designed to track the migration of IPCEF-expressing Th17 cells throughout the course of EAE disease to more fully assess the effect of IPCEF downregulation on pathogenic Th17 migration.
DISCUSSION
Th17 cells play key roles in autoimmune diseases, both in the initiation and maintenance of inflammation. Th17 cells are pathogenic in this context due to inflammatory damage inappropriately directed against self-tissue. Pathogenic Th17 cells are thus a target both for understanding and for treating autoimmune diseases. Studies of pathogenic Th17 cells up to this point, however, have mainly focused on the roles of IL-23 and IL-1β in differentiation and in vivo function. Our study addresses the role of the pattern recognition receptor TLR2 in inducing pathogenicity.
TLR2 is important for both innate and adaptive immunity, including regulating innate processes in autoimmune diseases such as rheumatoid arthritis (RA) and MS. For example, macrophages lose tolerance to TLR2 signaling in patients with primary progressive MS compared to people with relapsing-remitting MS (Fujiwara et al., 2018; Wasko et al., 2019). A similar effect for TLR2 and TLR4 signaling has been observed for macrophages in RA (Huang et al., 2007). In addition, TLR2 is more highly expressed in peripheral blood mononuclear cells (PBMCs) and monocytes from patients with MS and RA, respectively, compared to healthy controls (Hasheminia et al., 2014; He et al., 2012). Thus, TLR2 has emerged as a major influence on autoimmune inflammation and a potential therapeutic target. Few studies have addressed, however, the direct role of TLR2 on Th17 cells in autoimmune disease.
We have found that the activation of TLR2 in Th17(β) cells enhances proliferation in vitro, a phenomenon that is reversed when TLR2 is deleted after TCR activation (Figure 1). These experiments highlight the cell-intrinsic nature of TLR2 signaling in Th17 cells. Our previous study demonstrated a requirement of TLR2 on CD4+ T cells to induce severe EAE disease (Reynolds et al., 2010). We have now established that the ligation of TLR2 is sufficient to induce Th17 cells to become encephalitogenic and that TLR2-activated Th17 cells were surprisingly just as capable, if not more so, of inducing disease compared to IL-23 stimulation. However, our current studies used IFA/LPS rather than CFA to avoid off-target effects of TLR2 signaling, warranting investigation into methodologies to further compare pathogenic conditions in other systems.
Several studies have indicated that TLR2 can act as a co-stimulatory receptor to induce T cell proliferation, survival, and cytokine production (Komai-Koma et al., 2004; Liew et al., 2004; Jun et al., 2017; Cottalorda et al., 2006; Imanishi et al., 2020). TLR2 activation also affects the transcriptome of Th17 cells by inducing or suppressing several genes involved in lineage commitment and stability, as well as general proliferation and survival factors such as Ccnd2, Cdk6, and Bcl2 (Figures 3 and S3A; Table S1). GSEA demonstrated that TLR2 induces a pathogenic signature, somewhat similar to IL-23 and IL-1β stimulation (Figure 3C). Commonalities include positive regulation of genes such as Ahr, Irf4, Stat3, Runx3, and Fasn. Both TLR2 and IL-1β use the signaling adaptor MyD88 for their downstream signaling pathway, implying some potential overlap between TLR2 and IL-1β signaling programs in Th17 cells. While there are significant similarities between the molecular programs, there are also several genes targeted only by TLR2 or that varied in the degree of differential expression. Importantly, genes enriched in TLR2-activated Th17 cells are dissimilar to genes bound by FoxP3, complementing previous studies showing that TLR2 activation in Treg cells causes a loss of suppressive capability and increased IL-17 (Nyirenda et al., 2011; Lee, 2018). However, we also demonstrate (Figure 6) that TLR2 increases iTreg migration, which supports another study showing that TLR2 enhances Treg proliferation (Sutmuller et al., 2006) and highlights TLR2 as a possible contributor to the Th17 and iTreg cell dynamic in MS.
An important aspect of autoimmune disease is the requirement for inflammatory T cell migration to the site of cognate antigen. A recent study identified the role of IL-23 in enhancing Th17 cell motility through Rho-associated protein kinase (Álvarez-Salamero et al., 2020). We demonstrate increased migratory capacity in Th17(β) cells differentiated with PAM3CSK4, and also verified those results with IL-23, IL-1β, and IL-6 (Figure 6). Both conditions result in increased migration toward CCL20. TLR2 also enhanced migration toward CCL17, but not CCL19, in Th17(β) cells, which suggests that this signal is somewhat specific only for trafficking into inflamed tissues. TLR2-dependent increases in migratory potential do not appear to be a consequence of increased chemokine receptor expression despite moderate mRNA differences in CCR6 reported previously (Reynolds et al., 2010).
Outside of differences in chemokine receptor expression, we identified IPCEF as a candidate regulator of Th17 migration in EAE. Existing research on the scaffolding protein IPCEF is limited, but other studies demonstrated that IPCEF supports Rac activation and, subsequently, promotes migration (White et al., 2010). As a member of the Rho family, Rac contributes to the formation of lamellipodia and membrane ruffling at the leading edge while blocking actin depolymerization (Sakumura et al., 2005; Bros et al., 2019; Parri and Chiarugi, 2010). Loss of IPCEF in MDCK cells reduced the capacity of cells to form lamellipodia and to scatter (White et al., 2010). Importantly, Rac activation has been previously associated with CD4+ T cell migration (Arrieumerlou et al., 2000). Initial TCR engagement results in increased Rac activity and inhibition of RhoA, resulting in an initial lack of polarization and movement of CD4+ T cells (Cernuda-Morollón et al., 2010). However, we observed contradictory results in that TLR2 signaling increases migration while also downregulating IPCEF expression (Figures 3, 4, 5, and 6). Moreover, enforcing IPCEF expression sharply reduced the migratory capacity of pathogenic Th17 cells both in vitro and in vivo. TLR2 signaling or IPCEF overexpression induced more active Rac1 (Figure S6) in Th17(β) cells. Paradoxically, Th17 cells stimulated through TLR2 with or without ectopic IPCEF exhibited similar Rac1 activation, suggesting the involvement of additional molecules in the TLR2-IPCEF-Rac migratory axis. Interestingly, another cytohesin2 exchange factor contributing to Rac activation, Grasp/Tamalin, was not affected by TL2 activation (not shown), implying a unique role for IPCEF in TLR2-induced CD4+ T cell migration.
In addition to Th17 cells, we also analyzed the dynamics of IPCEF in Th1 and iTreg cells. Th1 cells did not readily accept the expression construct, suggesting that an overabundance of IPCEF may be detrimental to Th1 survival. Experiments are in progress to determine whether this is the case. Remarkably, iTreg cells express an abundance of IPCEF that rivals naive CD4+ T cells (Figure S5). Similar to Th17 cells, TLR2 ligation also drove IPCEF expression down in Treg cells, but the relative mRNA level remained substantially higher compared to other Th subsets. Moreover, TLR2 signaling was required for Tregs to migrate in vitro toward CCL1 and CCL19 while the enforcement of IPCEF did not affect migration (Figure 6). Thus, we observed differing roles of IPCEF in Th17 and Treg cells, and future studies will focus on the relationship between IPCEF and Rac activation in T helper cells.
The effect of IPCEF expression on CD4+ T cell migration is particularly relevant to neuroinflammatory diseases such as MS and neuromyelitis optica, in which blocking CD4+ T cell infiltration would have therapeutic benefit (Polman et al., 2006; Hou et al., 2019; Reboldi et al., 2009). We demonstrated the contribution of IPCEF in limiting Th17 cell infiltration into the CNS of EAE mice using an adoptive transfer model (Figure 7). IPCEF-overexpressing Th17 cells, while exhibiting similar expression of IL-17, IFN-γ, and GM-CSF, did not infiltrate into the CNS and cause disease upon transfer. While in vitro overexpression of IPCEF did not affect the viability or proliferation of differentiating Th17 cells, we cannot rule out a loss of viability or proliferation in IPCEF-overexpressing Th17 cells in vivo, especially for cells that have not migrated to their cognate antigen (MOG). We also must not discount the possibility of atypical migration to other tissues as a result of enforced IPCEF expression. Future studies will also be required to determine the role and/or necessity of IPCEF in Th17 cells that are not pathogenic for autoimmune inflammation. These studies will include assessing IPCEF function in Th17 cells residing in the gut lamina propria.
Overall, our study demonstrates that TLR2 activation induces pathogenicity in Th17 cells by inducing a pathogenic molecular program and that these pathogenic Th17 cells are capable of inducing autoimmune inflammation. Our observations described herein open up many avenues for further research, including a pathogenic paradigm whereby Th17 migration is regulated by modulating the expression of IPCEF. Most important, however, our observations reveal mechanisms related to the development of autoimmune inflammation that could be exploited for the treatment of such diseases.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joseph Reynolds (joseph.reynolds@rosalindfranklin.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA sequencing data presented in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GEO: GSE174469.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6, TLR2−/−, Rag1−/−, CD4CreERT2 and IL-17-GFP mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) on the C57BL/6 background. TLR2f/f mice on the C57BL/6 background were generously provided by Dr. Henry Boom at Case Western University. All experiments were carried out using mice 6–10 weeks old. Animals were maintained in SPF housing under the care of the Veterinary Staff at Rosalind Franklin University of Medicine and Science (RFUMS). In vitro experiments were completed using tissues from both female and male mice. All EAE experiments were conducted using female mice. The animal procedures described in this study were approved by the Institutional Animal Care and Use Committee at RFUMS.
Cell lines
The retroviral packaging cell line, PLAT-E (Cell BioLabs), was used to produce retroviral particles for T cell transduction. Cells were grown to a monolayer in DMEM supplemented with 10% FBS, 1% Penicillin-Streptomycin, and 1% L-glutamine.
METHOD DETAILS
T cell isolation and differentiation
For T cell differentiation, naive CD4+ T cells were isolated from secondary lymphoid organs of C57BL/6 mice or TLR2f/f mice as indicated by using EasySep Mouse CD4+ T Cell Isolation Kit (StemCell) and sorting as previously described for CD4+CD62LhiCD25−CD44− on a BD Biosciences FACS ARIAII (Flaherty and Reynolds, 2015). Naive CD4+ T cells were cultured for 5 days in 1ml of RPMI supplemented with 10% FBS, 1% Penicillin-Streptomycin, and 1% L-glutamine with plate bound 1 μg α-CD3 (BioXcell) and 1 μg α-CD28 (BioXcell) as well as 1ng TGFβ and 10ng IL-6 (R&D) (Th17(β)) or 20ng IL-1β (Peprotech), 20ng IL-6, and 25ng IL-23 (R&D) (Th17(IL-1/IL-23)). Th1 cells were polarized with 20 ng IL-12 (R&D) and 30 U hIL-2 (Peprotech). iTreg cells were polarized with 15 ng TGFβ and 30 U hIL-2. T cell cultures were stimulated with or without 2 μg Pam3Cys-Ser-(Lys)4-trihydrochloride (PAM3CSK4) or 5 μg Pam2CGDPKHPKSF (FSL-1) (InvivoGen). Cells were stimulated for flow cytometry analysis for 5hrs with PMA, ionomycin (Sigma-Aldrich), and brefeldin A (eBioscience), followed by intracellular cytokine staining for IL-17, IFN-γ, and GM-CSF (eBioscience) and analysis on a BD LSRII. For western blots, cells were lysed and probed for IPCEF1/Pip3E and actin (Sigma-Aldrich). Densitometry (integrated density values, IDV) of developed blots was completed using an AlphaImager (ProteinSimple).
For IPCEF overexpression studies, we cloned Ipcef1-1 into the PCMMP-MCS-IRES-eGFP vector. Confluent PLAT-E cells were then transfected with the vector using calcium phosphate. After 8hrs, the cell line media was changed and the cells were then incubated for 48h at 37°C/5% CO2. Supernatants containing the viral particles were then concentrated by centrifugation at 4°C/8,000 × g for 12–16 hours prior to T cell transduction. We transduced the construct containing Ipcef1-1 or the vector alone, empty vector (Mock), into T cells approximately 24hrs into differentiation by adding retrovirus supernatant and spinfecting the cells at 2,000 rpm for 1hr at 32°C in the presence of polybrene. The same procedure was also employed to express Cre recombinase in vitro.
qPCR
mRNA was isolated from cells lysed in TRIzol reagent (Thermo Fisher) following differentiation. cDNA was synthesized using Moloney murine leukemia virus system per manufacturer’s protocol (Thermo Fisher Scientific). qPCR was performed using SYBR Green on an Applied Biosystems 7500 Real Time System (Thermo Fisher Scientific). All gene expression values were normalized to the house-keeping genes Actb or Gapdh. Primer sequences are listed in Table S2.
RNA sequencing
For RNA sequencing, Th17 cells were differentiated as above for 3 days. Total RNA was extracted from naive CD4+ T cells and Th17 (β) +/− PAM3CSK4 conditions in TRIZoL reagent and was converted into strand-specific paired-end sequencing libraries using the TruSeq stranded total RNA kit (Illumina). Total RNA was rRNA-depleted, fragmented, converted to cDNA, adaptor-ligated with unique indices and PCR amplified. The final libraries were quantified and sequenced using paired-end 75bp chemistry on NextSeq 500 (Illumina). RNA-seq reads were aligned to UCSC mm9 with Bowtie2 and reads were counted with HTseq count (Dobin et al., 2013; Anders and Huber, 2010). Differential expression was calculated with DESeq2 and Gene set enrichment analysis (GSEA) was performed as previously described (Ghoreschi et al., 2010; Lee et al., 2012; Subramanian et al., 2005; Zheng et al., 2007).
Adoptive transfer EAE
IL-17-GFP mice and TLR2−/− mice were immunized with MOG35–55 emulsified in incomplete Freunds adjuvant (IFA) and 3 μg/ml LPS. 7 days later, IL-17A-GFP CD4+ T cells were isolated from spleen and lymph nodes by magnetic bead separation (StemCell) combined with CD4− APC cells isolated by Automacs (Miltenyi) from TLR2−/− mice. Cells were expanded in vitro with 3 μg/ml MOG35–55 in addition to 5ng/ml IL-23, 1 μg/ml PAM3CSK4, or no further addition. After 8 d expansion, GFP+ cells were sorted and transferred i.v. to Rag1−/− mice. MOG/IFA/LPS and 500ng/mouse pertussis toxin were administrated 1 and 2 days respectively following CD4+GFP+ transfer. Mice were scored based on paralysis as previously published (Reynolds et al., 2010) and monitored for weight loss. At peak disease, the mice were perfused with 0.6% heparin in sterile PBS. The brain, spinal cord, and spleen were isolated from individual mice. Isolated cells from spleens as well as combined brain and spinal cord were counted and analyzed for CD4, CD11b, CCR6, IL-17, IFN-γ, and GM-CSF expression (eBioscience) by flow cytometry.
Adoptive transfer EAE experiments with IPCEF overexpression were performed as before, however on day 3 of expansion the CD4+ cells were isolated by Automacs (Miltenyi) and transduced as described above with RV-mock or RV-IPCEF. After transduction, CD4+ T cells were added back to the culture with the CD4− APCs. Following expansion, the cells were sorted on positive transduction (GFP+) and transferred i.v. to Rag1−/− mice and the same EAE monitoring procedure and analysis was followed. At endpoint, splenocytes were plated at 3 × 106 cells/ml and re-stimulated with increasing concentrations of MOG35–55. After 3 days, supernatants were collected and analyzed for IL-17 and IFN-γ production by ELISA (BD Biosciences).
T cell migration assays
Following 4d differentiation, transduced CD4+ T cells were sorted and re-stimulated with plate bound α-CD3 in 0.5% FBS RPMI for 24hrs. The cells were then plated 0.5% FBS RPMI at 50K/well in a transwell (Abcam) containing 25ng/ml CCL20, CCL17, CCL1, or CCL19 (R&D) in the bottom chamber for 48hrs. The cells migrating into the bottom of the transwell plate were counted by an Accuri cytometer (BD Biosciences) in a blinded fashion by multiple individuals.
Active Rac1 pulldown
Th17 cells were differentiated as described in the presence or absence of PAM3CSK4. 1d after TCR activation, the cells were retrovirally transduced with either empty (RV-Mock) or IPCEF (RV-IPCEF) vector. After 4d, RV+ (GFP+) cells were sorted and lysed under non-denaturing conditions. Protein lysates were normalized to 500 μg total protein using a BCA assay (Pierce). Active Rac1 was immunoprecipitated using an Active Rac1 Detection Kit (Cell Signaling Technology) according to the vendor’s protocol. Briefly, 500 μg total protein from each sample was incubated with GST-PAK1-PBD followed by pull-down and elution from a glutathione resin column. The entirety of the elution fraction was then loaded onto a 10% SDS-PAGE gel and Western blow analysis was performed using an antibody specific for Rac1. Positive and negative controls were generated by lysing non-transduced Th17 cells and treating them with GTPγS or GDP, respectively, prior to active Rac1 pull-down.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism as indicated in the Figure legends. Significance was defined by FDR or p value determined by 1-way ANOVA, 2-way ANOVA, or Fisher’s exact test as indicated in the Figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD4 | Biolegend | Cat# 100431; RRID: AB_893329 |
| CD11B | Biolegend | Cat# 101205; RRID: AB_312788 |
| CCR6 | Biolegend | Cat# 129815; RRID: AB_1877244 |
| IL-17A | Biolegend | Cat# 506917; RRID: AB_893545 |
| IFNγ | Biolegend | Cat# 505809; RRID: AB_315403 |
| GM-CSF | Biolegend | Cat# 505405; RRID: AB_315381 |
| CD3 | Bio X Cell | Cat# BE0001-1, RRID: AB_1107634 |
| CD28 | Bio X Cell | Cat# BE0015-1; RRID: AB_1107624 |
| Bacterial and virus strains | ||
| PCMMP-MCS-IRES | (Kennedy and Sugden, 2003) | Addgene #36953; RRID: Addgene_36953 |
| Chemicals, peptides, and recombinant proteins | ||
| PAM3CSK4 | Invivogen | tlrl-pms |
| FSL-1 | Invivogen | tlrl-fsl |
| Deposited data | ||
| RNA sequencing datasets | GEO | GSE174469 |
| Experimental models: cell lines | ||
| PLAT-E cells | Cell Biolabs | Cat# RV-101; RRID: CVCL_B488 |
| Experimental models: organisms/strains | ||
| TLR2fl/fl mice, C57BL/6 background | Dr. Harry Boom | N/A |
| IL-17GFP mice, C57BL/6 background | Jackson | Cat# JAX:018472; RRID: IMSR_JAX:018472 |
| C57BL/6 mice | Jackson | Cat# JAX:000664; RRID: IMSR_JAX:000664 |
| CD4CreERT2, C57BL/6 background | Jackson | Cat# JAX:022356; RRID: IMSR_JAX:022356 |
| Rag1−/− mice, C57BL/6 background | Jackson | Cat# JAX:002216, RRID: IMSR_JAX:002216 |
| Oligonucleotides | ||
| See Table S2 for oligonucleotide information | N/A | N/A |
| Other | ||
| Active Rac1 Detection Kit | Cell Signaling Technology | 8815S |
| Cell Migration/Chemotaxis Assay Kit (96-well) | Abcam | ab235693 |
Highlights.
TLR2 signaling is sufficient to drive CD4+ T cell-mediated autoimmune inflammation
TLR2 activation drives an inflammatory transcriptional profile in Th17 cells
TLR2 ligation suppresses IPCEF expression in Th17 cells
Enforced IPCEF expression dampens the migratory potential of Th17 cells
ACKNOWLEDGMENTS
This work was supported in part by the US National Institutes of Health (5K22AI104941 and 1R01AI141596 to J.M.R.). We thank the Reynolds laboratory and the Center for Cancer Cell Biology, Immunology, and Infection at the Rosalind Franklin University of Medicine and Science. The graphical abstract was created with Biorender.com.
Footnotes
DECLARATION OF INTERESTS
Data generated from this study were used in part for a provisional patent application (K.E.M. and J.M.R.): MBHB 20-853-PRO2.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109303.
REFERENCES
- Álvarez-Salamero C, Castillo-González R, Pastor-Fernández G, Mariblanca IR, Pino J, Cibrian D, and Navarro MN (2020). IL-23 signaling regulation of pro-inflammatory T-cell migration uncovered by phosphoproteomics. PLoS Biol. 18, e3000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieumerlou C, Randriamampita C, Bismuth G, and Trautmann A (2000). Rac is involved in early TCR signaling. J. Immunol 165, 3182–3189. [DOI] [PubMed] [Google Scholar]
- Attar MA, Salem JC, Pursel HS, and Santy LC (2012). CNK3 and IPCEF1 produce a single protein that is required for HGF dependent Arf6 activation and migration. Exp. Cell Res 318, 228–237. [DOI] [PubMed] [Google Scholar]
- Bros M, Haas K, Moll L, and Grabbe S (2019). RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 8, E733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernuda-Morollón E, Millán J, Shipman M, Marelli-Berg FM, and Ridley AJ (2010). Rac activation by the T-cell receptor inhibits T cell migration. PLoS ONE 5, e12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, and Dong C (2009). Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30, 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, and McColl SR (2013). A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 24, 269–283. [DOI] [PubMed] [Google Scholar]
- Cottalorda A, Verschelde C, Marçais A, Tomkowiak M, Musette P, Uematsu S, Akira S, Marvel J, and Bonnefoy-Berard N (2006). TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur. J. Immunol 36, 1684–1693. [DOI] [PubMed] [Google Scholar]
- Dai J, Liu B, and Li Z (2009). Regulatory T cells and Toll-like receptors: what is the missing link? Int. Immunopharmacol 9, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias ASO, Sacramento PM, Lopes LM, Sales MC, Castro C, Araújo ACRA, Ornelas AMM, Aguiar RS, Silva-Filho RG, Alvarenga R, and Bento CAM (2019). TLR-2 and TLR-4 agonists favor expansion of CD4+ T cell subsets implicated in the severity of neuromyelitis optica spectrum disorders. Mult. Scler. Relat. Disord 34, 66–76. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman TH, Chauhan SK, Kodati S, Hua J, Chen Y, Omoto M, Sadrai Z, and Dana R (2013). The CCR6/CCL20 axis mediates Th17 cell migration to the ocular surface in dry eye disease. Invest. Ophthalmol. Vis. Sci 54, 4081–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, and Rostami A (2011). The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol 12, 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty S, and Reynolds JM (2015). Mouse Naı¨ve CD4+ T Cell Isolation and In vitro Differentiation into T Cell Subsets. J. Vis. Exp (98), 52739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Anstadt EJ, Flynn B, Morse K, Ng C, Paczkowski P, Zhou J, Mackay S, Wasko N, Nichols F, and Clark RB (2018). Enhanced TLR2 responses in multiple sclerosis. Clin. Exp. Immunol 193, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, and Cua DJ (2014). The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol 14, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al. (2010). Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasheminia SJ, Zarkesh-Esfahani SH, Tolouei S, Shaygannejad V, Shirzad H, and Hashemzadeh Chaleshtory M (2014). Toll like receptor 2 and 4 expression in peripheral blood mononuclear cells of multiple sclerosis patients. Iran. J. Immunol 11, 74–83. [PubMed] [Google Scholar]
- He Z, Shotorbani SS, Jiao Z, Su Z, Tong J, Liu Y, Shen P, Ma J, Gao J, Wang T, et al. (2012). HMGB1 promotes the differentiation of Th17 via upregulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis. Scand. J. Immunol 76, 483–490. [DOI] [PubMed] [Google Scholar]
- Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto, Yamaguchi T, Nomura T, Ito H, Nakamura T, et al. (2007). Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med 204, 2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol 12, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou M-M, Li Y-F, He L-L, Li X-Q, Zhang Y, Zhang S-X, and Li X-Y (2019). Proportions of Th17 cells and Th17-related cytokines in neuromyelitis optica spectrum disorders patients: a meta-analysis. Int. Immunopharmacol 75, 105793. [DOI] [PubMed] [Google Scholar]
- Huang Q, Ma Y, Adebayo A, and Pope RM (2007). Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 56, 2192–2201. [DOI] [PubMed] [Google Scholar]
- Imanishi T, Unno M, Kobayashi W, Yoneda N, Akira S, and Saito T (2020). mTORC1 Signaling Controls TLR2-Mediated T-Cell Activation by Inducing TIRAP Expression. Cell Rep. 32, 107911. [DOI] [PubMed] [Google Scholar]
- Jadidi-Niaragh F, and Mirshafiey A (2011). Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol 74, 1–13. [DOI] [PubMed] [Google Scholar]
- Janeway CA Jr., and Medzhitov R (2002). Innate immune recognition. Annu. Rev. Immunol 20, 197–216. [DOI] [PubMed] [Google Scholar]
- Jun JC, Jones MB, Oswald DM, Sim ES, Jonnalagadda AR, Kreisman LSC, and Cobb BA (2017). T cell-intrinsic TLR2 stimulation promotes IL-10 expression and suppressive activity by CD45RbHi T cells. PLoS ONE 12, e0180688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabelitz D (2007). Expression and function of Toll-like receptors in T lymphocytes. Curr. Opin. Immunol 19, 39–45. [DOI] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2007). TLR signaling. Semin. Immunol 19, 24–32. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, and Kawai T (2014). Toll-like receptor signaling pathways. Front. Immunol 5, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy G, and Sugden B (2003). EBNA-1, a bifunctional transcriptional activator. Mol. Cell. Biol 23, 6901–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH (2009). Migration and function of Th17 cells. Inflamm. Allergy Drug Targets 8, 221–228. [DOI] [PubMed] [Google Scholar]
- Komai-Koma M, Jones L, Ogg GS, Xu D, and Liew FY (2004). TLR2 is expressed on activated T cells as a costimulatory receptor. Proc. Natl. Acad. Sci. USA 101, 3029–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroenke MA, Carlson TJ, Andjelkovic AV, and Segal BM (2008). IL-12 and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J. Exp. Med 205, 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GR (2018). The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci 19, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, et al. (2012). Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol 13, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew FY, Komai-Koma M, and Xu D (2004). A toll for T cell costimulation. Ann. Rheum. Dis 63 (Suppl 2), ii76–ii78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAleer JP, Liu B, Li Z, Ngoi S-M, Dai J, Oft M, and Vella AT (2010). Potent intestinal Th17 priming through peripheral lipopolysaccharide-based immunization. J. Leukoc. Biol 88, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, and Cua DJ (2009). The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol 10, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawijn MC, Motta AC, Gras R, Shirinbak S, Maazi H, and van Oosterhout AJ (2013). TLR-2 activation induces regulatory T cells and long-term suppression of asthma manifestations in mice. PLoS ONE 8, e55307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyirenda MH, Sanvito L, Darlington PJ, O’Brien K, Zhang G-X, Constantinescu CS, Bar-Or A, and Gran B (2011). TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. J. Immunol 187, 2278–2290. [DOI] [PubMed] [Google Scholar]
- Parri M, and Chiarugi P (2010). Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, et al. ; AFFIRM Investigators (2006). A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med 354, 899–910. [DOI] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, and Sallusto F (2009). C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol 10, 514–523. [DOI] [PubMed] [Google Scholar]
- Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, and Dong C (2010). Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 32, 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, Martinez GJ, Chung Y, and Dong C (2012). Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc. Natl. Acad. Sci. USA 109, 13064–13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakumura Y, Tsukada Y, Yamamoto N, and Ishii S (2005). A molecular model for axon guidance based on cross talk between rho GTPases. Biophys. J 89, 812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, and Chi H (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med 208, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, and Adema GJ (2006). Toll-like receptor 2 controls expansion and function of regulatory T cells. J. Clin. Invest 116, 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateswarlu K (2003). Interaction protein for cytohesin exchange factors 1 (IPCEF1) binds cytohesin 2 and modifies its activity. J. Biol. Chem 278, 43460–43469. [DOI] [PubMed] [Google Scholar]
- Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, Lee YK, Weaver CT, Yagi R, and Lazarevic V (2014). The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity 40, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasko NJ, Kulak MH, Paul D, Nicaise AM, Yeung ST, Nichols FC, Khanna KM, Crocker S, Pachter JS, and Clark RB (2019). Systemic TLR2 tolerance enhances central nervous system remyelination. J. Neuroinflammation 16, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DT, McShea KM, Attar MA, and Santy LC (2010). GRASP and IPCEF promote ARF-to-Rac signaling and cell migration by coordinating the association of ARNO/cytohesin 2 with Dock180. Mol. Biol. Cell 21, 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L, Panopoulos AD, et al. (2008). CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol 181, 8391–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Takeuchi Y, and Hirota K (2019). The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol 41, 283–297. [DOI] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, et al. (2013). Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KE, Flaherty S, Woodman KM, Sharma-Walia N, and Reynolds JM (2017). Fatty acid synthase regulates the pathogenicity of Th17 cells. J. Leukoc. Biol 102, 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao RR, Yang XF, Dong J, Zhao YY, Wei X, Huang CX, Lian JQ, and Zhang Y (2015). Toll-like receptor 2 promotes T helper 17 cells response in hepatitis B virus infection. Int. J. Clin. Exp. Med 8, 7315–7323. [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, and Rudensky AY (2007). Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 445, 936–940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data presented in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GEO: GSE174469.