

Abstract
Familial juvenile hyperuricemic nephropathy (FJHN) is a rare autosomal dominant disease caused by mutations in the uromodulin (UMOD) gene. It is characterized by the development of gout, tubulointerstitial nephropathy, and end-stage renal disease. Here we report a case of FJHN that was diagnosed in early childhood in a boy with a novel gene mutation. At the age of 4 years, the patient was admitted with a diagnosis of purpura nephritis. He was discharged following symptom alleviation. However, hyperuricemia (7–9 mg/dL) and mild renal dysfunction [creatinine-estimated glomerular filtration rate (eGFR): 80–90 mL/min/1.73 m2] persisted after discharge. FJHN was suspected on the basis of a maternal family history of hyperuricemia, renal dysfunction, and dialysis. Direct sequence analysis performed at the age of 5 years revealed a novel missense mutation (c766T > G), p.Cys256Gly, in exon 3. Urate-lowering therapy was started, which provided good uric acid control (6.0 mg/dL). At the age of 8 years, persistent renal dysfunction was observed (eGFR: 80–90 mL/min/1.73 m2). Interestingly, cases of FJHN with c744C > G (p.Cys248Trp) mutations also exhibit a high incidence of juvenile onset, and identical disulfide bridges are considered responsible for the accumulation of mutant UMOD in the endoplasmic reticulum. Pediatricians should consider UMOD mutation analysis for families with autosomal dominant tubulointerstitial kidney disease (ADTKD) and a bland urinary sediment, even if hyperuricemia is mild. Also, sex and genotype are very important prognostic factors for ADTKD caused by UMOD mutations.
Keywords: Familial juvenile hyperuricemic nephropathy, Autosomal dominant tubulointerstitial kidney disease, Uromodulin, Disulfide bridges, Chaperone therapy
Introduction
Uromodulin (UMOD)-related autosomal dominant tubulointerstitial kidney disease (ADTKD-UMOD) is a rare monogenic disease advocated by KDIGO in 2015 [1]. It was previously known as familial juvenile hyperuricemic nephropathy (FJHN; OMIM #162, 000) [2], medullary cystic kidney disease type 2 (MCKD 2), and UMOD-associated kidney disease [3]. ADTKD-UMOD is characterized by early-onset excretory hyperuricemia, impaired urine-concentrating ability, tubulointerstitial nephropathy, and end-stage renal disease (ESRD) [4]. The symptoms of progressive renal impairment usually become apparent in adolescence, leading to ESRD within 10–20 years [5]. Moskowitz et al. reported that the mean age of ESRD development was 56 years [6]; thus, very few cases are diagnosed in the early years of their life. Here we report a case involving a young boy who exhibited hyperuricemia at the age of 4 years and showed a novel mutation at c.766 T > G (p.Cys256Gly) located in the domain of eight cysteine residues (D8C) in the UMOD gene.
Case report
A 4-year-old boy presented to our hospital with gross hematuria, proteinuria, and abdominal pain. Immunoglobulin A vasculitis with nephritis was suspected on admission. Physical examination revealed a height and weight of 96 cm and 14.0 kg, respectively. His blood pressure was normal (86/56 mmHg). Laboratory tests during hospitalization revealed hyperuricemia [uric acid (UA), 8.0 mg/dL; fractional excretion of UA, 3.6% (normal range 6–12%); UA clearance to creatinine clearance ratio 3.7% (normal range 5.5–11.1%)] and renal impairment [serum creatinine level 0.33 mg/dL; creatinine-estimated glomerular filtration rate (Cre-eGFR [7]), 84.41 mL/min/1.73m2], and a tubular disorder (urea-β2-microglobulin, 308 µg/L; urea-N-acetyl-β-glucosaminidase, 4.5 IU/L; fractional excretion of Na, 0.74%; fractional excretion of K, 8.1%). Serum electrolyte levels (including sodium, potassium, glucose, bicarbonate, and calcium) were normal. Ultrasonography showed no renal cysts or stones. There were no perinatal abnormalities and no previous history of medication. We identified a maternal history of hyperuricemia, renal impairment, and dialysis (Fig. 1). After several days of hospitalization, he was discharged as his clinical symptoms spontaneously alleviated with conservative treatment. However, at the age of 5 years, ADTKD-UMOD was suspected because of persistent hyperuricemia and mild renal dysfunction observed during subsequent outpatient visits. Given that 95% of UMOD mutations causing FJHN are known to exist in exons 3–4 [1, 9], we performed direct sequence analysis of exons 2–12 and found a novel missense mutation in exon 3; codon 766 was mutated from T to G (Fig. 2). A final diagnosis of FJHN was established. At the time of writing this report, the patient was 8 years old, and, fortunately, his UA level (6.0 mg/dL) was well controlled with allopurinol 10 mg/kg, while his eGFR was maintained at 80–90 mL/min/1.73 m2 without any worsening of renal dysfunction.
Fig. 1.
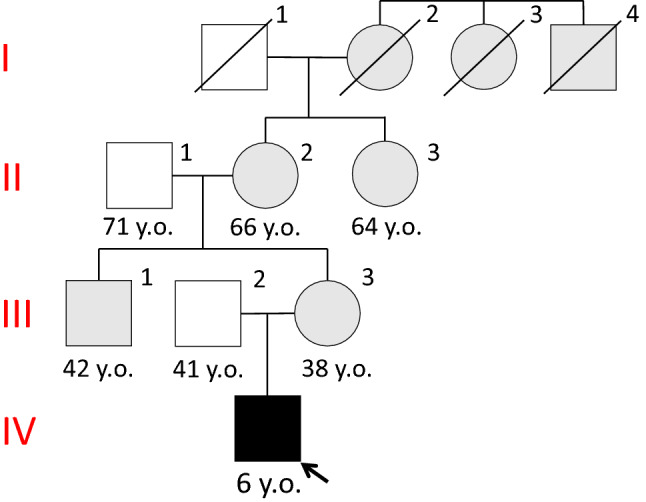
Family pedigree, I-2: dialysis since the age of 55 years. I-3,4: dialysis, II-1: gount II-2: hyperuricemia, chronic kidney disease, II-3: hyperuricemia, gout and dialysis since the age of 61 years, III-1: hyperuricemia III-3: hyperuricemia, renal dysfunction
Fig. 2.

EGF epidermal growth factor-like motifs, Cys cystenine, D8C domain of eight cysteines, ZP zona pellucida
Discussion
We identified a novel missense mutation (c766T > G), p.Cys256Gly, in exon 3 of the UMOD gene in a male patient with FJHN who developed hyperuricemia at the age of 4 years. His UA level was well controlled by allopurinol, and there was no progression of renal dysfunction at 8 years of age.
Autosomal dominant tubulointerstitial kidney disease-UMOD generally presents with hyperuricemia between 10 and 40 years of age, with progressive renal dysfunction leading to ESRD between 40 and 70 years of age [5, 6]. Hyperuricemia rarely occurs before the age of 10 years. Although the pathogenesis of ADTKD-UMOD remains unknown, the current possible pathogenesis and treatments are shown in Fig. 3, [9–14]. The UMOD gene encodes uromodulin, also known as Tamm–Horsfall protein, which is produced exclusively by the renal tubular cells in the thick ascending limb of the loop of Henle (TALH). Physiologically, UMOD is thought to be involved in defense mechanisms against infections in the urinary system and the suppression of water permeability in tubules. However, the biological function of this protein is incompletely understood. Most UMOD mutations that have been identified are missense mutations that mainly alter cysteine amino acids [6]. UMOD cannot be folded correctly by conversion cysteine because the cysteine residue is involved in disulfide bond formation. Accumulation of the misfolded, mutated UMOD in the endoplasmic reticulum (ER) potentially results in tubular cell dysfunction, apoptosis, and death, leading to tubular atrophy and interstitial fibrosis. It is thought that ER stress in TALH cells causes hyperuricemia and renal failure.
Fig. 3.

Proposed model of pathogenesis and treatment for FJHN. FJHN familial juvenile hyperuriccemic nephropathy, ER endoplasmic reticulum
Mutations in the uromodulin are thought to contain three epidermal growth factor (EGF)-like domains; the second and third domains contain a calcium-binding motif, D8C within a cysteine-rich region, and a zona pellucida (ZP)-like domain, which is responsible for the polymerization of extracellular proteins into helical filaments [8]. The coding region of exon 3 is from EGF1 to D8C (Fig. 2). The time to ESRD development is significantly associated with these UMOD domains. Patients with mutations in the D8C, EGF2, and EGF3 domains exhibit an increased risk of developing ESRD earlier than do patients with mutations in the Cys-Rich1 domain (hazard ratios, 4.37, 7.74, and 4.44, respectively) [6]. Moreover, the rate of progression to ESRD tends to be higher and faster for patients with mutations in D8C, EGF2, and EGF3 than in those with mutations in Cys-rich1 and Cys-rich2 [6]. It has also been reported that male patients are more likely to progress to ESRD than are female patients [6]. In the present case, the parents of the child did not consent to undergo genetic analysis. However, the patient's mother had developed renal dysfunction at the age of 38 years. In addition, he was male, showed a mutation in D8C, and developed hyperuricemia in childhood; therefore, he was at high risk of developing ESRD in the future. D8C contains eight cysteine residues that are predicted to form four pairs of disulfide crosslinks, with Cys 217 and Cys 223, Cys 248 and Cys 256, Cys 255 and Cys 267, and Cys 282 and Cys 287 having the potential to form disulfide bridges [15]. In a report of 11 cases with mutations at c.744 C > G (p.Cys248Trp), the incidence of juvenile onset was higher than that reported for cases with mutations at other amino acid sites. Moreover, disease onset was before the age of 10 years in 27% (n = 3) of the cases. One of these patients developed hyperuricemia at the age of 3 years and ESRD by the age of 6 years [16]. Interestingly, the present case showed a mutation at Cys256, which is a counterpart of Cys248. This finding is consistent with the accumulation of A caused by impaired disulfide bridges in D8C, as well as the risk of relatively early onset.
Although the alleviation of progression to renal failure by serum urate-lowering treatment remains a controversial topic [12], asymptomatic hyperuricemia associated with the use of a xanthine oxidase inhibitor is thought to reduce the risk of cardiovascular events and insulin resistance [17, 18]. In addition, the efficacy of kidney transplantation for ADTKD-UMOD was reported in 2007 [13], with no reoccurrence in the graft [13]. Therefore, early diagnosis would allow patients and their families to consider treatment options, including kidney transplantation. In the present case, because of the high risk of ESRD progression, it was necessary to consider a course of treatment that included renal transplantation while continuing the urate-lowering therapy.
Conclusion
In summary, we identified a novel mutation (c766T > G), p.Cys256Gly, in exon 3 of the UMOD gene in a boy with FJHN who developed hyperuricemia at the age of 4 years. Pediatricians should consider UMOD mutation analysis for families with ADTKD and a bland urinary sediment, even if hyperuricemia is mild. In addition, sex and genotype are very important prognostic factors for ADTKD-UMOD.
Acknowledgments
I would like to thank Dr. Norio Taniguchi of Tokyo Women’s Medical University for performing this genetic test.
Compliance with ethical standards
Conflict of interest
The authors have declared that no conflict of interest exists.
Informed consent
Informed consent for publication of this report was obtained from the patient.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Eckardt K-U, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, Deltas C, Hosking A, Kmoch S, Rampoldi L, Wiesener M. Kidney disease: improving global outcomes. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management—a KDIGO consensus report. Kidney Int. 2015;88:676–683. doi: 10.1038/ki.2015.28. [DOI] [PubMed] [Google Scholar]
- 2.Duncan H, Dixon AS. Gout, familial hyperuricaemia, and renal disease. Q J Med. 1960;29:127–135. [PubMed] [Google Scholar]
- 3.Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, Bleyer AJ. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39:882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bleyer AJ, Zivná M, Kmoch S. Uromodulin-associated kidney disease. Nephron Clin Pract. 2011;118:c31–c36. doi: 10.1159/000320889. [DOI] [PubMed] [Google Scholar]
- 5.Dahan K, Fuchshuber A, Adamis S, Smaers M, Kroiss S, Loute G, Cosyns JP, Hildebrandt F, Verellen-Dumoulin CH, Pirson Y. Familial juvenile hyperuricemic nephropathy and autosomal dominant medullary kidney cystic disease type 2: two facets of the same disease? J Am Soc Nephrol. 2001;12:2348–2357. doi: 10.1681/ASN.V12112348. [DOI] [PubMed] [Google Scholar]
- 6.Moskowitz JL, Piret SE, Lhotta K, Kitzler TM, Tashman AP, Velez E, Thakker RV, Kotanko P. Association between genotype and phenotype in uromodulin-associated kidney disease. Clin J Am Soc Nephrol. 2013;8:1349–1357. doi: 10.2215/CJN.11151012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uemura O, Nagai T, Ishikura K, Ito S, Hataya H, Gotoh Y, Fujita N, Akioka Y, Kaneko T, Honda M. Creatinine-based equation to estimate the glomerular filtration rate in Japanese children and adolescents with chronic kidney disease. Clin Exp Nephrol. 2014;18:626–633. doi: 10.1007/s10157-013-0856-y. [DOI] [PubMed] [Google Scholar]
- 8.Turner JJ, Stacey JM, Harding B, Kotanko P, Lhotta K, Puig JG, Roberts I, Torres RJ, Thakker RV. Uromodulin mutations cause familial juvenile hyperuricemic nephropathy. J Clin Endocrinol Metab. 2003;88:1398–1401. doi: 10.1210/jc.2002-021973. [DOI] [PubMed] [Google Scholar]
- 9.Liu M, Chen Y, Liang Y, Liu Y, Wang S, Hou P, Zhang H, Zhao M. Novel UMOD mutations in familial juvenile hyperuricemic nephropathy lead to abnormal uromodulin intracellular trafficking. Gene. 2013;531:363–369. doi: 10.1016/j.gene.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 10.Nasr SH, Lucia JP, Galgano SJ, Markowitz GS, D’Agati VD. Uromodulin storage disease. Kidney Int. 2008;73:971–976. doi: 10.1038/sj.ki.5002679. [DOI] [PubMed] [Google Scholar]
- 11.Bernascone I, Janas S, Ikehata M, Trudu M, Corbelli A, Schaeffer C, Rastaldi MP, Devuyst O, Rampoldi L. A transgenic mouse model for uromodulin-associated kidney diseases shows specific tubulointerstitial damage, urinary concentrating defect, and renal failure. Hum Mol Genet. 2010;19:2998–3010. doi: 10.1093/hmg/ddq205. [DOI] [PubMed] [Google Scholar]
- 12.Fairbanks LD, Cameron JS, Venkat-Raman G, Rigden SP, Rees L, Van'T Hoff W, Mansell M, Pattison J, Goldsmith DJ, Simmonds HA. Early treatment with allopurinol in familial juvenile hyerpuricaemic nephropathy (FJHN) ameliorates the long-term progression of renal disease. QJM. 2002;95:597–607. doi: 10.1093/qjmed/95.9.597. [DOI] [PubMed] [Google Scholar]
- 13.Labriola L, Ki D, Pirson Y. Outcome of kidney transplantation in familial juvenile hyperuricaemic nephropathy. Nephrol Dial Transplant. 2007;22:3070–3073. doi: 10.1093/ndt/gfm362. [DOI] [PubMed] [Google Scholar]
- 14.Kemter E, Sklenak S, Rathkolb B, de Angelis MH, Wolf E, Aigner B, Wanke R. No amelioration of uromodulin maturation and trafficking defect by sodium-4-phenylbutyrate in vivo: studies in mouse models of uromodulin-associated kidney disease. J Biol Chem. 2014;289:10715–10726. doi: 10.1074/jbc.M113.537035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H, Wu C, Zhao S, Guo J. Identification and characterization of D8C, a novel domain present in liver-specific LZP, uromodulin and glycoprotein 2, mutated in familial juvenile hyperuricaemic nephropathy. FEBS Lett. 2004;578:236–238. doi: 10.1016/j.febslet.2004.10.092. [DOI] [PubMed] [Google Scholar]
- 16.Wolf MT, Beck BB, Zaucke F, Kunze A, Misselwitz J, Ruley J, Ronda T, Fischer A, Eifinger F, Licht C, Otto E. The uromodulin C744G mutation causes MCKD2 and FJHN in children and adults and may be due to a possible founder effect. Kidney Int. 2007;71:574–581. doi: 10.1038/sj.ki.5002089. [DOI] [PubMed] [Google Scholar]
- 17.Goicoechea M, Garcia V, Verdalles U, Verde E, Macias N, Santos A, de Jose AP, Cedeño S, Linares T, Luño J. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis. 2015;65:543–549. doi: 10.1053/j.ajkd.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 18.Takir M, Kostek O, Ozkok A, Elcioglu OC, Bakan A, Erek A, Mutlu HH, Telci O, Semerci A, Odabas AR, Afsar B. Lowering uric acid with allopurinol improves insulin resistance and systemic inflammation in asymptomatic hyperuricemia. J Invest Med. 2015;63:924–929. doi: 10.1097/JIM.0000000000000242. [DOI] [PubMed] [Google Scholar]