

Significance
Primer-dependent transcription initiation—the use of RNA primers as initiating entities in transcription initiation—yields RNA products having a 5′-hydroxyl. Here, we show that primer-dependent initiation in vivo in Escherichia coli involves predominantly dinucleotide primers, involves any of the 16 possible dinucleotide primers, and depends on promoter sequences in, upstream, and downstream of the primer binding site. Crystal structures explain the structural basis of sequence dependence at the promoter position immediately upstream of the primer binding site, namely, interchain base stacking between the promoter template-strand nucleotide and primer 5′ nucleotide. Taken together, our findings provide a mechanistic and structural description of primer-dependent initiation in E. coli.
Keywords: RNA polymerase, primer, nanoRNA, promoter, RNA modification
Abstract
Chemical modifications of RNA 5′-ends enable “epitranscriptomic” regulation, influencing multiple aspects of RNA fate. In transcription initiation, a large inventory of substrates compete with nucleoside triphosphates for use as initiating entities, providing an ab initio mechanism for altering the RNA 5′-end. In Escherichia coli cells, RNAs with a 5′-end hydroxyl are generated by use of dinucleotide RNAs as primers for transcription initiation, “primer-dependent initiation.” Here, we use massively systematic transcript end readout (MASTER) to detect and quantify RNA 5′-ends generated by primer-dependent initiation for ∼410 (∼1,000,000) promoter sequences in E. coli. The results show primer-dependent initiation in E. coli involves any of the 16 possible dinucleotide primers and depends on promoter sequences in, upstream, and downstream of the primer binding site. The results yield a consensus sequence for primer-dependent initiation, YTSS−2NTSS−1NTSSWTSS+1, where TSS is the transcription start site, NTSS−1NTSS is the primer binding site, Y is pyrimidine, and W is A or T. Biochemical and structure-determination studies show that the base pair (nontemplate-strand base:template-strand base) immediately upstream of the primer binding site (Y:RTSS−2, where R is purine) exerts its effect through the base on the DNA template strand (RTSS−2) through interchain base stacking with the RNA primer. Results from analysis of a large set of natural, chromosomally encoded E. coli promoters support the conclusions from MASTER. Our findings provide a mechanistic and structural description of how TSS-region sequence hard-codes not only the TSS position but also the potential for epitranscriptomic regulation through primer-dependent transcription initiation.
In transcription initiation, the RNA polymerase (RNAP) holoenzyme binds promoter DNA by making sequence-specific interactions with core promoter elements and unwinds a turn of promoter DNA forming an RNAP-promoter open complex (RPo) containing a single-stranded “transcription bubble.” Next, RNAP selects a transcription start site (TSS) by placing the start-site nucleotide and the next nucleotide of the “template DNA strand” into the RNAP active-center product site (P site) and addition site (A site), respectively, and binding an initiating entity in the RNAP active-center P site (Fig. 1A). RNAPs can initiate transcription using either a primer-independent or primer-dependent mechanism (1–10). In primer-independent initiation, the initiating entity (typically a nucleoside triphosphate [NTP]) base pairs to the template-strand nucleotide in the RNAP active-center P site (TSS; Fig. 1A). In primer-dependent transcription initiation, the 3′ nucleotide of a two-, three-, or four-nucleotide RNA primer (di-, tri-, or tetra-nucleotide primer, respectively) base pairs to the template-strand nucleotide in the RNAP active-center P site, and the 5′ nucleotide of the primer base pairs to the template-strand nucleotide in the P−1, P−2, or P−3 site (TSS−1, TSS−2, and TSS−3, respectively; Fig. 1A).
Fig. 1.

Use of MASTER to monitor primer-independent and primer-dependent transcription initiation. (A) Binding of initiating entities to DNA template-strand nucleotides in primer-independent and primer-dependent transcription initiation. (Top) RPo. (Bottom) Enlarged view of initiating entities bound to template-strand nucleotides in the RNAP active center. Dark gray, RNAP; yellow, σ; blue, −10-element nucleotides; purple, TSS nucleotides; light gray, RNA nucleotides; pink, primer-binding nucleotides at positions TSS−1, TSS−2 or TSS−3; white boxes, DNA nucleotides; NT, nontemplate-strand nucleotides; T, template-strand nucleotides. P−3, P−2, P−1, and P, RNAP active-center initiating entity binding sites; A, RNAP active-center extending NTP binding site. Unwound transcription bubble in RPo indicated by raised and lowered nucleotides. (B) Analysis of primer-independent and primer-dependent initiation using MASTER. (Top) DNA fragment containing MASTER template library. Light green, randomized nucleotides in the promoter region; dark green, transcribed-region barcode. (Bottom) 5′ RNA-seq analysis of RNA products generated from the promoter library by primer-independent, NTP-dependent initiation (Rpp treatment) and primer-dependent initiaion (PNK treatment).
In Escherichia coli cells, primer-dependent transcription initiation occurs during stationary-phase growth and modulates the expression of genes involved in biofilm formation (9–11). RNAs generated by primer-dependent initiation in E. coli contain a 5′-end hydroxyl (5′-OH), indicating that the primers incorporated at the RNA 5′-end also contain a 5′-OH (9). Available data suggests that most primer-dependent initiation in E. coli involves use of dinucleotide primers, most frequently UpA and GpG (9–11). However, direct evidence that dinucleotides serve as the predominant initiating entity in primer-dependent initiation has not been presented. In addition, apart from the sequences complementary to the primer, the primer binding site, promoter-sequence determinants for primer-dependent initiation have not been defined.
Here, we adapt a massively parallel reporter assay to monitor primer-dependent initiation in E. coli. The results provide a complete inventory of the RNA 5′-end sequences generated by primer-dependent initiation in E. coli and define the critical promoter-sequence determinants for primer-dependent initiation. The results demonstrate that most, if not all, primer-dependent initiation in E. coli involves use of a dinucleotide as the initiating entity and identify a consensus sequence for primer-dependent initiation, YTSS−2NTSS−1NTSSWTSS+1, where TSS is the transcription start site, NTSS−1NTSS is the primer binding site, Y is pyrimidine, and W is A or T. We further demonstrate that sequence information at the position immediately upstream of the primer binding site resides exclusively in the template strand of the transcription bubble (RTSS−2, where R is purine). We report crystal structures of transcription-initiation complexes containing dinucleotide primers that reveal the structural basis for a purine at the template-strand position immediately upstream of the primer binding site (RTSS−2): namely, more extensive, and likely more energetically favorable, base stacking between the template-strand base and the primer 5′ base.
Results
Use of Massively Systematic Transcript End Readout to Monitor Primer-Dependent Initiation in E. coli.
To define, comprehensively, the promoter-sequence determinants for primer-dependent initiation in E. coli, we modified a massively parallel reporter assay previously developed in our laboratory, termed massively systematic transcript end readout (MASTER) (12, 13), in order to detect both primer-independent and primer-dependent initiation, to differentiate between primer-independent and primer-dependent initiation, and to define primer lengths in primer-dependent initiation (Fig. 1B).
MASTER involves construction of a promoter library that contains up to 411 (∼4,000,000) barcoded sequences, production of RNA transcripts from the promoter library, and analysis of RNA barcodes and RNA 5′-ends using high-throughput sequencing (5′ RNA-seq) to define, for each RNA product, the template that produced the RNA and the sequence of the RNA 5′-end (Fig. 1B) (12–14). The 5′ RNA-seq procedure used in MASTER relies on ligation of single-stranded oligonucleotide adaptors to RNAs containing a 5′-end monophosphate (5′-p) (13). In previous work, we treated RNAs with RNA 5′ pyrophosphohydrolase (Rpp), which converts a 5′-end triphosphate (5′-ppp) to a 5′-p; this procedure specifically enables detection of the 5′-ppp–bearing RNAs generated by primer-independent initiation (12, 14–16). Here, we treated RNAs in parallel with Rpp to detect RNAs generated by primer-independent initiation and with polynucleotide kinase (PNK), which converts a 5′-OH to a 5′-p, to detect RNAs generated by primer-dependent initiation (Fig. 1B). By comparing the results from Rpp and PNK reactions, we quantify, for each promoter sequence in the library, the relative efficiencies of primer-independent and primer-dependent initiation and primer lengths for primer-dependent initiation.
In the present work, we used a MASTER template library containing 410 (∼1,000,000) sequence variants at the positions 1 to 10 base pairs (bp) downstream of the −10 element of a consensus σ70-dependent promoter (placCONS-N10; Fig. 1B). The randomized segment of placCONS-N10 contains the full range of TSS positions for E. coli RNAP observed in previous work (i.e., TSS positions located 6, 7, 8, 9, and 10 bp downstream of the promoter −10 element; Fig. 2A). We introduced the placCONS-N10 library into E. coli, grew cells to stationary phase (the phase in which primer-dependent initiation has been observed in previous work; ref. 9), isolated total cellular RNA, and analyzed RNAs generated from each promoter sequence in the library by 5′ RNA-seq. The results provide complete inventories of RNA 5′-ends generated by primer-independent initiation and primer-dependent initiation in stationary-phase E. coli cells.
Fig. 2.

Distributions of 5′-end sequences for RNAs generated in primer-independent initiation and primer-dependent initiation in vivo and in vitro. (A) lacCONS-N10 library. Base pairs in the N10 region are numbered based on their position relative to the promoter −10 element. Colors as in Fig. 1B. (B and C) RNA 5′-end distribution histograms (mean ± SD, n = 3) for RNAs generated by primer-independent initiation (Top) or primer-dependent initiation (Bottom) in stationary-phase E. coli cells (B) or in vitro with the dinucleotide primer UpA (C). Dashed line indicates the mean 5′-end position (mean ± SD, n = 3). The relative number of reads for RNAs generated by primer-independent initiation versus primer-dependent initiation in stationary-phase E. coli cells is 5.8 ± 0.5 (mean ± SD, n = 3).
Primer-Dependent Initiation: 5′-End Positions.
Our results define distributions of 5′-end positions of the 5′-ppp RNAs generated by primer-independent initiation and the 5′-OH RNAs generated by primer-dependent initiation for transcription in stationary-phase E. coli cells (Fig. 2B).
The distributions of 5′-end positions for primer-independent initiation show 5′-end positions (TSS positions) ranging from 6 to 10 bp downstream of the promoter −10 element, with a mean 5′-end position ∼7.5 bp downstream of the promoter −10 element (Fig. 2 B, Top). The range, the mean, and the distribution shape closely match those previously observed for primer-independent initiation for cells in exponential phase (12).
The distributions of 5′-end positions for primer-dependent initiation show 5′-end positions ranging from 5 to 9 bp downstream of the promoter −10 element, with a mean 5′-end position ∼6.8 bp downstream of the promoter −10 element (Fig. 2 B, Bottom). The range, the mean, and the distribution shape closely match those for primer-independent initiation but with a ∼1-bp upstream shift.
Primer-Dependent Initiation: Primer Lengths.
Comparison of the 5′-end distributions for primer-independent initiation (Fig. 2 B, Top) versus primer-dependent initiation (Fig. 2 B, Bottom) indicates that, across all promoter sequences in the library, the 5′-end positions of RNAs generated by primer-independent initiation (mean position 7.54 ± 0.01 bp downstream of the −10 element) and RNAs generated by primer-dependent initiation (mean position 6.75 ± 0.05 bp downstream of the −10 element) differ by ∼1 bp (0.71 ± 0.06 bp). Following the logic of Fig. 1, based on the observed difference of almost exactly 1 bp in mean 5′-end position for primer-independent initiation versus primer-dependent initiation, we infer that primer length in primer-dependent initiation in stationary-phase E. coli cells is almost always 2 nt. Computational modeling using the distributions in Fig. 2B indicates that no more than ∼2.5% of the observed primer-dependent initiation could involve primer lengths greater than 2 nt (SI Appendix, Fig. S1A). Consistent with these inferences, comparison of distributions of RNA 5′-end positions for primer-independent initiation in vitro versus primer-dependent initiation in vitro with the dinucleotide primer UpA shows essentially the same ∼1-bp upstream shift in distribution range, mode, and mean (Fig. 2C and SI Appendix, Fig. S1B).
Primer-Dependent Initiation: Primer Sequences.
We next measured yields of 5′-OH RNAs generated by primer-dependent initiation with each of the 16 possible dinucleotide primers (Fig. 3A). The results show that primer-dependent initiation occurs with all 16 dinucleotide primers. The highest levels of primer-dependent initiation are observed with the dinucleotide primers UpA and GpG, which account for ∼27% and ∼17%, respectively, of 5′-OH RNAs generated across all promoters in the library (Fig. 3 A, Left). The other 14 dinucleotide primers each account for ∼1 to ∼8% of 5′-OH RNAs generated across all promoters in the library. Qualitatively similar results are obtained by analyzing RNA products from promoters in which the primer binding site is at positions 5 to 6, 6 to 7, 7 to 8, 8 to 9, and 9 to 10 bp downstream of the promoter −10 element (Fig. 3 A, Right). The demonstration that primer-dependent initiation occurs with all 16 dinucleotides in vivo is unique to this work, as is the demonstration that primer-dependent initiation occurs at the full range of TSS positions observed for primer-independent initiation in vivo (i.e., TSS positions located 6, 7, 8, 9, and 10 bp downstream of the promoter −10 element). The observation that UpA and GpG are preferentially used as primers in vivo is consistent with results of prior work (9, 10).
Fig. 3.

Promoter-sequence dependence of primer-dependent initiation: primer binding site. (A) Relative usage of dinucleotides in primer-dependent initiation in stationary-phase E. coli cells. Values represent the percentage of total 5′-OH RNAs generated using each of the 16 dinucleotide primers (mean, n = 3). Bold, dinucleotides preferentially used as primers. (B) Complementarity between the primer binding site and dinucleotide in primer-dependent initiation. (Top) Primer-dependent initiation involving template-strand complementarity to both 5′ and 3′ nucleotides of primer (TSS−1, TSS), template-strand complementarity to only the 3′ nucleotide of primer (TSS), template-strand complementarity to only the 5′ nucleotide of primer (TSS−1), or no template-strand complementarity to primer (none). Three vertical lines, complementarity; X, noncomplementarity. Other symbols and colors as in Fig. 1. (Bottom) Percentage of primer-dependent initiation involving complementarity to both 5′ and 3′ nucleotides of primer (TSS−1, TSS; pink), complementarity to only the 3′ nucleotide of primer (TSS; purple), or template-strand complementarity to only the 5′ nucleotide of primer or no template-strand complementarity to primer (TSS−1 or none; white) in stationary-phase E. coli cells (Left) or in vitro with the dinucleotide primer UpA (Right) (mean ± SD, n = 3).
Primer-Dependent Initiation: Promoter-Sequence Dependence, Primer Binding Site.
Analysis of the results for primer-dependent initiation, separately considering RNA products with 5′-ends complementary to the template and RNA products with 5′-ends noncomplementary to the template, shows that the overwhelming majority of primer-dependent initiation in stationary-phase E. coli cells occurs at primer binding sites that have perfect template-strand complementarity to the 5′ and 3′ nucleotides of the dinucleotide primer (93.3 ± 0.4%; Fig. 3 B, Bottom). This is true across the entire promoter library for each of the 16 possible dinucleotide primer sequences (73.9 ± 0.2 to 98.1 ± 0.01% of primer binding sites with perfect complementarity; Fig. 3 B, Bottom) and for each of the major primer binding-site positions (SI Appendix, Fig. S2). Most of the limited noncomplementarity observed involves the 5′ nucleotide of the dinucleotide primers CpG, UpG, CpU, and UpU (24.2 ± 0.4%, 21.1 ± 0.6%, 10.3 ± 0.6%, and 10.1 ± 0.9%, respectively; Fig. 3B and SI Appendix, Fig. S2). Consistent with these results, analysis of the same promoter library in vitro, assessing primer-dependent initiation with the dinucleotide primer UpA, shows that the overwhelming majority of primer-dependent initiation likewise occurs at primer binding sites that have perfect template-strand complementarity to the 5′ and 3′ nucleotides of the dinucleotide primer for each of the major primer binding-site positions (84.1 ± 0.6%; Fig. 3 B, Bottom Right and SI Appendix, Fig. S3). In vitro transcription experiments using heteroduplex templates (templates having noncomplementary transcription-bubble nontemplate-strand and template-strand sequences) and the dinucleotide primer UpA show that the strong preference for perfect template-strand complementarity to the 5′ and 3′ nucleotides of the dinucleotide primer reflects a requirement for Watson–Crick base pairing of template-strand nucleotides at positions TSS−1 and TSS with the 5′ and 3′ nucleotides of the dinucleotide RNA primer, respectively (SI Appendix, Fig. S4).
We conclude that primer-dependent initiation with a dinucleotide primer almost always involves a primer binding site having perfect template-strand complementarity to, and therefore able to engage in Watson–Crick base pairing with, the dinucleotide primer. This result is not completely unexpected. However, this point has not been demonstrated previously in vivo, and, in prior work, in vitro, with tetranucleotide primers (17), had indicated that perfect template-strand complementarity to the primer may not be necessary for primer-dependent initiation with longer primers.
Primer-Dependent Initiation: Promoter-Sequence Dependence, Sequences Flanking the Primer Binding Site.
The observed yields of 5′-OH RNA products from primer-dependent initiation in stationary-phase E. coli cells strongly correlate with the promoter sequences flanking the primer binding site (Fig. 4 and SI Appendix, Fig. S5). Levels of primer-dependent initiation depend on the identities of base pairs up to 7 bp upstream of the primer binding site and up to 3 bp downstream of the primer binding site. The base pair (nontemplate-strand base–template-strand base) at the position immediately upstream of the primer binding site, position TSS−2, makes the largest contribution to the sequence dependence of primer-dependent initiation. Levels of primer-dependent initiation are higher for promoters with a nontemplate-strand pyrimidine (C or T) and template-strand purine (A or G) at position TSS−2 (Fig. 4, Left). A strong preference for a Y–R base pair at position TSS−2 is observed at each of the major TSS positions (i.e., the positions 6, 7, and 8 bp downstream of the promoter −10 element; Fig. 4, Left). The results further show that the base pair at the position immediately downstream of the primer binding site, position TSS+1, makes the second largest contribution to the sequence dependence of primer-dependent initiation; levels of primer-dependent initiation are higher for promoters with a T:A or A:T base pair at position TSS+1 (Fig. 4, Left). A strong preference for T:A or A:T at position TSS+1 is observed when the TSS is 6 bp downstream of the promoter −10 element, and a weaker preference is observed when the TSS is 7 or 8 bp downstream of the promoter −10 element (Fig. 4, Left). Base pairs at positions 3, 4, 5, 6, and 7 bp upstream of the primer binding site (TSS−3, TSS−4, TSS−5, TSS−6, and TSS−7) and at positions 2 and 3 bp downstream of the primer binding site (TSS+2 and TSS+3), make small but significant contributions to levels of primer-dependent initiation (Fig. 4, Left). The results define a global consensus sequence for primer-dependent initiation: YTSS−2NTSS−1NTSSWTSS+1 (Y:RTSS−2N:NTSS−1N:NTSSW:WTSS+1), where TSS is the transcription start site, NTSS−1NTSS is the primer binding site, Y is pyrimidine, and W is A or T. The same—or essentially the same—consensus is observed for all 16 primer sequences and for each major primer binding-site position (SI Appendix, Fig. S5). Analysis of the same promoter library in vitro, assessing primer-dependent initiation with the dinucleotide primer UpA, yields the same consensus sequence, YTSS−2NTSS−1NTSSWTSS+1, and does so for each primer binding-site position (Fig. 4, Right and SI Appendix, Fig. S6).
Fig. 4.

Promoter-sequence dependence of primer-dependent initiation: sequences flanking the primer binding site. Sequence logos for primer-dependent initiation at TSS positions 7, 8, and 9 (corresponding to primer binding sites 6 to 7, 7 to 8, and 8 to 9, respectively) in stationary-phase E. coli cells (Left) or in vitro with the dinucleotide primer UpA (Right). The height of each base X at each position Y represents the relative log2 enrichment (averaged across replicates) of the percent 5′-OH RNAs expressed from promoter sequences containing nontemplate-strand X at position Y. Red, consensus nucleotides; black, nonconsensus nucleotides. Other symbols and colors as in Fig. 1.
In vitro transcription experiments assessing competition between primer-dependent transcription initiation with UpA and primer-independent transcription initiation with ATP show that primer-dependent initiation is ∼60 times more efficient than primer-independent initiation at a promoter conforming to the consensus sequence (TTSS−2TTSS−1ATSSTTSS+1; Fig. 5A) but is only ∼10 times more efficient at a promoter not having the consensus sequence (GTSS−2TTSS−1ATSSTTSS+1; Fig. 5A).
Fig. 5.

Promoter-sequence dependence of primer-dependent initiation in vitro: position TSS−2. (A) Relative efficiencies of primer-dependent initiation versus primer-independent initiation depends on promoter sequence at position TSS−2. (Top Left) Homoduplex DNA templates containing consensus or nonconsensus nucleotides for primer-dependent initiation at position TSS−2. (Bottom Left) Relative efficiencies of primer-dependent initiation with UpA versus primer-independent initiation with adenosine triphosphate (ATP) [(kcat/KM)UpA/(kcat/KM)ATP] and ratio of (kcat/KM)UpA/(kcat/KM)ATP for the indicated templates. (Right) Dependence of primer-dependent initiation on [UpA]/[ATP] ratio (mean ± SD, n = 3). Red, consensus nucleotides at position TSS−2. Unwound transcription bubble in RPo indicated by raised and lowered nucleotides. Other symbols and colors as in Fig. 1. (B) The template DNA strand carries sequence information at position TSS−2. (Top Left) DNA templates containing mismatches at position TSS−2. Templates contain a consensus nucleotide at position TSS−2 on only the nontemplate strand (T/CTSS−2 and T/XTSS−2), only the template strand (G/ATSS−2 and X/ATSS−2), or neither strand (X/CTSS−2 and G/XTSS−2). (Bottom Left) Relative efficiencies and efficiency ratios for the indicated heteroduplex templates. (Right) Dependence of primer-dependent initiation on [UpA]/[ATP] ratio (mean ± SD, n = 3). Red, consensus nucleotides at position TSS−2. Unwound transcription bubble in RPo indicated by raised and lowered nucleotides.
In vitro transcription experiments using heteroduplex templates and the dinucleotide primer UpA show that the sequence information responsible for the preference for Y:R at TSS−2 resides exclusively in the DNA template strand (Fig. 5B). Thus, in experiments with heteroduplex templates, primer-dependent initiation is reduced by replacement of the consensus nucleotide by a nonconsensus nucleotide or an abasic site on the DNA template strand but is not reduced by replacement of the consensus nucleotide by a nonconsensus nucleotide or an abasic site on the DNA nontemplate strand (Fig. 5B).
We conclude that primer-dependent initiation, in vivo and in vitro, depends not only on the sequence of the primer binding site but also on flanking sequences, with the preferred sequence being YTSS−2NTSS−1NTSSWTSS+1 (Y:RTSS−2N:NTSS−1N:NTSSW:WTSS+1).
Primer-Dependent Initiation: Chromosomal Promoters.
To assess whether the sequence preferences observed in the MASTER analysis apply also to natural promoters, we quantified primer-dependent initiation in stationary-phase E. coli cells at each of 93 promoters that use UpA as primer (SI Appendix, Table S1 and Fig. 6A). The results show the same sequence preferences at positions TSS-2 and TSS+1 observed in the MASTER analysis are observed in chromosomally-encoded promoters (Fig. 6A).
Fig. 6.
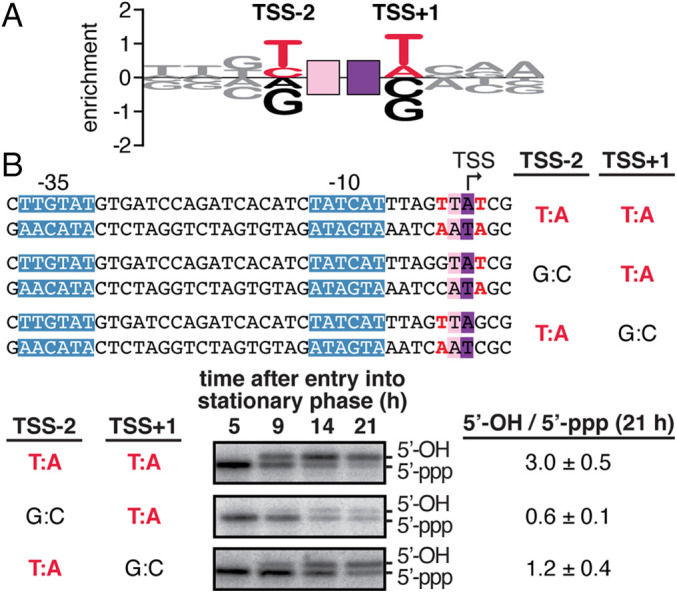
Promoter-sequence dependence of primer-dependent initiation: chromosomal promoters. (A) Sequence logo for primer-dependent initiation at TSS positions 7, 8, and 9 (corresponding to primer binding sites 6 to 7, 7 to 8, and 8 to 9, respectively) in stationary-phase E. coli cells for 93 natural, chromosomally encoded promoters that use UpA as a primer. The height of each base X at each position Y represents the relative log2 enrichment (averaged across replicates) of the percent 5′-OH RNAs expressed from promoter sequences containing nontemplate-strand base X at position Y. Red, consensus nucleotides; black, nonconsensus nucleotides. Other symbols and colors as in Fig. 1. (B) Promoter-sequence dependence of primer-dependent initiation at the E. coli bhsA promoter. (Top) Sequences of DNA templates containing wild-type and mutant derivatives of bhsA promoter. (Bottom) Primer extension analysis of 5′-end lengths of bhsA RNAs. In primer-dependent initiation with a dinucleotide primer, the RNA product acquires one additional nucleotide at the RNA 5′-end (Fig. 1). Gel shows radiolabeled cDNA products derived from primer-independent initiation (5′-ppp) and primer-dependent initiation (5′-OH) in stationary-phase E. coli cells. (Bottom Right) Ratios of primer-dependent initiation versus primer-independent initiation (mean ± SD, n = 4).
To assess directly the functional significance of the sequence preferences observed in the MASTER analysis and natural-promoter analysis, we constructed mutations at positions TSS−2 and TSS+1 of a natural promoter that uses UpA as primer (Fig. 6 B, Top) and assessed effects on function in stationary-phase E. coli cells (Fig. 6 B, Bottom). We observed that, at position TSS−2, the consensus base pair T:A is preferred over the nonconsensus base pair G:C by a factor of ∼5 (Fig. 6 B, Bottom), and, at position TSS+1, the consensus base pair T:A is preferred over the nonconsensus base pair G:C by a factor of ∼2.5 (Fig. 6 B, Bottom). We conclude that the sequence dependence for primer-dependent initiation defined using MASTER is also observed in natural, chromosomally encoded E. coli promoters.
Primer-Dependent Initiation: Structural Basis of Promoter Sequence Dependence.
To determine the structural basis of the preference for a template-strand purine nucleotide at position TSS−2 (RTSS−2) in primer-dependent initiation, we determined crystal structures of transcription-initiation complexes containing a template-strand purine nucleotide at position TSS−2 (Fig. 7). We first prepared crystals of Thermus thermophilus RPo using synthetic nucleotide scaffolds containing a template-strand purine nucleotide, A, at position TSS−2 and containing a template-strand primer binding site for either the dinucleotide primer used most frequently in primer-dependent initiation in vivo, UpA (refs. 9 and 10; Fig. 3A), or the dinucleotide primer used second most frequently in primer-dependent initiation in vivo, GpG (refs. 9 and 10; Fig. 3A). We next soaked the crystals either with UpA and CMPcPP or with GpG and CMPcPP to yield crystals of T. thermophilus RPo in complex with a dinucleotide primer and a nonreactive analog of an extending NTP. We then collected X-ray diffraction data, solved structures, and refined structures, obtaining structures of RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP at 2.8 Å resolution and RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP at 2.9 Å resolution (SI Appendix, Table S2 and Fig. 7).
Fig. 7.

Structural basis of promoter-sequence dependence of primer-dependent initiation at position TSS−2. Crystal structures of T. thermophilus RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP (Left) and T. thermophilus RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP (Right). (A) Experimental electron density (contoured at 2.5 σ; green mesh) and atomic model for the DNA template strand (yellow, red, blue, and orange for C, O, N, and P atoms), dinucleotide primer and CMPcPP (green, red, blue, and orange for C, O, N, and P atoms), RNAP active-center catalytic Mg2+(I) (violet sphere), and RNAP bridge helix (gray ribbon). (B) Contacts of RNAP residues (gray, red, and blue for C, O, and N atoms) with primer and RNAP active-center catalytic Mg2+(I). RNAP residues are numbered both as in T. thermophilus RNAP and as in E. coli RNAP (in parentheses). (C) Schematic summary of structures. Template-strand DNA (yellow); primer and CMPcPP (green); RNAP bridge helix (gray); RNAP active-center catalytic Mg2+(I) (violet). (D) Structural basis of promoter-sequence dependence at position TSS−2. Extensive interchain base stacking of template-strand purine, A or G, with the 5′ nucleotide of primer (upper row; red vertical dashed lines) and limited interchain base stacking of template-strand pyrimidine, C or T, and the 5′ nucleotide of primer (lower row). The interchain base-stacking patterns of template-strand A with primers UpA and GpG are as observed in structures of RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP and RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP (A–C); the interchain base-stacking pattern of template-strand T with primer GpG is as observed in structure of RPo[TTSS−2CTSS−1CTSS]-GpG-CMPcPP (SI Appendix, Fig. S7); the other interchain base-stacking patterns are modeled by analogy. Base atoms are shown as van der Waals surfaces. Colors are as in A.
For both RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP and RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP, experimental electron-density maps show unambiguous density for the dinucleotide primer in the RNAP active-center P−1 and P sites and for CMPcPP in the RNAP active-center A site (Fig. 7B). The dinucleotide primers make extensive interactions with RNAP, template-strand DNA, and CMPcPP. For each dinucleotide primer, the primer phosphate makes the same interactions with RNAP (residues β-H1237, β-K1065 and β-K1073; residues numbered as in E. coli RNAP) and the RNAP active-center catalytic Mg2+ as the primer phosphate in a previously reported structure of T. thermophilus RPo-GpA (ref. 18; Fig. 7 A–C). For each dinucleotide primer, the primer bases make Watson–Crick hydrogen bonds with template-strand nucleotides at TSS−1 and TSS and make an intrachain stacking interaction with the base of CMPcPP. Crucially, for each dinucleotide primer, the 5′ base of the primer makes an interchain stacking interaction with the base of the template-strand purine nucleotide at position TSS−2 (Fig. 7). Structural modeling indicates that this interchain base-stacking interaction can occur only when the template-strand nucleotide at position TSS−2 is a purine (Fig. 7D). Structural modeling further indicates that this interchain base-stacking interaction should stabilize the binding of the primer and CMPcPP to template-strand DNA, thereby facilitating primer-dependent initiation. Consistent with this structural modeling, a crystal structure of a complex obtained by soaking crystals of T. thermophilus RPo containing a template-strand pyrimidine nucleotide, T, at position TSS−2 with GpG and CMPcPP (RPo[TTSS−2CTSS−1CTSS]-GpG; 3.4 Å resolution; SI Appendix, Table S2) does not show significant interchain base-stacking with the template-strand at position TSS−2 and does not show binding of CMPcPP to template-strand DNA (SI Appendix, Fig. S7). Taken together, the crystal structures in Fig. 7 and SI Appendix, Fig. S7 define the structural basis of the preference for purine versus pyrimidine at template-strand position TSS−2 in primer-dependent transcription initiation: namely, interchain base stacking between the primer 5′ base and a purine at template-strand position TSS−2 facilitates binding of the primer and the extending NTP.
Discussion
Promoter-Sequence Dependence of Primer-Dependent Initiation: Mechanism and Structural Basis.
Our biochemical results show that primer-dependent initiation in stationary-phase E. coli almost always involves a dinucleotide primer (Fig. 2 and SI Appendix, Fig. S1), can involve any of the 16 possible dinucleotide primers (Fig. 3A), almost always involves a primer binding site complementary to both the 5′ and 3′ nucleotides of the dinucleotide primer (Fig. 3B and SI Appendix, Figs. S2–S4), depends on promoter sequences flanking the primer binding site (Figs. 4–6 and SI Appendix, Figs. S5 and S6), and exhibits the consensus sequence YTSS−2NTSS−1NTSSWTSS+1 (Y:RTSS−2N:NTSS−1N:NTSSW:WTSS+1; Figs. 4–6 and SI Appendix, Figs. S5 and S6), wherein the sequence information at positions TSS−2, TSS−1, and TSS is contained exclusively within the promoter template strand (Fig. 5 and SI Appendix, Fig. S4).
Our structural results show that the sequence preference for purine at template-strand position TSS−2 is a consequence of interchain base stacking of a purine at template-strand position TSS−2 with the 5′ nucleotide of a dinucleotide primer (Fig. 7 and SI Appendix, Fig. S7). The structural basis of the preference for purine versus pyrimidine at template-strand position TSS−2 in primer-dependent initiation is analogous to–and almost identical to–the previously described structural basis of the preference for purine versus pyrimidine at template-strand position TSS−1 in primer-independent initiation (19, 20). In the former case, interchain base stacking between the primer 5′ base in the RNAP active-center P-1 site and a purine at template-strand position TSS−2 facilitates binding of the primer and an extending NTP. In the latter case, interchain base stacking between the initiating NTP in the RNAP active-center P site and a purine at template-strand position TSS−1 facilitates binding of the initiating NTP and an extending NTP.
Promoter Sequences Upstream of the TSS Modulate the Chemical Nature of the RNA 5′-End.
Chemical modifications of the RNA 5′-end provide a layer of “epitranscriptomic” regulation influencing multiple aspects of RNA fate, including stability, processing, localization, and translation efficiency (21–24). Primer-dependent initiation provides one mechanism to alter the RNA 5′-end during transcription initiation. In primer-dependent initiation with a dinucleotide primer, the RNA product acquires a 5′ hydroxyl and acquires one additional nucleotide at the RNA 5′-end (Fig. 1). The presence of a 5′ hydroxyl has been shown to influence RNA stability in E. coli cells (25, 26). Thus, acquisition of a 5′ hydroxyl through primer-dependent initiation may provide a mechanism to modulate RNA stability in stationary phase. In an analogous manner, noncanonical initiating nucleotide (NCIN)-dependent initiation provides another mechanism to alter the RNA 5′-end during transcription initiation (27, 28). In NCIN-dependent initiation, the RNA product acquires an NCIN at the RNA 5′-end. NCIN-dependent initiation has been shown to occur with the oxidized and reduced forms of nicotinamide adenine dinucleotide (NAD), dephospho-coenzyme A (dpCoA), flavin adenine dinucleotide, uridine diphosphate N-acetylglucosamine, and dinucleoside tetraphosphates (Np4Ns) (27, 29–32). Furthermore, acquisition of a 5′ NAD through NCIN-dependent initiation has been shown to modulate RNA stability in both exponential-phase and stationary-phase E. coli cells (27).
All three modes of transcription initiation—primer-dependent, NCIN-dependent, and NTP-dependent—exhibit promoter-sequence dependence (refs. 12, 16, 27, and 32; Figs. 3–6 and SI Appendix, Figs. S2 and S6). All three modes of transcription initiation exhibit promoter consensus sequences that include base pairs upstream of the initiating-entity binding site (refs. 12, 16, 27, and 32; Figs. 4–6 and SI Appendix, Figs. S5 and S6). Crucially, the promoter consensus sequences upstream of the initiating-entity binding site for primer-dependent, NCIN-dependent (NAD, dpCoA, and Np4N), and NTP-dependent transcription initiation all are different: Y:RTSS−2, R:YTSS−1, and Y:RTSS−1, respectively (refs. 12, 16, 27, and 32; Figs. 4–6 and SI Appendix, Figs. S5 and S6). It follows that the sequence of the promoter TSS region hard-codes not only the TSS position but also the relative efficiencies of, and potentials for epitranscriptomic regulation through, primer-dependent, NCIN-dependent, and NTP-dependent transcription initiation.
Materials and Methods
Proteins.
The E. coli RNAP core enzyme used in transcription experiments was prepared from E. coli strain NiCo21(DE3) (New England Biolabs [NEB]) transformed with plasmid pIA900 (33) using culture and induction procedures, immobilized metal-ion affinity chromatography on Ni-NTA agarose, and affinity chromatography on Heparin HP as described in (34). E. coli σ70 was prepared from E. coli strain NiCo21 (DE3) transformed with plasmid pσ70-His using culture and induction procedures, immobilized metal-ion affinity chromatography on Ni-NTA agarose, and anion-exchange chromatography on Mono Q as described in ref. 35. A 10× RNAP holoenzyme was formed by mixing 0.5 μM RNAP core and 2.5 μM σ70 in 1× reaction buffer (40 mM Tris HCl, pH 7.5; 10 mM MgCl2; 150 mM KCl; 0.01% Triton X-100; and 1 mM dithiothreitol (DTT)).
The 5′ Rpp and T4 PNK were purchased from Epicentre and NEB, respectively.
Oligonucleotides.
Oligodeoxyribonucleotides (SI Appendix, Table S3) were purchased from Integrated DNA Technologies and were purified with standard desalting purification. UpA and GpG were purchased from Trilink Biotechnologies. ATP, guanosine triphosphate (GTP), cytidine triphosphate (CTP), and uridine triphosphate (UTP) were purchased from GE Healthcare Life Sciences.
Homoduplex and heteroduplex templates used in single-template in vitro transcription assays were generated by mixing 1.1 μM nontemplate-strand oligonucleotide with 1 μM template-strand oligonucleotide in 10 mM Tris (pH 8.0). Mixtures were heated to 90 °C for 10 min and slowly cooled to 40 °C (0.1 °C/s) using a Dyad PCR machine (Bio-Rad).
Plasmids.
Plasmid pBEN516 (9) contains sequences from −100 to +15 of the bhsA promoter fused to the tR′ terminator inserted between the HindIII and SalI sites of pACYC184 (NEB). Mutant derivatives of pBEN516 containing a G:C base pair at position TSS−2 (pKS494) or a G:C base pair at position +2 (pKS497) were generated using site-directed mutagenesis. Plasmid pPSV38 (9) contains a pBR322 origin, a gentamycin resistance gene (aaC1), and lacIq. Plasmid library pMASTER-lacCONS-N10 has been previously described (12).
Analysis of Primer-Dependent Initiation by MASTER.
Primer-dependent initiation in vitro: Transcription reaction conditions.
A linear DNA fragment containing placCONS-N10 generated as described in ref. 36 was used as a template for in vitro transcription assays (Figs. 2–4 and SI Appendix, Figs. S1–S3, S5, and S6). Transcription reactions (total volume = 100 μL) were performed by mixing 10 nM template DNA with 50 nM RNAP holoenzyme in E. coli RNAP reaction buffer (NEB) (40 mM Tris HCl, pH 7.5; 10 mM MgCl2; 150 mM KCl; 0.01% Triton X-100; and 1 mM DTT), 0.1 mg/mL bovine serum albumin (NEB), and 40 U murine RNase inhibitor (NEB). Reactions were incubated at 37 °C for 15 min to form open complexes. A single round of transcription was initiated by the addition of 1,000 μM ATP, 1,000 μM CTP, 1,000 μM UTP, 1,000 μM GTP, UpA (40 μM, 160 μM, or 640 μM), and 0.1 mg/mL heparin (Sigma-Aldrich). Reactions were incubated at 37 °C for 15 min and stopped by the addition of 0.5 M EDTA (pH 8) to a final concentration of 50 mM (for each replicate, two 100-μL transcription reactions were performed separately and combined after the addition of EDTA). Nucleic acids were recovered by ethanol precipitation, reconstituted in 25 μL nuclease-free water, mixed with 25 μL 2× RNA loading dye (95% formamide, 25 mM EDTA, 0.025% sodium dodecyl sulfate (SDS), 0.025% xylene cyanol, and 0.025% bromophenol blue), and separated by electrophoresis on 10% 7-M urea slab gels (Invitrogen) equilibrated and run in 1× Tris/Borate/EDTA buffer (TBE). The gel was stained with SYBR gold nucleic acid gel stain (Invitrogen) and bands visualized on an ultraviolet (UV) transilluminator, and RNA products ∼150 nt in length were excised from the gel. The excised gel slice was crushed, 300 μL 0.3 M NaCl in 1× Tris/EDTA buffer was added, and the mixture was incubated at 70 °C for 10 min. Eluted RNAs were collected using a Spin-X column (Corning). After the first elution, the crushed gel fragments were collected, and the elution procedure was repeated; nucleic acids were collected, pooled with the first elution, isolated by isopropanol precipitation, and resuspended in 25.5 μL RNase-free water (Invitrogen). Reactions were performed in triplicate.
Primer-dependent initiation in stationary-phase E. coli cells: Cell growth.
Three independent 25-mL cell cultures of E. coli MG1655 cells (gift of A. Hochschild, Harvard Medical School, Boston, MA) containing placCONS-N10 and pPSV38 were grown in Lysogeny Broth (LB) (Millipore) containing chloramphenicol (25 μg/mL), gentamicin (10 μg/mL), and IPTG (1 mM) in a 125-mL DeLong flask (Bellco Glass) shaken at 220 rpm at 37 °C until late stationary phase (∼21 h after entry into stationary phase; final optical density at 600 nm of ∼3.5). The 2-mL aliquots of cell suspensions were placed in 2-mL tubes, and cells were collected by centrifugation (1 min; 21,000 × g; 20 °C). Supernatants were removed, and cells were stored at −80 °C.
Primer-dependent initiation in stationary-phase E. coli cells: RNA isolation.
RNA was isolated from frozen cell pellets as described in ref. 12. Cell pellets were resuspended in 600 μL TRI Reagent solution (Molecular Research Center), incubated at 70 °C for 10 min, and centrifuged (10 min; 21,000 × g; 4 °C) to remove insoluble material. The supernatant was transferred to a fresh tube, ethanol was added to a final concentration of 60.5%, and the mixture was applied to a Direct-zol spin column (Zymo Research). DNase I (Zymo Research) treatment was performed on-column according to the manufacturer’s recommendations. RNA was eluted from the column using nuclease-free water heated to 70 °C (3 × 30 μL elutions; total volume of eluate = 90 μL). RNA was treated with 2 U TURBO DNase (Invitrogen) at 37 °C for 1 h, samples were extracted with acid phenol:chloroform (Ambion), and RNA was recovered by ethanol precipitation and resuspended in RNase-free water. A MICROBExpress Kit (Invitrogen) was used to remove ribosomal RNAs (rRNAs) from ∼36 μg of recovered RNA, and rRNA-depleted RNA was isolated by ethanol precipitation and resuspended in 40 μL RNase-free water.
Enzymatic treatment of RNA products.
For RNAs isolated from E. coli, 3 μg rRNA-depleted RNA was used in each reaction. RNAs isolated from in vitro transcription reactions were split into four equal portions and used in each reaction.
Rpp treatment (total reaction volume = 30 μL): RNA products were mixed with 20 U Rpp and 40 U RNaseOUT (Invitrogen) in 1× Rpp reaction buffer (50 mM Hepes-KOH, pH 7.5; 100 mM NaCl; 1 mM EDTA; 0.1% β-mercaptoethanol; and 0.01% Triton X-100) and incubated at 37 °C for 1 h. Reactions were extracted with acid phenol:chloroform, RNA was recovered by ethanol precipitation and resuspended in 10.5 μL RNase-free water.
PNK treatment (total reaction volume = 50 μL): RNA products were mixed with 20 U PNK, 40 U RNaseOUT, and 1 mM ATP (NEB) in 1× PNK reaction buffer (70 mM Tris⋅HCl, pH 7.6; 10 mM MgCl2; and 5 mM DTT) and incubated at 37 °C for 1 h. Processed RNAs were recovered using Qiagen’s RNeasy MinElute kit (following the manufacturer’s recommendations, with the exception that RNAs were eluted from the column using 200 μL nuclease-free water heated to 70 °C). RNA was recovered by ethanol precipitation and resuspended in 10.5 μL RNase-free water.
Rpp and PNK treatment: RNA products were mixed with 20 U PNK, 40 U RNaseOUT, and 1 mM ATP in 1× PNK reaction buffer (total reaction volume = 50 μL) and incubated at 37 °C for 1 h. Processed RNAs were recovered using Qiagen’s RNeasy MinElute kit (following the manufacturer’s recommendations, with the exception that RNAs were eluted from the column using 25 μL nuclease-free water heated to 70 °C). Recovered RNA products were mixed with 20 U Rpp and 40 U RNaseOUT in 1× Rpp reaction buffer (total reaction volume = 30 μL) and incubated at 37 °C for 1 h. Reactions were extracted with acid phenol:chloroform, and RNA was recovered by ethanol precipitation and resuspended in 10.5 μL RNase-free water.
“Mock” PNK treatment (total reaction volume = 50 μL): RNA products were mixed with 40 U RNaseOUT and 1 mM ATP in 1× PNK reaction buffer and incubated at 37 °C for 1 h. Reactions were extracted with acid phenol:chloroform, and RNA was recovered by ethanol precipitation and resuspended in 10.5 μL RNase-free water.
“Mock” Rpp treatment (total reaction volume = 30 μL): RNA products were mixed with 40 U RNaseOUT in 1× Rpp reaction buffer (total reaction volume = 30 μL) and incubated at 37 °C for 1 h. Reactions were extracted with acid phenol:chloroform, and RNA was recovered by ethanol precipitation and resuspended in 10.5 μL RNase-free water.
5'-adaptor ligation.
To enable quantitative comparisons between samples, we performed the 5′-adaptor ligation step using barcoded 5′-adaptor oligonucleotides as described in ref. 16. For RNA products isolated from stationary-phase E. coli cells, oligonucleotide i105 was used for RNAs processed by Rpp, oligonucleotide i106 was used for RNAs processed with PNK, oligonucleotide i107 was used for RNAs processed with both Rpp and PNK, and oligonucleotide i108 was used for unprocessed RNAs (mock PNK-treated). For RNA products isolated from in vitro reactions, oligonucleotide i105 was used for RNAs processed by Rpp, oligonucleotide i106 was used for unprocessed RNAs (mock Rpp-treated), oligonucleotide i107 was used for RNAs processed by PNK, and oligonucleotide i108 was used for unprocessed RNAs (mock PNK-treated).
Processed RNA products isolated from stationary-phase E. coli cells (in 10.5 μL nuclease-free water) were combined with 1 mM ATP (NEB), 40 U RNaseOUT, 1× T4 RNA ligase buffer (NEB), and 10 U T4 RNA ligase 1 (NEB) and 1 μM 5′ adaptor oligonucleotide (total reaction volume = 20 μL) and incubated at 37 °C for 2 h. Reactions were then supplemented with 1× T4 RNA ligase buffer, 1 mM ATP, polyethylene glycol (PEG) 8000 (10% final), 5 U T4 RNA ligase 1, and 20 U RNaseOUT (total reaction volume = 30 μL) and further incubated at 16 °C for 16 h. Processed RNA products isolated from in vitro reactions (in 10.5 μL nuclease-free water) were combined with PEG 8000 (10% final concentration), 1 mM ATP, 40 U RNaseOUT, 1× T4 RNA ligase buffer, 10 U T4 RNA ligase 1, and 1 μM 5′ adaptor oligonucleotide (total reaction volume = 30 μL) and incubated at 16 °C for 16 h.
Ligation reactions were stopped by the addition of 30 μL 2× RNA loading dye and heated at 95 °C for 5 min. For each replicate, the four ligation reactions were combined and separated by electrophoresis on 10% 7-M urea slab gels (equilibrated and run in 1× TBE). Gels were incubated with SYBR gold nucleic acid gel stain, and bands were visualized with UV transillumination. For RNAs isolated from stationary-phase E. coli cells, products migrating above the 5′-adapter oligonucleotide were gel purified and resuspended in 50 μL nuclease-free water. For RNAs generated in vitro, products ∼150 nt in length were gel purified and resuspended in 16 μL nuclease-free water. The 5′-adaptor-ligated RNAs were used for analysis of primer-dependent initiation from placCONS-10 (this section) and for analysis of primer-dependent initiation from natural, chromosomally encoded promoters (next section).
First-strand complementary DNA synthesis.
For RNAs isolated from stationary-phase E. coli cells, 25 μL 5′-adaptor–ligated RNAs were mixed with 1.5 μL s128A oligonucleotide (3 μM) and 3.5 μL nuclease-free water. The 30-μL mixture was incubated at 65 °C for 5 min, cooled to 4 °C, and combined with 20 μL solution containing 10 μL 5× First-Strand buffer (Invitrogen), 2.5 μL10 mM dNTP mix (NEB), 2.5 μL 100 mM DTT, 2.5 μL 40 U/μL RNaseOUT, and 2.5 μL 100 U/μL SuperScript III reverse transcriptase (Invitrogen) for a final reaction volume of 50 μL. Reactions were incubated at 25 °C for 5 min, 55 °C for 60 min, and 70 °C for 15 min and then kept at 25 °C. Next, 5.4 μL 1M NaOH was added, reactions were incubated at 95 °C for 5 min and kept at 10 °C, and 4.5 μL 1.2M HCl was added followed by 60 μL2× RNA loading dye.
For RNAs isolated from in vitro reactions, 16 μL 5′-adaptor–ligated RNAs were mixed with 0.5 μL s128A oligonucleotide (1.5 μM). The 16.5-μL mixture was incubated at 65 °C for 5 min, cooled to 4 °C, and combined with 13.5 μL solution containing 6 μL 5× First-Strand buffer, 1.5 μL 10 mM dNTP mix, 1.5 μL 100 mM DTT, 1 μL 40 U/μL RNaseOUT, 1.5 μL 100 U/μL SuperScript III reverse transcriptase, and 2 μL nuclease-free water for a final reaction volume of 30 μL. Reactions were incubated at 25 °C for 5 min, 55 °C for 60 min, and 70 °C for 15 min and then cooled to 25 °C. Next, 10 U RNase H (NEB) was added, reactions were incubated at 37 °C for 15 min, and 31 μL 2× RNA loading dye was added.
Nucleic acids were separated by electrophoresis on 10% 7-M urea slab gels (equilibrated and run in 1× TBE). Gels were incubated with SYBR gold nucleic acid gel stain, bands were visualized with UV transillumination, and species ∼80 to ∼150 nt in length were gel purified and resuspended in 20 μL nuclease-free water.
Complementary DNA amplification.
Complementary DNA (cDNA) derived from RNA products generated in vitro or in vivo was diluted with nuclease-free water to a concentration of ∼109 molecules/μL. A total of 2 μL diluted cDNA solution was used as a template for emulsion PCR reactions containing Illumina index primers using a Micellula DNA Emulsion and Purification Kit (EURx). The Illumina PCR forward primer and Illumina index primers from the TruSeq Small RNA Sample Prep Kits were used. The emulsion was broken, and DNA was purified according to the manufacturer’s recommendations. Amplicons were gel purified on 10% TBE slab gels (Invitrogen; equilibrated in 1× TBE), recovered by isopropanol precipitation, and reconstituted in 13 μL nuclease-free water.
High-throughput sequencing.
Barcoded libraries were pooled and sequenced on an Illumina NextSeq platform in high-output mode using custom sequencing primer s1115.
Sample serial numbers.
Samples KS112 through KS114 are cDNA derived from RNA products generated in stationary-phase E. coli cells treated with 1) both PNK and Rpp (PNK + Rpp), 2) Rpp only (Rpp), 3) PNK only (PNK), or 4) neither PNK nor Rpp (mock). Samples KS86 through KS97 are cDNA derived from RNA products generated in vitro in the presence of no UpA (KS86 through KS88), 40 μM UpA (KS89 through KS91), 160 μM UpA (KS92 through KS94), or 640 μM UpA (KS95 through KS97) treated with 1) Rpp only (Rpp), 2) PNK only (PNK), or 3) neither PNK nor Rpp (mock).
Data analysis: Separation of RNA 5'-end sequences by enzymatic treatment, promoter sequence, and promoter position.
RNA 5′-end sequences were associated with an enzymatic treatment using the 4-nt barcode sequence acquired upon ligation of the 5′-adaptor as described in ref. 16. RNA 5′-end sequences were associated with a placCONS promoter sequence using transcribed-region barcode assignments derived from the analysis of sample Vv945 described in ref. 14. RNA 5′-end sequences that could be aligned to their template of origin with no mismatches were used for results presented in Figs. 2, 3A, and 4 and SI Appendix, Figs. S1, S5, S6, and S8. RNA 5′-end sequences with mismatches at the first and/or second base of the 5′-end were also included for results shown in Fig. 3B and SI Appendix, Figs. S2 and S3. All logos were created using Logomaker (37). A detailed description of the data analysis is provided in the SI Appendix, Supplemental Text.
Analysis of Primer-Dependent Initiation from Chromosomally Encoded E. coli Promoters.
cDNA library construction and sequencing.
Cell growth, RNA isolation, enzymatic treatments, and 5′-adaptor ligations (Fig. 6A and SI Appendix, Table S1) were performed using procedures identical to those for analysis of primer-dependent initiation by MASTER. A total of 25 μL 5′-adaptor–ligated RNAs were mixed with 5 μL 18.5 μM (1.86 μM final) of an oligonucleotide pool consisting of a mixture of 93 gel-purified oligodeoxyribonucleotides each having a 5′-end sequence identical to the reverse transcription primer contained in Illumina TruSeq Small RNA Sample Prep Kits and a 3′-end sequence complementary to a chromosomally encoded E. coli promoter that uses UpA as primer (SI Appendix, Table S3). The mixture was incubated at 65 °C for 5 min, kept at 4 °C, combined with 20 μL solution containing 10 μL 5× First-Strand buffer (Invitrogen), 2.5 μL 10 mM dNTP mix (NEB), 2.5 μL 100 mM DTT, 2.5 μL 40 U/μL RNaseOUT, and 2.5 μL 100 U/μL SuperScript III Reverse Transcriptase (Invitrogen) for a final reaction volume of 50 μL. Reactions were incubated at 25 °C for 5 min, 55 °C for 60 min, and 70 °C for 15 min and then cooled to 25 °C. Next, 5.4 μL 1M NaOH was added, and reactions were incubated at 95 °C for 5 min and cooled to 10 °C; 4.5 μL 1.2M HCl was added followed by 60 μL 2× RNA loading dye. Nucleic acids were separated by electrophoresis on 10% 7-M urea slab gels (equilibrated and run in 1× TBE). Gels were incubated with SYBR gold nucleic acid gel stain, bands were visualized with UV transillumination, and species ∼80 to ∼150 nt in length were gel purified and resuspended in 20 μL nuclease-free water.
cDNA amplification and high-throughput sequencing was performed using procedures identical to those for analysis of primer-dependent initiation by MASTER. Serial numbers for these samples are KS118 through KS120.
Data analysis: Chromosomal promoter sequence logo.
Sequencing reads were associated with one of the four reaction conditions based on the identity of the 4-nt barcode sequence (Fig. 6A). RNA 5′-end sequences that could be aligned to the chromosomally encoded promoter from which they were expressed with no mismatches were used for results presented in Fig. 6A. The number of 5′-end sequences emanating from each position up to four bases upstream and downstream of the UpA binding site (TSS−5 to TSS+4, where UpA binds positions TSS−1, TSS) was determined for each enzymatic treatment. To represent these data as a sequence logo, we first estimated the fraction of transcripts that had 5′-OH ends at TSS−1. For each promoter sequence , this was computed using
where
and where and denote the number of reads initiating from position (which ranges over positions from TSS−5 to TSS+4) observed for promoter in the PNK + Rpp and Rpp treatments, respectively. These values were then averaged across three replicates, and a sequence logo reflecting the average log2 value of these ratios was rendered using mean-centered character heights given by
where takes the value 1 if base occurs at position in sequence and is 0 otherwise.
Single-Template In Vitro Transcription Assays.
A total of 10 nM linear template was mixed with 50 nM RNAP holoenzyme in transcription buffer and incubated at 37 °C for 15 min to form open complexes (Fig. 5 and SI Appendix, Fig. S4). 1,000 μM ATP and increasing concentrations of UpA (0, 10, 40, 160, and 640 μM) were added along with 10 μM nonradiolabeled UTP plus 6 mCi [α32P]-UTP (PerkinElmer). Upon addition of nucleotides, reactions were incubated at 37 °C for 10 min to allow for product formation. Reactions were stopped by the addition of an equal volume of gel-loading buffer (95% formamide, 25 mM EDTA, 0.025% SDS, 0.025% xylene cyanol, and 0.025% bromophenol blue).
Samples were run on 20% TBE-Urea polyacrylamide gels. Bands were quantified using ImageQuant software. Observed values of UpApU/(pppApU + UpApU) were plotted versus [UpA]/[ATP] on a semilog plot (Sigmaplot). Nonlinear regression was used to fit the data to the equation: y = (ax)/(b+x), where y is UpApU/(pppApU + UpApU), x is [UpA]/[ATP], and a and b are regression parameters. The resulting fit yields the value of x for which y = 0.5. The relative efficiency (kcat/KM)UpA/(kcat/KM)ATP is equal to 1/x.
Analysis of Primer-Dependent Initiation from the E. coli bhsA Promoter.
Culture growth and cell harvesting.
Plasmids pBEN516, pKS494, or pKS497 were introduced into E. coli MG1655 cells (Fig. 6B). Plasmid-containing cells were grown in 25 mL LB-containing chloramphenicol (25 μg/mL) in a 125-mL DeLong flask (Bellco Glass) at 37 °C and harvested 5, 9, 14, or 21 h after cells had entered stationary phase (optical density at 600 nm of ∼3.3, ∼3.1, ∼2.9, and ∼2.6, respectively). Cell suspensions were removed to 2-mL microcentrifuge tubes (Axygen), cells were collected by centrifugation (21,000 rpm; 30 s; 4 °C), and cell pellets were stored at −80 °C.
RNA isolation.
Cells were resuspended in 0.6 mL TRI-Reagent and incubated at 70 °C for 10 min, and the cell lysate was centrifuged to remove insoluble material (10 min; 21,000 × g; 4 °C). The supernatant was transferred to a fresh tube, ethanol was added to a final concentration of 60.5%, and the mixture was applied to a Direct-zol spin column. DNase I treatment was performed on-column according to the manufacturer’s recommendations. RNA was eluted from the column using nuclease-free water that had been heated to 70 °C (3 × 30 μL elutions; total volume of eluate = 90 μL). RNA was treated with 2 U TURBO DNase at 37 °C for 1 h to remove residual DNA. Samples were extracted with acid phenol:chloroform, and RNA was recovered by ethanol precipitation and resuspended in RNase-free water.
Primer extension analysis.
Assays were performed essentially as described in ref. 10. A total of 10 μg RNA was combined with 3 μM primer k711 (5′-radiolabeled using PNK and [γ32P]-ATP). The RNA–primer mixture was heated to 95 °C for 10 min, slowly cooled to 40 °C (0.1 °C/s), incubated at 40 °C for 10 min, and cooled to 4 °C using a thermal cycler (Biorad). Next, 10 U avian myeloblastosis virus reverse transcriptase (NEB) was added, and reactions were incubated at 55 °C for 60 min, heated to 90 °C for 10 min, cooled to 4 °C for 30 min, and mixed with 10 μL 2× RNA loading buffer (95% formamide; 0.5 mM EDTA, pH 8; 0.025% SDS; 0.0025% bromophenol blue; and 0.0025% xylene cyanol). Nucleic acids were separated by electrophoresis on 8%, 7-M urea slab gels (equilibrated and run in 1× TBE), and radiolabeled products were visualized by storage phosphor imaging. Band assignments were made by comparison to a DNA-sequence ladder prepared using primer k711 and pBEN516 as templates (Affymetrix Sequenase DNA sequencing kit, version 2).
Structure Determination.
The nucleic-acid scaffold for assembly of T. thermophilus RPo was prepared from synthetic oligonucleotides (Sangon Biotech) by an annealing procedure (Fig. 7 and SI Appendix, Table S2 and Fig. S7) (95 °C, 5 min followed by 2 °C step cooling to 25 °C) in 5 mM Tris⋅HCl (pH 8.0), 200 mM NaCl, and 10 mM MgCl2 (nontemplate strand for all structures: 5′-TATAATGGGAGCTGTCACGGATGCAGG-3′; template strand for RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP: 5′-CCTGCATCCGTGAGTAAAG-3′; template strand for RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP: 5′-CCTGCATCCGTGAGCCAAG-3′; template strand for RPo[TTSS−2CTSS−1CTSS]-GpG-CMPcPP: 5′-CCTGCATCCGTGAGCCTAG-3′).
For each structure, T. thermophilus RPo was reconstituted by mixing T. thermophilus RNAP holoenzyme purified as described in ref. 18 and the nucleic-acid scaffold at a 1:1.2 molar ratio and incubating for 1 h at 22 °C. Crystals of T. thermophilus RPo were obtained and handled essentially as in ref. 18. The primer (UpA or GpG) and CMPcPP were subsequently soaked into RPo crystals by addition of 0.2 μL 100 mM primer (UpA or GpG) and 0.2 μL 50 mM CMPcPP in RNase-free water to the crystallization drops (2 μL) and incubation for 30 min at 22 °C. Crystals were transferred in a stepwise fashion to reservoir solution (0.2 M KCl; 0.05 M MgCl2; 0.1 M Tris⋅HCl, pH 7.9; 9% PEG 4000) containing 0.5%, 1%, 5%, 10%, and 17.5% (vol/vol) (2R,3R)-(-)-2,3-butanediol and cooled in liquid nitrogen.
Diffraction data were collected at Shanghai Synchrotron Radiation Facility beamlines 17U and processed using HKL2000 (38). The structures were solved by molecular replacement with Phaser MR in Phenix using one molecule of RNAP holoenzyme from the structure of T. thermophilus RPo (PDB:4G7H) as the search model (18, 39). Early-stage rigid-body refinement of the RNAP molecule revealed good electron-density signals for the primer (UpA or GpG) and CMPcPP. Cycles of iterative model building with Coot and refinement with Phenix were performed (40, 41). The models of the primer (UpA or GpG) and CMPcPP were built into the map at a later stage of refinement.
The final model of RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP was refined to Rwork and Rfree of 0.205 and 0.245, respectively. The final model of RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP was refined to Rwork and Rfree of 0.187 and 0.252, respectively. The final model of RPo[TTSS−2CTSS−1CTSS]-GpG was refined to Rwork and Rfree of 0.194 and 0.246, respectively.
Quantification and Statistical Analysis.
The number of replicates and statistical test procedures are in the figure legends.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China Grant 31822001 (Yu Zhang) and NIH Grants GM124976 (P.S.), GM133777 (J.B.K.), GM041376 (R.H.E.), and GM118059 (B.E.N.).
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
See online for related content such as Commentaries.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2106388118/-/DCSupplemental.
Data Availability
Sequencing reads have been deposited in the NIH/National Center for Biotechnology Information Sequence Read Archive under the study accession number PRJNA718578. Source code and documentation are provided at https://www.github.com/jbkinney/20_nickels. Structures of RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP, RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP, and RPo[TTSS−2CTSS−1CTSS]-GpG have been deposited in the Protein Data Bank under accession numbers 7EH0, 7EH1, and 7EH2, respectively.
References
- 1.Hoffman D. J., Niyogi S. K., RNA initiation with dinucleoside monophosphates during transcription of bacteriophage T4 DNA with RNA polymerase of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 70, 574–578 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minkley E. G., Pribnow D., Transcription of the early region of bacteriophage T7: Selective initiation with dinucleotides. J. Mol. Biol. 77, 255–277 (1973). [DOI] [PubMed] [Google Scholar]
- 3.Dausse J. P., Sentenac A., Fromageot P., Interaction of RNA polymerase from Escherichia coli with DNA. Analysis of T7 DNA early-promoter sites. Eur. J. Biochem. 57, 569–578 (1975). [DOI] [PubMed] [Google Scholar]
- 4.Di Nocera P. P., Avitabile A., Blasi F., In vitro transcription of the Escherichia coli histidine operon primed by dinucleotides. Effect of the first histidine biosynthetic enzyme. J. Biol. Chem. 250, 8376–8381 (1975). [PubMed] [Google Scholar]
- 5.Smagowicz W. J., Scheit K. H., Primed abortive initiation of RNA synthesis by E. coli RNA polymerase on T7 DNA. Steady state kinetic studies. Nucleic Acids Res. 5, 1919–1932 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smagowicz W. J., Scheit K. H., The properties of ATP-analogs in initiation of RNA synthesis catalyzed by RNA polymerase from E coli. Nucleic Acids Res. 9, 2397–2410 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruetsch N., Dennis D., RNA polymerase. Limit cognate primer for initiation and stable ternary complex formation. J. Biol. Chem. 262, 1674–1679 (1987). [PubMed] [Google Scholar]
- 8.Goldman S. R., et al., NanoRNAs prime transcription initiation in vivo. Mol. Cell 42, 817–825 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vvedenskaya I. O., et al., Growth phase-dependent control of transcription start site selection and gene expression by nanoRNAs. Genes Dev. 26, 1498–1507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Druzhinin S. Y., et al., A conserved pattern of primer-dependent transcription initiation in Escherichia coli and Vibrio cholerae revealed by 5′ RNA-seq. PLoS Genet. 11, e1005348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nickels B. E., A new way to start: NanoRNA-mediated priming of transcription initiation. Transcription 3, 300–304 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vvedenskaya I. O., et al., Massively systematic transcript end readout, “MASTER”: Transcription start site selection, transcriptional slippage, and transcript yields. Mol. Cell 60, 953–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vvedenskaya I. O., Goldman S. R., Nickels B. E., Analysis of bacterial transcription by “massively systematic transcript end readout,” MASTER. Methods Enzymol. 612, 269–302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winkelman J. T., et al., Multiplexed protein-DNA cross-linking: Scrunching in transcription start site selection. Science 351, 1090–1093 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vvedenskaya I. O., et al., Interactions between RNA polymerase and the core recognition element are a determinant of transcription start site selection. Proc. Natl. Acad. Sci. U.S.A. 113, E2899–E2905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vvedenskaya I. O., et al., CapZyme-seq comprehensively defines promoter-sequence determinants for RNA 5′ capping with NAD<sup/>. Mol. Cell 70, 553–564.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nudler E., Avetissova E., Korzheva N., Mustaev A., Characterization of protein-nucleic acid interactions that are required for transcription processivity. Methods Enzymol. 371, 179–190 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., et al., Structural basis of transcription initiation. Science 338, 1076–1080 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gleghorn M. L., Davydova E. K., Basu R., Rothman-Denes L. B., Murakami K. S., X-ray crystal structures elucidate the nucleotidyl transfer reaction of transcript initiation using two nucleotides. Proc. Natl. Acad. Sci. U.S.A. 108, 3566–3571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Basu R. S., et al., Structural basis of transcription initiation by bacterial RNA polymerase holoenzyme. J. Biol. Chem. 289, 24549–24559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shuman S., RNA capping: Progress and prospects. RNA 21, 735–737 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jäschke A., Höfer K., Nübel G., Frindert J., Cap-like structures in bacterial RNA and epitranscriptomic modification. Curr. Opin. Microbiol. 30, 44–49 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Ramanathan A., Robb G. B., Chan S. H., mRNA capping: Biological functions and applications. Nucleic Acids Res. 44, 7511–7526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Höfer K., Jäschke A., Epitranscriptomics: RNA modifications in bacteria and archaea. Microbiol. Spectr. 6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Celesnik H., Deana A., Belasco J. G., Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell 27, 79–90 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giliberti J., O’Donnell S., Etten W. J., Janssen G. R., A 5′-terminal phosphate is required for stable ternary complex formation and translation of leaderless mRNA in Escherichia coli. RNA 18, 508–518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bird J. G., et al., The mechanism of RNA 5′ capping with NAD+, NADH and desphospho-CoA. Nature 535, 444–447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barvík I., Rejman D., Panova N., Šanderová H., Krásný L., Non-canonical transcription initiation: The expanding universe of transcription initiating substrates. FEMS Microbiol. Rev. 41, 131–138 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Julius C., Yuzenkova Y., Bacterial RNA polymerase caps RNA with various cofactors and cell wall precursors. Nucleic Acids Res. 45, 8282–8290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bird J. G., et al., Highly efficient 5′ capping of mitochondrial RNA with NAD+ and NADH by yeast and human mitochondrial RNA polymerase. eLife 7, e42179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luciano D. J., Levenson-Palmer R., Belasco J. G., Stresses that raise Np4A levels induce protective nucleoside tetraphosphate capping of bacterial RNA. Mol. Cell 75, 957–966.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luciano D. J., Belasco J. G., Np4A alarmones function in bacteria as precursors to RNA caps. Proc. Natl. Acad. Sci. U.S.A. 117, 3560–3567 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svetlov V., Artsimovitch I., Purification of bacterial RNA polymerase: Tools and protocols. Methods Mol. Biol. 1276, 13–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Artsimovitch I., Svetlov V., Murakami K. S., Landick R., Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J. Biol. Chem. 278, 12344–12355 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Marr M. T., Roberts J. W., Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science 276, 1258–1260 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Winkelman J. T., Chandrangsu P., Ross W., Gourse R. L., Open complex scrunching before nucleotide addition accounts for the unusual transcription start site of E. coli ribosomal RNA promoters. Proc. Natl. Acad. Sci. U.S.A. 113, E1787–E1795 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tareen A., Kinney J. B., Logomaker: Beautiful sequence logos in Python. Bioinformatics 36, 2272–2274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otwinowski Z., Minor W., Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 39.McCoy A. J., et al., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Adams P. D., et al., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing reads have been deposited in the NIH/National Center for Biotechnology Information Sequence Read Archive under the study accession number PRJNA718578. Source code and documentation are provided at https://www.github.com/jbkinney/20_nickels. Structures of RPo[ATSS−2ATSS−1TTSS]-UpA-CMPcPP, RPo[ATSS−2CTSS−1CTSS]-GpG-CMPcPP, and RPo[TTSS−2CTSS−1CTSS]-GpG have been deposited in the Protein Data Bank under accession numbers 7EH0, 7EH1, and 7EH2, respectively.