

Abstract
Premise
An informatics approach was used for the construction of an Axiom genotyping array from heterogeneous, high‐throughput sequence data to assess the complex genome of loblolly pine (Pinus taeda).
Methods
High‐throughput sequence data, sourced from exome capture and whole genome reduced‐representation approaches from 2698 trees across five sequence populations, were analyzed with the improved genome assembly and annotation for the loblolly pine. A variant detection, filtering, and probe design pipeline was developed to detect true variants across and within populations. From 8.27 million variants, a total of 642,275 were evaluated and 423,695 of those were screened across a range‐wide population.
Results
The final informatics and screening approach delivered an Axiom array representing 46,439 high‐confidence variants to the forest tree breeding and genetics community. Based on the annotated reference genome, 34% were located in or directly upstream or downstream of genic regions.
Discussion
The Pita50K array represents a genome‐wide resource developed from sequence data for an economically important conifer, loblolly pine. It uniquely integrates independent projects that assessed trees sampled across the native range. The challenges associated with the large and repetitive genome are addressed in the development of this resource.
Keywords: exome capture, genomic selection, genotype array, genotyping‐by‐sequencing (GBS), loblolly pine, Pinus taeda, variant detection
Loblolly pine (Pinus taeda L.) is a long‐lived, diploid, outcrossing conifer of the family Pinaceae. It is the most important timber tree in its native range of the southeastern United States, where it is grown for pulpwood, timber, and as a biofuel source (Prestemon and Abt, 2002). Improvement of economically important traits such as tree height, stem volume, wood quality, and resistance to fusiform rust disease through traditional breeding requires more than 15 years per cycle (Isik and McKeand, 2019). These complex traits vary substantially between populations (Lauer et al., 2020). Marker‐assisted selection (MAS) describes a technique in which a small collection of variants (typically single‐nucleotide polymorphisms [SNPs]), linked to known quantitative trait loci (QTL), are used to predict genetic merit. This has been applied successfully in several crop systems and is ideal when traits are controlled by few loci of large effect, and when markers are tightly linked to (or within) the loci of interest (Spindel and McCouch, 2016). However, MAS is limited in its ability to predict complex traits controlled by many unidentified small‐effect loci as is often seen in growth traits, drought tolerance, and wood quality of forest trees. The alternative approach, genomic selection (GS), utilizes genome‐wide variants to predict the breeding value for individuals with unobserved phenotype (Spindel and McCouch, 2016; Xu et al., 2020). Genome‐wide markers capture both major and minor contributing alleles and can be used to maintain genetic diversity in the selected populations (Goddard and Hayes, 2007; Olatoye et al., 2019). With sufficient variant density, the robust statistical models available today are ideal for large‐scale application (Resende et al., 2012; Bhat et al., 2016). This is promising for species with long generation times and traits that are costly to measure. GS has been successfully applied in many breeding programs, including livestock, legumes, and maize (Resende et al., 2012; Bhat et al., 2016; Chen et al., 2018; Thistlethwaite et al., 2019; Beaulieu et al., 2020; Lenz et al., 2020). There is an increasing interest in GS for forest tree breeding, as genotyping and high‐throughput sequencing platforms have become more efficient and less expensive (Grattapaglia et al., 2018; Ukrainetz and Mansfield, 2019).
Genomic selection requires that a large population is genotyped with a sufficient set of markers such that every QTL influencing a trait is in linkage disequilibrium with at least one marker in the panel (VanRaden, 2020). The variants must be dispersed throughout the genome with high enough density to ensure appropriate linkage with the loci influencing the desired traits (Spindel and McCouch, 2016; Xu et al., 2020). Genotyping arrays have been used for both MAS and GS because they allow rapid assessment of a large number of individuals for thousands of loci. If designed from an appropriately diverse population, the array will represent a set of variants that should be polymorphic in most study populations. Array design requires substantial informatic investment for their initial design but does not require programmatic expertise or specialized hardware to obtain the variant calls. Because the markers on the array are based on specific probes, access to a high‐quality reference genome dramatically improves the accuracy of the design process. Exome sequencing and genotyping‐by‐sequencing (GBS) are alternative resequencing approaches that can also leverage a reference genome to identify polymorphisms in a high‐throughput manner (Bhat et al., 2016; Lu et al., 2016; Acosta et al., 2019). Although GBS can be successful in conifers (Calleja‐Rodriguez et al., 2020), the depth of sequencing required to capture loci across thousands of individuals in a species with a large genome remains cost‐prohibitive. Although both approaches may be subject to sampling/selection bias, this is more likely in arrays where variants are pre‐determined (Heslot et al., 2013).
The most significant challenge in creating a genotyping array for any conifer is the large and repetitive genome. The initial assembly of the loblolly pine genome was generated entirely through short‐read Illumina data (Neale et al., 2014). This assembly (v1.01) yielded a total of 22.5 Gbp spread across 2,158,326 scaffolds (N50 of 75 kbp, 28% BUSCO completeness v.4.0.2 embryophyta v.10). The second version (v2.0), assembled in 2017, leveraged 267 Gbp of genomic long reads (PacBio), resulting in a 22.1 Gbp genome represented by 1,762,655 scaffolds (N50 of 107 kbp, 36% BUSCO completeness v.4.0.2 embryophyta v.10) (Zimin et al., 2017). Finally, v2.01 raised the N50 to 111 kbp (1,489,469 scaffolds, 37.8% BUSCO completeness v.4.0.2 embryophyta v.10) by scaffolding the genome with 70,064 PacBio Iso‐Seq transcripts. Despite these improvements, the v2.01 assembly remains fragmented as a consequence of the short‐read inputs and repetitive content that exceeds 85% of the genome (Wegrzyn et al., 2014). The structural annotation of the coding region is challenged by the prevalence of pseudogenes, which represent five times more of the genome sequence than functional genes (Wegrzyn et al., 2014). Finally, the high density of polymorphisms (Brown et al., 2004) further complicates the selection of viable polymorphisms for a genotyping array that can be used broadly. Array creation for GS must therefore balance the need for accurate markers, a high‐density genome‐wide distribution of these polymorphisms, and an avoidance of probe misalignment that can result in off‐target variants.
Early genotyping arrays for loblolly pine successfully identified QTLs for valued economic traits including wood formation and disease resistance (Eckert et al., 2010; Resende et al., 2012). Construction of these arrays, however, pre‐dated the loblolly pine genome assembly and used expressed sequence tags (ESTs). ESTs are generated from cDNA and therefore only represent transcribed sequences. This greatly limits the available polymorphisms and may not provide the genome‐wide distribution needed for GS. In addition, the ability to properly design probes in the absence of genomic alignments of the transcriptomic targets remains a challenge. For this same reason, the potential for off‐target probe hybridization could not be estimated or resolved. The first Illumina Infinium array had just over 5000 EST‐derived markers and was used to genotype more than 7000 loblolly and slash pine trees (Eckert et al., 2010). For most populations, the array had a conversion rate (defined as percentage of successful polymorphic markers; De La Vega et al., 2005) of just under 60% (Eckert et al., 2010; Chhatre et al., 2013; Cumbie et al., 2020). Recently, the first genome‐scale loblolly pine Axiom array was designed from a small population (10 megagametophyte samples) and applied to 359 unrelated individuals from the Allele Discovery of Economic Pine Traits II project (ADEPT2) (Cumbie et al., 2011). Among 635,000 variants tested, approximately 13% (84,700) were polymorphic and high quality in the population assessed. This speaks to the challenges of large polymorphic genomes and small discovery panels (Telfer et al., 2019; De La Torre et al., 2019; Perry et al., 2020). Arrays designed for other conifers incorporated a variety of transcriptomic resources with varying support from recently sequenced reference genomes. An Axiom array for Douglas‐fir (Howe et al., 2020), designed from transcriptomes (and previously validated probes), was assessed via alignments to the Douglas‐fir reference genome and reported a 40.8% conversion rate. In another example, four species of European pine (Perry et al., 2020) (42% conversion rate) were represented on a single Axiom array that was designed from transcriptomes of the target species. The loblolly pine reference genome and transcriptome assemblies helped to eliminate probes that overlapped estimated intron boundaries and also provided an estimate of multiple‐hit alignments. Other recent approaches in Axiom array development for species with limited genome resources used RNA‐seq (Mishima et al., 2018) or a combination of RNA and restriction site–associated DNA sequencing (RAD‐seq) (Silva et al., 2020) to both generate EST contigs and identify SNPs and coding regions for probe design. These arrays had moderate conversion rates (<50%). Comparatively, arrays constructed for three spruce species (Norway spruce, black spruce, and white spruce) (Pavy et al., 2013, 2016; Azaiez et al., 2018) achieved a higher conversion rate (64–96%). These generally applied stricter informatic thresholds for the discovery and filtering of candidate variants, and the array technology required longer probes (Illumina iSelect technology 50‐mer vs. Thermo Fisher Axiom technology 35‐mer). Taken together, these reinforce the benefit of reference genomes in designing probes and filtering for repetitive content, intronic regions, and potential off‐target hybridization that can lead to low conversion. For a loblolly pine array, the reference genome will standardize the various genomic data used to identify variants and to produce genome‐wide markers for GS implementation.
Here we describe the bioinformatic process that integrated heterogeneous genomic resources and identified high‐quality variants for loblolly pine. The resulting “Pita50K” array uses the Thermo Fisher Axiom technology. This was organized through the Conifer SNP Consortium, which represents six coordinated genotyping projects across 14 species (Bernhardsson et al., 2020; Howe et al., 2020) and is aimed at improving tree breeding worldwide. The Pita50K resource utilized a diverse population of trees gathered across five studies covering range‐wide genetic variation for loblolly pine. To overcome the challenges associated with a massive, fragmented, and polymorphic genome, we opted for probe selection that minimizes variants in flanking sequences and off‐target hybridization, thereby reducing the potential for erroneous genotypes. Using the new structural annotation of the loblolly pine genome, we annotated variants proximal to genes and in regions of accessible chromatin. Finally, a screening array was used to estimate population genetic parameters for variant candidates to enable a data‐driven approach for marker selection. With this, we created a selection technology for loblolly pine that overcomes the challenges of genome size, repetitive content, and polymorphic density. Future applications of this array include creation of an updated linkage map, a more contiguous reference genome, and more powerful GS programs for loblolly pine and taxonomically related species.
METHODS
Genomic data sources for loblolly pine
The final loblolly pine genotyping array (Pita50K) was designed from five population studies implemented with high‐throughput sequencing and reduced‐representation strategies. All alignments and resulting variants were based on early versions of the loblolly pine genome (i.e., v1.0, v1.01, and v2.0). Many of the original studies, while independently designed, were part of the Pine Integrated Network: Education, Mitigation, and Adaptation project (PINEMAP; http://www.pinemap.org). Combined, they assessed 2698 unique trees representing much of the native range of loblolly pine (Table 1, Fig. 1) (Farjat et al., 2017b). The investigations provided different genomic sources and were sequenced at a range of depths. The first of two exome capture studies, conducted at the University of Florida (Acosta et al., 2019), included 24 distinct, naturally distributed populations across 11 U.S. states. Exome capture was performed on haploid megagametophyte tissue using 54,773 120‐mer RNA probes designed from loblolly ESTs. Sequencing was performed on an Illumina HiSeq2000 (2 × 100 nucleotides [nt]; Illumina, San Diego, California, USA) to a depth of approximately 30× and initially called 67,071 SNPs. The second exome capture study, led by a team at Texas A&M University (Lu et al., 2016), represented 375 clonally propagated samples from 12 U.S. states in the ADEPT2 population. Exome capture was performed via the NimbleGen SeqCap EZ method (Roche Sequencing and Life Science, Indianapolis, Indiana, USA) that targeted over 196,000 exons from the v1.01 reference. The needle samples were sequenced to a depth of approximately 30× by Illumina HiSeq 2500 (2 × 125 nt) and originally called 2.82 million SNPs. Two data sets contained genomic sequence from double‐digest RAD sequencing (ddRADseq [Peterson et al., 2012]; PstI and MspI restriction enzymes were used in both studies). The first, conducted at North Carolina State University (NCSU), contained 1536 phloem (diploid) samples sourced from the Plantation Selection Seed Source Study (PSSSS), a set of field trials of 140 families planted in 20 locations across 11 U.S. states (Farjat et al., 2017a). These samples were collected from a field site near Oliver, Georgia, and the libraries were sequenced on an Illumina HiSeq2000 (2 × 100 nt) across 16 lanes. Average sequence coverage at sites with at least one aligning read was approximately 15× (sample standard deviation of 6×). The second study, conducted at Virginia Polytechnic Institute and State University (VTech), represented 752 individuals from a subset of the same PSSSS families. Samples were sequenced on an Illumina HiSeq 2500 (2 × 100 nt) and demultiplexed with Stacks (v1.44) (Catchen et al., 2013). Coverage was approximately 22× (sample standard deviation of 5×). Both ddRADseq studies were originally aligned against the v1.01 reference genome. Finally, one whole‐genome sequencing data set representing 10 megagametophytes derived from individuals from seven states within the native range of the species was included (De La Torre et al., 2019). Sequencing was performed on an Illumina HiSeq 3000 (2 × 150 nt) to a depth of 10× and was aligned to the v2.0 reference genome (Bowtie2 alignment and variants called with SAMTools/BEDTools [Li et al., 2009; Quinlan and Hall, 2010]) to call more than 455 million SNPs.
TABLE 1.
Summary of populations and variant filters.
| Information category | Cohort | |||||
|---|---|---|---|---|---|---|
| Exome capture of 375 trees | Exome capture of 24 trees | ddRAD of 1536 trees | ddRAD of 753 trees | WGS of 10 trees | Illumina Infinium array | |
| Tissue and ploidy | Needle (diploid) | Megagametophyte (haploid) |
Phloem (diploid) |
Phloem (diploid) | Megagametophyte (haploid) | Megagametophyte (haploid) |
| Sequencing platform (estimated coverage) | Illumina HiSeq 2500 (30×) | Illumina HiSeq 2000 (30×) | Illumina HiSeq 2000 (15×) | Illumina HiSeq 2500 (22×) | Illumina HiSeq 3000 (>10×) | Probes designed from Sanger resequenced ESTs |
| Total reads aligned to reference (%) | 91% | 98% | 35% | 75% | >99% | NA |
| Total strict quality variants a | 7,702,804 | 1,516,877 | 261,768 | 1,105,218 | 1,546,311 | 1181 aligned, 1840 unaligned |
| Total pre‐screening variants b | 109,602 | 86,200 | 268,154 | 34,388 | 156,456 | 1178 aligned, 1656 unaligned |
| Total post‐screening variants c | 28,518 | 7642 | 27,657 | 6009 | 17,973 | 108 aligned, 1209 unaligned |
| Total variants on final array (Pita50K) d | 13,962 | 4432 | 15,635 | 3398 | 10,854 | 36 aligned, 919 unaligned |
ddRAD = double‐digest RAD sequencing; NA = not applicable; WGS = whole genome sequencing.
Total number of strict quality variants: 8,272,630.
Total number of pre‐screening variants: 642,275.
Total number of post‐screening variants: 84,845.
Total number of variants on the final array: 46,439.
FIGURE 1.
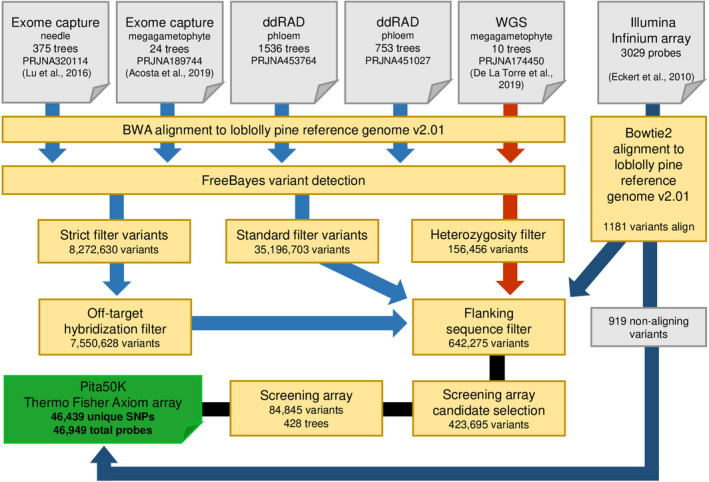
Informatic workflow describing the Pita50K array design. Four types of genomic data across six data sets were used in array design. Genomic reads from exome capture, ddRADseq, and whole genome sequencing (WGS) studies were aligned to the Pinus taeda reference genome, and variants were called with two thresholds. Probes designed around strict quality variants of exome capture and ddRADseq studies were assessed for potential off‐target hybridization through k‐mer to genome alignment scores. Variants from the WGS study (and later all variants from haploid megagametophyte tissue) that were heterozygous were removed. Previously successful Illumina Infinium array probes that align to the genome were assessed alongside the candidate variants for polymorphisms within flanking regions of probes. Passing probes were scored by Thermo Fisher Scientific, and recommended probes were further filtered via a screening array to create the final Pita50K Thermo Fisher Axiom array. This array contains 919 probes from the Illumina Infinium array that did not align to the reference genome and 36 that did align.
Variant detection and selection
The Illumina short reads from all genomic sources were quality controlled by Sickle (v1.33; Joshi and Fass, 2011) with quality and length thresholds set to 30Q and 50 bp, respectively. The reduced set of trimmed reads were aligned to the loblolly pine reference genome (v2.01, 568,339 scaffolds >3000 nt) with BWA‐MEM (v0.7.15; Li, 2013). This aligner was chosen for its ability to index large genomes and its robust strategy to contend with the anticipated gaps and polymorphisms. Alignment rates varied by genomic source (Table 1). In the exome capture and whole genome sequencing (WGS) cohorts, an average of >90% of reads aligned per sample. In the VTech and NCSU ddRADseq cohorts, average alignment rates per sample were lower at 75% and 35%. The NCSU samples possessed markedly greater adapter contamination and low‐complexity reads, resulting in overall lower alignment success. No individual NCSU samples were eliminated due to low alignment rates, as all samples were affected and there were no clear outliers.
Variant discovery was performed with FreeBayes (v1.0.2; Garrison and Marth, 2012) on the five source cohorts independently prior to creating a merged data set. FreeBayes was chosen because the Bayesian framework is designed to detect rare variants (increased sensitivity), consider multi‐mapping reads, and also accommodate variable ploidy (haploid and pooled designs). For the standard quality data set, variant discovery required minimum coverage of 8, minimum alternative allele count of 2, base quality of 15, and no threshold for mapping quality. The strict quality variants required a minimum coverage of 10, alternative coverage of 6, mapping quality of 20, and base quality of 25. Both runs allowed SNPs and indels, but no complex events, and required a minimum within‐sample minor allele frequency of 0.2. Population‐level allele frequency was not considered for further filtering of alleles due to pooling effects across the variable genomic data sources used. All variants were annotated with SnpEff (v4.3q; Cingolani et al., 2012) using a custom database of the loblolly reference genome and annotation (v2.01). The genome annotation of the loblolly pine assembly v2.01 represents 51,571 genes (48,238 multi‐exonic, 3513 mono‐exonic). The annotated coding regions have an estimated BUSCO (Simão et al., 2015) v.4.0.2 completeness of 41.7% and 47% when assessed with lineages Embryophyta v.10 and Viridiplantae v.10, respectively. Regions of open chromatin were identified in the reference genotype (N201010) using the ATAC‐seq protocol (Buenrostro et al., 2013), with unexpanded immature foliage as the source of isolated nuclei for six replicate reactions. The six libraries were sequenced first using Illumina MiSeq (3.43 million to 4.36 million pairs of 76 nt reads/sample) for quality‐control purposes, then using Illumina NextSeq (85.95 million to 95.38 million pairs of 76 nt reads/sample), to yield a total of 43 Gbp of raw sequence data. Reads were filtered using the BBMap suite of tools (B. Bushnell, https://sourceforge.net/projects/bbmap/) to remove reads from organellar genomes, and aligned to the v2.01 assembly using BWA‐MEM (v0.7.15). The short reads were pooled together, and genome regions less than 140 nt in length that were detected in four or more of the six replicate reactions were considered “open chromatin” for purposes of filtering and annotating candidate variants.
To assess the potential for off‐target probe hybridization, probes are broken down into k‐mers and aligned to the genome (Appendix S1A). For k‐mer alignment testing, probes consisting of 35‐nt flanks around strict quality variants were generated in reference sequence (71 nt total). Each probe was then split into four 18‐mer sequences with the central variant represented twice. If the variant was an indel, only the first reference nucleotide was used. The genome was subsequently parsed with an 18‐nt sliding window and matched to the list of probe 18‐mers. Each probe was scored by the sum of alignment counts for each of its 18‐mers. Variants with a score above the mean of 286 alignments were removed. Heterozygosity filtering was performed on the whole genome–sequenced megagametophyte (haploid) samples because these calls could only result from misalignment or sequencing errors. Strict quality variants with at least one heterozygous call across all 10 samples were excluded. From the previously designed Illumina Infinium array, a total of 3029 successful variants and their reported flanking regions were available. Because these were originally designed from transcriptomic resources (ESTs), they were assessed against the genome for the first time. These 3029 variants were reduced to 3021 probes that contained at least 70 nucleotides of flanking sequence (35 nucleotides on either side of the variant as required by the Axiom platform). The probe sequences with the variant coded as the appropriate International Union of Pure and Applied Chemistry (IUPAC) code were aligned to the loblolly pine reference genome (v2.01) with Bowtie2 (v2.3.3.1; Langmead and Salzberg, 2012). Bowtie2 was chosen for its support of ambiguous bases in the aligned sequence. For probes that aligned, success was determined by querying the resulting alignment files (71 matches indicated a non‐gapped alignment). Variants from all studies were combined and probes were re‐generated in a similar manner (2 × 35 nt), but with IUPAC codes assigned to identify variants of both strict and standard quality in the flanking regions. Probes were removed if polymorphisms of any quality occurred in both flanking arms. If polymorphisms occurred in only one arm, the probe was trimmed before that site.
Screening array
To guide the selection of variants using population‐based criteria, a screening phase was executed. To create the screening array, the filtered set of 642,275 variants were scored by Thermo Fisher Scientific through their custom informatic assessment. This assessment categorized each variant as “recommended,” “neutral,” “not recommended,” or “not possible” for probe development. These categories corresponded to a number of metrics derived from alignments of each probe sequence against the loblolly pine v2.01 reference genome, which downgraded probes in highly repetitive regions as well as probes with interfering polymorphisms outside the focal variant. All variants in the “recommended” and “neutral” categories were automatically retained for the screening array. For variants in the “not recommended” category, a linear index incorporating several alignment metrics was used to rank the variants. These metrics included “polycount” (the number of instances where 24 bp flanking the variant match other sequences on the list, but the alleles differ), “wobble count” (the number of interfering polymorphisms within 24 bp), and “wobble distance” (the distance between the focal variant and the nearest interfering polymorphism). An index was produced for each strand of each probe. The linear index took the form
| (1) |
Where I represents the index value, q represents the wobble distance, and p represents the summation of the polycount and wobble count. This index places a positive weight on the wobble distance and a negative weight on the counts. The index was used to sort the list of “not recommended” markers in decreasing index order.
A total of 423,695 variants were included on the assay, using a panel of 424 samples (388 diploid samples and 36 haploid megagametophyte samples). A total of 84,845 variants were labeled as “polyhighres,” “nominorhom,” or “callratebelowthreshold” in the Axiom Analysis Suite (Thermo Fisher Scientific, 2020) software. The average homozygosity of megagametophyte samples was 0.9. Therefore, variants with more than 10% heterozygous genotypes across the 36 haploid megagametophyte samples were removed. Genotype frequencies were estimated using the diploid samples only. Variants that failed a chi‐squared test for Hardy–Weinberg equilibrium as determined by a Bonferroni‐corrected P value of <5.89e‐07 were removed. From the remaining markers, variants in the “callratebelowthreshold” category with only two genotype clusters were filtered. Next, from the remaining markers, any sites with >10% missing data were filtered. Finally, variants with a minor allele frequency of <0.01 were removed. In an attempt to identify variants segregating according to Mendelian ratios in families, the haploid samples were reanalyzed with the variants passing the filters described above. For each variant, the genotype of the six parents was ascertained. For each variant with a heterozygous call in the parents, the six offspring megagametophytes of the respective parent were tested for Mendelian segregation. However, in a sample this small, it is feasible to observe only one genotype among the six offspring; therefore, greater weight was placed on heterozygosity among these samples. Variants with more than one heterozygous result out of its six tests were removed. In an effort to maximize the number of features on the array, the final list was sorted such that variants with complementary nucleotides (A/T, G/C) were given the lowest priority, as these require more than one probe.
To estimate the ability of the selected markers to capture known genetic relationships among individuals, a comparison was made between A, the additive (or average) numerator relationship matrix estimated from the pedigree (Henderson, 1975), and G, the genomic relationship matrix estimated from the marker data. This subset represented only the diploid samples with known pedigree and the markers that were polymorphic on these samples. Missing data were imputed using mean imputation via custom in‐house R scripts (R Core Team, 2020). For the purpose of this comparison, G matrix was calculated as in VanRaden (2008):
| (2) |
With M representing a (375 × 49,014) marker matrix with values of 0, 1, or 2 representing minor allele counts. P is a (375 × 49,014) matrix composed of doubled minor allele frequencies. The denominator, , represents the variance of allele frequencies and scales G to be analogous with A (VanRaden, 2008). We used the difference between A and G, represented by matrix C, to gauge the ability of the markers to capture known relationships.
RESULTS
Variant detection and selection
The samples included here were sequenced using very different approaches (WGS, ddRADseq, and exome capture) and used less contiguous versions of the loblolly pine reference genome. As such, variation in sequence depth required that we realign the reads from each sample to the most recent reference. Variant discovery prioritized finding strong heterozygous polymorphisms in individual samples while tracking additional lower‐confidence variation in the sequence. To assess genomic heterozygosity and select optimal variants for array design, two versions of variant detection were performed on the exome capture and ddRADseq cohorts. First, a standard quality run was performed on the aligned reads to determine base‐level polymorphisms. A stricter second run was subsequently performed to identify a subset of high‐quality variants for probe design (Fig. 1, Table 1). The number of strict and standard quality variants relates to genomic source, number of individual trees, and the fraction of reads that map to the reference genome. Exome capture data sets from 375 and 24 trees produce approximately 7.7 million and 1.5 million strict quality variants, respectively. The lower‐coverage ddRADseq data sets of 1536 (35% mapping rate) and 753 trees (75% mapping rate) generated approximately 260,000 and 1.1 million strict quality variants, respectively. Variants from the previous Illumina Infinium array (Eckert et al., 2010) were also included. These 3029 probes (≤50‐bp flanks surrounding the target genotype) were generated from EST contigs and therefore lacked reference genome coordinates. To include them, probes that contained at least 70 nucleotides of flanking sequence (3021 of the 3029) were aligned to the reference genome. A total of 1294 variants aligned (43%), but only 1181 (39%) aligned without gaps and could be assigned reference genome coordinates. The low alignment success and high prevalence of gaps may be the result of exceptional variation in the probe regions, fragmentation of the reference, and assembly errors. However, all 3021 Illumina Infinium probes with sufficient sequence were included as candidates for the screening array because they previously converted in loblolly pine populations. The set of well‐aligned probes (1181) was assessed with the filtering criteria applied to all other variants with physical positions in the genome. The final merged set of alleles contained 35,196,703 bi‐allelic variants with 8,272,630 candidate variants of strict quality across 2688 unique individuals. The variants from the ddRADseq and exome sequencing were coded to include 35 nucleotides of reference flanking sequence on either side of the variant to represent final probe sequence length. These probes were assessed for off‐target hybridization to filter variants based on the 18‐mer alignment frequency (Appendix S2A). A total of 33,087,264 18‐mers comprised the set of probes, with 24,875,817 being unique. The average 18‐mer mapped 14.6 times to the genome, demonstrating the expansive repetitive content and potential for off‐target hybridization (Appendix S2B). The full length of the probe was assessed on the sum genome alignments of each 18‐mer that comprised the flanking sequence (Appendix S2C). The minimum score for a probe would therefore be four if each of the four probe 18‐mers mapped to the genome uniquely. The mean probe score was 286 (median 8; maximum 4,667,554). Probes with a score above the mean were removed, resulting in a filtered set of 7,744,534 variants from 2608 unique individuals.
The 10 whole genome re‐sequenced haploid (megagametophyte) samples were included to increase the pool of genome‐wide variants (De La Torre et al., 2019). These samples were not included in the k‐mer testing, but flanking sequence content was subject to downstream filtering steps. Polymorphisms were called and filtered in an identical manner to the previous cohorts and provided 1,546,311 strict and 7,702,804 standard quality variants. These variants were further filtered to remove heterozygous calls as the haploid megagametophyte tissue source should not be heterozygous. The heterozygous calls in these samples represented 64% of strict quality sites. These likely arose from alignment errors (multi‐mapping) stemming from the genome’s repetitive and pseudogene content. Strict quality variants that were heterozygous in at least one haploid sample were removed, resulting in 557,204 retained variants. Further heterozygosity filtering on the exome capture cohorts (also haploid tissue sourced) was performed later in the screening array and removed approximately 10% of variants from these samples. When combined with the other cohorts, there were 8,101,034 unique variants across 2618 unique individuals. The number of variants contributed from each data set (Table 1) is dependent on the number of samples, genomic data source, and sequence depth. Strict quality variant filtering required a minimum coverage of 10 and a minimum alternate allele coverage of 6, which limits identifying variants from WGS samples and lower‐coverage sources.
Probe creation and annotation
Designing probes with polymorphic sites in the flanking sequence is known to decrease binding affinity during genotyping (Benovoy et al., 2008). To mitigate this, the 8.1 million filtered probe flank sequences were annotated with polymorphisms from both the strict and standard quality variant data set. Probes containing at least one flanking arm without variants of any type or quality were kept. The other arm could contain variant sites but was trimmed before the first variant. This dramatically reduced the set of passing probes to 642,275 (622,443 SNPs and 19,832 indels) from 2618 unique individuals. A total of 1656 of these probes were from the Illumina Infinium array that did not align to the v2.01 genome and 1178 were from those that did align. All 642,275 variants were passed to Thermo Fisher Scientific for scoring, using the v2.01 reference genome. To additionally assess the proximity of these probes to genes, variants were annotated with SnpEff (Fig. 2A, Appendix S3). This resulted in 18.4% of the total variants situated in or around 19,001 unique genes (less than 5000 nt upstream or downstream of a gene). The largest of these categories were intron variants (6.3%, variants within an intron including splice sites), upstream or downstream variants (6.5%, within 5000 nt upstream or downstream of a gene), and synonymous variants (3.6%). A total of 5% of variants were annotated as having a high or moderate impact on a predicted protein product. High‐impact variants are predicted to be greatly disruptive to the protein product (e.g., truncation, frameshift, splice site variant), whereas moderate‐impact variants may disrupt protein functionality (e.g., missense variant, inframe indel). In total, synonymous or missense variants occurred within the coding region in 2798 unique genes.
FIGURE 2.
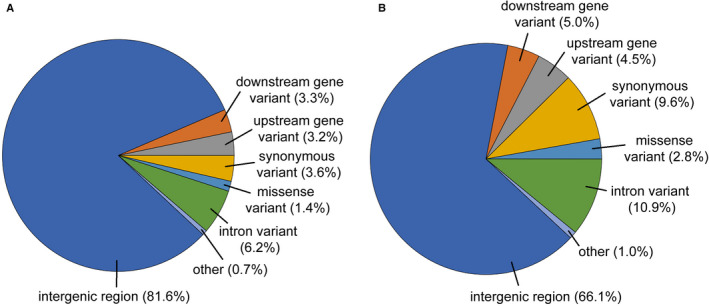
Proximity and function of pre‐screening candidate variants and final array variants to genes. (A) Annotation of 642,275 candidate probes prior to scoring by Thermo Fisher Scientific and screening array selection. Results do not include 1656 Illumina Infinium array probes that did not have reference gene coordinates. Rare functions were grouped as “other.” All annotation categories and effects are available in Appendices S3 and S7. (B) Annotation of the final array probes. Results do not include 919 Illumina Infinium array probes that do not have reference genome coordinates.
Screening array
The 642,275 variants for manufacture on the screening array were scored and included 58,155 variants in the “recommended” category, 99,982 variants in the “neutral” category, 475,585 variants in the “not recommended” category, and 8551 variants in the “not possible” category. A subset of these markers was used to gauge the relationship between the bioinformatic scoring categories and previously obtained empirical data, because they were successfully converted to array SNPs as part of the previously successful Illumina Infinium array project (Eckert et al., 2010). Out of the 3279 markers, 595 were labeled as “recommended,” 588 were labeled as “neutral,” and 2096 were labeled as “not recommended.” This suggests that the bioinformatic scoring categories used here may have little utility in predicting the conversion success of markers in complex genomes and reiterates the need for empirical data when evaluating variants in such species. Due to the presence of markers with homologous alleles that require more than one probe, the final manufactured screening array contained a total of 423,695 variants from the original list of 642,275 (Appendix S4). The preliminary output of the Axiom Analysis Suite software, provided as a service by Thermo Fisher Scientific, labeled 27,336 variants as “polymorphic, high‐resolution with three clusters,” 32,277 variants as “polymorphic, high‐resolution with two clusters,” and 25,232 variants as “call rate below threshold” (Appendix S4). Here, the word “cluster” refers to a group of genotype calls having a similar signal intensity based on the colors associated with the two alleles of a given marker on the array. The number of clusters for a microarray variant is the number of distinct genotypes that a marker yields. The screening array pipeline described above in the Methods took as input these 84,845 markers, and filtered 5991, 16,050, 4786, 1963, 4409, and 1228 variants in steps 1 through 6, respectively. The conversion rate was 97%, 44%, and 38% for variants in the scoring categories “polymorphic, high‐resolution with three clusters,” “polymorphic, high‐resolution with two clusters,” and “call rate below threshold,” respectively. This suggests that two‐cluster markers should still be considered in outcrossing species with large effective population sizes such as conifers, where the site‐frequency spectrum has a well‐known bias toward low‐frequency variants. No minor‐allele homozygotes were observed for close to 30% of the selected variants. The variant filtering procedure using the screening array increased the average marker‐based heterozygosity by 80%, and nearly doubled the average polymorphic information content of selected markers relative to the non‐selected markers (Appendices S2, S5). The site‐frequency spectrum showed a large shift toward moderate allele frequencies relative to the non‐selected markers (Appendix S6), which should be considered in downstream applications where omission of rare variants affects methodological performance (e.g., demographic model fitting). This underscores the fundamental importance of empirical population‐based data in variant selection for genotyping arrays, particularly in conifer species with a large effective population size. In its final form, the production array contained 46,439 unique features (45,197 SNPs and 1242 indels) from 2423 unique individuals. The final number of markers was lower than the number of variants in the original list (50,418) for two reasons: (1) variants with complementary nucleotides require more than one probe, and (2) stochastic production issues during array manufacture resulted in less than 100% of the surface area of the chip being available for probe synthesis.
Final genotyping array
The final Pita50K production array contained 36 aligned and 919 non‐aligned Illumina Infinium variants. Of the 45,520 probes with reference genome coordinates, 33.9% were annotated in or near genes, which is markedly higher than the 18.4% in the 642,275 candidates on the pre‐screening array (Fig. 2B, Appendix S7). Similarly, the largest of these categories were intron variants (10.9%), upstream or downstream variants (9.5%), and synonymous variants (9.6%). A reduced fraction of 2.9% were annotated as having a medium or high impact on the protein product. The type of initial genomic resource did not heavily bias the proportion of variants contributed in or near genic regions. ddRAD data sets provided 36.7% of variants in the final array and 28.4% of those annotated in/near genes. Together, a total of 5854 unique genic regions (including variants upstream and downstream of a gene) are represented, with 5688 variants occurring within the coding region of 2796 unique genes. A total of 10,469 variants on the final array are within regions of open chromatin. The 45,520 probes cover 2,553,757 unique nucleotides on 20,682 unique scaffolds in the 22.5 Gbp v2.01 genome assembly that comprises 1,762,655 scaffolds.
The genomic relationship matrix computed from these markers captured relationships between individuals previously thought to be unrelated (Appendix S8). The mean of the distribution of C matrix was −0.035, indicating that the markers were largely in agreement with the pedigree information, but also suggesting previously unknown relationships between individuals. This result demonstrates the value of genome‐wide markers in capturing putative ancestral relationships that are missed by the pedigree or errors in pedigree records. A small number of samples showed large disagreements between the pedigree and the marker information; in at least two cases, this represented cross‐contamination between adjacent wells of a DNA plate. In a small number of other cases, pedigree recording errors were deemed the most likely explanation for the discrepancies.
DISCUSSION
As evident through the completion of the first 11 gymnosperm genomes in the past six years, sequencing and assembling large genomes is now accessible (Zimin et al., 2017). Among these conifer genomes, only one is organized into chromosome‐scale scaffolds and all remain challenging to annotate (Scott et al., 2020). Despite this, exome capture and other reduced representation approaches are now commonly used to interrogate these mega‐genomes. Their complexity, paired with long generation times, makes them an ideal candidate for GS. At the same time, the diverse (polymorphic) outcrossing populations and the repetitive nature of conifer genomes present unique challenges. The Pita50K array represents the first pine resource that is designed across several independent high‐throughput genomics studies. The diverse populations represented by these studies increased the representation of genetic variation across the species range. Compared to the arrays that have come before it, the Pita50K array has the added advantage of a comprehensive screening design that will guarantee success in future applications. Unlike recent approaches that primarily leveraged transcriptomes or exome capture, Pita50K includes variants both within and around genes along with substantially further upstream or downstream variants that may capture important regulatory elements. Finally, the use of an improved reference genome provides insight into the limitations of using only gene‐targeting data sets. The exome capture sources in this study provided over nine million high‐quality variants but only within 45.7% of all predicted genes and only 43% of EST‐derived probes aligned to the reference genome. Exome bait design resulting in off‐target or failed genome alignments has been observed in other pine species (Suren et al., 2016; Telfer et al., 2019).
While the Thermo Fisher Axiom technology provides a cost‐competitive approach for genotyping at scale, the use of shorter probes is a considerable hurdle for repetitive plant genomes. Although longer probes provide greater sequence complexity and reduce off‐target alignments, the ubiquity of polymorphisms in flanking sequences (the largest filter barrier in constructing this array) may further limit the number of viable probes. The bioinformatic approach presented in the construction of this array considers the unique aspects of the reduced representation approaches, ploidy of the original samples, off‐target probe potential, and polymorphic nature of the populations to provide high‐quality variants. Compared to previous array constructions for pines, the use of a reference genome greatly enhanced the pool of potential high‐quality variants for the screening array. Limiting the scope to genic regions would have reduced the final size of the array or required lower‐quality thresholds for variants. The high frequency of problematic heterozygous calls from haploid tissue and poor alignments of probes from the set of successful variants evaluated on the first array remain a challenge. Above all, we demonstrated that a pedigree‐informed screening array is imperative to the final selection process and should be used with the informatic recommendations provided here.
AUTHOR CONTRIBUTIONS
M.C. and E.L. led the writing of the manuscript with support from J.L.W. and input from all authors. F.I. and J.L.W. conceived and supervised the project. M.C. and E.L. performed the primary computational analysis with support from J.B., S.Z., S.M., and C.J. J.A., L.T., A.E., R.W.W., C.L., J.H., and M.M. contributed the genomic resources and associated information, and consulted on the final array design. S.Z. and S.M. provided analysis on the structural annotation of the genome. J.A., L.T., C.L., J.H., and M.M. provided the genomic resources used in array design. All authors reviewed the results and approved the final version of the manuscript.
Supporting information
APPENDIX S1. Distributions of 18‐mer genome mapping rates and probe composition. (A) Each of the 8,272,630 candidate probes built around high‐quality variants are broken into four 18‐mer sequences with the central variant represented twice in reference bases. An 18‐mer sliding window queries the reference genome (v2.01) for 18‐mer alignment rates. A probe’s four 18‐mer genome alignment counts are then summed to create the probe’s total mapping score. (B) The distribution of mapping rates for probe 18‐mers (barred at <1000) is shown. A majority of 18‐mers map to the genome multiple times, which could result in off‐target probe hybridization. (C) The distribution of probe mapping scores summed from each probe’s four 18‐mers from (B). Probes with a score greater than the mean (286) are eliminated.
APPENDIX S2. Marker‐based heterozygosity (H) increased by 80% for selected SNPs relative to non‐selected SNPs. A histogram and overlaid density plot for marker‐based heterozygosity (H) is shown for non‐selected SNPs (blue) and selected SNPs (orange). Vertical dashed lines show the mean for both groups.
APPENDIX S3. Full SnpEff annotation of 642,275 variants with reference genome coordinates before scoring by Thermo Fisher Scientific and in the final array. A total of 117,783 variants (18.4%) are annotated near/in genes.
APPENDIX S4. Bioinformatic scoring categories for screening array variants provided by the Axiom Analysis Suite, based on a number of metrics including cluster quality, signal intensity, number of distinct clusters, and amount of missing data.
APPENDIX S5. Polymorphic information content (PIC) nearly doubled for selected SNPs relative to non‐selected SNPs. A histogram and overlaid density plot for PIC is shown for non‐selected SNPs (blue) and selected SNPs (orange). Vertical dashed lines show the mean for both groups.
APPENDIX S6. A folded site frequency spectrum of selected markers showed a marked shift toward intermediate‐frequency SNPs relative to non‐selected markers. A histogram and overlaid density plot for minor allele frequency (MAF) is shown for non‐selected SNPs (blue) and selected SNPs (orange). Vertical dashed lines show the mean for both groups.
APPENDIX S7. Full SnpEff annotation of the 45,520 variants with reference genome coordinates in the final array. A total of 15,424 variants (33.9%) are annotated near/in genes.
APPENDIX S8. Distribution of deviations between the additive numerator relationship matrix (A) and the genomic relationship matrix (G). The distribution shows the efficiency of molecular markers in capturing ancestral relationships among individuals.
Acknowledgments
This work was supported by the U.S. Department of Agriculture National Institute of Food and Agriculture (NIFA; grants #2011‐68002‐30185 [Pine Integrated Network: Education, Mitigation, and Adaptation] and #2015‐05832 [Towards Genomic Selection in Forest Trees]) and the North Carolina State University Cooperative Tree Improvement Program. We thank the Institute for Systems Genomics, Computational Biology Core at the University of Connecticut for high‐performance computing resources.
Caballero, M. , Lauer E., Bennett J., Zaman S., McEvoy S., Acosta J., Jackson C., et al. 2021. Toward genomic selection in Pinus taeda: Integrating resources to support array design in a complex conifer genome. Applications in Plant Sciences 9(6): e11439.
Contributor Information
Jill L. Wegrzyn, Email: jill.wegrzyn@uconn.edu.
Fikret Isik, Email: fisik@ncsu.edu.
Data Availability
All genomic sequences used in this study are available from the National Center for Biotechnology Information (NCBI; BioProjects PRJNA451027, PRJNA453764, PRJNA320114, PRJNA189744, and PRJNA174450). Detailed steps including scripts, software versions, and intermediate results are available on GitLab (https://gitlab.com/PlantGenomicsLab/loblolly‐probe‐creation).
LITERATURE CITED
- Acosta, J. J. , Fahrenkrog A. M., Neves L. G., Resende M. F. R., Dervinis C., Davis J. M., Holliday J. A., and Kirst M.. 2019. Exome resequencing reveals evolutionary history, genomic diversity, and targets of selection in the conifers Pinus taeda and Pinus elliottii . Genome Biology and Evolution 11(2): 508–520. 10.1093/gbe/evz016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez, A. , Pavy N., Gérardi S., Laroche J., Boyle B., Gagnon F., Mottet M.‐J., et al. 2018. A catalog of annotated high‐confidence SNPs from exome capture and sequencing reveals highly polymorphic genes in Norway spruce (Picea abies). BMC Genomics 19(1): 942. 10.1186/s12864-018-5247-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu, J. , Nadeau S., Ding C., Celedon J. M., Azaiez A., Ritland C., Laverdière J.‐P., et al. 2020. Genomic selection for resistance to spruce budworm in white spruce and relationships with growth and wood quality traits. Evolutionary Applications 13(10): 2704–2722. 10.1111/eva.13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovoy, D. , Kwan T., and Majewski J.. 2008. Effect of polymorphisms within probe–target sequences on olignonucleotide microarray experiments. Nucleic Acids Research 36(13): 4417–4423. 10.1093/nar/gkn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardsson, C. , Zan Y., Chen Z., Ingvarsson P., and Wu H.. 2020. Development of a highly efficient 50K SNP genotyping array for the large and complex genome of Norway spruce (Picea abies L. Karst) by whole genome re‐sequencing and its transferability to other spruce species. Molecular Ecology Resources 21: 880–896. 10.1111/1755-0998.13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat, J. A. , Ali S., Salgotra R. K., Mir Z. A., Dutta S., Jadon V., Tyagi A., et al. 2016. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Frontiers in Genetics 7(December): 221. 10.3389/fgene.2016.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R. , Gill G. P., Kuntz R. J., Langley C. H., and Neale D. B.. 2004. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proceedings of the National Academy of Sciences, USA 101(42): 15255–115260. 10.1073/pnas.0404231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro, J. D. , Giresi P. G., Zaba L. C., Chang H. Y., and Greenleaf W. J.. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nature Methods 10(12): 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calleja‐Rodriguez, A. , Pan J., Funda T., Chen Z., Baison J., Isik F., Abrahamsson S., and Wu H. X.. 2020. Evaluation of the efficiency of genomic versus pedigree predictions for growth and wood quality traits in Scots pine. BMC Genomics 21(1): 796. 10.1186/s12864-020-07188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. , Hohenlohe P. A., Bassham S., Amores A., and Cresko W. A.. 2013. Stacks: An analysis tool set for population genomics. Molecular Ecology 22(11): 3124–3140. 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z.‐Q. , Baison J., Pan J., Karlsson B., Andersson B., Westin J., García‐Gil M. R., and Wu H. X.. 2018. Accuracy of genomic selection for growth and wood quality traits in two control‐pollinated progeny trials using exome capture as the genotyping platform in Norway spruce. BMC Genomics 19(December): 946. 10.1186/s12864-018-5256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatre, V. E. , Byram T. D., Neale D. B., Wegrzyn J. L., and Krutovsky K. V.. 2013. Genetic structure and association mapping of adaptive and selective traits in the east Texas loblolly pine (Pinus taeda L.) breeding populations. Tree Genetics & Genomes 9(5): 1161–1178. 10.1007/s11295-013-0624-x. [DOI] [Google Scholar]
- Cingolani, P. , Platts A., Wang L. L., Coon M., Nguyen T., Wang L., Land S. J., et al. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6(2): 80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumbie, W. P. , Eckert A., Wegrzyn J., Whetten R., Neale D., and Goldfarb B.. 2011. Association genetics of carbon isotope discrimination, height and foliar nitrogen in a natural population of Pinus taeda L. Heredity 107(2): 105–114. 10.1038/hdy.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumbie, W. P. , Huber D. A., Steel V. C., Rottmann W., Cannistra C., Pearson L., and Cunningham M.. 2020. Marker associations for fusiform rust resistance in a clonal population of loblolly pine (Pinus taeda L.). Tree Genetics & Genomes 16(6): 86. 10.1007/s11295-020-01478-4. [DOI] [Google Scholar]
- De La Torre, A. R. , Puiu D., Crepeau M. W., Stevens K., Salzberg S. L., Langley C. H., and Neale D. B.. 2019. Genomic architecture of complex traits in loblolly pine. New Phytologist 221(4): 1789–1801. 10.1111/nph.15535. [DOI] [PubMed] [Google Scholar]
- De La Vega, F. M. , Lazaruk K. D., Rhodes M. D., and Wenz M. H.. 2005. Assessment of two flexible and compatible SNP genotyping platforms: TaqMan® SNP Genotyping Assays and the SNPlexTM Genotyping System. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 573(1): 111–135. 10.1016/j.mrfmmm.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Eckert, A. J. , J. van Heerwaarden , Wegrzyn J. L., Nelson C. D., Ross‐Ibarra J., González‐Martínez S. C., and Neale D. B.. 2010. Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., Pinaceae). Genetics 185(3): 969–982. 10.1534/genetics.110.115543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farjat, A. E. , Chamblee A. K., Isik F., Whetten R. W., and McKeand S. E.. 2017a. Variation among loblolly pine seed sources across diverse environments in the southeastern United States. Forest Science 63(1): 39–48. 10.5849/forsci.15-107. [DOI] [Google Scholar]
- Farjat, A. , Reich B. J., Guinness J., Whetten R., McKeand S., and Isik F.. 2017b. Optimal seed deployment under climate change using spatial models: Application to loblolly pine in the southeastern US. Journal of the American Statistical Association 112(519): 909–920. 10.1080/01621459.2017.1292179. [DOI] [Google Scholar]
- Garrison, E. , and Marth G.. 2012. Haplotype‐based variant detection from short‐read sequencing. ArXiv:1207.3907 [Preprint] [posted 17 July 2012]. Available at http://arxiv.org/abs/1207.3907 [accessed 1 June 2021].
- Goddard, M. E. , and Hayes B. J.. 2007. Genomic selection. Journal of Animal Breeding and Genetics 124(6): 323–330. 10.1111/j.1439-0388.2007.00702.x. [DOI] [PubMed] [Google Scholar]
- Grattapaglia, D. , Silva‐Junior O. B., Resende R. T., Cappa E. P., Müller B. S. F., Tan B., Isik F., et al. 2018. Quantitative genetics and genomics converge to accelerate forest tree breeding. Frontiers in Plant Science 9(November): 1693. 10.3389/fpls.2018.01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, C. R. 1975. Rapid method for computing the inverse of a relationship matrix. Journal of Dairy Science 58(11): 1727–1730. 10.3168/jds.S0022-0302(75)84776-X. [DOI] [Google Scholar]
- Heslot, N. , Rutkoski J., Poland J., Jannink J.‐L., and Sorrells M. E.. 2013. Impact of marker ascertainment bias on genomic selection accuracy and estimates of genetic diversity. PLoS ONE 8(9): e74612. 10.1371/journal.pone.0074612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, G. T. , Jayawickrama K., Kolpak S. E., Kling J., Trappe M., Hipkins V., Ye T., et al. 2020. An Axiom SNP genotyping array for Douglas‐fir. BMC Genomics 21(1): 9. 10.1186/s12864-019-6383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isik, F. , and McKeand S. E.. 2019. Fourth cycle breeding and testing strategy for Pinus taeda in the NC State University Cooperative Tree Improvement Program. Tree Genetics & Genomes 15(5): 70. 10.1007/s11295-019-1377-y. [DOI] [Google Scholar]
- Joshi, N. A. , and Fass J. N.. 2011. Sickle: A sliding‐window, adaptive, quality‐based trimming tool for FastQ Files (Version 1.33). Available at https://github.com/najoshi/sickle [accessed 2 June 2021].
- Langmead, B. , and Salzberg S. L.. 2012. Fast Gapped‐Read Alignment with Bowtie 2. Nature Methods 9(4): 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer, E. , Sims A., McKeand S., and Isik F.. 2020. Genetic parameters and genotype‐by‐environment interactions in regional progeny tests of Pinus taeda L. in the southern USA. Forest Science 67: 60–71. 10.1093/forsci/fxaa035. [DOI] [Google Scholar]
- Lenz, P. R. N. , Nadeau S., Mottet M.‐J., Perron M., Isabel N., Beaulieu J., and Bousquet J.. 2020. Multi‐trait genomic selection for weevil resistance, growth, and wood quality in Norway spruce. Evolutionary Applications 13(1): 76–94. 10.1111/eva.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. ArXiv:1303.3997 [Preprint] [posted 16 March 2013, updated 23 May 2013]. Available at http://arxiv.org/abs/1303.3997 [accessed 1 June 2021].
- Li, H. , Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, M. , Krutovsky K. V., Nelson C. D., Koralewski T. E., Byram T. D., and Loopstra C. A.. 2016. Exome genotyping, linkage disequilibrium and population structure in loblolly pine (Pinus taeda L.). BMC Genomics 17(1): 730. 10.1186/s12864-016-3081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima, K. , Hirao T., Tsubomura M., Tamura M., Kurita M., Nose M., Hanaoka S., et al. 2018. Identification of novel putative causative genes and genetic marker for male sterility in Japanese cedar (Cryptomeria japonica D.Don). BMC Genomics 19(1): 277. 10.1186/s12864-018-4581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, D. B. , Wegrzyn J. L., Stevens K. A., Zimin A. V., Puiu D., Crepeau M. W., Cardeno C., et al. 2014. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biology 15(3): R59. 10.1186/gb-2014-15-3-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olatoye, M. O. , Clark L. V., Wang J., Yang X., Yamada T., Sacks E. J., and Lipka A. E.. 2019. Evaluation of genomic selection and marker‐assisted selection in Miscanthus and energycane. Molecular Breeding 39(12): 171. 10.1007/s11032-019-1081-5. [DOI] [Google Scholar]
- Pavy, N. , Gagnon F., Rigault P., Blais S., Deschênes A., Boyle B., Pelgas B., et al. 2013. Development of high‐density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and Nordic congeners. Molecular Ecology Resources 13(2): 324–336. 10.1111/1755-0998.12062. [DOI] [PubMed] [Google Scholar]
- Pavy, N. , Gagnon F., Deschênes A., Boyle B., Beaulieu J., and Bousquet J.. 2016. Development of highly reliable in silico SNP resource and genotyping assay from exome capture and sequencing: An example from black spruce (Picea mariana). Molecular Ecology Resources 16(2): 588–598. 10.1111/1755-0998.12468. [DOI] [PubMed] [Google Scholar]
- Perry, A. , Wachowiak W., Downing A., Talbot R., and Cavers S.. 2020. Development of a single nucleotide polymorphism array for population genomic studies in four European pine species. Molecular Ecology Resources 20(6): 1697–1705. 10.1111/1755-0998.13223. [DOI] [PubMed] [Google Scholar]
- Peterson, B. K. , Weber J. N., Kay E. H., Fisher H. S., and Hoekstra H. E.. 2012. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non‐model species. PLoS ONE 7(5): e37135. 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestemon, J. P. , and Abt R. C.. 2002. Southern Forest Resource Assessment highlights: The southern timber market to 2040. Journal of Forestry 100(7): 16–22. 10.1093/jof/100.7.16. [DOI] [Google Scholar]
- Quinlan, A. R. , and Hall I. M.. 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . 2020. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Website https://www.R‐project.org/ [accessed 2 June 2021]. [Google Scholar]
- Resende, M. F. R. , Muñoz P., Resende M. D. V., Garrick D. J., Fernando R. L., Davis J. M., Jokela E. J., et al. 2012. Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 190(4): 1503–1510. 10.1534/genetics.111.137026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, A. D. , Zimin A. V., Puiu D., Workman R., Britton M., Zaman S., Caballero M., et al. 2020. A reference genome sequence for giant sequoia. G3: Genes, Genomes, Genetics 10(11): 3907–3919. 10.1534/g3.120.401612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, P. I. T. , Silva‐Junior O. B., Resende L. V., Sousa V. A., Aguiar A. V., and Grattapaglia D.. 2020. A 3K Axiom SNP array from a transcriptome‐wide SNP resource sheds new light on the genetic diversity and structure of the iconic subtropical conifer tree Araucaria angustifolia (Bert.) Kuntze. PLoS ONE 15(8): e0230404. 10.1371/journal.pone.0230404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse R. M., Ioannidis P., Kriventseva E. V., and Zdobnov E. M.. 2015. BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31(19): 3210–3212. 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Spindel, J. E. , and McCouch S. R.. 2016. When more is better: How data sharing would accelerate genomic selection of crop plants. New Phytologist 212(4): 814–826. 10.1111/nph.14174. [DOI] [PubMed] [Google Scholar]
- Suren, H. , Hodgins K. A., Yeaman S., Nurkowski K. A., Smets P., Rieseberg L. H., Aitken S. N., and Holliday J. A.. 2016. Exome capture from the spruce and pine giga‐genomes. Molecular Ecology Resources 16(5): 1136–1146. 10.1111/1755-0998.12570. [DOI] [PubMed] [Google Scholar]
- Telfer, E. , Graham N., Macdonald L., Li Y., Klápště J., Resende M. Jr., Gomide Neves L., et al. 2019. A high‐density exome capture genotype‐by‐sequencing panel for forestry breeding in Pinus radiata . PLoS ONE 14(9): e0222640. 10.1371/journal.pone.0222640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermo Fisher Scientific . 2020. Axiom Analysis Suite v5.1 User Guide. Affymetrix, Santa Clara, California, USA. [Google Scholar]
- Thistlethwaite, F. R. , Ratcliffe B., Klápště J., Porth I., Chen C., Stoehr M. U., and El‐Kassaby Y. A.. 2019. Genomic selection of juvenile height across a single‐generational gap in Douglas‐fir. Heredity 122(6): 848–863. 10.1038/s41437-018-0172-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukrainetz, N. K. , and Mansfield S. D.. 2019. Assessing the sensitivities of genomic selection for growth and wood quality traits in lodgepole pine using Bayesian models. Tree Genetics & Genomes 16(1): 14. 10.1007/s11295-019-1404-z. [DOI] [Google Scholar]
- VanRaden, P. M. . 2008. Efficient methods to compute genomic predictions. Journal of Dairy Science 91(11): 4414–4423. 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- VanRaden, P. M. 2020. Symposium Review: How to implement genomic selection. Journal of Dairy Science 103(6): 5291–5301. 10.3168/jds.2019-17684. [DOI] [PubMed] [Google Scholar]
- Wegrzyn, J. L. , Liechty J. D., Stevens K. A., Wu L.‐S., Loopstra C. A., Vasquez‐Gross H. A., Dougherty W. M., et al. 2014. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 196(3): 891–909. 10.1534/genetics.113.159996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Liu X., Fu J., Wang H., Wang J., Huang C., Prasanna B. M., et al. 2020. Enhancing genetic gain through genomic selection: From livestock to plants. Plant Communications 1(1): 100005. 10.1016/j.xplc.2019.100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin, A. V. , Stevens K. A., Crepeau M. W., Puiu D., Wegrzyn J. L., Yorke J. A., Langley C. H., et al. 2017. An improved assembly of the loblolly pine mega‐genome using long‐read single‐molecule sequencing. GigaScience 6(1): giw016. 10.1093/gigascience/giw016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. Distributions of 18‐mer genome mapping rates and probe composition. (A) Each of the 8,272,630 candidate probes built around high‐quality variants are broken into four 18‐mer sequences with the central variant represented twice in reference bases. An 18‐mer sliding window queries the reference genome (v2.01) for 18‐mer alignment rates. A probe’s four 18‐mer genome alignment counts are then summed to create the probe’s total mapping score. (B) The distribution of mapping rates for probe 18‐mers (barred at <1000) is shown. A majority of 18‐mers map to the genome multiple times, which could result in off‐target probe hybridization. (C) The distribution of probe mapping scores summed from each probe’s four 18‐mers from (B). Probes with a score greater than the mean (286) are eliminated.
APPENDIX S2. Marker‐based heterozygosity (H) increased by 80% for selected SNPs relative to non‐selected SNPs. A histogram and overlaid density plot for marker‐based heterozygosity (H) is shown for non‐selected SNPs (blue) and selected SNPs (orange). Vertical dashed lines show the mean for both groups.
APPENDIX S3. Full SnpEff annotation of 642,275 variants with reference genome coordinates before scoring by Thermo Fisher Scientific and in the final array. A total of 117,783 variants (18.4%) are annotated near/in genes.
APPENDIX S4. Bioinformatic scoring categories for screening array variants provided by the Axiom Analysis Suite, based on a number of metrics including cluster quality, signal intensity, number of distinct clusters, and amount of missing data.
APPENDIX S5. Polymorphic information content (PIC) nearly doubled for selected SNPs relative to non‐selected SNPs. A histogram and overlaid density plot for PIC is shown for non‐selected SNPs (blue) and selected SNPs (orange). Vertical dashed lines show the mean for both groups.
APPENDIX S6. A folded site frequency spectrum of selected markers showed a marked shift toward intermediate‐frequency SNPs relative to non‐selected markers. A histogram and overlaid density plot for minor allele frequency (MAF) is shown for non‐selected SNPs (blue) and selected SNPs (orange). Vertical dashed lines show the mean for both groups.
APPENDIX S7. Full SnpEff annotation of the 45,520 variants with reference genome coordinates in the final array. A total of 15,424 variants (33.9%) are annotated near/in genes.
APPENDIX S8. Distribution of deviations between the additive numerator relationship matrix (A) and the genomic relationship matrix (G). The distribution shows the efficiency of molecular markers in capturing ancestral relationships among individuals.
Data Availability Statement
All genomic sequences used in this study are available from the National Center for Biotechnology Information (NCBI; BioProjects PRJNA451027, PRJNA453764, PRJNA320114, PRJNA189744, and PRJNA174450). Detailed steps including scripts, software versions, and intermediate results are available on GitLab (https://gitlab.com/PlantGenomicsLab/loblolly‐probe‐creation).