

Abstract
RNA undergoes extensive biochemical modification following transcription. In addition to RNA splicing, transcripts are processed by a suite of enzymes that alter the chemical structure of different nucleobases. Broadly termed as “RNA editing,” these modifications impart significant functional changes to translation, localization, and stability of individual transcripts within the cell. These changes are dynamic and required for a number of critical cellular processes, and dysregulation of these pathways is responsible for several disease states. Accurately detecting, measuring, and mapping different RNA modifications across the transcriptome is vital to understanding their broader functions as well as leveraging these events as diagnostic biomarkers. Here, we review recent advances in profiling several types of RNA modifications, with particular emphasis on adenosine-to-inosine (A-to-I) and N6-methyladenosine (m6A) RNA editing. We especially highlight approaches that utilize proteins to detect or enrich modified RNA transcripts prior to sequencing, and we summarize recent insights yielded from these techniques.
Keywords: RNA editing, epitranscriptomics, A-to-I editing, m6A
Introduction
RNA is a key information-carrying biomolecule that controls cellular function in all living organisms. In addition to capping, polyadenylation, and splicing steps that yield mature mRNA,1 individual nucleobases within transcripts can also be modified by a number of enzymes. These processes are now described as “RNA editing” or “epitranscriptomic modifications” (Fig. 1),2–5 and they affect nearly all types of RNA and encompasses a significant and increasing number of known modifications. Figure 1b displays a select group of these modified bases, and while there are many (>100) additional modifications known and more continue to be discovered, these highlighted alterations are the most prevalent in the human transcriptome and significantly influence RNA coding and function.2–4 Adenosines in particular are frequently edited in humans, and different modifications are installed by several enzyme classes. Adenosine-to-inosine (A-to-I) conversions are catalyzed by adenosine deaminases acting on RNA (ADARs) (Fig. 1b), which in turn alter the base pairing properties of the nucleobase. Similar to epigenetic modifications in DNA, methylation of certain nucleotides in RNA is emerging as a widespread cellular event for tuning RNA function and regulating gene expression.6 In particular, N6-methyladenosine (m6A, Fig. 1b) has been identified as the most frequent RNA editing event in humans,7–9 and appears to both enhance protein translation10 and promote phase-separation of different transcripts into cytosolic granules.11–12 Additional methylation of m6A at the 2’ OH position (m6Am) is also prevalent, and is enriched at the 5’ termini of mRNAs.7 Uridines can also be converted into pseudouridine (Ψ), and while originally discovered as a common modification in both transfer RNA (tRNA) and ribosomal rRNA (rRNA),13 these edits have now been detected in thousands of human mRNAs.14–15 Similarly, cytidines can be converted to N4-acetylcytidine (ac4C),16–17 5-hydroxylmethylcytidine (hm5C),18 and 5-methylcytidine (m5C),19 with each of these modifications playing key roles in translational quality control and RNA stability. The discovery of these modifications represents an exciting new frontier in RNA biology, and determining their precise cellular functions will likely yield significant insight into basic cellular physiology. However, elucidating these roles has presented a significant challenge for the field and required creative strategies to detect and map these events. Toward this end, many groups have utilized natural protein machinery which displays exquisite molecular recognition properties. In this review, we discuss several key protein-based technologies for characterizing RNA editing events. While uridine and cytidine modification are biologically important, detection and enrichment of these modifications is typically achieved using chemical strategies, and these methods are described in greater detail in other reviews.3–4 Because of their prevalence and utilization of protein-based platforms for detection and enrichment, in this review we focus primarily on the two major adenosine modifications in RNA. In particular, we describe inosine (I) and N6-methyladenosine (m6A), and highlight recent technological developments for their recognition as well as new biological insights gained from these methods.
Figure 1. RNA editing introduces chemical and structural alterations to different nucleobases.
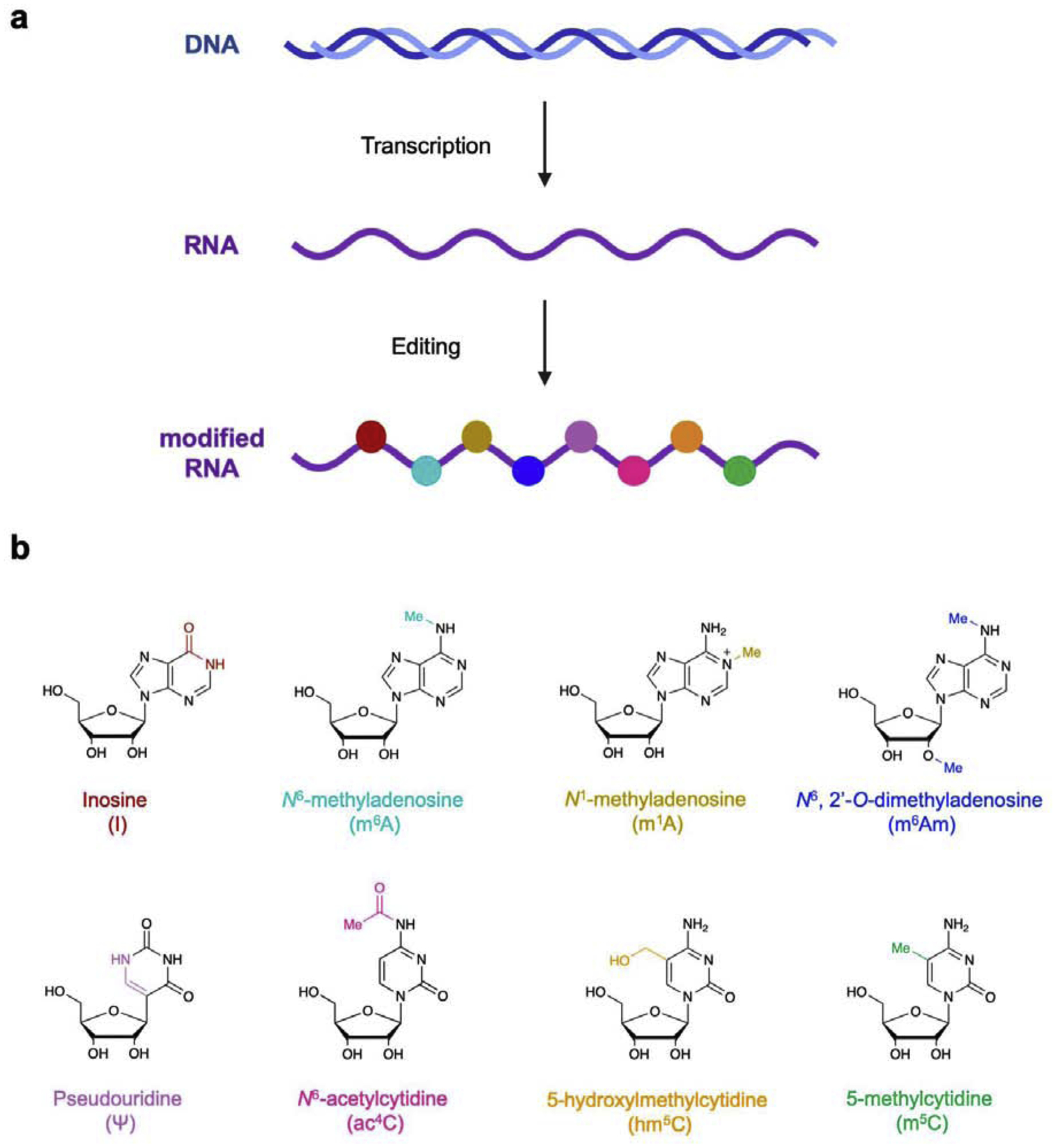
a) General schematic of RNA editing as a post-transcriptional processing step. b) Selected chemical structures of several known modified ribonucleosides, with key molecular changes highlighted in color. Reproduced with permission.5
Adenosine-to-Inosine (A-to-I) RNA Editing
Inosine in RNA is formed through a deamination mechanism (Fig. 2a), and ADAR1 and ADAR2 are the primary enzymes responsible for catalyzing A-to-I editing in humans.20–21 Although a third enzyme, ADAR3, has also been identified and is highly expressed in the brain, this isoform lacks catalytic editing activity and may play indirect roles in regulating A-to-I editing levels.22–23 To date, a significant number of A-to-I sites have been catalogued in the human transcriptome (>16 million),24–25 and several apparent functional roles have emerged. Because inosine base pairs with cytosine and thus results in a functional A→G transition (Fig. 2b), these alterations can directly modify codon sequences within mRNAs and subsequently impart amino acid substitutions in proteins. mRNA editing is especially prevalent in the brain,26–29 where several ion channel receptor mRNAs undergo precise A-to-I modification, which creates structural changes in the resulting proteins to adjust ion permeability and membrane potential.30–31 Although these sites impart significant functional change and there is great interest in leveraging A-to-I mRNA editing for therapeutic protein recoding,32–36 the vast majority of editing actually occurs in repetitive, non-coding RNA sequences.24–25 ADAR enzymes have strong affinity for long RNA duplexes,20 and widespread Alu elements throughout the transcriptome form long inverted repeats which are extensively edited.37–38 Unmodified dsRNA within the cytosol is also typically indicative of viral infection and is quickly recognized by a suite of sensor proteins that activate the innate immune system.39 Alu editing accounts for millions of A-to-I sites,24–25 and their ubiquitous presence is now thought to help mark host RNA as “self” and regulate these inflammatory pathways.20–21, 40–42 Together, A-to-I RNA editing plays crucial roles in cellular development, metabolism, and overall physiology, and dysregulation of this process is associated with neurological disorders, immune disease, and several cancer types.29, 43–45
Figure 2. Adenosine-to-Inosine RNA editing and current protein-based analytical platforms.
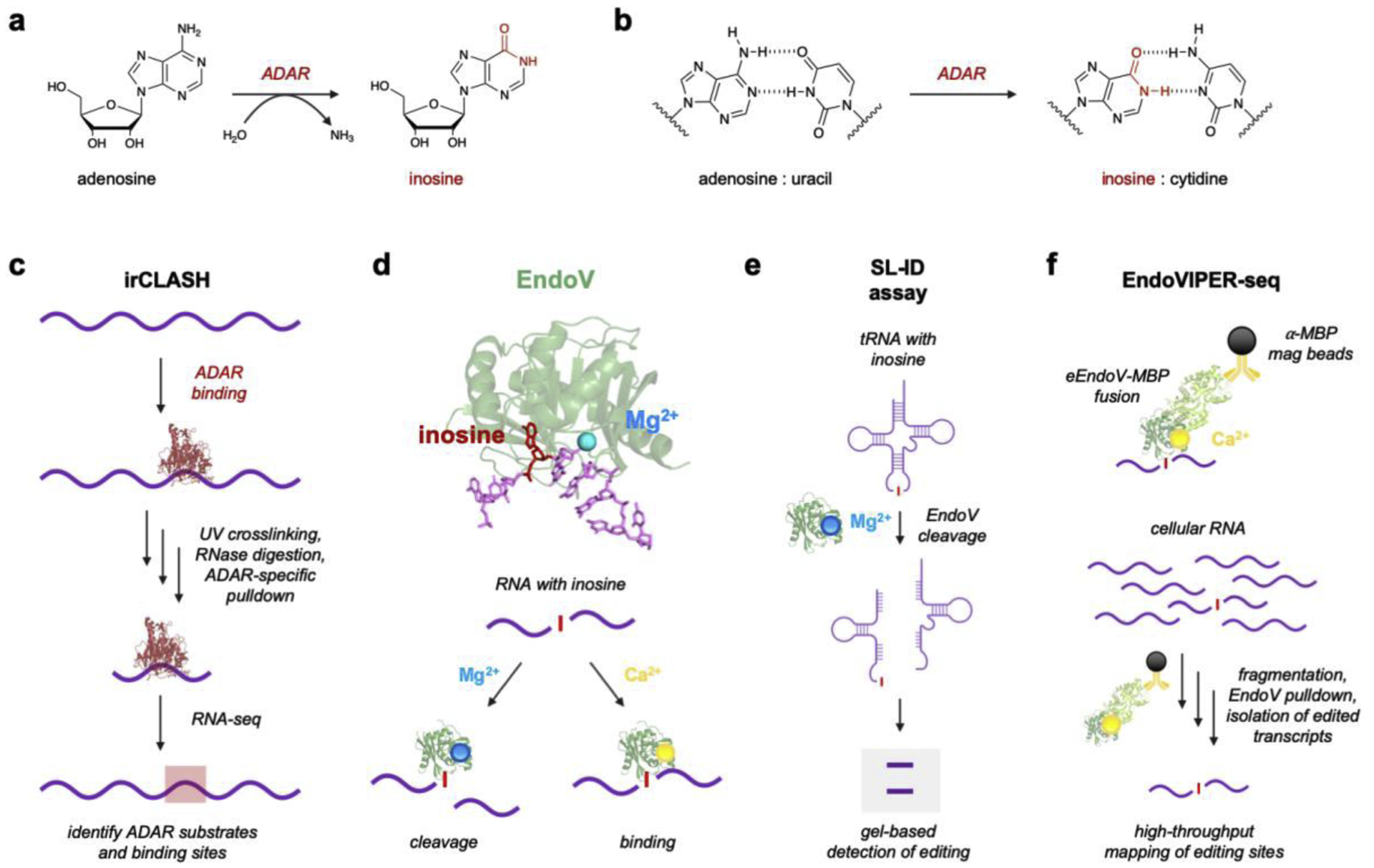
a) A-to-I RNA editing is catalyzed by ADARs via a deamination reaction. b) Resulting inosines base pair with cytidine rather than uracil. c) irCLASH enables large-scale identification of RNA binding sites for different ADAR enzymes. RNA-protein complexes are crosslinked and unbound RNA is digested. Immunoprecipitation (IP) of ADAR-RNA complexes enables sequencing of bound RNA regions. d) Endonuclease V (EndoV) is an inosine-specific ribonuclease. The crystal structure of EndoV (green) from T. maritima (PDB 2W35) is shown complexed with both Mg2+ (blue) and a ssDNA (purple) containing inosine (red). EndoV activity can be modulated by supplementing the enzyme with either Mg2+ for cleavage or Ca2+ for RNA binding. Figure reproduced with permission.53 d) EndoV’s natural cleavage ability with Mg2+ can be used to detect inosine in tRNAs using splinted ligation-based inosine detection (SL-ID). f) EndoV can also be supplemented with Ca2+ and used to bind A-to-I edited transcripts, which was developed into EndoVIPER-seq (EndoV immunoprecipitation enrichment sequencing). Figure reproduced with permission.53
Because of this importance, there is great interest in developing technologies to robustly map A-to-I sites and determine their biological significance. Although biochemical characterization of ADAR enzymes has provided insight into editing mechanisms, it remains unclear why some A’s are edited over others or how editing rates at individual sites are regulated. Moreover, several fundamental questions remain as to how editing is differentially regulated between different cell types and at varying stages of development. A powerful workflow to overcome these knowledge gaps was recently developed by Song and coworkers to map ADAR binding sites in RNA with both high resolution and throughput.46 Their method, termed irCLASH (infrared crosslinking, ligation, and sequencing of hybrids), identifies ADAR binding sites by crosslinking the enzymes to RNA and then isolating and sequencing these regions (Fig. 2c). Interestingly, this revealed that ADARs bind ~50 bp footprints in tandem pairs on long RNA duplexes, corroborating earlier notions that ADAR dimerization is critical for activity.47–48 Because irCLASH can isolate specific ADAR isoforms, this method also elucidated key binding differences for both ADAR1 and ADAR2, revealing unique signatures for both protein-recoding (predominantly ADAR2) and Alu-type (ADAR1) editing sites. This study was also notable for mapping ADAR3 binding sites, which have been notoriously difficult to characterize because the enzyme does not introduce detectable editing sites in RNA. Interestingly, irCLASH datasets demonstrated that ADAR3 binding sites significantly overlapped with ADAR2 regions, suggesting it may compete with ADAR2 for these editing substrates to negatively regulate overall editing levels.46
A fortunate advantage in studying A-to-I editing is that the introduced base pairing change is easily detected by high-throughput RNA-sequencing (RNA-seq) at single nucleotide resolution (Fig. 2b). However, despite tens of millions of potential editing sites, these events are quite rare in the context of total cellular RNA (~0.01 − <0.0001% of all nucleotides),24–25 and it has been technically challenging to comprehensively map these events using RNA-seq. Our lab as well as other groups have explored inosine chemical labeling as a means to avoid this limitation altogether,49–52 and while acrylonitrile or acrylamide derivatives are feasible for labeling simple RNA substrates, these reagents also display off-target labeling with other nucleobases and are generally intractable for use with complex RNA samples. While antibodies have proven to be effective reagents for recognition of a variety of modified RNA nucleotides, to date there are no inosine-specific antibodies available. Thus, our lab and others have looked to Nature for a solution to this molecular recognition challenge, harnessing the exquisite ability of Endonuclease V (EndoV) to recognize and bind inosine-containing RNAs.53–56 EndoV is present in almost all organisms and appears to have originally evolved in prokaryotes to cleave and repair inosine lesions in DNA arising from spontaneous oxidative damage.57 However, these prokaryotic EndoV isoforms still recognize inosine in both DNA and RNA, and this property was recently utilized for detecting A-to-I editing in transfer RNAs (tRNAs).56 In particular, the A34 position in the anticodon loop of tRNAs in prokaryotes (tRNAArgACG) and eukaryotes (tRNAArgACG, tRNAAlaAGC, tRNAProAGG, tRNAThrAGU, tRNAValAAC, tRNASerAGA, tRNALeuAAG, and tRNAIleAAU) is converted to inosine, in turn facilitating wobble base pairing and correct translation.58 While reverse transcription and Sanger sequencing are feasible for detecting this modification, these assays are especially difficult with tRNAs, as they are small, highly structured, and extensively modified. Using a prokaryotic EndoV homolog from Thermotoga maritima, Torres and coworkers overcame these limitations and developed the splinted ligation-based inosine detection (SL-ID) assay, which represented a major advance for directly measuring A-to-I editing status in the tRNA repertoire. This report validated SL-ID in several classes of tRNA, demonstrated assay robustness and selectivity in total RNA from different species, and further resolved prior ambiguity by confirming that tRNAAlaAGC indeed undergoes A-to-I editing in higher mammals and is critical for cellular function.
Although EndoV appears to naturally utilize Mg2+ for RNA cleavage,53–54 supplementing the enzyme with Ca2+ facilitates inosine recognition and binding, but does not support cleavage (Fig. 2d).59 Because of the relative scarcity of A-to-I edited transcripts in cellular RNA, we recently leveraged this property to enrich inosine-containing RNAs prior to sequencing.55 Using EndoV from E. coli (eEndoV), we first tested binding activity towards short RNA strands in the presence of Ca2+, and demonstrated that the enzyme has low nanomolar affinity for inosine in RNA.55 Because the recombinant eEndoV is commercially available as a fusion to a maltose-binding protein (MBP) affinity tag, we were then able to capture and isolate edited transcripts using magnetic beads functionalized with anti-MBP antibody (Fig. 2f). We termed our method EndoVIPER (Endonuclease V immunoprecipitation enrichment) and demonstrated its ability to selectively enrich A-to-I edited transcripts from cellular RNA, roughly doubling the amount of newly identified editing sites compared to standard RNA-seq. Given these promising results, ongoing work in our group aims to explore other EndoV isoforms for enrichment, as well as deploying the enzyme in other immunodetection assays for measuring A-to-I prevalence and specific signatures. Despite its utility as a detection platform, the biological functions of EndoV in humans remain mysterious. Several groups independently demonstrated that human EndoV is an inosine-specific ribonuclease and cleaves A-to-I edited RNAs in vitro,53–54 but recapitulating this activity in vivo has yielded confounding results in that enzyme levels do not correlate with overall inosine abundance in RNA.60 It is likely that EndoV activity is regulated or suppressed in some way, and RNA structure may play a key role in determining which transcripts are degraded by EndoV. Although it is easy to modulate EndoV activity in vitro by changing Ca2+ and Mg2+ concentrations in different buffers, it is also interesting to speculate whether ion switching is occurring in the cytosol to control EndoV activity. In any case, EndoV has emerged as a powerful molecular recognition platform for characterizing A-to-I editing, and determining its natural biological functions in humans is an area of high interest in the RNA editing field, as it is likely to yield critical information for understanding epitranscriptomics and its relationship to RNA metabolism.
It is increasingly clear that A-to-I editing is a crucial RNA modification. While the vast majority of these sites occur in repetitive regions, a small number of protein-recoding events produce significant biological consequences.24–25 Despite their importance, limitations of existing methods continue to make the detection these rare sites problematic. It is our opinion that many of these events remain undiscovered, and that the use of techniques that improve the efficiency and sensitivity of A-to-I site identification, including EndoVIPER55 or microfluidics-based multiplex PCR sequencing,61 will likely improve our ability to identify new candidate sites. Surprisingly, there have also been very few studies focusing specifically on A-to-I editing in small non-coding RNAs, including microRNAs and small interfering RNAs, which have been demonstrated to both undergo editing as well as influence overall cellular behavior.21, 62 We similarly predict that high-resolution protein-based detection platforms will also identify crucial editing sites in these transcript classes. Although not as impactful on an individual basis, repetitive Alu editing sites across the transcriptome also collectively contribute to overall tissue development and immune system regulation.20–21, 40–42 While potential functional roles have been hypothesized for the distinct global editing patterns observed between different cell and tissue types, the biological significance of these differences remain somewhat mysterious. ADAR expression is also poorly correlated with overall inosine content in cellular RNA,25 and there is great interest in identifying mechanisms that regulate both ADAR activity and overall editing levels. Sampling a large number of biological contexts will likely reveal additional editing patterns and hint at context-specific functions and regulatory mechanisms, but the present requirement for lengthy and high-cost RNA-seq limits our ability to efficiently ex-plore these settings. Similarly, transcriptome-level A-to-I signatures are also emerging as a key biomarker for several diseases,29, 43–45 but there are few methods available that can cheaply and rapidly detect these changes. Protein-based platforms that do not rely on sequencing are well poised for addressing both of these major pursuits, enabling facile characterization of global editing changes as well as rapid diagnostic detection of disease-relevant epitranscriptomic signatures.
N6-methyladenosine (m6A)
The most widespread and abundant RNA modification in humans is N6-methyladenosine (m6A),7, 10 which is introduced by a large protein complex that includes methyltransferases METTL3 and METTL14 (Fig. 3a). m6A is typically installed within “DRACH” consensus sequence motifs (D=G/A/U, R=G/A, H=A/U/C), and is highly enriched at stop codons and 3’ untranslated regions in mRNAs.9 Accumulating evidence suggests that m6A enhances stability and translation of edited transcripts, which is facilitated by a suite of “reader” proteins that contain a YT521-B homology (YTH) domain.10, 63 m6A also appears to play a critical role in the cellular stress response, and YTH readers preferentially bind and sequester methylated RNAs into liquid-like, “membrane-less” granules to protect transcripts from environmental insults as well as silence their activity.12, 64 In contrast to A-to-I editing, m6A is also reversible by demethylase enzymes FTO and ALKBH5 (Fig. 3a),65–66 suggesting this modification enables fine-tuning of mRNA activity within the cell. Unsurprisingly, m6A is quickly emerging as a critical and dynamic modification for a variety of cellular processes, including metabolism,3, 67 stem cell differentiation,68 and immune responses.69
Figure 3. Current protein-based platforms for detecting and profiling N6-methyladenosine (m6A) in RNA.
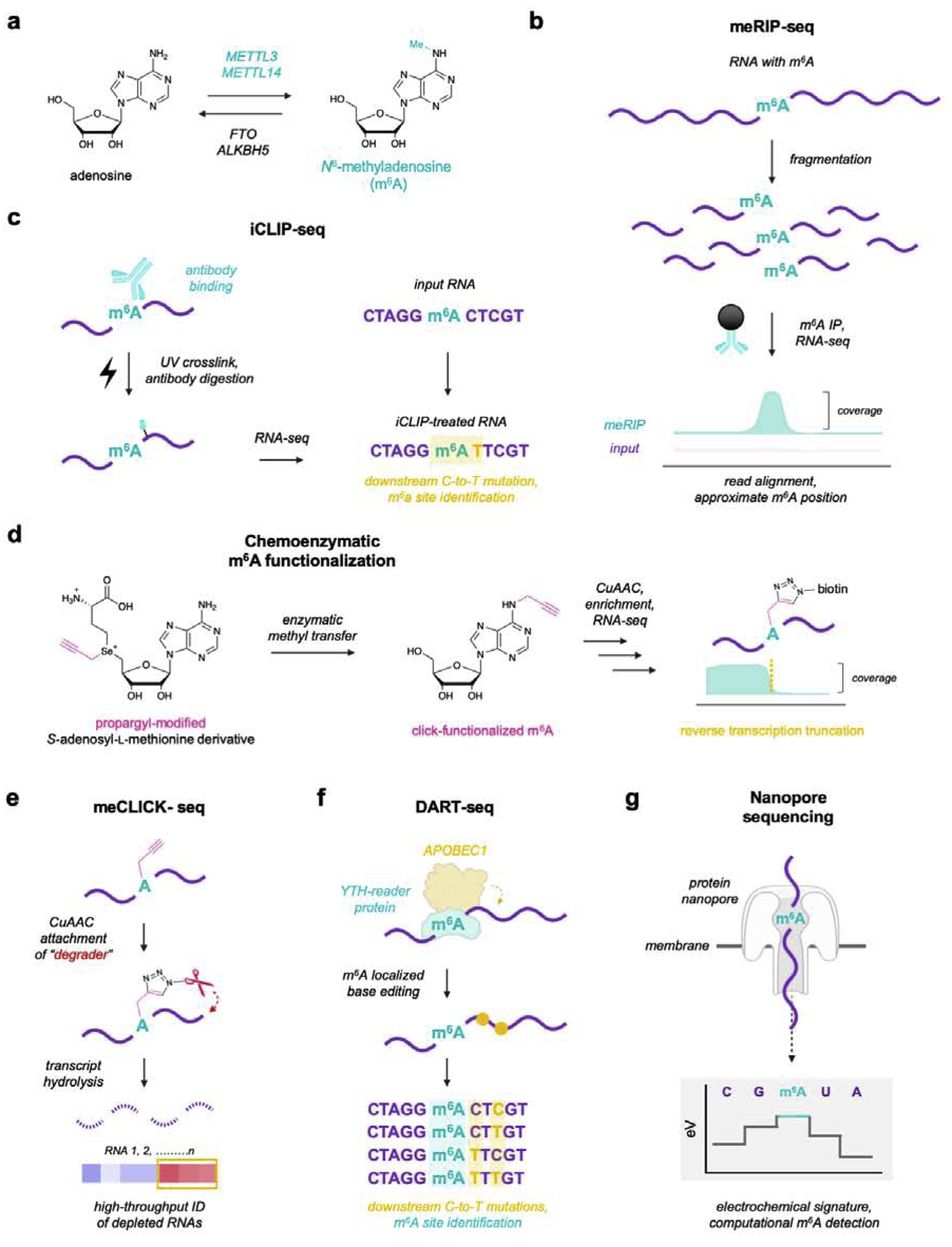
a) m6A is a reversible modification in RNA, installed by methyltransferases METTL3 and METTL14 and removed by demethylases FTO and ALKBH5. b) One of the most common methods for detecting m6A relies on meRIP-seq, in which cellular RNA is sheared into 100–200 nt fragments and immunoprecipitated with an m6A-specific antibody. Compiled reads from both input and meRIP-enriched RNA can be used to identify and approximate m6A locations. c) To achieve single-nucleotide resolution, the iCLIP workflow uses a UV-crosslinking and digestion step to produce a detectable mutation downstream of m6A sites. d) METTL3 and METTL14 utilize S-adenosyl-L-methionine, which can be derivatized with an alkyne handle to click-functionalize m6A sites and enable both enrichment and detection. e) meClick-seq leverages alkyne-functionalized m6A to attach a chemical degrader, allowing for monitoring of depletion to identify edited transcripts. f) DART-seq utilizes a YTH-APOBEC fusion protein to add C-to-U(T) edits near m6A sites and facilitate detection. g) RNA modifications, including m6A, can be directly detected in Nanopore sequencing by measuring small electrochemical changes arising from differential channel blockage by nucleobases.
In contrast to A-to-I editing, the m6A modification does not introduce a base pairing change or a detectable sequencing signature, and has thus proven highly difficult to identify in RNA. Isolation of monoclonal antibodies specific for m6A provided a crucial enabling technology for mapping modification sites in RNA. Termed “meRIP-seq” (m6A-specific methylated RNA immunoprecipitation sequencing), this method first randomly hydrolyzes mRNA into ~100–200 nt fragments, which are then enriched using anti-m6A antibody-functionalized beads. By comparing input (non-enriched) and meRIP-seq samples, m6A positions can be approximated by identifying coverage peaks in sequencing tracks (Fig. 3b). While meRIP-seq enabled the first large-scale studies of m6A prevalence in the human transcriptome and identified >10,000 putative editing sites across thousands of transcripts,8–9 this technique is also significantly limited in both accuracy and resolution, and can only estimate m6A locations within a 100–200 nt window. To overcome this, Linder and colleagues developed iCLIP-seq, wherein they UV-crosslinked and digested the bound antibody to introduce a covalent adduct on the RNA molecule and produce a C-to-T mutational signature one nucleotide downstream of each m6A site (Fig. 3c).70 This provided greater accuracy and single-nucleotide resolution detection of m6A sites, in turn facilitating better identification of the m6A consensus sequences.
Although these immunodetection techniques have been widely adopted for mapping m6A locations, there is growing controversy surrounding the accuracy and reproducibility of these assays. In particular, evidence suggests that these antibodies recognize multiple types of RNA methylation (such as m6Am),7 and there is considerable performance variability between antibodies acquired from different vendors.71 Concerningly, a large-scale meta-analysis also found that few published meRIP-seq assays were performed using more than one replicate measurement, and datasets between different labs often exhibited poor overlap, even when using the same cell lines and RNA material.72
To avoid these problems, several antibody-independent assays for mapping m6A sites have been developed. In particular, several groups have designed reverse transcription-based assays to detect RNA methylation at single nucleotide resolution. In these assays, natural or engineered polymerases are used to synthesize complementary DNA from a transcript of interest, producing detectable mutations at methylated sites. These techniques have been used to great benefit for detecting m6A as well as m1A in RNA,73–75 producing high-resolution editing maps and unprecedented scale. However, these enzymes can also exhibit low or unpredictable mutation frequencies, often necessitating extensive optimization or directed evolution strategies for practical implementation. Alternatively, the Rentmeister group has pioneered a distinct chemoenzymatic detection approach, wherein m6A sites can be functionalized with a click chemistry-compatible alkyne handle (Fig. 3d). Because methyltransferases rely on S-adenosyl-L-methionine (SAM) as a donor for m6A formation, the group demonstrated that an alkyne-functionalized, seleno-SAM derivative enabled selective installation of these handles at putative m6A sites.76 This approach is elegant in that the introduced alkyne handle enables enrichment of edited transcripts using an azide-modified biotin, and this large moiety effectively terminates reverse transcription at these sites, enabling precise detection of m6A positions with single nucleotide resolution. Excitingly, these SAM derivatives are well tolerated by cells and can be derivatized to induce chemoenzymatic installation of azides, alkyl chains, and photoprotecting groups,76–77 enabling exquisite detection and control over m6A-modified transcripts in cellulo. Inspired by this approach, Mikutis and coworkers demonstrated that these alkyne-tagged m6As could also be targeted with an azide-functionalized chemical “degrader,” enabling m6A-modified RNAs to be detected at high-throughput by monitoring transcript depletion (Fig. 3e).78 This method proved especially valuable for low abundance targets that do not always enrich well in traditional workflows, and enabled the discovery that intronic polyadenylation sites are a key methylation event important for mRNA splicing. Another bio-inspired technology developed by the Meyer group, termed deamination adjacent to RNA modification targets sequencing (DART-seq), takes advantage of natural proteins in two clever ways - 1) a YTH domain that specifically recognizes m6A in RNA is fused to 2) an APOBEC1 cytidine deaminase enzyme that, similarly to ADAR, introduces a detectable mutation signature downstream to a candidate m6A site (Fig. 3f).79 While simple, this platform avoids many of the pitfalls associated with antibody-based assays, including compatibility with very low amounts of input RNA (<10 ng) and the ability to detect dynamic m6A changes over time. Importantly, because it does not require fragmentation, this approach can also be used with long-read sequencing to detect m6A distributions in single mRNA molecules.
All of the previously discussed approaches, both for A-to-I and m6A detection, typically utilize second-generation Illumina sequencing technologies for downstream analysis. While these platforms are highly accurate and offer single nucleotide resolution, they also rely on reverse transcription and PCR amplification prior to sequencing and thus functionally erase modifications such as m6A that do not introduce base pairing changes. As highlighted earlier, this has spurred the development of approaches to induce these signatures, and while innovative, they are also inherently indirect and subject to errors, preventing facile and comprehensive epitranscriptomic profiling. An interesting alternative to these shortcomings is nanopore sequencing, which can directly detect nucleic acid sequences and modifications by threading the strand through a protein nanopore and measuring small electrochemical current changes generated from differential ion blockage by each passing nucleobase.80 Recognizing the power of this technology, work led by Novoa utilized a machine-learning approach to develop a computational algorithm that can recognize the minute differences between A and m6A and precisely detect the location of the modified nucleotide in a native RNA molecule (Fig. 3f).81 Unfortunately, nanopore sequencing is still limited in fidelity, and this platform typically requires a combination of computational filtering and highly repetitive sequencing of the same strand in order to produce accurate reads. Despite current performance limitations, it is our opinion that these technical challenges will be overcome in the near future, and we predict that nanopore sequencing has potential to become the predominant method-of-choice for directly detecting and characterizing RNA modifications.
Conclusions
RNA modification represents a critical layer of control for regulating transcriptional activity and tuning overall cellular function. A large number of modifications (>100) have now been discovered in RNA, the majority of which are completely unmapped and have no obvious biological functions. Central to addressing these knowledge gaps are the development of robust technologies to detect the location and prevalence of these modifications throughout the transcriptome and measure changes in their abundance in different biological settings. Moreover, the ability to elucidate changes in modification patters in various disease states could lead to the development of powerful new diagnostic tools. Two of the most abundant and impactful RNA modifications, A-to-I and N6-methyladenosine RNA editing, have become much more well understood through the development and use of enzyme- and antibody-based assays for high-throughput analysis and detection. In particular, these platforms utilize bio-inspired approaches to leverage the high-performance molecular recognition properties of natural enzymes and reader proteins, and together have enabled substantial increases in our understanding of these epitranscriptomic marks. However, significant effort is still needed to refine and improve these assays as well as deploy them toward other RNA modifications. Additionally, utilizing these platforms in a variety of biological settings is anticipated to contribute to a better understand human cellular physiology and its relationship with disease mechanisms.
Supplementary Material
Acknowledgements
Work described in this review was supported by the National Institutes of Health (R01GM116991 and R21 GM134564-01 to J.M.H.). This study was also in part supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Licatalosi DD; Darnell RB, RNA processing and its regulation: global insights into biological networks. Nature Reviews Genetics 2010, 11 (1), 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shafik A; Schumann U; Evers M; Sibbritt T; Preiss T, The emerging epitranscriptomics of long noncoding RNAs. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms 2016, 1859 (1), 59–70. [DOI] [PubMed] [Google Scholar]
- 3.Peer E; Rechavi G; Dominissini D, Epitranscriptomics: regulation of mRNA metabolism through modifications. Current opinion in chemical biology 2017, 41, 93–98. [DOI] [PubMed] [Google Scholar]
- 4.Grozhik AV; Jaffrey SR, Epitranscriptomics: Shrinking maps of RNA modifications. Nature 2017, 551 (7679), 174–177. [DOI] [PubMed] [Google Scholar]
- 5.Li X; Xiong X; Yi C, Epitranscriptome sequencing technologies: decoding RNA modifications. Nature methods 2017, 14 (1), 23–31. [DOI] [PubMed] [Google Scholar]
- 6.Shi H; Wei J; He C, Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Molecular cell 2019, 74 (4), 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li K; Cai J; Zhang M; Zhang X; Xiong X; Meng H; Xu X; Huang Z; Peng J; Fan J, Landscape and regulation of m6A and m6Am methylome across human and mouse tissues. Molecular cell 2020, 77 (2), 426–440. e6. [DOI] [PubMed] [Google Scholar]
- 8.**.Dominissini D; Moshitch-Moshkovitz S; Schwartz S; Salmon-Divon M; Ungar L; Osenberg S; Cesarkas K; Jacob-Hirsch J; Amariglio N; Kupiec M, Topology of the human and mouse m 6 A RNA methylomes revealed by m 6 A-seq. Nature 2012, 485 (7397), 201. [DOI] [PubMed] [Google Scholar]
- 9.**.Meyer KD; Saletore Y; Zumbo P; Elemento O; Mason CE; Jaffrey SR, Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149 (7), 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]; Together, these two studies (refs 8 and 9) represent the first uses of m6A-targeting antibodies for large-scale meRIP-seq identifcation of methylation sites in the human transcriptome.
- 10.Mao Y; Dong L; Liu X-M; Guo J; Ma H; Shen B; Qian S-B, m 6 A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nature communications 2019, 10 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arguello AE; DeLiberto AN; Kleiner RE, RNA chemical proteomics reveals the N6-methyladenosine (m6A)-regulated protein–RNA interactome. Journal of the American Chemical Society 2017, 139 (48), 17249–17252. [DOI] [PubMed] [Google Scholar]
- 12.Fu Y; Zhuang X, m 6 A-binding YTHDF proteins promote stress granule formation. Nature Chemical Biology 2020, 16, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X; Ma S; Yi C, Pseudouridine: the fifth RNA nucleotide with renewed interests. Current Opinion in Chemical Biology 2016, 33, 108–116. [DOI] [PubMed] [Google Scholar]
- 14.Carlile TM; Rojas-Duran MF; Zinshteyn B; Shin H; Bartoli KM; Gilbert WV, Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515 (7525), 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlile TM; Martinez NM; Schaening C; Su A; Bell TA; Zinshteyn B; Gilbert WV, mRNA structure determines modification by pseudouridine synthase 1. Nature chemical biology 2019, 15 (10), 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin G; Xu M; Zou M; Duan S, The Processing, Gene Regulation, Biological Functions, and Clinical Relevance of N4-Acetylcytidine on RNA: A Systematic Review. Molecular Therapy-Nucleic Acids 2020, 20, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao W; Zhou Y; Cui Q; Zhou Y, PACES: prediction of N4-acetylcytidine (ac4C) modification sites in mRNA. Scientific reports 2019, 9 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delatte B; Wang F; Ngoc LV; Collignon E; Bonvin E; Deplus R; Calonne E; Hassabi B; Putmans P; Awe S, Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 2016, 351 (6270), 282–285. [DOI] [PubMed] [Google Scholar]
- 19.Squires JE; Preiss T, Function and detection of 5-methylcytosine in eukaryotic RNA. Epigenomics 2010, 2 (5), 709–715. [DOI] [PubMed] [Google Scholar]
- 20.Bass BL, RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 2002, 71, 817–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishikura K, A-to-I editing of coding and non-coding RNAs by ADARs. Nature reviews Molecular cell biology 2016, 17 (2), 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C-X; CHO D-SC; WANG Q; LAI F; CARTER KC; NISHIKURA K, A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6 (5), 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oakes E; Anderson A; Cohen-Gadol A; Hundley HA, Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. Journal of Biological Chemistry 2017, 292 (10), 4326–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansi L; Tangaro MA; Lo Giudice C; Flati T; Kopel E; Schaffer AA; Castrignanò T; Chillemi G; Pesole G; Picardi E, REDIportal: millions of novel A-to-I RNA editing events from thousands of RNAseq experiments. Nucleic Acids Research 2020, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan MH; Li Q; Shanmugam R; Piskol R; Kohler J; Young AN; Liu KI; Zhang R; Ramaswami G; Ariyoshi K, Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550 (7675), 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li JB; Church GM, Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nature neuroscience 2013, 16 (11), 1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakurai M; Ueda H; Yano T; Okada S; Terajima H; Mitsuyama T; Toyoda A; Fujiyama A; Kawabata H; Suzuki T, A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome research 2014, 24 (3), 522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Behm M; Öhman M, RNA editing: a contributor to neuronal dynamics in the mammalian brain. Trends in Genetics 2016, 32 (3), 165–175. [DOI] [PubMed] [Google Scholar]
- 29.Hwang T; Park C-K; Leung AK; Gao Y; Hyde TM; Kleinman JE; Rajpurohit A; Tao R; Shin JH; Weinberger DR, Dynamic regulation of RNA editing in human brain development and disease. Nature neuroscience 2016, 19 (8), 1093. [DOI] [PubMed] [Google Scholar]
- 30.Wright AL; Vissel B, The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Frontiers in molecular neuroscience 2012, 5, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sommer B; Köhler M; Sprengel R; Seeburg PH, RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67 (1), 11–19. [DOI] [PubMed] [Google Scholar]
- 32.Montiel-Gonzalez MF; Quiroz JFD; Rosenthal JJ, Current strategies for site-directed RNA editing using ADARs. Methods 2019, 156, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marina RJ; Brannan KW; Dong KD; Yee BA; Yeo GW, Evaluation of Engineered CRISPR-Cas-Mediated Systems for Site-Specific RNA Editing. Cell Reports 2020, 33 (5), 108350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merkle T; Merz S; Reautschnig P; Blaha A; Li Q; Vogel P; Wettengel J; Li JB; Stafforst T, Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nature biotechnology 2019, 37 (2), 133–138. [DOI] [PubMed] [Google Scholar]
- 35.Rauch S; He E; Srienc M; Zhou H; Zhang Z; Dickinson BC, Programmable RNA-guided RNA effector proteins built from human parts. Cell 2019, 178 (1), 122–134. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qu L; Yi Z; Zhu S; Wang C; Cao Z; Zhou Z; Yuan P; Yu Y; Tian F; Liu Z, Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nature biotechnology 2019, 37 (9), 1059–1069. [DOI] [PubMed] [Google Scholar]
- 37.Kim DD; Kim TT; Walsh T; Kobayashi Y; Matise TC; Buyske S; Gabriel A, Widespread RNA editing of embedded alu elements in the human transcriptome. Genome research 2004, 14 (9), 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roth SH; Levanon EY; Eisenberg E, Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nature methods 2019, 1–8. [DOI] [PubMed] [Google Scholar]
- 39.Ruan J; Cao Y; Ling T; Li P; Wu S; Peng D; Wang Y; Jia X; Chen S; Xu A, DDX23, an evolutionary conserved dsRNA sensor, participates in innate antiviral responses by pairing with TRIF or MAVS. Frontiers in immunology 2019, 10, 2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mannion NM; Greenwood SM; Young R; Cox S; Brindle J; Read D; Nellåker C; Vesely C; Ponting CP; McLaughlin PJ, The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell reports 2014, 9 (4), 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishizuka JJ; Manguso RT; Cheruiyot CK; Bi K; Panda A; Iracheta-Vellve A; Miller BC; Du PP; Yates KB; Dubrot J, Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2018, 565 (7737), 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung H; Calis JJ; Wu X; Sun T; Yu Y; Sarbanes SL; Thi VLD; Shilvock AR; Hoffmann H-H; Rosenberg BR, Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 2018, 172 (4), 811–824. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maas S; Kawahara Y; Tamburro KM; Nishikura K, A-to-I RNA editing and human disease. RNA biology 2006, 3 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slotkin W; Nishikura K, Adenosine-to-inosine RNA editing and human disease. Genome medicine 2013, 5 (11), 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han L; Diao L; Yu S; Xu X; Li J; Zhang R; Yang Y; Werner HMJ; Eterovic AK; Yuan Y; Li J; Nair N; Minelli R; Tsang YH; Cheung LWT; Jeong KJ; Roszik J; Ju Z; Woodman SE; Lu Y; Scott KL; Li JB; Mills GB; Liang H, The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 2015, 28 (4), 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.**.Song Y; Yang W; Fu Q; Wu L; Zhao X; Zhang Y; Zhang R, irCLASH reveals RNA substrates recognized by human ADARs. Nature Structural & Molecular Biology 2020, 27 (4), 351–362. [DOI] [PubMed] [Google Scholar]; An influential report that developed a new type of technology for mapping ADAR binding sites in human RNA.
- 47.Cho D-SC; Yang W; Lee JT; Shiekhattar R; Murray JM; Nishikura K, Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. Journal of Biological Chemistry 2003, 278 (19), 17093–17102. [DOI] [PubMed] [Google Scholar]
- 48.Thuy-Boun AS; Thomas JM; Grajo HL; Palumbo CM; Park S; Nguyen LT; Fisher AJ; Beal PA, Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic acids research 2020, 48 (14), 7958–7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knutson SD; Ayele TM; Heemstra JM, Chemical Labeling and Affinity Capture of Inosine-Containing RNAs Using Acrylamidofluorescein. Bioconjugate chemistry 2018, 29 (9), 2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knutson SD; Korn MM; Johnson RP; Monteleone LR; Dailey DM; Swenson CS; Beal PA; Heemstra JM, Chemical Profiling of A-to-I RNA Editing Using a Click-Compatible Phenylacrylamide. Chemistry-A European Journal 2020, 26 (44), 9874–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sakurai M; Yano T; Kawabata H; Ueda H; Suzuki T, Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol 2010, 6 (10), 733–40. [DOI] [PubMed] [Google Scholar]
- 52.Li Y; Göhl M; Ke K; Vanderwal CD; Spitale RC, Identification of Adenosine-to-Inosine RNA Editing with Acrylonitrile Reagents. Organic letters 2019, 21 (19), 7948–7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morita Y; Shibutani T; Nakanishi N; Nishikura K; Iwai S; Kuraoka I, Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nature communications 2013, 4, 2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vik ES; Nawaz MS; Andersen PS; Fladeby C; Bjørås M; Dalhus B; Alseth I, Endonuclease V cleaves at inosines in RNA. Nature communications 2013, 4, 2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.**.Knutson SD; Arthur RA; Johnston HR; Heemstra JM, Selective enrichment of A-to-I edited transcripts from cellular RNA using Endonuclease V. Journal of the American Chemical Society 2020, 142 (11), 5241–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first use of an inosine-specific endonuclease for enriching A-to-I edited transcripts prior to RNA-seq.
- 56.Torres AG; Wulff TF; Rodriíguez-Escribaà, M.; Camacho, N.; Ribas de Pouplana, L. s., Detection of Inosine on transfer RNAs without a reverse transcription reaction. Biochemistry 2018, 57 (39), 5641–5647. [DOI] [PubMed] [Google Scholar]
- 57.Wu J; Samara NL; Kuraoka I; Yang W, Evolution of Inosine-Specific Endonuclease V from Bacterial DNase to Eukaryotic RNase. Molecular cell 2019, 76 (1), 44–56. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Torres AG; Piñeyro D; Filonava L; Stracker TH; Batlle E; de Pouplana LR, A-to-I editing on tRNAs: biochemical, biological and evolutionary implications. FEBS letters 2014, 588 (23), 4279–4286. [DOI] [PubMed] [Google Scholar]
- 59.Hitchcock TM; Gao H; Cao W, Cleavage of deoxyoxanosine-containing oligodeoxyribonucleotides by bacterial endonuclease V. Nucleic acids research 2004, 32 (13), 4071–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kong XY; Vik ES; Nawaz MS; Berges N; Dahl TB; Vågbø C; Suganthan R; Segers F; Holm S; Quiles-Jiménez A, Deletion of Endonuclease V suppresses chemically induced hepatocellular carcinoma. Nucleic acids research 2020, 48 (8), 4463–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang R; Li X; Ramaswami G; Smith KS; Turecki G; Montgomery SB; Li JB, Quantifying RNA allelic ratios by microfluidic multiplex PCR and sequencing. Nature methods 2014, 11 (1), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kawahara Y; Zinshteyn B; Sethupathy P; Iizasa H; Hatzigeorgiou AG; Nishikura K, Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315 (5815), 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao W; Adhikari S; Dahal U; Chen Y-S; Hao Y-J; Sun B-F; Sun H-Y; Li A; Ping X-L; Lai W-Y, Nuclear m6A reader YTHDC1 regulates mRNA splicing. Molecular cell 2016, 61 (4), 507–519. [DOI] [PubMed] [Google Scholar]
- 64.Gao Y; Pei G; Li D; Li R; Shao Y; Zhang QC; Li P, Multivalent m 6 A motifs promote phase separation of YTHDF proteins. Cell research 2019, 29 (9), 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao X; Yang Y; Sun B-F; Shi Y; Yang X; Xiao W; Hao Y-J; Ping X-L; Chen Y-S; Wang W-J, FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell research 2014, 24 (12), 1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng G; Dahl JA; Niu Y; Fedorcsak P; Huang C-M; Li CJ; Vågbø CB; Shi Y; Wang W-L; Song S-H, ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular cell 2013, 49 (1), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhong X; Yu J; Frazier K; Weng X; Li Y; Cham CM; Dolan K; Zhu X; Hubert N; Tao Y, Circadian clock regulation of hepatic lipid metabolism by modulation of m6A mRNA methylation. Cell reports 2018, 25 (7), 1816–1828. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Batista PJ; Molinie B; Wang J; Qu K; Zhang J; Li L; Bouley DM; Lujan E; Haddad B; Daneshvar K, m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell stem cell 2014, 15 (6), 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paramasivam A; Priyadharsini JV; Raghunandhakumar S, Implications of m6A modification in autoimmune disorders. Cellular & Molecular Immunology 2019, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Linder B; Grozhik AV; Olarerin-George AO; Meydan C; Mason CE; Jaffrey SR, Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature methods 2015, 12 (8), 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matsuzawa S; Wakata Y; Ebi F; Isobe M; Kurosawa N, Development and validation of monoclonal antibodies against N6-methyladenosine for the detection of RNA modifications. PloS one 2019, 14 (10), e0223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.*.McIntyre AB; Gokhale NS; Cerchietti L; Jaffrey SR; Horner SM; Mason CE, Limits in the detection of m 6 A changes using MeRIP/m 6 A-seq. Scientific reports 2020, 10 (1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]; A timely analysis and comparison of current m6A datasets, highlighting both the value and the potential drawbacks of these assays.
- 73.Harcourt EM; Ehrenschwender T; Batista PJ; Chang HY; Kool ET, Identification of a selective polymerase enables detection of N6-methyladenosine in RNA. Journal of the American Chemical Society 2013, 135 (51), 19079–19082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aschenbrenner J; Werner S; Marchand V; Adam M; Motorin Y; Helm M; Marx A, Engineering of a DNA polymerase for direct m6A sequencing. Angewandte Chemie International Edition 2018, 57 (2), 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou H; Rauch S; Dai Q; Cui X; Zhang Z; Nachtergaele S; Sepich C; He C; Dickinson BC, Evolution of a reverse transcriptase to map N 1-methyladenosine in human messenger RNA. Nature methods 2019, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.**.Hartstock K; Nilges BS; Ovcharenko A; Cornelissen NV; Püllen N; Lawrence-Dörner AM; Leidel SA; Rentmeister A, Enzymatic or in vivo installation of propargyl groups in combination with click chemistry for the enrichment and detection of methyltransferase target sites in RNA. Angewandte Chemie International Edition 2018, 57 (21), 6342–6346. [DOI] [PubMed] [Google Scholar]; The first major new assay developed after meRIP-seq that can detect m6A in RNA that does not rely on antibody binding or enrichment.
- 77.Ovcharenko A; Weissenboeck FP; Rentmeister A, Tag-free internal RNA labeling and photocaging based on mRNA methyltransferases. Angewandte Chemie International Edition 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lopes Bernardes G; Mikutis S, meCLICK-Seq-a substrate-hijacking and RNA degradation strategy for the study of RNA methylation. ACS Central Science 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.**.Meyer KD, DART-seq: an antibody-free method for global m 6 A detection. Nature methods 2019, 16, 1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]; A highly innovative new approach to mapping m6A sites by using both YTH reader proteins and a base-editing enzyme.
- 80.Garalde DR; Snell EA; Jachimowicz D; Sipos B; Lloyd JH; Bruce M; Pantic N; Admassu T; James P; Warland A, Highly parallel direct RNA sequencing on an array of nanopores. Nature methods 2018, 15 (3), 201. [DOI] [PubMed] [Google Scholar]
- 81.Liu H; Begik O; Lucas MC; Ramirez JM; Mason CE; Wiener D; Schwartz S; Mattick JS; Smith MA; Novoa EM, Accurate detection of m 6 A RNA modifications in native RNA sequences. Nature communications 2019, 10 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.