

Abstract
It is well established that diabetes and its associated hyperglycemia negatively impact retinal function, yet we know little about the role played by augmented flux through the hexosamine biosynthetic pathway (HBP). This offshoot of the glycolytic pathway produces UDP-N-acetyl-glucosamine, which serves as the substrate for post-translational O-linked modification of proteins in a process referred to as O-GlcNAcylation. HBP flux and subsequent protein O-GlcNAcylation serve as nutrient sensors, enabling cells to integrate metabolic information to appropriately modulate fundamental cellular processes including gene expression. Here we summarize the impact of diabetes on retinal physiology, highlighting recent studies that explore the role of O-GlcNAcylation-induced variation in mRNA translation in retinal dysfunction and the pathogenesis of diabetic retinopathy (DR). Augmented O-GlcNAcylation results in wide variation in the selection of mRNAs for translation, in part, due to O-GlcNAcylation of the translational repressor 4E-BP1. Recent studies demonstrate that 4E-BP1 plays a critical role in regulating O-GlcNAcylation-induced changes in the translation of the mRNAs encoding vascular endothelial growth factor (VEGF), a number of important mitochondrial proteins, and CD40, a key costimulatory molecule involved in diabetes-induced retinal inflammation. Remarkably, 4E-BP1/2 ablation delays the onset of diabetes-induced visual dysfunction in mice. Thus, pharmacological interventions to prevent the impact of O-GlcNAcylation on 4E-BP1 may represent promising therapeutics to address the development and progression of DR. In this regard, we discuss the potential interplay between retinal O-GlcNAcylation and the ocular renin-angiotensin system as a potential therapeutic target of future interventions.
Keywords: diabetes, retinopathy, O-GlcNAcylation, mRNA translation, renin-angiotensin system, oxidative stress, inflammation
1. DIABETIC RETINOPATHY
1.1. Introduction to Diabetes-induced Retinal Dysfunction
Diabetic retinopathy (DR) is the most frequent cause of new cases of blindness among adults aged 20–74 in developed countries. DR is a neurovascular complication that is observed in individuals with type 1 or type 2 diabetes. Almost everyone with type 1 diabetes, and most people with type 2 diabetes, develop some form of visual deficit within two decades of disease onset [1]. Furthermore, studies by multiple investigators have reported retinal defects [2, 3] and impaired visual function [4, 5] in pre-diabetic patients, as well as individuals with relatively normal glucose tolerances [6–8]. Despite decades of study, there remains a critical gap in our understanding of the initiating molecular events that lead to loss of retinal homeostasis and visual dysfunction in diabetes and prediabetes. As a result, there is a lack of effective therapeutic interventions to prevent the functional decline in vision that is observed in most diabetic individuals. Herein we review the impact of diabetes on retinal physiology, highlighting the role of O-GlcNAcylation-induced variation in mRNA translation in retinal dysfunction and the pathogenesis of DR. We also discuss the potential interplay between retinal O-GlcNAcylation and the ocular renin-angiotensin system as it was recently demonstrated that inhibition of the angiotensin-converting enzyme suppresses retinal protein O-GlcNAcylation via a signaling axis that is initiated by Ang1–7 and the Mas receptor.
1.2. Retinal Anatomy & Physiology
Located in the posterior segment of the eye, the retina is responsible for converting light into an electrochemical signal to relay an image to the brain. The retina contains ~50 distinct cell types arranged into three layers of nerve cell bodies interspersed by two plexiform layers of synapses [9]. In the classical model of phototransduction, photons of light are absorbed by photosensitive pigments in the outer segments of the rods and cones (Fig. 1). This leads to hyperpolarization of photoreceptors, and in turn, electrochemical stimulation of second-order neurons, including primarily bipolar and horizontal cells whose cell bodies reside in the inner nuclear layer (INL) [10, 11]. The signal is subsequently relayed to amacrine and retinal ganglion cells (RGCs) via either excitatory or inhibitory connections [12]. Axons of RGCs form the nerve fiber layer (NFL) that enters the optic nerve and ultimately synapses with the brain [13].
Fig. (1). Diagram of the retinal neurovasculature.
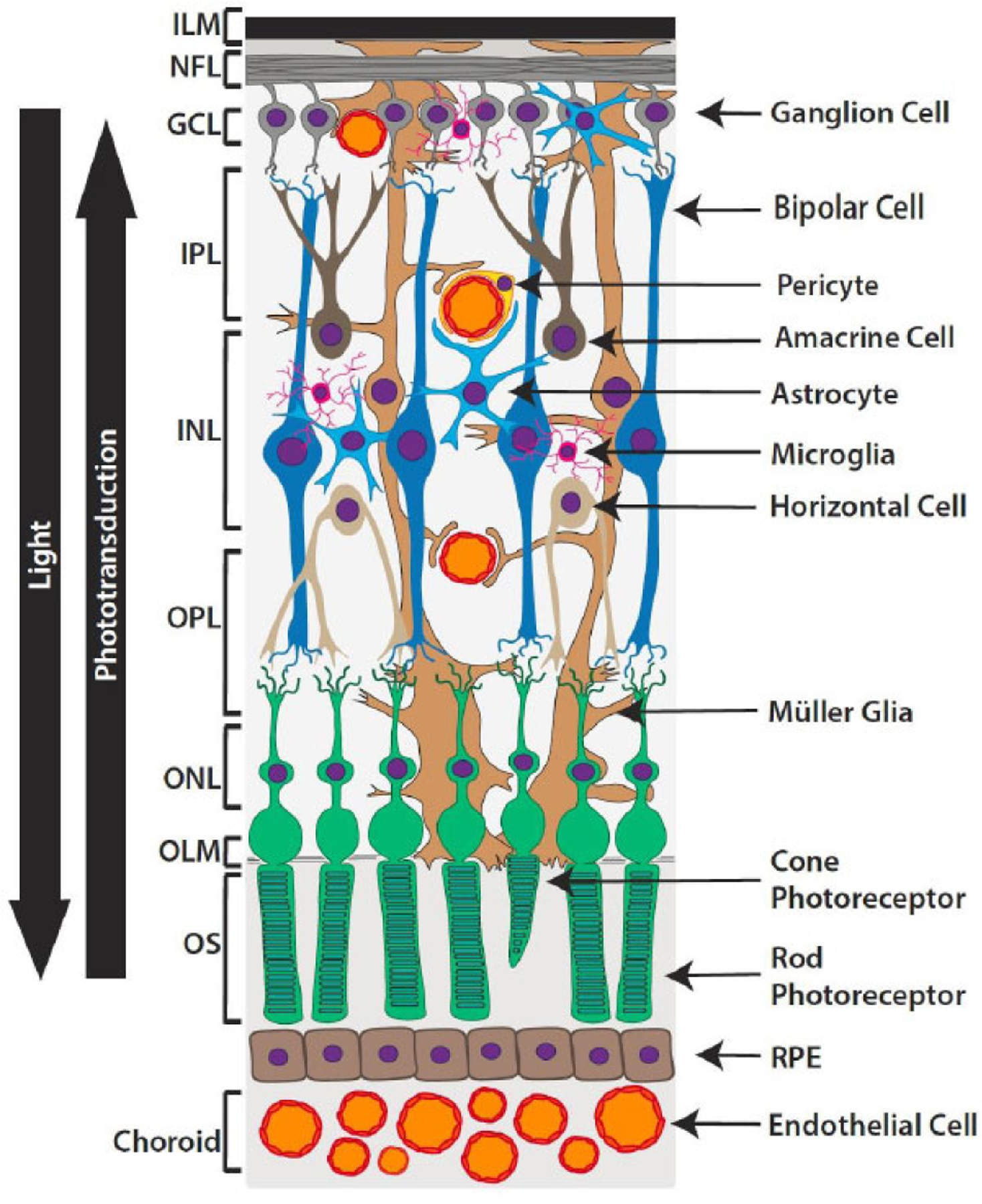
The retina contains a diverse array of cell types arranged into 9 neurosensory layers: 1. inner limiting membrane (ILM), 2. nerve fiber layer (NFL), 3. ganglion cell layer (GCL), 4. Inner plexiform layer (IPL), 5. inner nuclear layer (INL), 6. outer plexiform layer (OPL), 7. outer nuclear layer (ONL), 8. outer limiting membrane (OLM), 9. outer segments (OS) of photoreceptors. Situated just outside the neurosensory layers, a retinal pigmented epithelium (RPE) monolayer and underlying vasculature known as the choroid, provides oxygen and nourishment. Photons of light travel through the inner retinal layers and are absorbed by visual pigments in the outer segments of rods and cones. Phototransduction relays this input from the photoreceptors to bipolar and horizontal cells, with the signal subsequently being passed to amacrine and ganglion cells. The axon of a ganglion cell leaves the retina to ultimately transmit the message to the brain.
The retina is among the highest energy-consuming tissues, with sensory neurons depending almost entirely on oxidative metabolism [14]. Situated just outside the neurosensory layers, a retinal pigmented epithelium (RPE) monolayer and underlying vasculature known as the choroid provides oxygen and nutrients to the outer retina. Three layers of capillary networks are located in the NFL (superficial vascular plexus) and along both the inner and outer border of the INL (intermediate and deep capillary plexus, respectively). Stellate-shaped astrocytes are mostly confined to the NFL; however, the presence and distribution of astrocytes are also correlated with the distribution of retinal blood vessels [15, 16]. This correlation reflects the close association astrocytes have with endothelial cells of the retinal vasculature. Astrocytes exert supportive functions for the retinal vasculature: ensheathing blood vessels, producing trophic factors (including vascular endothelial growth factor (VEGF)), and maintaining the integrity of the blood retinal barrier (BR-B) [17, 18]. As an immune-privileged site, the retina relies on resident microglia to respond to inflammatory insults. Microglia represent the retinal macrophage population; they undergo rapid morphological and functional transformation into phagocytes upon activation by a stimulus. Additional roles for retinal microglial homeostasis include control of neuronal apoptosis, the guidance of retinal vasculature growth, and aid in forming neuronal connections [19–21].
The most common glial cell type in the retina are Müller cells, which are situated transversally to all nuclear and plexiform layers of the retina. Longitudinal cytoplasmic projections of Müller cells contact retinal blood vessels, neurons, and other glial cells. Müller cells play a critical role in supporting and maintaining homeostasis of the entire retina. The function of Müller cells can generally be grouped into three categories: 1) metabolic support and nutrient synthesis, 2) handling of neurotransmitters and ions, and 3) maintenance of the BRB [22, 23]. Structurally, Müller cells help to minimize intraretinal light scattering by providing light a direct path to focus onto photoreceptors, thereby improving visual acuity [24]. Given the intimate associations formed with other retinal cell types and their geographic domination, Müller cells are critical for maintaining healthy retinal function and homeostasis.
1.3. Clinical Manifestations of DR
The intimate associations formed between neuronal, endothelial, and glial cells of the retina support a model wherein different retinal cell types cooperate to form a properly functioning neurovascular unit [25–27]. It has long been accepted that DR adversely impacts the retinal microvasculature. More recently, the concept has evolved to suggest a more accurate representation of diabetic pathophysiology in the retina that encompasses defects in the retinal neurovascular unit [28, 29]. DR manifests clinically in the form of retinal hemorrhaging, neovascularization, leakage of fluid into the retina, and formation of cotton wool spots and hard exudates. However, loss of neurovascular coupling, gliosis, neuroinflammation, and neurodegeneration, can precede and even predict visible signs of microvascular disease in diabetic patients [30, 31].
It is estimated that 42% of patients with type 1 diabetes and 8% of patients with type 2 diabetes have a vision-threatening form of DR [32, 33]. While diabetic macular edema (DME) is most closely associated with visual acuity loss; impaired contrast sensitivity, visual field deficits, and diminished color vision can develop earlier in disease progression and cause significant difficulties, particularly in low light conditions [26]. Thus, it is critical to better understand the early molecular events that occur prior to the overt vascular manifestations of DR that are visible via fundus examination (Fig. 2). Within the first months of disease onset, adaptive biochemical changes in the retina include a reduction biosynthetic and electrical activity, likely to reset the normally high metabolic activity of the retina [26, 34]. Müller cells become reactive and exhibit morphological and functional alterations shortly after the development of diabetes, as they attempt to counter-regulate the changing metabolic environment [23]. Their activation in response to mounting deleterious stimuli is referred to as gliosis. Microglial activation also contributes to the pathogenesis of retinal degenerative disease through the release of pro-inflammatory [35]. After prolonged hyperglycemia, adaptive changes begin to fail, and retinal neurons and pericytes are lost to apoptosis [36, 37]. Impairment of the retinal neurovasculature manifests clinically in the form of non-proliferative DR (NP-DR). Over time, the disintegration of the retinal neurovascular unit worsens, resulting in the most severe stage of the disease, proliferative DR (PDR), which is characterized by pre-retinal neovascularization [26]. Ultimately, vision loss occurs from either unrestrained proliferation of new blood vessels or from leakage of fluid through the retinal vasculature (i.e. DME) [38].
Fig. (2). Model for the impact of diabetes on retinal neurovasculature leading to a decline in visual function.
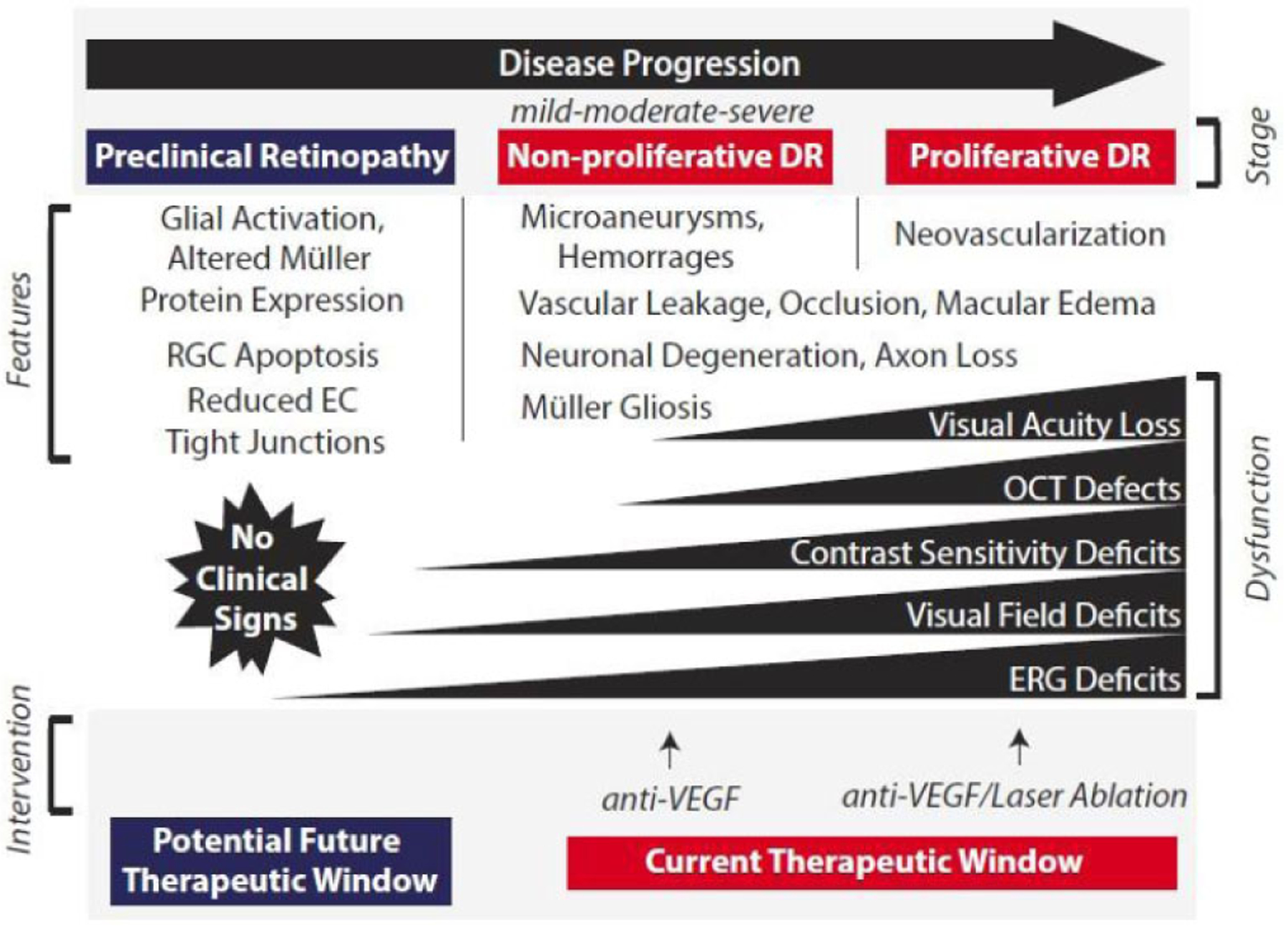
Adaptive and maladaptive changes in the neuroglial precede the microvascular dysfunction and neovascularization that characterizes the later stages of DR progression. Macular edema is most closely associated with vision loss, however, defects in visual function likely develop prior to overt clinical signs of disease. There is presently an absence of interventions that prevent the onset of disease or arrest its progression prior to a clinical diagnosis based on the detection of microvascular defects. DR, diabetic retinopathy; EC, endothelial cell; RGC, retinal ganglion cell; OCT, optical coherence tomography; ERG, electroretinogram; VEGF, vascular endothelial growth factor.
1.4. Current Interventions and the Molecular Pathogenesis of DR
Current therapeutic interventions for PDR or DME include laser-mediated ablation of the peripheral retina, surgical vitrectomy, or intraocular injections of steroids and VEGF inhibitors. Laser treatments and surgical approaches are accompanied by significant side effects (including diminished peripheral and night vision) and ultimately fail to alter the molecular events that cause the development of complications. More recently, VEGF inhibitors have emerged as the first clinical approach to address the molecular cause of DR. VEGF plays a central role in mediating the breakdown of the blood-retinal barrier, and examination of vitreous fluid from the eyes of patients with PDR demonstrates an increase in VEGF [39, 40]. Anti-VEGF therapies have dramatically improved treatment outcomes for DR; however, they reduce the severity of retinopathy in less than one-third of eyes treated for two years [41]. This limitation is likely due to the relatively poor correlation between VEGF levels and the extent of retinal edema [42], implying that additional factors are likely to play a role in disease progression. Indeed, the molecular pathophysiology of DR likely encompasses a complex interaction that includes pro-angiogenic signaling pathways, oxidative stress, inflammation, and endoplasmic reticulum (ER) stress [43–45].
A major underlying factor in the development and progression of DR is hyperglycemia [46, 47]. The Diabetes Control and Complications Trial (DCCT) for type 1 diabetes and the United Kingdom Prospective Diabetes Trial (UKPDS) for type 2 diabetes demonstrated that intensive glycemic control delays both the onset and progression of DR [48, 49]. An instigating factor of hyperglycemia-induced diabetic complications is hypothesized to be the overproduction of superoxide, a type of reactive oxygen species (ROS) produced by the electron transport chain of the mitochondria [50]. Although hyperglycemia systemically affects the body, differential effects may be seen in specific cell types such as retinal endothelium or mesangial cells of the kidney because they fail to restrict glucose transport into the cell in a hyperglycemic environment [51, 52]. Consequently, the large influx and subsequent oxidation of glucose increase the voltage gradient across the electron transport chain. Eventually, oxidative phosphorylation activity reaches a threshold, and the electron transport chain becomes bottlenecked, favoring the formation of superoxide [47, 53]. Downstream effects of ROS include activation of Protein Kinase C, which has several purported mechanisms of influencing blood flow in the retina, including elevated expression of VEGF [50, 54]. Additionally, downstream metabolites formed consequentially of ROS can feed into the polyol pathway which oxidizes glucose into sorbitol (consuming NADH and contributing to ROS susceptibility in the process) [55]. Finally, aberrant ROS production resulting from hyperglycemia can increase flux through the hexosamine biosynthetic pathway (HBP). The HBP is responsible for the production of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), which serves as the substrate for the enzymatic glycosylation of the hydroxyl side chains of serine and threonine residues of proteins known as O-GlcNAcylation [56].
2. PROTEIN O-GLCNACYLATION
2.1. O-GlcNAcylation as a Nutrient Sensor
First described in 1984, O-GlcNAcylation is one of the most abundant post-translational modifications within the nucleocytoplasmic compartment [57]. O-GlcNAcylation is more reminiscent of protein phosphorylation than other classical forms of glycosylation, as it is a single monosaccharide (N-acetyl glucosamine) that is not modified by elongation or branching. Rather, O-GlcNAc modifications are dynamically (and reversibly) cycled in response to environmental stimuli by two proteins: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). OGT catalyzes the addition of the O-GlcNAc moiety while the hydrolase OGA catalyzes its removal. Traditional N-linked glycosylation occurs in the lumen of the ER and Golgi body [58]. However, proteins may be O-GlcNAcylated in either the cytoplasm or nucleus. O-GlcNAc modifications are widely abundant (~1,500 unique proteins [59]) and critical for fundamental cellular processes including gene expression, cell cycle control, apoptosis, inflammation, and nutrient sensing [60].
Global O-GlcNAcylation levels are highly sensitive to nutrient status within the cell, as the HBP requires from nearly every major metabolic pathway-glucose, amino acid, nucleotide, and fatty acid (Fig. 3). Typically, the HBP is a minor metabolic pathway, accounting for only 2–5% of glucose metabolism [61]. However, an overabundance of nutrients, as seen with diabetes and over-nutrition, can drastically augment HBP flux leading to enhanced protein O-GlcNAcylation. The HBP ultimately metabolizes the glycolytic intermediate fructose-6-phosphate (F-6-P) into the O-GlcNAc donor UDP-GlcNAc. Glutamine:fructose-6-phosphate aminotransferase (GFAT) catalyzes the first and rate-determining step in this pathway, which involves the irreversible transfer of the amino group from glutamine and the isomerization of F-6-P to produce glucosamine-6-phosphate. Variation in GFAT isoform expression and activity are critical determinants of protein O-GlcNAcylation in a given cell. Two GFAT isoforms are differentially expressed across tissues and unlike GFAT1, GFAT2 is insensitive to feedback inhibition from UDP-GlcNAc [62]. Thus, tissues with a predominance of GFAT2 are more likely to overcommit glucose to the HBP. Consistent with studies demonstrating that GFAT2 is the predominant isoform in the central nervous system [63], GFAT2 is likely the primary isoform responsible for committing F-6-P to the HBP in the retina. Notably, isoform-specific expression of the mRNA encoding GFAT2 is dramatically increased in the retina of mice fed a high-fat diet as compared to control chow [64].
Fig. (3). Glucose is metabolized by the hexosamine biosynthetic pathway to facilitate protein O-GlcNAcylation.
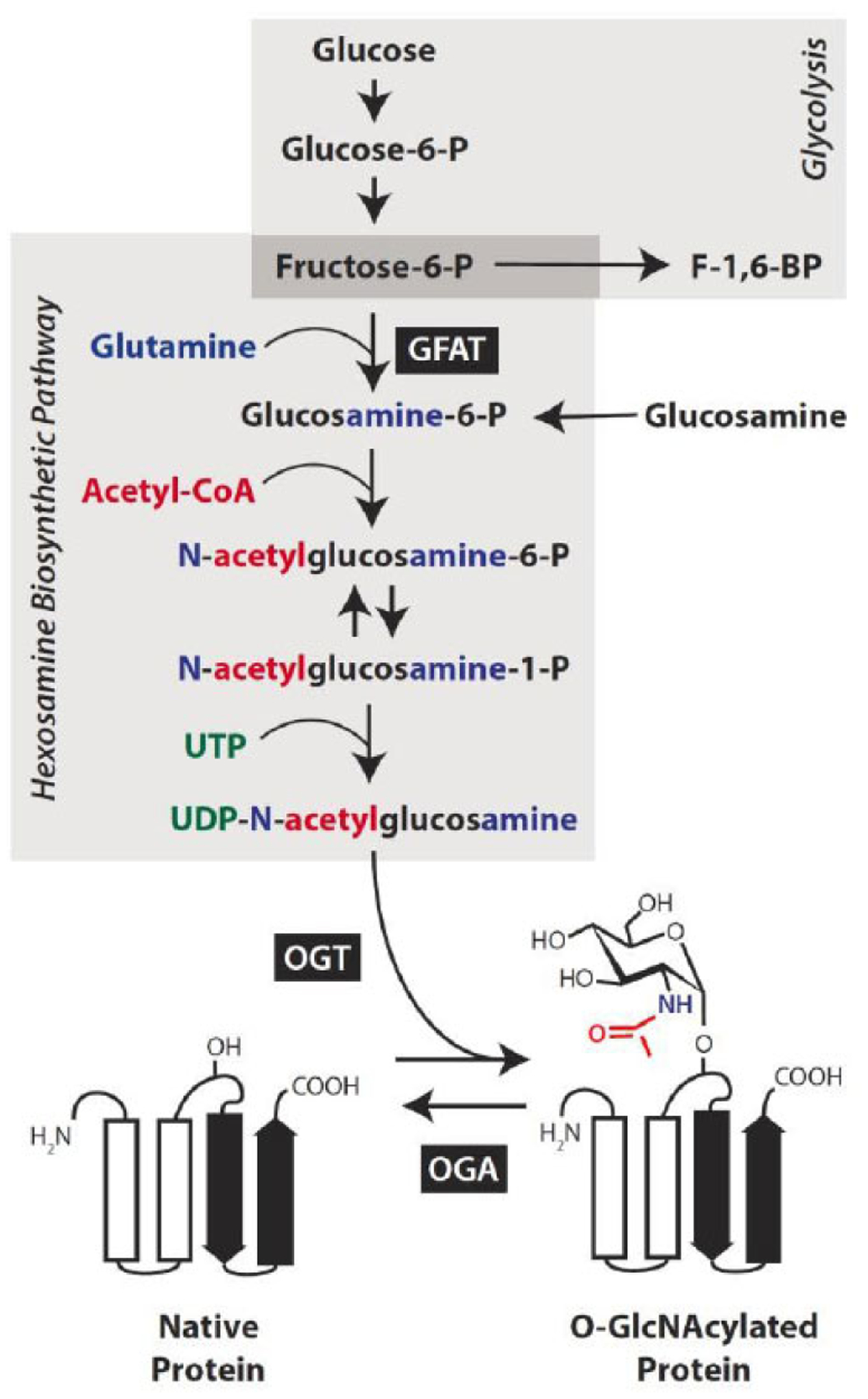
In addition to glucose, the synthesis of uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc) requires input from major metabolic pathways, including amino acid (blue), fatty acid (red), and nucleotide (green) metabolism. The first and rate-limiting step in this nutrient sensitive pathway involves the isomerization of fructose-6-phosphate to produce glucosamine-6-phosphate and is catalyzed by the enzyme glutamin-fructose-6-phosphate amidotransferase (GFAT). Alternatively, glucosamine enters the pathway downstream of this rate-limiting step. Protein O-GlcNAcylation involves the enzymatic O-linked addition of the GlcNAc moiety via the Ser/Thr hydroxyl side chain of a nucleocytoplasmic protein (shown as ribbon diagram) and is catalyzed by the enzyme O-GlcNAc transferase (OGT). The enzyme O-GlcNAcase (OGA) catalyzes the removal of the O-GlcNAc modification.
2.2. O-GlcNAcylation-induced Abnormalities in DR
Several studies have confirmed O-GlcNAcylation is abnormally elevated in a variety of tissues in both diabetic humans and animals [61, 65–67]. Retinal protein O-GlcNAcylation is increased in both streptozotocin (STZ)-induced diabetic mice [68] and mice fed an HFD [64]. These results are consistent with other reports indicatingthat retinal protein O-GlcNAcylation is elevated in 6-week-old diabetic Akita mice [69] and in the db/db mouse model of type 2 diabetes, in which O-GlcNAcylated retinal proteins localize to the ganglion cell layer, retinal pigmented epithelium, and inner plexiform layers [70].
Abnormalities in O-GlcNAc cycling are associated with glucose toxicity in diabetic individuals [71]. Additionally, the global elevation of O-GlcNAcylation, induced by inhibiting OGA can cause insulin resistance, even in the absence of hyperglycemia [72]. In addition to poor glycemic control, impaired insulin signaling is a primary agent responsible for the development of DR [34, 46, 73, 74]. OGT binds the insulin receptor substrate 1 (IRS1), where it catalyzes the addition of O-GlcNAc modifications on downstream effectors of the insulin signaling pathway. Direct O-GlcNAcylation of IRS1 attenuates the pro-survival effects of insulin thereby dampening insulin-stimulated Akt activation [72, 75–78]. Furthermore, enhancing intracellular O-GlcNAcylation levels via glucosamine (a metabolite that enters the HBP downstream of GFAT (Fig. 3) impairs activation of critical pro-survival proteins downstream of IRS1, including phosphatidylinositol-4,5-bisphosphate-3-kinase (PI3K), Akt, glycogen synthase kinase 3 (GSK3), and the transcription factor forkhead box protein O1a (FOXO1a) [79]. Systemic insulin administration reduces the rate of apoptosis in retinas of diabetic mice [36]. Furthermore, either systemic or local delivery of insulin prevents loss of retinal insulin receptor kinase activity [34]. However, exposure to high glucose cell culture medium and glucosamine attenuates the neuroprotective action of insulin in R28 cells, a model of retinal neurons [80]. Therefore, it is unclear if in vivo insulin administration and subsequent improvements in glucose homeostasis are sufficient to overcome hyperglycemia-induced retinal protein O-GlcNAcylation.
Aside from the neuroretina, enhanced protein O-GlcNAcylation also causes deleterious consequences for the retinal endothelial, epithelial, and glial cells. Abnormal O-GlcNAc cycling can induce early cell death of retinal pericytes, one of the earliest signs of DR [81]. Protein O-GlcNAcylation is elevated in both retinal endothelial cells and pericytes in Akita mice and in a model of oxygen-induced ischemic retinopathy during the neovascularization phase [69]. In cells exposed to the hyperglycemic culture medium, protein O-GlcNAcylation is higher in retinal pericytes as compared to retinal endothelial cells or astrocytes [81]. Hyperglycemic cell culture conditions also induce proliferation and migration of retinal endothelial cells with minimal apoptotic effects [81, 82]. Furthermore, enhanced protein O-GlcNAcylation in retinal endothelial cells contributes to angiogenic factor release and enhanced proliferation and migration [69, 70]. Elevation of intracellular O-GlcNAc is sufficient to upregulate VEGF in multiple retinal epithelial cell lines [83]. In an astrocyte cell line, O-GlcNAc modification of the cell cycle regulator p27 inhibits astrocyte migration while glucosamine-induced flux through the HBP causes acute activation of Akt and consequently increased ER stress [84, 85].
Retinal Müller cells are a particularly susceptible target of the glucotoxic effects of hyperglycemia. Müller cells are the only cell type in the retina to express GLUT2 transporters, which possess a high Michaelis constant (Km ~17 mM [86];) and a high maximum velocity for transporting glucose [87]. By contrast, GLUT1, 3, and 4 exhibit a much higher affinity for glucose (Km ~5 mM), and therefore saturated transport at much lower blood glucose concentrations. Moreover, MIO-M1 Müller cells exhibit predominant expression of the GFAT2 isoform [64]. Thus, Muller glia exposed to hyperglycemic conditions have a particularly high capacity for shifting glucose molecules into the HBP. Recent evidence suggests that Müller cells are also particularly sensitive to dyslipidemia associated with type 2 diabetes [88, 89]. GFAT2 expression and O-GlcNAcylation levels are elevated in Müller cells exposed to the saturated fatty acid palmitate or the ceramide analog Cer6 [64]. Such findings are consistent with the nutrient-sensing role of the HBP that requires input of the fatty acid metabolite acetyl-CoA for UDP-GlcNAc synthesis (Fig. 3).
2.3. O-GlcNAcylation and Transcription
Protein O-GlcNAcylation plays a key role in nutrient regulation of gene expression. Many early studies investigating protein O-GlcNAcylation support a model wherein the O-GlcNAc modification alters the transcriptional activity, localization, and binding partners of multiple transcription factors [90–93]. Nearly all RNA polymerase II transcription factors are O-GlcNAc modified, and assembly of the pre-initiation complex necessary for transcription initiation requires O-GlcNAcylation of the C-terminal domain of RNA polymerase II [94–97]. OGA itself localizes to transcription start sites to catalyze the removal of the O-GlcNAc moiety from the C-terminal domain of RNA polymerase II in order for transcription elongation to proceed [98]. O-GlcNAc also plays an epigenetic role as it regulates both ubiquitination and methylation of histones [99–101]. A detailed overview of O-GlcNAc-mediated regulation of gene expression at the level of transcription has been reviewed [95].
Several transcription factors that play a role in the pathogenesis of DR are subject to O-GlcNAc modification. Specificity protein 1 (Sp1) is a transcription factor that facilitates upregulation of the VEGF gene in response to hyperglycemia. In retinal endothelial and pigmented epithelial cells in culture, O-GlcNAcylation of Sp1 resulted in upregulation of VEGF mRNA and protein [83]. Notably, depletion of cellular OGT prevents glucose-induced changes in levels of VEGF [83]. Similarly, O-GlcNAcylated Sp1 also mediates hyperglycemia-induced intracellular cell adhesion molecule 1 (ICAM-1), an important inflammatory molecule expressed in retinal endothelial cells [102]. NF-κB is a pleiotropic transcription factor with multiple roles in development, apoptosis, and inflammation [103]. Activation of NF-κB is associated with experimental and clinical diabetes [104]. After two months of streptozotocin-induced diabetes, O-GlcNAcylated NF-κB is enhanced in retinas from diabetic mice as compared to non-diabetic controls [68]. In fact, the level of O-GlcNAcylated NF-κB is elevated in apoptotic RGCs of diabetic mice [68]. These results indicate that diabetes-induced O-GlcNAcylation of NF-κB induces the activation of the transcription factor and contributes to RGC cell death that is associated with diabetes. Another important mediator of cell death is pro-apoptotic transcription factor p53. Primary cultures of retinal pericytes exposed to hyperglycemic culture medium display elevated levels of O-GlcNAc modified and total p53 [81]. Pharmacological inhibition of both GFAT and OGT attenuates the hyperglycemia-induced increase in total p53. Given these results, it is plausible that O-GlcNAcylation enhances the stability of p53, possibly by interfering with its proteasome-mediated degradation.
While there are several descriptive studies interrogating the role of O-GlcNAc in transcription, functional significance of O-GlcNAc modification in retinal gene transcription in the diabetic milieu remains to be fully elucidated. The limited studies mentioned herein suggest that O-GlcNAcylation of certain transcription factors affects their transcriptional activity. Further detailed studies identifying a potential molecular mechanism by which this regulation happens are needed to develop a more robust understanding of how nutrient-sensing via HBP flux and subsequent protein O-GlcNAcylation control gene expression at the level of transcription.
2.4. O-GlcNAcylation and mRNA Translation
Gene expression is ultimately the sum of events, including transcription, translation, and mRNA and protein degradation, that regulate cellular protein content. Translational control is a particularly important step in gene expression for short-term adaptation to changing metabolic conditions [105] and is thus likely to be a fundamental mechanism used in the early adaptive response of retinal cells to diabetes. In fact, mRNAs containing sequence elements associated with translational control are most enriched in the gene categories of synaptic transmission, angiogenesis, and cell adhesion [106], which are all key processes disrupted in the retina by diabetes. Mounting evidence suggests that the augmented protein O-GlcNAcylation level acts as a molecular switch that causes a fundamental shift in gene expression. In fact, many proteins are O-GlcNAcylated co-translationally in a manner that prevents their proteasomal degradation [107]. In retinal pericytes exposed to hyperglycemic culture conditions, O-GlcNAcylated proteins are most heavily represented in the functional category Protein Synthesis and Processing [81]. Nearly half of the ~80 ribosomal proteins are O-GlcNAcylated, and both OGT and OGA are tightly bound to cytosolic ribosomes [108]. In order to determine the extent to which O-GlcNAcylation affects retinal gene expression, next-generation sequencing was used to evaluate total mRNA abundances, and ribosome profiling was used to determine which mRNAs were actively being translated into protein. When protein O-GlcNAcylation levels are augmented, ~19% of the retinal transcriptome exhibits variation in ribosome density [109].
The rate at which a given mRNA is translated is limited by the recruitment of ribosomal subunits during the initiation phase (Fig. 4). The 5ʹ-end of all eukaryotic mRNAs is capped by an m7GTP structure. The cap-binding complex eIF4F (a heterotrimer composed of a helicase (eIF4A), a scaffolding protein (eIF4G), and a cap-binding protein (eIF4E)) assembles at the 5ʹ-cap structure, and the eIF4F·mRNA complex recruits the small ribosomal subunit [110]. Interaction between initiation factors eIF4E and eIF4G is critical for cap-dependent translation initiation and is regulated by the eIF4E binding proteins (4E-BPs), which are direct targets of the master kinase mammalian target of rapamycin complex 1 (mTORC1). In addition, ~10% of mRNAs contain highly-structured nucleotide sequence elements, such as internal ribosomal entry sites (IRES), that enable ribosome recruitment independent of the 5ʹ-cap, i.e. cap-independent translation initiation [111]. These sequences form complex structural elements that potentially hinder cap-dependent translation, due to the high unfolding energy required to facilitate 5´-UTR scanning. However, these same structural elements can facilitate translation initiation under adverse physiological conditions. Whereas eIF4E inhibits cap-independent translation by increasing competition for initiation factors [112], sequestration of eIF4E by 4E-BP1 creates a cellular environment that potentially favors cap-independent translation. Importantly, eIF4E is the only canonical initiation factor differentially utilized in cap-dependent translation, but not cap-independent translation [113].
Fig. (4). Diabetes-induced O-GlcNAcylation of the translational repressor 4E-BP1 mediates a shift from cap-dependent to cap-independent mRNA translation.
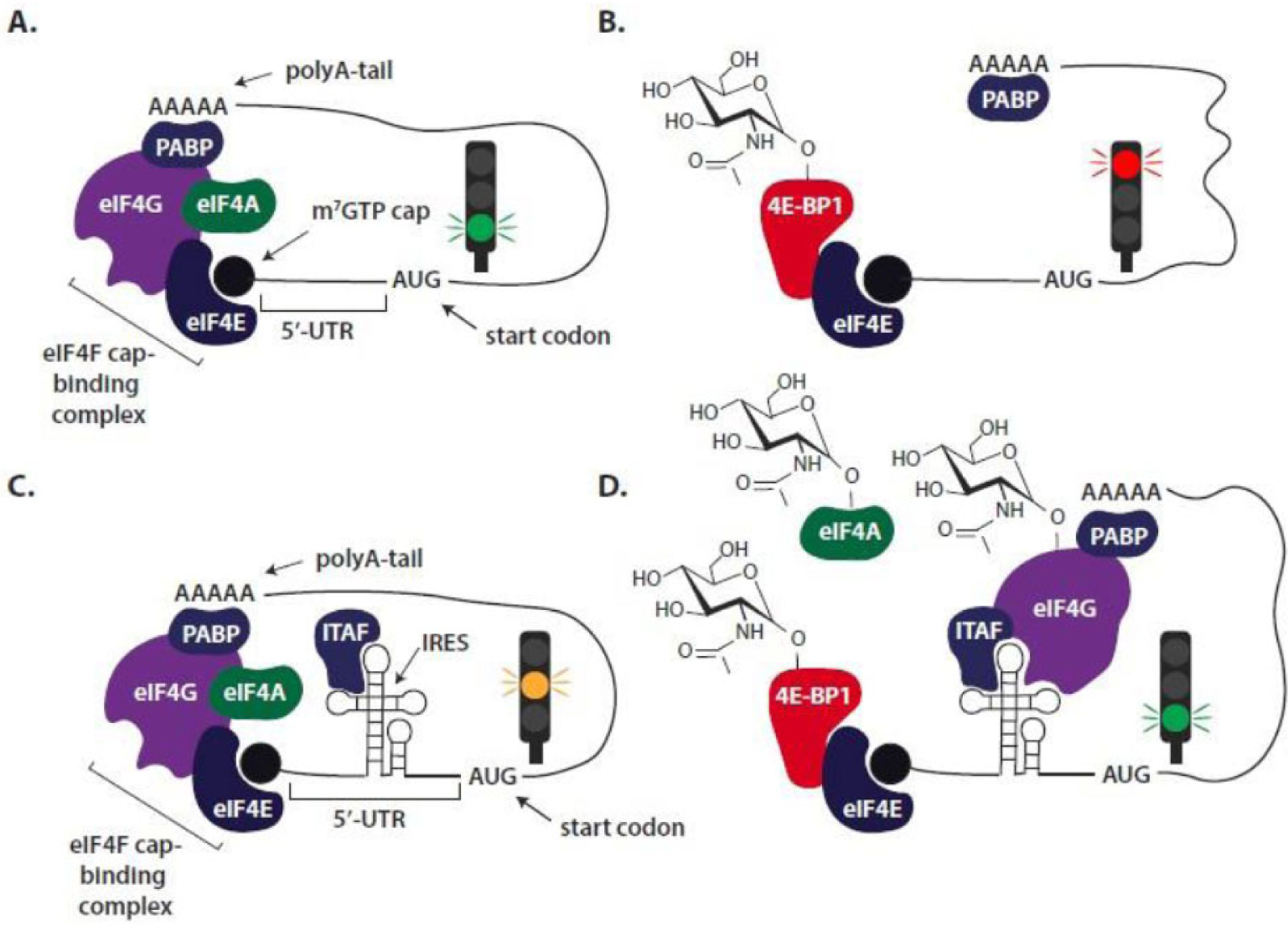
Translation initiation represents a key rate-controlling step in gene expression. All eukaryotic mRNAs include an m7GTP cap that is recognized by the cap-binding protein eIF4E. According to the traditional model of cap-dependent mRNA translation, recruitment of ribosomes is initiated by the assembly of the eIF4F complex consisting of eIF4E, eIF4G and eIF4A at the m7GTP cap (A.). An interaction between eIF4G and polyA-binding protein (PABP), which associates with the 3´-polyA tail, leads to circularization of the mRNA and facilitates translation initiation. Sequestration of eIF4E by 4E-BP1 prevents eIF4F assembly and represses cap-dependent translation (B). A small percentage of mRNAs, including the mRNA that encodes VEGF, contain unique sequence elements in their 5´-untranslated region (UTR) such as internal ribosome entry sites (IRES). These complex structural elements potentially hinder cap-dependent translation (C.). However, diabetes-induced sequestration of eIF4E by 4E-BP1 creates a cellular environment that favors cap-independent translation (D.). While there is significant diversity in the mechanisms utilized by IRES, cap-independent translation of specific mRNAs may be facilitated by IRES-transacting factors (ITAF), as well as O-GlcNAcylation of eIF4A and eIF4G.
Diabetes is associated with both an increase in retinal 4E-BP1 protein O-GlcNAcylation [61, 65–67] and enhanced binding of 4E-BP1 to eIF4E [114]. Increasing O-GlcNAcylation levels by OGA inhibition promotes 4E-BP1 interaction with eIF4E, independent of a change in 4E-BP1 phosphorylation status [115]. In rodents, retinal 4E-BP1 protein expression is elevated shortly after the onset of diabetes-induced hyperglycemia or OGA inhibition [114, 116]. Pharmacological induction of glucosuria in diabetic mice to lower serum glucose concentrations reduces O-GlcNAcylation of 4E-BP1 in the retina and normalizes 4E-BP1 expression [116]. This hyperglycemia- or O-GlcNAc-mediated induction of 4E-BP1 expression is attributed to a reduced rate of degradation [116]. Unlike 4E-BP2, 4E-BP1 contains a PEST motif that is associated with targeting proteins for degradation through the ubiquitin-proteasome pathway [117]. Disrupting a key phosphorylation site at Thr-82 within the PEST motif of 4E-BP1 prevents polyubiquitination and degradation of the protein [116]. That same study demonstrated a critical role for 4E-BP1/2 in the pathology that leads to diabetes-induced vision loss. Remarkably, in the retina of mice deficient for 4E-BP1/2, diabetes-induced visual dysfunction is delayed [116]. This protective effect of 4E-BP1/2 ablation on visual function is likely mediated by preventing a diabetes-induced change in mRNA translation. Indeed, 4E-BP1 O-GlcNAcylation has been implicated in promoting the translation of mRNAs that encode key proteins involved in pro-angiogenic signaling, oxidative stress, and inflammation, which are all critical in the pathogenesis of DR.
As described above, the role of VEGF as a therapeutic target for DR is well established, however the specific mechanisms that upregulate VEGF protein levels in DR are less well explored. While VEGF expression is often thought of as being regulated transcriptionally (i.e. by hypoxia-inducible factor 1alpha), more recent studies indicate that it is also regulated at the level of mRNA translation [118–120]. The VEGF mRNA transcript has an exceptionally long G-C base pair rich 5´-untranslated region (UTR) that contains alternative start sites and stop codons in frame with the classical start site. Two independent IRES sequences within the UTR control start codon selection [121] and allow the VEGF mRNA to be translated independently of its 5´-cap [118]. A series of studies support that hyperglycemic conditions promote VEGF mRNA translation via 4E-BP1 action [114, 122, 123]. In both the retina of diabetic mice and in cells exposed to hyperglycemic culture conditions, 4E-BP1/2 is necessary for upregulated VEGF expression [114]. In cells exposed to culture media containing either increased glucose concentrations or glucosamine, the activity of the VEGF 5´-UTR is enhanced, and 4E-BP1/2 deletion is sufficient to prevent increased activity of the VEGF 5´-UTR upon exposure to hyperglycemic conditions [122, 123].
4E-BP-dependent translational control is also implicated in the development of diabetes-induced oxidative stress in the retina. Mitochondrial dysfunction and oxidative stress are critical factors in the development and progression DR. In the whole retina, translation of mRNAs encoding ~50 mitochondrial proteins is sensitive to OGA inhibition, and at least, a subset of these changes (e.g. suppressed translation of SOD2) required 4E-BP1/2 [109]. O-GlcNAcylation alters the translation of mRNAs encoding mitochondrial proteins involved in oxidative phosphorylation, mitochondria-specific tRNA synthetases, and subunits of the translocase of the inner membrane (TIM)/translocase of outer membrane (TOM) complex. The functional outcome of BP1/2-dependent variation in mRNA translation is an increase in mitochondrial superoxide levels [109]. Whereas diabetic wild-type mice exhibit an increase in retinal ROS levels, 4E-BP1/2-deficient mice do not.
Bioinformatic analysis demonstrates that retinal mRNAs exhibiting altered translation in response to enhanced O-GlcNAcylation are most heavily represented in the “acute-phase response” pathway, which includes a range of pro-inflammatory cytokines, as well as the co-stimulatory molecule cluster of differentiation 40 (CD40) [109]. CD40 expression in retinal Müller glia is necessary to induce expression of other critical immunomodulatory molecules, including NOS2 (nitric oxide synthase 2) [124]. In a RiboTag mouse model [125] that expresses a hemagglutinin (HA)-tagged variant of ribosomal protein Rpl22 specifically in Müller cells, diabetes or administration of an OGA inhibitor promotes CD40 mRNA translation in Müller cells (Dierschke et al, 2020). In Müller cells in culture, expression of a 4E-BP1 variant that constitutively sequesters eIF4E is sufficient to promote cap-independent CD40 mRNA translation driven by the CD40 5´-UTR (Dierschke et al, 2020). Cd40 mRNA translation, CD40 protein expression, and Nos2 mRNA abundance are upregulated in the retina of wild-type diabetic mice, but not in the retina of diabetic 4E-BP1/2-deficient mice (Dierschke et al, 2020). Collectively, these findings support a model wherein O-GlcNAc-mediated sequestration of eIF4E by 4E-BP1 serves to promote retinal inflammation.
Other core translation initiation factors have also been reported as being O-GlcNAc modified. eIF2α is a major mediator of the unfolded protein response (UPR), a mechanism induced by ER stress that inhibits global translation initiation, with the exception of a few specific, stress-related mRNAs. O-GlcNAcylation of eIF2α inhibits its phosphorylation at a critical serine residue necessary to carry out the UPR, and thereby prevents ER stress-induced apoptosis [126]. O-GlcNAcylation of eIF4G enhances its stability [127]. In human HEK293T cells, O-GlcNAcylation of eIF4G promotes its interaction with poly(A)-binding protein (PABP) and poly(A) mRNA, which facilitates the interaction of the 5ʹ-cap and 3ʹ -end of mRNA to form a closed loop and stimulate translation by enabling terminating ribosomes to come into close proximity with the start codon [128, 129]. In MEF cells, the O-GlcNAc modification on eIF4G facilitates the preferential translation of stress mRNAs during the heat shock response, a signaling pathway that results in rapid induction of genes encoding molecular chaperones necessary for recovery from cellular damages [130]. Heat stress-induced eIF4G O-GlcNAcylation not only suppresses cap-dependent translation, but simultaneously enables eIF4G to preferentially bind specific mRNAs and initiate translation via a cap-independent mechanism [131]. Li and colleagues also demonstrated that the core translation factor eIF4A, an ATP-dependent RNA helicase, is also O-GlcNAc modified [128]. O-GlcNAcylation of eIF4A disrupts assembly of the eIF4F complex and inhibits the duplex unwinding activity of eIF4A, thereby resulting in an overall inhibition in protein synthesis and cellular proliferation [128].
Given the differential effects of O-GlcNAcylation on core translation initiation proteins, it is likely that the modification exists to fine-tune protein synthesis in response to various biological cues and fluctuations in nutrient status. Protein synthesis is a tightly regulated process that is fundamental to cell growth and proliferation. While many studies have begun to uncover the role the nutrient- and stress-responsive post-translational O-GlcNAc modification may play in mRNA translation, more studies are necessary to fully appreciate this mechanism whereby the cell integrates nutritional status with the fundamental cellular process of synthesizing proteins.
3. THE RENIN-ANGIOTENSIN SYSTEM (RAS)
3.1. A Role for the Ocular RAS in DR
The pathogenesis of DR encompasses dysregulation of a multitude of signaling pathways. However, recent evidence indicates dysregulation of the renin-angiotensin system (RAS) may play a prominent role in the disruption of the retinal neurovascular unit [132]. The RAS is classically defined by its systemic physiological functions, but it is becoming increasingly evident that local, tissue-specific RAS systems play an important role in disease pathologies, including DR. The RAS is one of the oldest studied hormone systems and is well known for its role in systemic vascular control and fluid homeostasis. The retina is one of several tissues that possess a local RAS, and it is well-established that all components of the RAS are expressed in the retina, including angiotensinogen, prorenin, renin, angiotensin I (Ang I), angiotensin II (Ang II), angiotensin-converting enzyme (ACE), Ang II type 1 receptor (AT1R), and Ang II type 2 receptor (AT2R) (reviewed in [133]). Secreted renin serves to cleave the pro-peptide angiotensinogen into Ang I (Fig. 5). ACE cleaves Ang I into the primary effector peptide of the RAS, Ang II. Ang II stimulates vasoconstriction, aldosterone synthesis and subsequent sodium resorption, fibrosis, and inflammation, mediated through binding to the AT1R G protein-coupled receptor [134]. The mechanisms induced by AT2R binding are less well understood; although, it is thought that activation of AT2R may mediate opposing actions of AT1R, such as stimulating vasodilation [135].
Fig. (5). Angiotensin-(1–7) signaling attenuates protein O-GlcNAcylation in the retina.
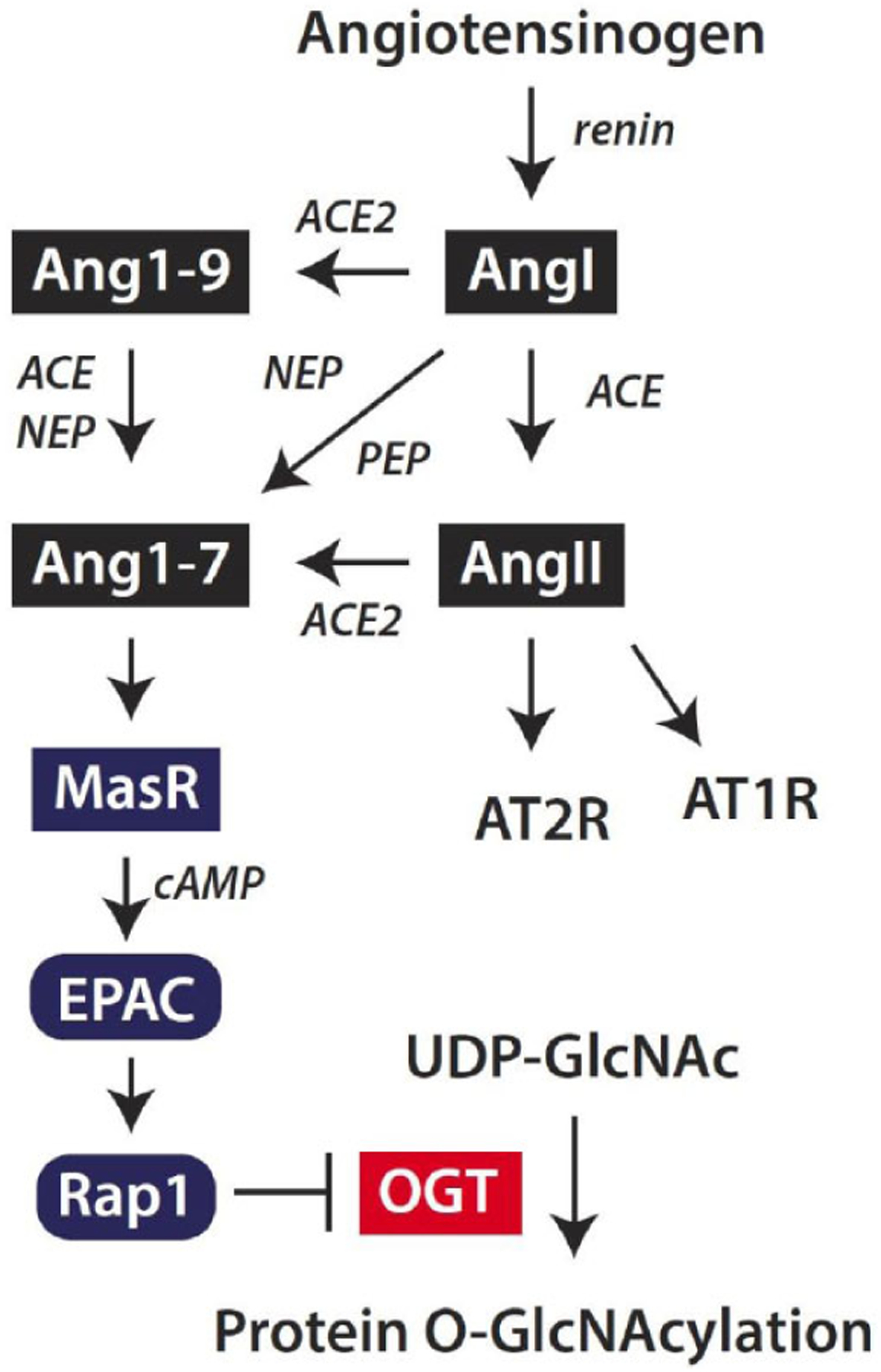
The classic renin-angiotensin system is initiated by cleavage of the pro-peptide angiotensinogen by renin to form Angiotensin I (AngI). AngI may be cleaved by angiotensin-converting enzyme (ACE) to generate angiotensin II (AngII), which acts via AT1 and AT2 receptors (AT1R and AT2R, respectively). Alternatively, Ang II may be cleaved by ACE2 to generate Angiotensin-(1–7) (Ang1–7). ACE2 also cleaves AngI to generate Ang1–9, which may be degraded by ACE or neutral endopeptidase (NEP) to produce Ang1–7. Finally, AngI may be degraded by endopeptidases such as NEP or prolyl endopeptidase (PEP) to directly form Ang1–7. Ang1–7 acts via the Mas receptor (MasR) to enhance cyclic AMP (cAMP) levels and suppress protein O-GlcNAcylation in retinal cells. Ang1–7 acts to suppress OGT activity via a signaling axis that includes the exchange protein activated by cAMP (EPAC) and Rap1.
The role of Ang II in a healthy retina is likely related to the regulation of retinal blood vessel tonicity, glial function, and neuronal function [136]. In DR, Ang II formation is elevated, potentiates VEGF-mediated angiogenesis, and increases retinal blood vessel permeability (reviewed in [133]). Interest in further understanding the role of Ang II and other components of the RAS increased after clinical studies revealed that blocking either Ang II signaling with ARBs (Angiotensin Receptor Blockers) or preventing Ang II formation with ACE inhibitors (ACEi) reduced the risk for both incidence and progression of DR [137]. These intriguing clinical results prompted a question that would be best answered on a molecular level: how do ARBs and ACEi mediate beneficial effects in the retina?
The hypothesis that was subsequently developed to answer that question involved the action of the more recently discovered arm of the RAS that is mediated by the effector peptide angiotensin- [1–7] (Ang1–7). Ang1–7 is generated by the cleavage of Ang II by the enzyme ACE2 or by the cleavage of Ang I by various endopeptidases. Ang I may be alternatively degraded by ACE2 to generate angiotensin- [1–9], which in turn may be cleaved by neutral endopeptidase or ACE to generate Ang1–7. Ang1–7 binds to the G protein-coupled receptor Mas to elicit systemic vasodilatory and antihypertensive effects. A handful of recent studies have provided evidence that some of the positive cardiometabolic effects observed in rodents administered RAS blockers are due to increased circulating levels of Ang1–7 [138–140]. In the case of ACEi, the basis for this rise in Ang1–7 is due to decreased degradation of Ang1–7 to angiotensin- [1–5] by ACE and increased Ang I resulting from a reduction in Ang II formation [141, 142]. Importantly, the positive effects of Ang1–7 thus far reported are independent of changes in blood pressure [143, 144].
Pharmacological blockade of the RAS also has beneficial effects on visual function. Administration of the ACEi perindopril in diabetic rodents improved both photoreceptoral and inner retinal function comparable to that of nondiabetic rodents [145]. ACEi also reduces VEGF expression and vascular permeability in the retinas of diabetic rodents [146, 147]. Systemic administration of the ACEi captopril enhances the expression of Ang1–7 in the retina with a concomitant decrease in diabetes-induced Ang II expression [148]. Intravitreal delivery of adenoviral encoded Ang1–7 also ameliorates hallmark characteristics of DR, including diabetes-induced retinal vascular permeability, the formation of acellular capillaries, infiltration of inflammatory cells, and oxidative damage in diabetic rodents [149]. These pioneering descriptive studies have done much to demonstrate the existence of an ocular RAS and have established a convincing line of evidence that targeting the ACE2/Ang1–7/Mas axis of the RAS in the retina could have promising therapeutic effects in ameliorating the deleterious consequences of DR.
3.2. Evidence for Interplay between Protein O-GlcNAcylation and the RAS
A number of studies provide evidence for the interplay between protein O-GlcNAcylation and the RAS. However, little is known about the dynamic between these two pathways in DR. In fact, the impact of O-GlcNAc modified proteins on ocular RAS signaling is completely unexplored. In mesangial and proximal tubular cells, exposure to either hyperglycemic culture conditions or glucosamine stimulates angiotensinogen gene expression [150–152]. Other studies suggest that enhanced protein O-GlcNAcylation modulates hypertrophic pathways associated with cardiovascular dysfunction in diabetes. Activation of the HBP in cardiomyocytes isolated from diabetic db/db mice resulted in an attenuated hypertrophic response upon exposure to Ang II as compared to cardiomyocytes isolated from nondiabetic control mice [153]. Inhibiting the HBP partially restored the hypertrophic signaling response in diabetic cardiomyocytes [153]. In adipose tissue, attenuation of protein O-GlcNAcylation resulted in a concomitant decrease in angiotensinogen, AT1R, OGT, and GFAT transcripts [154]. Similarly, infusion of glucosamine increased the expression of angiotensinogen in adipose tissue [155]. Taken together, these studies provide evidence to support a role for O-GlcNAc-mediated regulation of local and systemic RAS. However, studies examining specific signaling cascades that generate these results are lacking.
A recent study provides evidence that the ocular RAS modulates protein O-GlcNAcylation in the retina [156]. In the retina of mice fed an HFD, retinal protein O-GlcNAcylation is enhanced relative to chow-fed control mice [64]. However, when mice fedan HFD were administered the ACEi captopril, retinal protein O-GlcNAcylation is attenuated relative to mice fed an HFD alone [156]. Thus, ACE inhibition is sufficient to attenuate retinal protein O-GlcNAcylation. This observation supports a previous report that pharmacological blockade of the RAS prevented the diabetes-induced increase in global O-GlcNAcylation in the kidney [157]. Administration of both captopril and A779, a Mas receptor antagonist, reverses the captopril-mediated attenuation of HFD-induced retinal protein O-GlcNAcylation, indicating that binding of Ang1–7 to the Mas receptor is necessary for the effect to occur [156]. In Müller cells in culture, Ang1–7 signals via a cAMP/EPAC (Exchange Proteins Activated by cAMP)/Rap1 axis to prevent an increase in O-GlcNAcylation (Fig. 5). Surprisingly, the suppressive effect of this signaling axis on O-GlcNAcylation is not overcome by glucosamine supplementation, indicating that suppression is mediated downstream of the typical rate-limiting enzyme GFAT [156]. Indeed, OGT activity is attenuated in Müller cells exposed to Ang1–7 [156]. Given the integral role of both protein O-GlcNAcylation and the RAS in DR, defining the specific molecular events associated with the intersection of these pathways may represent novel therapeutic targets for DR.
CONCLUSIONS
The retina has the unique function of transmitting visual information to the brain to elicit a behaviorally useful response. Retinal glial, endothelial, and neuronal cells cooperate to form a retinal neurovascular unit. The retina has one of the highest metabolic demands of the body, and therefore is particularly susceptible to hyperglycemia-mediated tissue damage. Early adaptive changes of the retina to the diabetic metabolic environment involve attenuation of biosynthetic pathways. However, common signaling pathways in the retina regulate both biosynthesis and cell survival (283, 310), so a loss of biosynthetic activity is part of a larger disruption of retinal metabolism that results in retinal cell death as adaptive changes become maladaptive. This process most likely occurs in diabetic patients before impairments in the retinal vasculature are clinically visible. At this early stage of the disease, defects in the neuroretina, such as electroretino-graphic responses and visual field sensitivity, manifest (311, 312). Therefore, there is a need to understand what mediates the early metabolic adaptations the retina undergoes, and to understand how those alterations ultimately become maladaptive.
The global re-programming of retinal metabolism is mediated, in part, via increased flux through the HBP and consequent enhancement in protein O-GlcNAcylation. The HBP exists to integrate nutritional signals and fine-tune cellular responses to biological cues. The retina is particularly dependent upon such fine-tune regulation given the sophisticated coordination required of the retinal neurovascular unit that must exist to visually process the outside world. Recent studies demonstrate that enhanced retinal protein O-GlcNAcylation primarily alters gene expression at the level of mRNA translation. Further evidence supports the hypothesis that O-GlcNAcylation enhances the interaction between 4E-BP1 and eIF4E to promote angiogenic cytokine production, enhance mitochondrial superoxide production, and initiate retinal inflammation. Importantly, augmented O-GlcNAcylation may be attenuated via Ang1–7/cAMP-mediated attenuation of OGT activity. While further studies are needed, addressing the defect in retinal protein O-GlcNAcylation may prove to be beneficial in preventing the development of visual dysfunction in diabetes.
FUTURE INSIGHTS.
Many descriptive studies have identified O-GlcNAc modified transcription factors, but studies investigating the effects of this modification on protein synthesis are lacking. Therefore, O-GlcNAc-mediated control of gene expression at the level of mRNA translation is a potentially underappreciated phenomenon that could have far-reaching clinical implications. Indeed, O-GlcNAc-mediated control of gene expression in the retina could contribute noteworthy findings to the wider field of retinal physiology. To build upon this foundation of O-GlcNAc-mediated control of gene expression via 4E-BP, future studies should focus on mechanisms underlying the translation of mRNAs that contain internal, highly-structured elements in the context of diabetes and enhanced O-GlcNAcylation in the retina. Further defining these mechanisms and the RNA binding proteins involved in the regulation of protein synthesis, a better understanding of which messages get translated into a retinal protein will aid in developing novel therapies that can target early biochemical defects in the retina. Clinically available interventions for DR are not universally effective at reversing nor preventing vision loss. These therapeutic interventions for DR and associated diabetes-related ocular pathologies are targeted at consequences of the disease, rather than underlying molecular abnormalities. Therefore, while further mechanistic studies are needed, addressing abnormalities in retinal protein O-GlcNAcylation may prove to be beneficial in preventing the development of visual dysfunction in diabetes.
ACKNOWLEDGEMENTS
The authors thank Drs. Alistair Barber and Scot Kimball for critically evaluating the manuscript.
FUNDING
This research was supported by the American Diabetes Association Pathway to Stop Diabetes Grant 1-14-INI-04 and National Eye Institute grant R01 EY029702 (to M.D.D.).
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest, financial or otherwise.
Publisher's Disclaimer: DISCLAIMER: The above article has been published in Epub (ahead of print) on the basis of the materials provided by the author. The Editorial Department reserves the right to make minor modifications for further improvement of the manuscript.
REFERENCES
- [1].Fong DS, Aiello LP, Ferris FL III, Klein R. Diabetic retinopathy. Diabetes Care 2004; 27(10): 2540–53. 10.2337/diacare.27.10.2540 [DOI] [PubMed] [Google Scholar]
- [2].Lott ME, Slocomb JE, Shivkumar V, et al. Impaired retinal vasodilator responses in prediabetes and type 2 diabetes. Acta Ophthalmol 2013; 91(6): e462–9. 10.1111/aos.12129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zaleska-Żmijewska A, Piątkiewicz P, Śmigielska B, et al. Retinal Photoreceptors and Microvascular Changes in Prediabetes Measured with Adaptive Optics (rtx1™): A Case-Control Study. J Diabetes Res 2017; 2017: 4174292. 10.1155/2017/4174292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Klein R, Barrett-Connor EL, Blunt BA, Wingard DL. Visual impairment and retinopathy in people with normal glucose tolerance, impaired glucose tolerance, and newly diagnosed NIDDM. Diabetes Care 1991; 14(10): 914–8. 10.2337/diacare.14.10.914 [DOI] [PubMed] [Google Scholar]
- [5].Karadeniz S, Kir N, Yilmaz MT, et al. Alteration of visual function in impaired glucose tolerance. Eur J Ophthalmol 1996; 6(1): 59–62. 10.1177/112067219600600112 [DOI] [PubMed] [Google Scholar]
- [6].Chan JY, Cole E, Hanna AK. Diabetic nephropathy and proliferative retinopathy with normal glucose tolerance. Diabetes Care 1985; 8(4): 385–90. 10.2337/diacare.8.4.385 [DOI] [PubMed] [Google Scholar]
- [7].Harrower AD, Clarke BF. Diabetic retinopathy with normal glucose tolerance. Br J Ophthalmol 1976; 60(6): 459–63. 10.1136/bjo.60.6.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hutton WL, Snyder WB, Vaiser A, Siperstein MD. Retinal microangiopathy without associated glucose intolerance. Transactions - American Academy of Ophthalmology and Otolaryngology. American Academy of Ophthalmology and Otolaryngology; 1972; 76: 968–80. [PubMed] [Google Scholar]
- [9].Masland RH. The fundamental plan of the retina. Nat Neurosci 2001; 4(9): 877–86. 10.1038/nn0901-877 [DOI] [PubMed] [Google Scholar]
- [10].Masland RH. The neuronal organization of the retina. Neuron 2012; 76(2): 266–80. 10.1016/j.neuron.2012.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gollisch T, Meister M. Eye smarter than scientists believed: neural computations in circuits of the retina. Neuron 2010; 65(2): 150–64. 10.1016/j.neuron.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Flores-Herr N, Protti DA, Wässle H. Synaptic currents generating the inhibitory surround of ganglion cells in the mammalian retina. J Neurosci 2001; 21(13): 4852–63. 10.1523/JNEUROSCI.21-13-04852.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bruesch SR, Arey LB. The number of myelinated and unmyelinated fibers in the optic nerve of vertebrates - Bruesch. J Comp Neurol 1942; 77: 361. 10.1002/cne.900770310 [DOI] [Google Scholar]
- [14].Niven JE, Laughlin SB. Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol 2008; 211(Pt 11): 1792–804. 10.1242/jeb.017574 [DOI] [PubMed] [Google Scholar]
- [15].Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. Glia-neuron interactions in the mammalian retina. Prog Retin Eye Res 2016; 51: 1–40. 10.1016/j.preteyeres.2015.06.003 [DOI] [PubMed] [Google Scholar]
- [16].Watanabe T, Raff MC. Retinal astrocytes are immigrants from the optic nerve. Nature 1988; 332(6167): 834–7. 10.1038/332834a0 [DOI] [PubMed] [Google Scholar]
- [17].Stone J, Itin A, Alon T, et al. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 1995; 15(7 Pt 1): 4738–47. 10.1523/JNEUROSCI.15-07-04738.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ozaki H, Seo MS, Ozaki K, et al. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am J Pathol 2000; 156(2): 697–707. 10.1016/S0002-9440(10)64773-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bessis A, Béchade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia 2007; 55(3): 233–8. 10.1002/glia.20459 [DOI] [PubMed] [Google Scholar]
- [20].Provis JM. Development of the primate retinal vasculature. Prog Retin Eye Res 2001; 20(6): 799–821. 10.1016/S1350-9462(01)00012-X [DOI] [PubMed] [Google Scholar]
- [21].Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012; 74(4): 691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Coughlin BA, Feenstra DJ, Mohr S. Müller cells and diabetic retinopathy. Vision Res 2017; 139: 93–100. 10.1016/j.visres.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Subirada PV, Paz MC, Ridano ME, et al. A journey into the retina: Müller glia commanding survival and death. Eur J Neurosci 2018; 47(12): 1429–43. 10.1111/ejn.13965 [DOI] [PubMed] [Google Scholar]
- [24].Agte S, Junek S, Matthias S, et al. Müller glial cell-provided cellular light guidance through the vital guinea-pig retina. Biophys J 2011; 101(11): 2611–9. 10.1016/j.bpj.2011.09.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Metea MR, Newman EA. Signalling within the neurovascular unit in the mammalian retina. Exp Physiol 2007; 92(4): 635–40. 10.1113/expphysiol.2006.036376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gardner TW, Davila JR. The neurovascular unit and the pathophysiologic basis of diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol 2017; 255(1): 1–6. 10.1007/s00417-016-3548-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Antonetti DA, Barber AJ, Bronson SK,et al. JDRF Diabetic Retinopathy Center Group. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 2006; 55(9): 2401–11. 10.2337/db05-1635 [DOI] [PubMed] [Google Scholar]
- [28].Simó R, Stitt AW, Gardner TW. Neurodegeneration in diabetic retinopathy: does it really matter? Diabetologia 2018; 61(9): 1902–12. 10.1007/s00125-018-4692-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Barber AJ, Baccouche B. Neurodegeneration in diabetic retinopathy: Potential for novel therapies. Vision Res 2017; 139: 82–92. 10.1016/j.visres.2017.06.014 [DOI] [PubMed] [Google Scholar]
- [30].Adams AJ, Bearse MA Jr. Retinal neuropathy precedes vasculopathy in diabetes: a function-based opportunity for early treatment intervention? Clin Exp Optom 2012; 95(3): 256–65. 10.1111/j.1444-0938.2012.00733.x [DOI] [PubMed] [Google Scholar]
- [31].Han Y, Bearse MA Jr, Schneck ME, Barez S, Jacobsen CH, Adams AJ. Multifocal electroretinogram delays predict sites of subsequent diabetic retinopathy. Invest Ophthalmol Vis Sci 2004; 45(3): 948–54. 10.1167/iovs.03-1101 [DOI] [PubMed] [Google Scholar]
- [32].Kempen JH, O’Colmain BJ, Leske MC, et al. Eye Diseases Prevalence Research Group. The prevalence of diabetic retinopathy among adults in the United States. Arch Ophthalmol 2004; 122(4): 552–63. 10.1001/archopht.122.4.552 [DOI] [PubMed] [Google Scholar]
- [33].Roy MS, Klein R, O’Colmain BJ, Klein BE, Moss SE, Kempen JH. The prevalence of diabetic retinopathy among adult type 1 diabetic persons in the United States. Arch Ophthalmol 2004; 122(4): 546–51. 10.1001/archopht.122.4.546 [DOI] [PubMed] [Google Scholar]
- [34].Reiter CE, Wu X, Sandirasegarane L, et al. Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin. Diabetes 2006; 55(4): 1148–56. 10.2337/diabetes.55.04.06.db05-0744 [DOI] [PubMed] [Google Scholar]
- [35].Langmann T Microglia activation in retinal degeneration. J Leukoc Biol 2007; 81(6): 1345–51. 10.1189/jlb.0207114 [DOI] [PubMed] [Google Scholar]
- [36].Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 1998; 102(4): 783–91. 10.1172/JCI2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kern TS, Tang J, Mizutani M,et al. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci 2000; 41(12): 3972–8. [PubMed] [Google Scholar]
- [38].Aiello LP. Angiogenic pathways in diabetic retinopathy. N Engl J Med 2005; 353(8): 839–41. 10.1056/NEJMe058142 [DOI] [PubMed] [Google Scholar]
- [39].Adamis AP, Miller JW, Bernal MT, et al. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol 1994; 118(4): 445–50. 10.1016/S0002-9394(14)75794-0 [DOI] [PubMed] [Google Scholar]
- [40].Aiello LP, Avery RL, Arrigg PG,et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994; 331(22): 1480–7. 10.1056/NEJM199412013312203 [DOI] [PubMed] [Google Scholar]
- [41].Bressler NM, Varma R, Suñer IJ, et al. RIDE and RISE Research Groups. Vision-related function after ranibizumab treatment for diabetic macular edema: results from RIDE and RISE. Ophthalmology 2014; 121(12): 2461–72. 10.1016/j.ophtha.2014.07.008 [DOI] [PubMed] [Google Scholar]
- [42].Kita T, Clermont AC, Murugesan N, et al. Plasma kallikrein-kinin system as a VEGF-independent mediator of diabetic macular edema. Diabetes 2015; 64(10): 3588–99. 10.2337/db15-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pusparajah P, Lee LH, Abdul Kadir K Molecular Markers of Diabetic Retinopathy: Potential Screening Tool of the Future? Front Physiol 2016; 7: 200. 10.3389/fphys.2016.00200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Behl T, Kaur I, Kotwani A Implication of oxidative stress in progression of diabetic retinopathy. Surv Ophthalmol 2016; 61(2): 187–96. 10.1016/j.survophthal.2015.06.001 [DOI] [PubMed] [Google Scholar]
- [45].Semeraro F, Cancarini A, dell’Omo R, Rezzola S, Romano MR, Costagliola C Diabetic Retinopathy: Vascular and Inflammatory Disease. J Diabetes Res 2015; 2015: 582060. 10.1155/2015/582060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lind M, Odén A, Fahlén M, Eliasson B The true value of HbA1c as a predictor of diabetic complications: simulations of HbA1c variables. PLoS One 2009; 4(2): e4412. 10.1371/journal.pone.0004412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brownlee M The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005; 54(6): 1615–25. 10.2337/diabetes.54.6.1615 [DOI] [PubMed] [Google Scholar]
- [48].Nathan DM, Genuth S, Lachin J, et al. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329(14): 977–86. 10.1056/NEJM199309303291401 [DOI] [PubMed] [Google Scholar]
- [49].Group UPDS. UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352(9131): 837–53. 10.1016/S0140-6736(98)07019-6 [DOI] [PubMed] [Google Scholar]
- [50].Brownlee M Biochemistry and molecular cell biology of diabetic complications. Nature 2001; 414(6865): 813–20. 10.1038/414813a [DOI] [PubMed] [Google Scholar]
- [51].Kaiser N, Sasson S, Feener EP, et al. Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes 1993; 42(1): 80–9. 10.2337/diab.42.1.80 [DOI] [PubMed] [Google Scholar]
- [52].Heilig CW, Concepcion LA, Riser BL, Freytag SO, Zhu Cortes P. Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J Clin Invest 1995; 96(4): 1802–14. 10.1172/JCI118226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 1997; 416(1): 15–8. 10.1016/S0014-5793(97)01159-9 [DOI] [PubMed] [Google Scholar]
- [54].Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 2010; 106(8): 1319–31. 10.1161/CIRCRESAHA.110.217117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J 1999; 13(1): 23–30. 10.1096/fasebj.13.1.23 [DOI] [PubMed] [Google Scholar]
- [56].Wells L, Hart GW. O-GlcNAc turns twenty: functional implications for post-translational modification of nuclear and cytosolic proteins with a sugar. FEBS Lett 2003; 546(1): 154–8. 10.1016/S0014-5793(03)00641-0 [DOI] [PubMed] [Google Scholar]
- [57].Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem 1984; 259(5): 3308–17. [PubMed] [Google Scholar]
- [58].Copeland RJ, Bullen JW, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab 2008; 295(1): E17–28. 10.1152/ajpendo.90281.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hahne H, Sobotzki N, Nyberg T, et al. Proteome wide purification and identification of O-GlcNAc-modified proteins using click chemistry and mass spectrometry. J Proteome Res 2013; 12(2): 927–36. 10.1021/pr300967y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zachara NE, Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta 2006; 1761(5–6): 599–617. 10.1016/j.bbalip.2006.04.007 [DOI] [PubMed] [Google Scholar]
- [61].Buse MG. Hexosamines, insulin resistance, and the complications of diabetes: current status. Am J Physiol Endocrinol Metab 2006; 290(1): E1–8. 10.1152/ajpendo.00329.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hu Y, Riesland L, Paterson AJ, Kudlow JE. Phosphorylation of mouse glutamine-fructose-6-phosphate amidotransferase 2 (GFAT2) by cAMP-dependent protein kinase increases the enzyme activity. J Biol Chem 2004; 279(29): 29988–93. 10.1074/jbc.M401547200 [DOI] [PubMed] [Google Scholar]
- [63].Oki T, Yamazaki K, Kuromitsu J, Okada M, Tanaka I. cDNA cloning and mapping of a novel subtype of glutamine:fructose-6-phosphate amidotransferase (GFAT2) in human and mouse. Genomics 1999; 57(2): 227–34. 10.1006/geno.1999.5785 [DOI] [PubMed] [Google Scholar]
- [64].Dai W, Dierschke SK, Toro AL, Dennis MD. Consumption of a high fat diet promotes protein O-GlcNAcylation in mouse retina via NR4A1-dependent GFAT2 expression. Biochim Biophys Acta Mol Basis Dis 2018; 1864(12): 3568–76. 10.1016/j.bbadis.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Clark RJ, McDonough PM, Swanson E, et al. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem 2003; 278(45): 44230–7. 10.1074/jbc.M303810200 [DOI] [PubMed] [Google Scholar]
- [66].Konrad RJ, Kudlow JE. The role of O-linked protein glycosylation in beta-cell dysfunction. Int J Mol Med 2002; 10(5): 535–9. [PubMed] [Google Scholar]
- [67].Walgren JL, Vincent TS, Schey KL, Buse MG. High glucose and insulin promote O-GlcNAc modification of proteins, including alpha-tubulin. Am J Physiol Endocrinol Metab 2003; 284(2): E424–34. 10.1152/ajpendo.00382.2002 [DOI] [PubMed] [Google Scholar]
- [68].Kim SJ, Yoo WS, Choi M, Chung I, Yoo JM, Choi WS. Increased O-GlcNAcylation of NF-κB Enhances Retinal Ganglion Cell Death in Streptozotocin-induced Diabetic Retinopathy. Curr Eye Res 2016; 41(2): 249–57. 10.3109/02713683.2015.1006372 [DOI] [PubMed] [Google Scholar]
- [69].Gurel Z, Sieg KM, Shallow KD, Sorenson CM, Sheibani N. Retinal O-linked N-acetylglucosamine protein modifications: implications for postnatal retinal vascularization and the pathogenesis of diabetic retinopathy. Mol Vis 2013; 19: 1047–59. [PMC free article] [PubMed] [Google Scholar]
- [70].Xu C, Liu G, Liu X, Wang F. O-GlcNAcylation under hypoxic conditions and its effects on the blood-retinal barrier in diabetic retinopathy. Int J Mol Med 2014; 33(3): 624–32. 10.3892/ijmm.2013.1597 [DOI] [PubMed] [Google Scholar]
- [71].Yki-Järvinen H, Daniels MC, Virkamäki A, Mäkimattila S, De-Fronzo RA, McClain D. Increased glutamine:fructose-6-phosphate amidotransferase activity in skeletal muscle of patients with NIDDM. Diabetes 1996; 45(3): 302–7. 10.2337/diab.45.3.302 [DOI] [PubMed] [Google Scholar]
- [72].Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci USA 2002; 99(8): 5313–8. 10.1073/pnas.072072399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Reiter CE, Sandirasegarane L, Wolpert EB, et al. Characterization of insulin signaling in rat retina in vivo and ex vivo. Am J Physiol Endocrinol Metab 2003; 285(4): E763–74. 10.1152/ajpendo.00507.2002 [DOI] [PubMed] [Google Scholar]
- [74].Barber AJ, Nakamura M, Wolpert EB, et al. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem 2001; 276(35): 32814–21. 10.1074/jbc.M104738200 [DOI] [PubMed] [Google Scholar]
- [75].Andreozzi F, D’Alessandris C, Federici M, et al. Activation of the hexosamine pathway leads to phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 and impairs the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin insulin biosynthetic pathway in RIN pancreatic beta-cells. Endocrinology 2004; 145(6): 2845–57. 10.1210/en.2003-0939 [DOI] [PubMed] [Google Scholar]
- [76].Yang X, Ongusaha PP, Miles PD,et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature 2008; 451(7181): 964–9. 10.1038/nature06668 [DOI] [PubMed] [Google Scholar]
- [77].Whelan SA, Dias WB, Thiruneelakantapillai L, Lane MD, Hart GW. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked β-N-acetylglucosamine in 3T3-L1 adipocytes. J Biol Chem 2010; 5204–11. 10.1074/jbc.M109.077818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Patti ME, Virkamäki A, Landaker EJ, Kahn CR, Yki-Järvinen H. Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance of early postreceptor insulin signaling events in skeletal muscle. Diabetes 1999; 48(8): 1562–71. 10.2337/diabetes.48.8.1562 [DOI] [PubMed] [Google Scholar]
- [79].D’Alessandris C, Andreozzi F, Federici M, et al. Increased O-glycosylation of insulin signaling proteins results in their impaired activation and enhanced susceptibility to apoptosis in pancreatic beta-cells. FASEB J 2004; 18(9): 959–61. 10.1096/fj.03-0725fje [DOI] [PubMed] [Google Scholar]
- [80].Nakamura M, Barber AJ, Antonetti DA,et al. Excessive hexosamines block the neuroprotective effect of insulin and induce apoptosis in retinal neurons. J Biol Chem 2001; 43748–55. 10.1074/jbc.M108594200 [DOI] [PubMed] [Google Scholar]
- [81].Gurel Z, Zaro BW, Pratt MR, Sheibani N. Identification of O-GlcNAc modification targets in mouse retinal pericytes: implication of p53 in pathogenesis of diabetic retinopathy. PLoS One 2014; 9(5): e95561. 10.1371/journal.pone.0095561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Huang Q, Sheibani N. High glucose promotes retinal endothelial cell migration through activation of Src, PI3K/Akt1/eNOS, and ERKs. Am J Physiol Cell Physiol 2008; 295(6): C1647–57. 10.1152/ajpcell.00322.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Donovan K, Alekseev O, Qi X, Cho W, Azizkhan-Clifford J. O-GlcNAc modification of transcription factor Sp1 mediates hyperglycemia-induced VEGF-A upregulation in retinal cells. Invest Ophthalmol Vis Sci 2014; 55(12): 7862–73. 10.1167/iovs.14-14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Matthews JA, Belof JL, Acevedo-Duncan M, Potter RL. Glu-cosamine-induced increase in Akt phosphorylation corresponds to increased endoplasmic reticulum stress in astroglial cells. Mol Cell Biochem 2007; 298(1–2): 109–23. 10.1007/s11010-006-9358-5 [DOI] [PubMed] [Google Scholar]
- [85].Mao X, Zhang D, Tao T, et al. O-GlcNAc glycosylation p27(kip1) promotes astrocyte migration and functional recovery after spinal cord contusion. Exp Cell Res 2015; 339(2): 197–205. 10.1016/j.yexcr.2015.11.007 [DOI] [PubMed] [Google Scholar]
- [86].Johnson JH, Newgard CB, Milburn JL, Lodish HF, Thorens B. The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J Biol Chem 1990; 265(12): 6548–51. [PubMed] [Google Scholar]
- [87].Watanabe T, Mio Y, Hoshino FB, Nagamatsu S, Hirosawa K, Nakahara K. GLUT2 expression in the rat retina: localization at the apical ends of Müller cells. Brain Res 1994; 655(1–2): 128–34. 10.1016/0006-8993(94)91606-3 [DOI] [PubMed] [Google Scholar]
- [88].Capozzi ME, Giblin MJ, Penn JS. Palmitic acid induces Muller cell inflammation that is potentiated by co-treatment with glucose. Sci Rep 2018; 8(1): 5459. 10.1038/s41598-018-23601-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Capozzi ME, McCollum GW, Cousins DB, Penn JS. Linoleic acid is a diabetes-relevant stimulator of retinal inflammation in human retinal Muller cells and microvascular endothelial cells. J Diabetes Metab 2016; 7(12): 7. 10.4172/2155-6156.1000718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature 1998; 393(6686): 684–8. 10.1038/31474 [DOI] [PubMed] [Google Scholar]
- [91].Zachara NE. The roles of O-linked β-N-acetylglucosamine in cardiovascular physiology and disease. Am J Physiol Heart Circ Physiol 2012; 302(10): H1905–18. 10.1152/ajpheart.00445.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Comer FI, Hart GW. O-GlcNAc and the control of gene expression. Biochim Biophys Acta 1999; 1473(1): 161–71. 10.1016/S0304-4165(99)00176-2 [DOI] [PubMed] [Google Scholar]
- [93].Issad T, Kuo M. O-GlcNAc modification of transcription factors, glucose sensing and glucotoxicity. Trends Endocrinol Metab 2008; 19(10): 380–9. 10.1016/j.tem.2008.09.001 [DOI] [PubMed] [Google Scholar]
- [94].Nagel AK, Ball LE. Intracellular protein O-GlcNAc modification integrates nutrient status with transcriptional and metabolic regulation. Adv Cancer Res 2015; 126: 137–66. 10.1016/bs.acr.2014.12.003 [DOI] [PubMed] [Google Scholar]
- [95].Hart GW. Nutrient regulation of signaling and transcription. J Biol Chem 2019; 294(7): 2211–31. 10.1074/jbc.AW119.003226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA. Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. J Biol Chem 2012; 287(28): 23549–61. 10.1074/jbc.M111.330910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lewis BA, Burlingame AL, Myers SA. Human RNA Polymerase II Promoter Recruitment in Vitro Is Regulated by O-Linked N-Acetylglucosaminyltransferase (OGT). J Biol Chem 2016; 291(27): 14056–61. 10.1074/jbc.M115.684365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Resto M, Kim BH, Fernandez AG, Abraham BJ, Zhao K, Lewis BA. O-GlcNAcase Is an RNA Polymerase II Elongation Factor Coupled to Pausing Factors SPT5 and TIF1β. J Biol Chem 2016; 291(43): 22703–13. 10.1074/jbc.M116.751420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wang P, Peng C, Liu X, et al. OGT mediated histone H2B S112 GlcNAcylation regulates DNA damage response. J Genet Genomics 2015; 42(9): 467–75. 10.1016/j.jgg.2015.07.002 [DOI] [PubMed] [Google Scholar]
- [100].Leturcq M, Lefebvre T, Vercoutter-Edouart AS. O-GlcNAcylation and chromatin remodeling in mammals: an up-to-date overview. Biochem Soc Trans 2017; 45(2): 323–38. 10.1042/BST20160388 [DOI] [PubMed] [Google Scholar]
- [101].Fujiki R, Hashiba W, Sekine H, et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 2011; 480(7378): 557–60. 10.1038/nature10656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhang Y, Qu Y, Niu T, Wang H, Liu K. O-GlcNAc modification of Sp1 mediates hyperglycaemia-induced ICAM-1 up-regulation in endothelial cells. Biochem Biophys Res Commun 2017; 484(1): 79–84. 10.1016/j.bbrc.2017.01.068 [DOI] [PubMed] [Google Scholar]
- [103].Baltimore D NF-κB is 25. Nat Immunol 2011; 12(8): 683–5. 10.1038/ni.2072 [DOI] [PubMed] [Google Scholar]
- [104].Zhao Y, Krishnamurthy B, Mollah ZU, Kay TW, Thomas HE. NF-κB in type 1 diabetes. Inflamm Allergy Drug Targets 2011; 10(3): 208–17. 10.2174/187152811795564046 [DOI] [PubMed] [Google Scholar]
- [105].Liu Y, Beyer A, Aebersold R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016; 165(3): 535–50. 10.1016/j.cell.2016.03.014 [DOI] [PubMed] [Google Scholar]
- [106].Wellensiek BP, Larsen AC, Stephens B, et al. Genome-wide profiling of human cap-independent translation-enhancing elements. Nat Methods 2013; 10(8): 747–50. 10.1038/nmeth.2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Powers ET. Translation: An O-GlcNAc stamp of approval. Nat Chem Biol 2015; 11(5): 307–8. 10.1038/nchembio.1777 [DOI] [PubMed] [Google Scholar]
- [108].Zeidan Q, Wang Z, De Maio A, Hart GW. O-GlcNAc cycling enzymes associate with the translational machinery and modify core ribosomal proteins. Mol Biol Cell 2010; 21(12): 1922–36. 10.1091/mbc.e09-11-0941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dierschke SK, Miller WP, Favate JS, et al. O-GlcNAcylation alters the selection of mRNAs for translation and promotes 4E-BP1-dependent mitochondrial dysfunction in the retina. J Biol Chem 2019; 294(14): 5508–20. 10.1074/jbc.RA119.007494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Pestova TV, Kolupaeva VG, Lomakin IB, et al. Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci USA 2001; 98(13): 7029–36. 10.1073/pnas.111145798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].López-Lastra M, Rivas A, Barría MI. Protein synthesis in eukaryotes: the growing biological relevance of cap-independent translation initiation. Biol Res 2005; 38(2–3): 121–46. 10.4067/S0716-97602005000200003 [DOI] [PubMed] [Google Scholar]
- [112].Svitkin YV, Herdy B, Costa-Mattioli M, Gingras AC, Raught B, Sonenberg N. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol Cell Biol 2005; 25(23): 10556–65. 10.1128/MCB.25.23.10556-10565.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Pestova TV, Hellen CU, Shatsky IN. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol 1996; 16(12): 6859–69. 10.1128/MCB.16.12.6859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Schrufer TL, Antonetti DA, Sonenberg N, Kimball SR, Gardner TW, Jefferson LS. Ablation of 4E-BP1/2 prevents hyperglycemia-mediated induction of VEGF expression in the rodent retina and in Muller cells in culture. Diabetes 2010; 59(9): 2107–16. 10.2337/db10-0148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Dennis MD, Schrufer TL, Bronson SK, Kimball SR, Jefferson LS. Hyperglycemia-induced O-GlcNAcylation and truncation of 4E-BP1 protein in liver of a mouse model of type 1 diabetes. J Biol Chem 2011; 286(39): 34286–97. 10.1074/jbc.M111.259457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Miller WP, Mihailescu ML, Yang C, et al. The Translational Repressor 4E-BP1 Contributes to Diabetes-Induced Visual Dysfunction. Invest Ophthalmol Vis Sci 2016; 57(3): 1327–37. 10.1167/iovs.15-18719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci 1996; 21(7): 267–71. 10.1016/S0968-0004(96)10031-1 [DOI] [PubMed] [Google Scholar]
- [118].Bornes S, Prado-Lourenco L, Bastide A, et al. Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress. Circ Res 2007; 100(3): 305–8. 10.1161/01.RES.0000258873.08041.c9 [DOI] [PubMed] [Google Scholar]
- [119].Bastide A, Karaa Z, Bornes S,et al. An upstream open reading frame within an IRES controls expression of a specific VEGF-A isoform. Nucleic Acids Res 2008; 36(7): 2434–45. 10.1093/nar/gkn093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Morris MJ, Negishi Y, Pazsint C, Schonhoft JD, Basu S. An RNA G-quadruplex is essential for cap-independent translation initiation in human VEGF IRES. J Am Chem Soc 2010; 17831–9. 10.1021/ja106287x [DOI] [PubMed] [Google Scholar]
- [121].Huez I, Créancier L, Audigier S, Gensac MC, Prats AC, Prats H. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Mol Cell Biol 1998; 18(11): 6178–90. 10.1128/MCB.18.11.6178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Dennis MD, Shenberger JS, Stanley BA, Kimball SR, Jefferson LS. Hyperglycemia mediates a shift from cap-dependent to cap-independent translation via a 4E-BP1-dependent mechanism. Diabetes 2013; 62(7): 2204–14. 10.2337/db12-1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Dennis MD, Kimball SR, Fort PE, Jefferson LS. Regulated in development and DNA damage 1 is necessary for hyperglycemia-induced vascular endothelial growth factor expression in the retina of diabetic rodents. J Biol Chem 2015; 290(6): 3865–74. 10.1074/jbc.M114.623058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Portillo JC, Lopez Corcino Y, Miao Y,et al. CD40 in Retinal Müller Cells Induces P2X7-Dependent Cytokine Expression in Macrophages/Microglia in Diabetic Mice and Development of Early Experimental Diabetic Retinopathy. Diabetes 2017; 66(2): 483–93. 10.2337/db16-0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci USA 2009; 106(33): 13939–44. 10.1073/pnas.0907143106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Jang I, Kim HB, Seo H,et al. O-GlcNAcylation of eIF2α regulates the phospho-eIF2α-mediated ER stress response. Biochim Biophys Acta 2015; 1853(8): 1860–9. 10.1016/j.bbamcr.2015.04.017 [DOI] [PubMed] [Google Scholar]
- [127].Jo S, Lockridge A, Alejandro EU. eIF4G1 and carboxypeptidase E axis dysregulation in O-GlcNAc transferase-deficient pancreatic β-cells contributes to hyperproinsulinemia in mice. J Biol Chem 2019; 294(35): 13040–50. 10.1074/jbc.RA119.008670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Li X, Zhu Q, Shi X, et al. O-GlcNAcylation of core components of the translation initiation machinery regulates protein synthesis. Proc Natl Acad Sci USA 2019; 116(16): 7857–66. 10.1073/pnas.1813026116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Gallie DR. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev 1991; 5(11): 2108–16. 10.1101/gad.5.11.2108 [DOI] [PubMed] [Google Scholar]
- [130].Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell 2010; 40(2): 253–66. 10.1016/j.molcel.2010.10.006 [DOI] [PubMed] [Google Scholar]
- [131].Zhang X, Shu XE, Qian SB. O-GlcNAc modification of eIF4GI acts as a translational switch in heat shock response. Nat Chem Biol 2018; 14(10): 909–16. 10.1038/s41589-018-0120-6 [DOI] [PubMed] [Google Scholar]
- [132].Phipps JA, Dixon MA, Jobling AI, et al. The renin-angiotensin system and the retinal neurovascular unit: A role in vascular regulation and disease. Exp Eye Res 2019; 187: 107753. 10.1016/j.exer.2019.107753 [DOI] [PubMed] [Google Scholar]
- [133].Choudhary R, Kapoor MS, Singh A, Bodakhe SH. Therapeutic targets of renin-angiotensin system in ocular disorders. J Curr Ophthalmol 2016; 29(1): 7–16. 10.1016/j.joco.2016.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Culman J, Höhle S, Qadri F, et al. Angiotensin as neuromodulator/neurotransmitter in central control of body fluid and electrolyte homeostasis. Clin Exp Hypertens 1995; 17(1–2): 281–93. 10.3109/10641969509087071 [DOI] [PubMed] [Google Scholar]
- [135].Chung O, Kühl H, Stoll M, Unger T. Physiological and pharmacological implications of AT1 versus AT2 receptors. Kidney Int Suppl 1998; 67: S95–9. 10.1046/j.1523-1755.1998.06719.x [DOI] [PubMed] [Google Scholar]
- [136].Fletcher EL, Phipps JA, Ward MM, Vessey KA, Wilkinson-Berka JL. The renin-angiotensin system in retinal health and disease: Its influence on neurons, glia and the vasculature. Prog Retin Eye Res 2010; 29(4): 284–311. 10.1016/j.preteyeres.2010.03.003 [DOI] [PubMed] [Google Scholar]
- [137].Wang B, Wang F, Zhang Y, et al. Effects of RAS inhibitors on diabetic retinopathy: a systematic review and meta-analysis. Lancet Diabetes Endocrinol 2015; 3(4): 263–74. 10.1016/S2213-8587(14)70256-6 [DOI] [PubMed] [Google Scholar]
- [138].Campbell DJ, Lawrence AC, Towrie A, Kladis A, Valentijn AJ. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension 1991; 18(6): 763–73. 10.1161/01.HYP.18.6.763 [DOI] [PubMed] [Google Scholar]
- [139].Kohara K, Brosnihan KB, Ferrario CM. Angiotensin(1–7) in the spontaneously hypertensive rat. Peptides 1993; 14(5): 883–91. 10.1016/0196-9781(93)90063-M [DOI] [PubMed] [Google Scholar]
- [140].Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1–7). Hypertension 2002; 40(5): 774–9. 10.1161/01.HYP.0000035396.27909.40 [DOI] [PubMed] [Google Scholar]
- [141].Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1–7) by angiotensin-converting enzyme. Hypertension 1998; 31(1 Pt 2): 362–7. 10.1161/01.HYP.31.1.362 [DOI] [PubMed] [Google Scholar]
- [142].Roks AJ, van Geel PP, Pinto YM, et al. Angiotensin-(1–7) is a modulator of the human renin-angiotensin system. Hypertension 1999; 34(2): 296–301. 10.1161/01.HYP.34.2.296 [DOI] [PubMed] [Google Scholar]
- [143].Guimaraes PS, Oliveira MF, Braga JF, et al. Increasing angiotensin-(1–7) levels in the brain attenuates metabolic syndrome-related risks in fructose-fed rats. Hypertension 2014; 63(5): 1078–85. 10.1161/HYPERTENSIONAHA.113.01847 [DOI] [PubMed] [Google Scholar]
- [144].Williams IM, Otero YF, Bracy DP, Wasserman DH, Biaggioni I, Arnold AC. Chronic Angiotensin-(1–7) improves insulin sensitivity in high-fat fed mice independent of blood pressure. Hypertension 2016; 67(5): 983–91. 10.1161/HYPERTENSIONAHA.115.06935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Bui BV, Armitage JA, Tolcos M, Cooper ME, Vingrys AJ. ACE inhibition salvages the visual loss caused by diabetes. Diabetologia 2003; 46(3): 401–8. 10.1007/s00125-003-1042-7 [DOI] [PubMed] [Google Scholar]
- [146].Gilbert RE, Kelly DJ, Cox AJ, et al. Angiotensin converting enzyme inhibition reduces retinal overexpression of vascular endothelial growth factor and hyperpermeability in experimental diabetes. Diabetologia 2000; 43(11): 1360–7. 10.1007/s001250051539 [DOI] [PubMed] [Google Scholar]
- [147].Funatsu H, Yamashita H, Noma H, Mimura T, Sakata K, Hori S. Risk evaluation of outcome of vitreous surgery for proliferative diabetic retinopathy based on vitreous level of vascular endothelial growth factor and angiotensin II. Br J Ophthalmol 2004; 88(8): 1064–8. 10.1136/bjo.2003.032656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Senanayake PD, Bonilha VL, W Peterson J, et al. Retinal angiotensin II and angiotensin-(1–7) response to hyperglycemia and an intervention with captopril. J Renin Angiotensin Aldosterone Syst 2018; 19(3): 1470320318789323. 10.1177/1470320318789323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Verma A, Shan Z, Lei B, et al. ACE2 and Ang-(1–7) confer protection against development of diabetic retinopathy. Mol Ther 2012; 20(1): 28–36. 10.1038/mt.2011.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Singh R, Singh AK, Alavi N, Leehey DJ. Mechanism of increased angiotensin II levels in glomerular mesangial cells cultured in high glucose. J Am Soc Nephrol 2003; 14(4): 873–80. 10.1097/01.ASN.0000060804.40201.6E [DOI] [PubMed] [Google Scholar]
- [151].Hsieh TJ, Fustier P, Zhang SL, et al. High glucose stimulates angiotensinogen gene expression and cell hypertrophy via activation of the hexosamine biosynthesis pathway in rat kidney proximal tubular cells. Endocrinology 2003; 144(10): 4338–49. 10.1210/en.2003-0220 [DOI] [PubMed] [Google Scholar]
- [152].Vidotti DB, Casarini DE, Cristovam PC, Leite CA, Schor N, Boim MA. High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am J Physiol Renal Physiol 2004; 286(6): F1039–45. 10.1152/ajprenal.00371.2003 [DOI] [PubMed] [Google Scholar]
- [153].Marsh SA, Dell’Italia LJ, Chatham JC. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 2011; 40(3): 819–28. 10.1007/s00726-010-0699-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Tanaka T, Sohmiya K, Kono T, et al. Thiamine attenuates the hypertension and metabolic abnormalities in CD36-defective SHR: uncoupling of glucose oxidation from cellular entry accompanied with enhanced protein O-GlcNAcylation in CD36 deficiency. Mol Cell Biochem 2007; 299(1–2): 23–35. 10.1007/s11010-005-9032-3 [DOI] [PubMed] [Google Scholar]
- [155].Einstein FH, Fishman S, Bauman J, et al. Enhanced activation of a “nutrient-sensing” pathway with age contributes to insulin resistance. FASEB J 2008; 22(10): 3450–7. 10.1096/fj.08-109041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Dierschke SK, Toro AL, Barber AJ, Arnold AC, Dennis MD. Angiotensin-(1–7) Attenuates Protein O-GlcNAcylation in the Retina by EPAC/Rap1-Dependent Inhibition of O-GlcNAc Transferase. Invest Ophthalmol Vis Sci 2020; 61(2): 24. 10.1167/iovs.61.2.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Gellai R, Hodrea J, Lenart L, et al. Role of O-linked N-acetylglucosamine modification in diabetic nephropathy. Am J Physiol Renal Physiol 2016; 311(6): F1172–81. 10.1152/ajprenal.00545.2015 [DOI] [PubMed] [Google Scholar]