

Abstract
Background:
Fibrous dysplasia/McCune-Albright syndrome (FD/MAS) is a rare mosaic disorder of Gαs activation. Fibroblast Growth Factor 23 (FGF23)-mediated hypophosphatemia is a feature of FD/MAS that has been associated with poor skeletal outcomes. Standard therapy includes oral phosphorus and vitamin D analogs; however, treatment is limited by potential adverse renal and gastrointestinal effects. Burosumab is a monoclonal antibody to FGF23 approved to treat patients with X-linked hypophosphatemia and tumor-induced osteomalacia. There is currently no safety or efficacy data to support burosumab use in patients with FD/MAS.
Case Description:
An 8-year-old boy with severe FD/MAS presented with persistent hypophosphatemia and skeletal complications despite conventional treatment with oral phosphate and calcitriol. He was started on burosumab and achieved sustained normalization of serum phosphorus and marked improvement in alkaline phosphatase levels. This was accompanied by an encouraging clinical response, including decreased bone pain, improved muscle strength, and improved ambulation. No adverse effects of burosumab therapy were observed.
Conclusions:
This is the first reported case of burosumab treatment in a patient with FD/MAS. The encouraging biochemical and clinical response in this patient highlights the need for future studies to explore the safety and efficacy of burosumab in the FD/MAS pediatric population.
Keywords: FGF23, hypophosphatemia, rickets, osteomalacia, McCune-Albright syndrome
1. INTRODUCTION
Fibrous dysplasia/McCune-Albright syndrome (FD/MAS) is a rare, mosaic disorder of Gαs activation presenting with a variable combination of typical skin hyperpigmentation, hyperfunctioning endocrinopathies, and fibrous dysplasia (FD) of bone [1]. FD lesions arise from impaired differentiation of multipotent skeletal stem cells, resulting in abnormal bone prone to fractures, deformities, and pain [2]. Inappropriate FGF23 production from pathogenic variant-bearing bone cells results in renal phosphate wasting [3 4]. The mechanism of Fibroblast Growth Factor-23 (FGF23) production in FD/MAS is poorly understood, but appears to be related to effects of Gαs in abnormally differentiated osteoprogenitor cells [5]. While most patients with FD/MAS have elevated circulating FGF23 levels, increased cleavage of intact FGF23 to its inactive fragments frequently protects against the development of frank hypophosphatemia [4 5]. However, because FGF23 levels are correlated with overall FD tissue burden, frank hypophosphatemia may occur in patients with extensive skeletal involvement [6].
Similar to other disorders of FGF23 excess, management of hypophosphatemia in FD/MAS has traditionally focused on repletion with oral phosphate and vitamin D analogs [5]. While this regimen is effective and well-tolerated in many patients, some may experience potentially dose-limiting side effects, including gastrointestinal intolerance, hypercalciuria and adverse renal effects [7]. Burosumab, a human recombinant monoclonal antibody to FGF23, represents a new, targeted treatment approach. Multiple clinical trials have demonstrated safe, efficacious use of burosumab in X-linked hypophosphatemia and tumor-induced osteomalacia, leading to improved serum phosphorus levels and skeletal outcomes without adverse gastrointestinal or renal effects [8–11]. Based on these studies, burosumab recently received approval from the Food and Drug Administration and European Medicines Agency for treatment of X-linked hypophosphatemia and tumor-induced osteomalacia [12]. While the underlying pathophysiology of FGF23 excess is shared between these conditions, there is no literature regarding the use of burosumab in FD/MAS.
2. CASE PRESENTATION
A 7-year-2 month old boy with FD/MAS presented for consideration of burosumab therapy due to FGF23-mediated hypophosphatemia and skeletal complications, which were not controlled with conventional therapy. The patient was diagnosed with FD/MAS at 1 month of age, after hyperpigmented macules were noted on his face, chest, abdomen, back and buttocks, with typical features including irregular borders and location respecting the midline of the body (Fig 1C). He was born small for gestational age and had early feeding difficulties with severe gastroesophageal reflux, and laboratory evaluation revealed non-autoimmune hyperthyroidism, confirming a clinical diagnosis of MAS [6 13]. At age 14 months his parents reported signs of bone pain, including crying and reluctance to bear weight on his lower extremities. Radiographic evaluation revealed diffuse FD involving most of his skeleton and rachitic changes (Fig 1A, D&E). Laboratory workup confirmed a low serum phosphorus of 2.0 mg/dL (normal age-related range 4.5-6.5) and elevated alkaline phosphatase of 2,367 U/L (normal age-related range 100-350)[14]. He was subsequently found to have an inappropriately high-normal serum FGF23 level of 230 RU/mL (normal <290), confirming the diagnosis of FGF23-mediated hypophosphatemia. The patient was initiated on oral phosphorus and calcitriol supplements with improvement in serum phosphorus levels (Fig 2), however treatment was complicated by gastrointestinal intolerance and malabsorption. Ongoing feeding difficulties necessitated placement of a gastrostomy-jejunostomy tube at age 6 months. Abdominal ultrasound and endoscopy revealed no obvious abnormalities. He received intensive feeding therapy and was able to transition to an oral diet by age 4 years, but continued to have intermittent vomiting and loose stools, which were exacerbated by phosphorus supplements. The patient was maintained on calcitriol therapy and monitored with PTH and urine calcium levels, however he developed intermittent hypercalciuria and bilateral nephrocalcinosis. Over the next several years phosphorus levels ranged 3.1-3.8 mg/dL, while on treatment with up to 55 mg/kg of phosphorus and 80 ng/kg of calcitriol daily.
Figure 1.
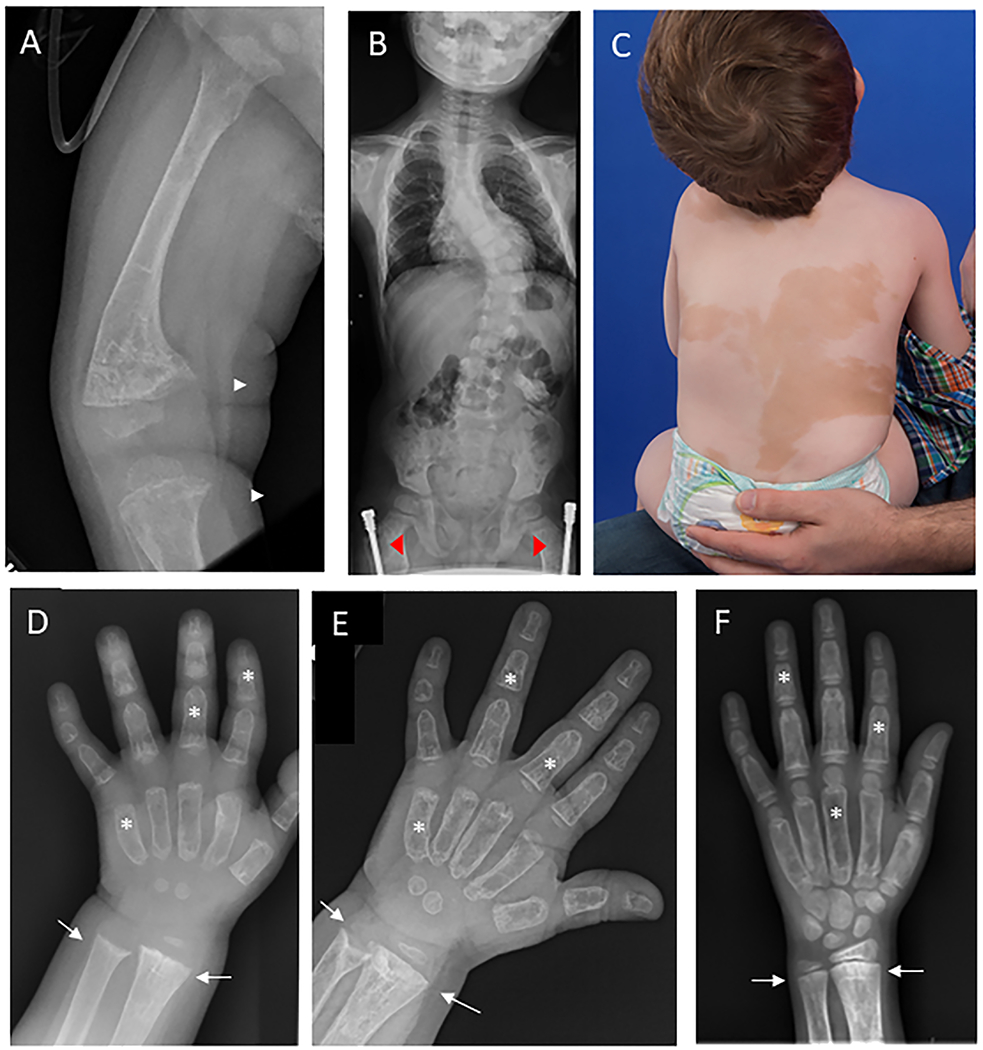
Representative clinical images. A) Radiograph at the time of hypophosphatemia diagnosis shows severe anterolateral femoral bowing and widened, frayed metaphyses at the distal femur and proximal tibia (white arrows), consistent with active rickets. The bones are diffusely involved with fibrous dysplasia, as evidenced by homogeneous “ground glass” lucency and severe cortical thinning. B) Spinal radiograph shows moderate-severe thoracolumbar scoliosis with a Cobb angle of approximately 40 degrees. Note the presence of bilateral femoral implants (red arrowheads), placed for correction of fractures and deformities. C) Photograph shows large areas of skin hyperpigmentation with features typical of McCune-Albright syndrome, including irregular borders and distribution reflecting along the midline of the body. D) hand radiograph at the time of hypophosphatemia diagnosis shows metaphyseal flaring and widening at the distal radial and ulnar metaphyses (white arrows). Note the presence of diffuse fibrous dysplasia involving all bones of the hand, leading to “ground glass” lucency and widening of the metacarpals and phalanges (white stars). E) Radiograph taken six months after starting conventional treatment demonstrates improvement in rachitic change, however there is evidence of persistent metaphyseal cupping in both the radius and ulna. F) Hand radiograph after 5 months of burosumab shows no evidence of rachitic changes at the distal radius and ulna..
Figure 2.
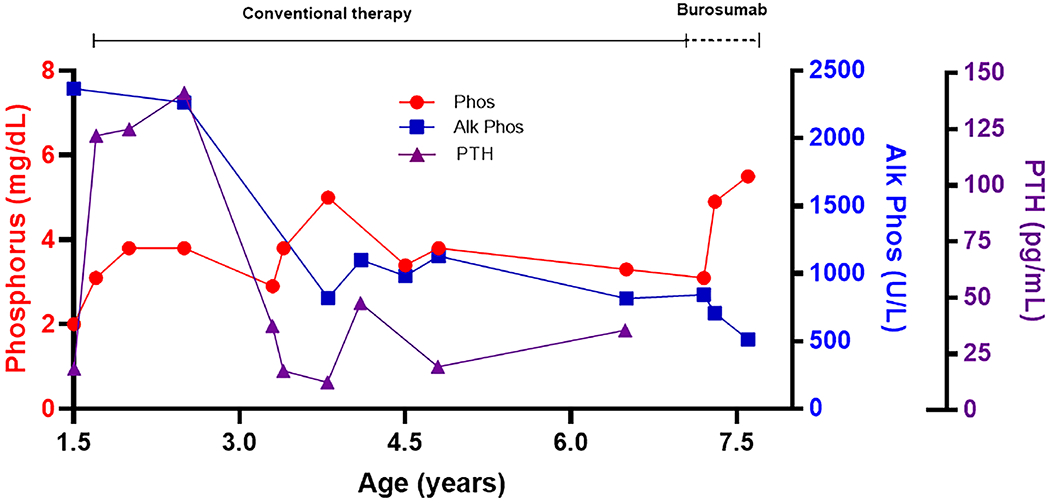
Biochemical response. A graph shows changes in serum phosphorus (normal range age 5-13 years: 3.7-5.4 U/L), alkaline phosphatase (normal range age 1-10 years: 142-335 U/L), and parathyroid hormone (normal range 12 months-10 years: 11-59 pg/mL) levels during conventional therapy and treatment with burosumab.
Throughout childhood he developed complications of severe FD. His first fracture occurred at age 2, and by presentation at age 7 years 2 months he had suffered 17 fractures, including bilateral femurs, tibias, ulnas, and radii, requiring surgical intervention and intermittent use of assistive ambulation devices. Additional sequelae included bowing deformities of his lower extremities (Fig 1A) and severe thoracolumbar scoliosis treated with bracing (Fig 1B). He continued to experience chronic bone pain, which impacted his daily activities. Zoledronate was initiated at age 4 years, however after his first infusion he developed electrolyte abnormalities including hyperchloremic acidosis, hypokalemia, and proteinuria, concerning for potential bisphosphonate-induced Fanconi syndrome [15]. His renal tubular function resolved over the following weeks, and additional bisphosphonate treatment was not attempted.
Additional MAS endocrinopathies included hyperthyroidism diagnosed at age 4 months, initially managed with methimazole followed by thyroidectomy at age 4 years. Post-surgical hypothyroidism was well-controlled with thyroid hormone replacement. Growth hormone excess was diagnosed at age 3 years and was well-controlled with monthly octreotide injections. Precocious puberty was diagnosed at age 5 years and well-controlled with letrozole and bicalutamide.
Because of concerns about the potential contribution of persistent hypophosphatemia to skeletal morbidity and pain, the patient was initiated on burosumab at age 7 years 2 months. Oral phosphorus and calcitriol were stopped and a serum phosphorus level one week later was confirmed low at 3.1 mg/dL (normal age-related range 3.6-5.8) (Fig 2). He received two 1.1 mg/kg subcutaneous injections dosed 2 weeks apart, and labs performed 4 weeks after his second injection showed a mid-normal phosphorus level of 4.9 mg/dL. Treatment was initiated off-label as a clinically indicated intervention. Of note, the burosumab dose chosen based on the recommended 0.8 mg/kg rounded up to the nearest 10 mg, which resulted in a starting dose of 20 mg (1.1 mg/kg) in this 19 kg child. He was subsequently maintained on biweekly burosumab with sustained normalization of serum phosphate and improvement in alkaline phosphatase (Fig 2). A bone age exam obtained after 17 months of therapy revealed no rachitic changes (Fig 1F). After several months of burosumab the patient reported clinical improvement in bone pain, strength, and stamina. He has had no fractures in the 17 months since starting burosumab, has had no additional surgeries, and has not required use of assistive ambulation devices. No adverse effects of burosumab were observed.
3. DISCUSSION
FGF23-mediated hypophosphatemia is an important contributor to skeletal morbidity in patients with FD/MAS. In addition to typical pain and rachitic complications, studies have demonstrated an association between hypophosphatemia and FD-related fractures [16], progressive scoliosis [17], and skull-based deformities [18]. Because FD tissue is inherently poorly mineralized and structurally unsound, this may reflect an increased vulnerability to the deleterious effects of hypophosphatemia compared to typical bone. This has important potential clinical implications, because it is conceivable that FD-related complications may arise in patients with mild hypophosphatemia even in the absence of active metaphyseal changes [1].
Conventional therapy for hypophosphatemia has limitations which can create particular difficulties in patients with FD/MAS. Because FGF23 impairs 1α-hydroxylase activity, administration of active vitamin D analogs is necessary to maintain mineral metabolism and prevent secondary hyperparathyroidism. However, because these analogs increase calcium absorption, they carry a dose-related risk of hypercalciuria and renal complications [7 19]. Normalization of serum phosphorus is therefore not a practical goal with conventional treatment, which may place patients with FD/MAS at continued risk of skeletal complications from persistent hypophosphatemia. Oral phosphate supplements are well-known gastrointestinal irritants [7 19], which can be problematic in patients with FD/MAS who are at increased risk for gastrointestinal disease [20 21]. In this patient, chronic malabsorption with symptoms of vomiting and diarrhea limited his tolerability to phosphorus supplements. Treatment with calcitriol was also limited by the development of nephrocalcinosis, which was associated with intermittent hypercalciuria and decreased PTH levels. The patient reported in this case thus experienced both renal and gastrointestinal complications with conventional therapy, with persistent hypophosphatemia despite maximal conventional management.
Given these limitations, burosumab is an intuitive choice for management of hypophosphatemia in FD/MAS. In the patient reported in this case, serum phosphorus was maintained in the middle of the normal range, which may potentially explain his favorable clinical response. However, additional research into the safety and efficacy of burosumab is needed before it can be used routinely in patients with FD/MAS. The potential for FD tissue effects is an important consideration; it is unknown whether burosumab may impact skeletal stem cell proliferation or other drivers of FD lesion growth and activity. In addition, studies are needed to determine if burosumab may have beneficial effects on lesional mineralization, an inherent feature of FD even in the absence of frank hypophosphatemia [2]. The high financial cost of burosumab should also be considered. At present, use of burosumab in FD should be limited to clinical trials, and as compassionate use for patients with significant skeletal complications who cannot be managed with conventional therapy.
Monitoring the efficacy of hypophosphatemia treatment in FD/MAS also presents unique challenges. Lower extremity bowing deformities are an established complication of X-linked hypophosphatemia, and prevention of these deformities is a primary treatment goal [7 19]. Studies similarly support that hypophosphatemia is a contributor to skeletal deformities in FD/MAS, and optimal metabolic management is likely to decrease their severity and progression. However, deformities in FD are likely driven primarily by structural instability and can occur even in the absence of hypophosphatemia. Linear growth velocity is complicated by these deformities and other MAS endocrinopathies, making it another unreliable measure of control. In patients with X-linked hypophosphatemia and tumor-induced osteomalacia, serum bone turnover markers correlate with osteomalacia and are useful in monitoring treatment efficacy [7 19 22]. However, FD lesions demonstrate inherent high bone turnover, resulting in elevated formation and resorption markers that correlate with overall disease burden [2 23]. As seen in the patient reported here, a relative decrease in turnover markers may potentially occur with improved hypophosphatemia management, however they cannot be used as consistent indicators of efficacy. Monitoring hypophosphatemia treatment in patients with FD/MAS is therefore complex and must include multiple domains, including serum and urine markers, radiographic metaphyseal appearance, and assessment of bone pain, functional parameters, and skeletal outcomes.
Muscle weakness is common in patients with FD/MAS and may impact mobility [24]; it is possible that hypophosphatemia may exacerbate weakness and ambulation issues. The reported subjective improvements in strength and ambulation suggest that burosumab may have had functional benefits this patient. However additional studies in patients with FD/MAS including formal musculoskeletal assessment are needed.
4. CONCLUSION
This case demonstrates the beneficial effect of burosumab in a child with severe FD/MAS who was not controlled with conventional therapy. Given the pathophysiologic role of FGF23 excess in FD, these findings are likely to be replicated in other patients and support the need for further investigation of burosumab in the FD/MAS population.
Funding:
This work was funded in part by the Intramural Research Program of the NIH, NIDCR.
Declarations of competing interest:
NIDCR receives funding from Ultragenyx Pharmaceuticals, Inc and Amgen, Inc for studies related to this work.
Abbreviations:
- FD
fibrous dysplasia
- MAS
McCune-Albright syndrome
- FGF23
fibroblast growth factor 23
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Boyce AM, Collins MT. Fibrous Dysplasia/McCune-Albright Syndrome: A Rare, Mosaic Disease of Galpha s Activation. Endocr Rev 2020;41(2) doi: 10.1210/endrev/bnz011[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riminucci M, Liu B, Corsi A, et al. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol 1999;187(2):249–58 doi: [published Online First: Epub Date]∣. [DOI] [PubMed] [Google Scholar]
- 3.Riminucci M, Collins MT, Fedarko NS, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 2003;112(5):683–92 doi: 10.1172/jci18399[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharyya N, Wiench M, Dumitrescu C, et al. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res 2012;27(5):1132–41 doi: 10.1002/jbmr.1546[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyce AM, Bhattacharyya N, Collins MT. Fibrous dysplasia and fibroblast growth factor-23 regulation. Curr Osteoporos Rep 2013;11(2):65–71 doi: 10.1007/s11914-013-0144-5[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis 2012;7 Suppl 1(Suppl 1):S4 doi: 10.1186/1750-1172-7-s1-s4[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect 2014;3(1):R13–30 doi: 10.1530/ec-13-0103[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carpenter TO, Whyte MP, Imel EA, et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N Engl J Med 2018;378(21):1987–98 doi: 10.1056/NEJMoa1714641[published Online First: Epub Date]∣. [DOI] [PubMed] [Google Scholar]
- 9.Imel EA, Glorieux FH, Whyte MP, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet 2019;393(10189):2416–27 doi: 10.1016/s0140-6736(19)30654-3[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Insogna KL, Briot K, Imel EA, et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J Bone Miner Res 2018;33(8):1383–93 doi: 10.1002/jbmr.3475[published Online First: Epub Date]∣. [DOI] [PubMed] [Google Scholar]
- 11.Jan de Beur SM, Miller PD, Weber TJ, et al. Burosumab for the Treatment of Tumor-Induced Osteomalacia. J Bone Miner Res 2020. doi: 10.1002/jbmr.4233[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crysvita [package insert]. Novato, CA: Ultragenyx Pharmaceuticals Inc. 2020 [Google Scholar]
- 13.Javaid MK, Boyce A, Appelman-Dijkstra N, et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis 2019;14(1):139 doi: 10.1186/s13023-019-1102-9[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.K/DOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Children with Chronic Kidney Disease. American Journal of Kidney Diseases 2005;46 [Google Scholar]
- 15.Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008;74(11):1385–93 doi: 10.1038/ki.2008.356[published Online First: Epub Date]∣. [DOI] [PubMed] [Google Scholar]
- 16.Leet AI, Chebli C, Kushner H, et al. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res 2004;19(4):571–7 doi: 10.1359/jbmr.0301262[published Online First: Epub Date]∣. [DOI] [PubMed] [Google Scholar]
- 17.Berglund JA, Tella SH, Tuthill KF, et al. Scoliosis in Fibrous Dysplasia/McCune-Albright Syndrome: Factors Associated With Curve Progression and Effects of Bisphosphonates. J Bone Miner Res 2018;33(9):1641–48 doi: 10.1002/jbmr.3446[published Online First: Epub Date]∣. [DOI] [PubMed] [Google Scholar]
- 18.Pan KS, Heiss JD, Brown SM, Collins MT, Boyce AM. Chiari I Malformation and Basilar Invagination in Fibrous Dysplasia: Prevalence, Mechanisms, and Clinical Implications. J Bone Miner Res 2018;33(11):1990–98 doi: 10.1002/jbmr.3531[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen LH, Rahbek ET, Beck-Nielsen SS, Christesen HT. Treatment of hypophosphataemic rickets in children remains a challenge. Dan Med J 2014;61(7):A4874. [PubMed] [Google Scholar]
- 20.Robinson C, Estrada A, Zaheer A, et al. Clinical and Radiographic Gastrointestinal Abnormalities in McCune-Albright Syndrome. J Clin Endocrinol Metab 2018;103(11):4293–303 doi: 10.1210/jc.2018-01022[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood LD, Noë M, Hackeng W, et al. Patients with McCune-Albright syndrome have a broad spectrum of abnormalities in the gastrointestinal tract and pancreas. Virchows Arch 2017;470(4):391–400 doi: 10.1007/s00428-017-2086-2[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Florenzano P, Hartley IR, Jimenez M, Roszko K, Gafni RI, Collins MT. Tumor-Induced Osteomalacia. Calcif Tissue Int 2021;108(1):128–42 doi: 10.1007/s00223-020-00691-6[published Online First: Epub Date]∣. [DOI] [PubMed] [Google Scholar]
- 23.Florenzano P, Pan KS, Brown SM, et al. Age-Related Changes and Effects of Bisphosphonates on Bone Turnover and Disease Progression in Fibrous Dysplasia of Bone. J Bone Miner Res 2019;34(4):653–60 doi: 10.1002/jbmr.3649[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paul SM, Gabor LR, Rudzinski S, et al. Disease severity and functional factors associated with walking performance in polyostotic fibrous dysplasia. Bone 2014;60:41–7 doi: 10.1016/j.bone.2013.11.022[published Online First: Epub Date]∣. [DOI] [PMC free article] [PubMed] [Google Scholar]