

Abstract
Compelling evidence supports vascular contributions to cognitive impairment and dementia (VCID) including Alzheimer’s disease (AD), but the underlying pathogenic mechanisms and treatments are not fully understood. Cis P-tau is an early driver of neurodegeneration resulting from traumatic brain injury, but its role in VCID remains unclear. Here, we found robust cis P-tau despite no tau tangles in patients with VCID and in mice modeling key aspects of clinical VCID, likely because of the inhibition of its isomerase Pin1 by DAPK1. Elimination of cis P-tau in VCID mice using cis-targeted immunotherapy, brain-specific Pin1 overexpression, or DAPK1 knockout effectively rescues VCID-like neurodegeneration and cognitive impairment in executive function. Cis mAb also prevents and ameliorates progression of AD-like neurodegeneration and memory loss in mice. Furthermore, single-cell RNA sequencing revealed that young VCID mice display diverse cortical cell type–specific transcriptomic changes resembling old patients with AD, and the vast majority of these global changes were recovered by cis-targeted immunotherapy. Moreover, purified soluble cis P-tau was sufficient to induce progressive neurodegeneration and brain dysfunction by causing axonopathy and conserved transcriptomic signature found in VCID mice and patients with AD with early pathology. Thus, cis P-tau might play a major role in mediating VCID and AD, and antibody targeting it may be useful for early diagnosis, prevention, and treatment of cognitive impairment and dementia after neurovascular insults and in AD.
INTRODUCTION
Compelling evidence supports vascular contributions to cognitive impairment and dementia (VCID) including Alzheimer’s disease (AD) (1, 2). VCID can be broadly caused by reduced cerebral blood flow due to vascular pathology or dysfunction and is traditionally referred to as vascular dementia (VaD) (1, 2). However, despite insights into links of vascular factors to progressive neurodegeneration and memory loss, the underlying pathogenic mechanisms are unclear and there is no disease-modifying treatment to slow the progression. A neuropathological hallmark of AD (3, 4) and chronic traumatic encephalopathy (CTE) associated with traumatic brain injury (TBI) (5) is neurofibrillary tangles (NFTs) made of hyperphosphorylated tau. Tau hyperphosphorylation disrupts its normal function to bind microtubules, leading to axonopathy including impaired axonal micro-tubules and transport; to compromised neuronal and synaptic function; and to increased propensity for tau oligomerization, aggregation, and tangle formation (3, 4). Ischemic stroke induces some limited tau changes, and tau knockout (KO) reduces stroke-induced acute brain damage in mice (6, 7). However, because there is no NFT in VCID in humans or animal models, VCID is not generally considered as a tauopathy and little is known about the role of tau in the progression from neurovascular insults to neurodegeneration (1, 2).
We have identified a unique phosphorylation-specific proline isomerase, Pin1, that inhibits neurodegeneration in AD by converting the phosphorylated Thr231-Pro motif in tau (P-tau) from cis to trans conformation in cell, animal models, and human AD tissues (8–11). Using recently developed cis-trans conformation–specific monoclonal antibodies, we have identified that cis, but not trans, P-tau is induced upon hypoxic neuronal stress and after TBI in humans and mouse models, with its concentration depending on injury severity and frequency, and correlating well with axonal injury and clinical outcome (12, 13). Unlike physiologic trans isomer, cis P-tau fails to bind or stabilize microtubules and resists protein degradation or dephosphorylation (14) so that it disrupts axonal microtubule network and mitochondrial transport, resulting in axonopathy, spreads to other neurons, and leads to neuron death (12). This process, which we termed “cistauosis,” occurs long before tau oligomerization and tangle formation but can be blocked by cis P-tau monoclonal antibody (cis mAb), which enters cells via Fc receptors and targets nondegradable cis P-tau for Tripartite motif-containing protein (TRIM21)-mediated proteasome degradation (12, 15). Moreover, eliminating cis P-tau with cis mAb in mice after severe or repetitive TBI prevents the development of cistauosis, neuropathology, and brain dysfunction, including CTE-like degeneration after repetitive TBI (12, 13). Others also found that high P-tau (pT231) concentration in the blood correlated well with poor clinical outcome in patients with acute and chronic TBI (16). Thus, cis P-tau is likely the causal molecular link between TBI and CTE, and cis mAb offers a promising therapy for treating tauopathies (12–14, 17), with humanized cis mAb being currently evaluated in clinical trials (18). However, it remains unclear whether cis P-tau plays any role in the progression from neurovascular insults to neurodegeneration and whether cis mAb has efficacy in treating VCID. Here, we uncover that cis P-tau underlies VCID and AD, and can be effectively targeted by immunotherapy in mice.
RESULTS
Cis P-tau is induced in patients with VaD and BCAS mice modeling key aspects of clinical VCID
To explore the role of cis P-tau in VCID, we first examined cis P-tau in human VaD brains. As expected, verified VaD human brains (table S1) displayed robust markers consistent with demyelination (P < 0.003 for Luxol fast blue staining and P < 0.007 for 2′,3′-cyclic nucleotide 3′-phosphodiesterase staining; fig. S1, A and B) and neuroinflammation [P < 0.01 for glial fibrillary acidic protein (GFAP) staining and P = 0.02 for ionized calcium-binding adaptor molecule 1 (Iba1) staining; fig. S1, C and D]. None of nine human VaD brains had any obvious NFTs, in contrast to the patients with AD or mixed AD and VaD pathology (Fig. 1, A to C, and fig. S1, E and F). However, we detected robust cis P-tau signals in the cingulate cortex overlying the corpus callosum in large sections of brains using immunostaining with near-infrared detection (Fig. 1, D and E). Immunostaining with confocal microscopy further confirmed robust cis P-tau signals in all nine patients with VaD, particularly in the same subcortical regions, with little signals in all eight healthy controls (Fig. 1, F and G, and fig. S1G). Cis P-tau was localized to axons (Fig. 1H and fig. S1H), within the surrounding myelin sheath (Fig. 1I), in oligodendrocytes (fig. S1I) and in microvascular endothelial cells (fig. S1J), consistent with the findings that phosphorylated tau has been detected in oligodendrocytes (19) and lung vascular endothelial cells (20). Thus, robust cis P-tau is detected in human VaD brains.
Fig. 1. Robust cis P-tau in axons with no evidence of tau tangles in patients with VaD and BCAS mice.
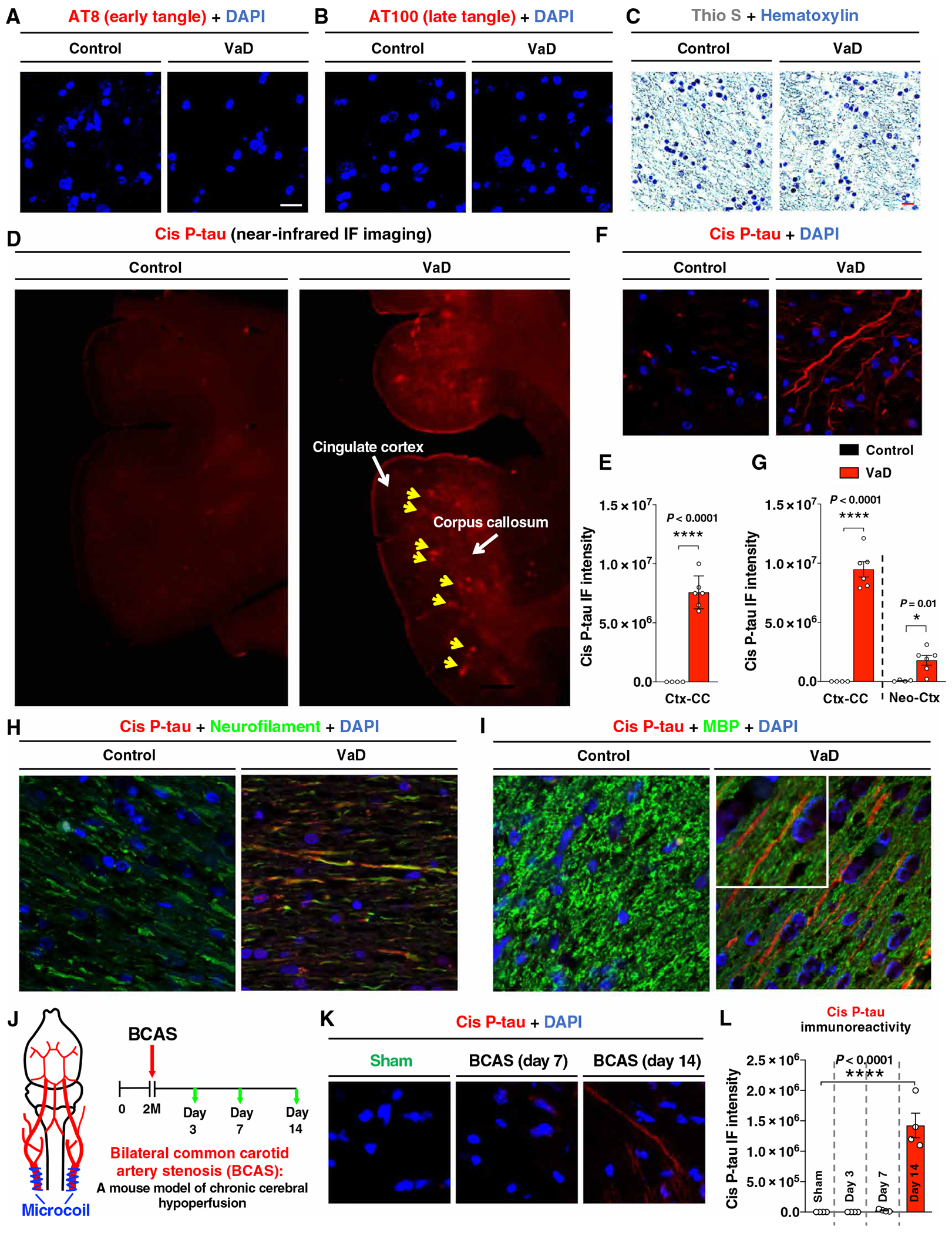
(A to C) Immunofluorescence (IF) staining of brain sections from patients with VaD and age-matched controls with AT8 antibody for early tangle-like structures (A), AT100 antibody for late tangle-like structures (B), and with thioflavin S for NFT-like structure (C). DAPI, 4API, with thioflavin S for NF. (D and E) Near-infrared (NIR) fluorescence imaging of cis P-tau in brain sections from patients with VaD and age-matched controls (D). Yellow arrows indicated the cortex overlying corpus callosum. Quantitation and statistics for the staining intensity in the cingulate cortex overlying corpus callosum (CCtx-CC) are shown in (E). (F and G) IF staining of cis P-tau in the CCtx-CC and neocortex (Neo-Ctx) from patients with VaD and age-matched controls with cis P-tau mAb. A representative image is shown in (F). Quantitation and statistics for the staining intensity is shown in (G). (H and I) IF costaining of cis P-tau with neurofilament (H) or MBP (I) in the CCtx-CC and CC from patients with VaD. Insets show higher magnifications. (J to L) Two-month-old WT mice were subjected to sham or BCAS operation using external microcoils in blue to reduce cerebral blood flow by ~50% (J), followed by IF of cis P-tau in the cortex overlying corpus callosum (Ctx-CC) at different post-surgery time points (green arrows) (K and L). The data were presented as means ± SEM. The P values in (E) and (G) were calculated using unpaired two-tailed Student’s t test, and the P values in (L) were calculated using one-way analysis of variance (ANOVA) with post hoc Dunnett’s multiple comparison test. *P < 0.05 and ****P < 0.0001.
Because these human VaD brains are at late stages of VCID, critical questions are whether cis P-tau is induced after vascular insufficiency and plays any role in VCID. Because chronic cerebral hypoperfusion is a major vascular factor in VCID (2), we examined cis P-tau in the bilateral common carotid artery stenosis (BCAS) mouse model, where external microcoils (0.18 mm in diameter) were permanently placed around both common carotid arteries (Fig. 1J) to chronically reduce parenchymal cerebral blood flow by ~50%, especially in the subcortical brain region (21, 22). This model has been widely used to mimic key aspects of clinical VCID (21, 22). BCAS surgery significantly induced cis P-tau, especially in the cortex overlying the corpus callosum (Ctx-CC) at day 14 (P < 0.0001; Fig. 1, K and L). Thus, cis P-tau is induced in both patients with VaD and BCAS mice modeling VCID.
Cis mAb rescued VCID-like neurodegeneration and cognitive impairment in BCAS mice
Because VCID-like pathology and brain dysfunction in executive function usually occur at 14 days and become obvious at 28 days after BCAS surgery (21, 22), cis P-tau might affect the pathological and functional outcomes. To test this possibility, we eliminated cis P-tau in BCAS mice using cis mAb, which targets non-degradable cis P-tau for TRIM21-mediated proteasome degradation (12, 14, 15), followed by the assessment of VCID-like pathologies and executive functions (Fig. 2A). Cis mAb treatment largely inhibited cis P-tau induction without affecting total endogenous tau (Fig. 2, B and C, and fig. S2, A and B), leading to the rescue of phenotypes that have been shown in BCAS mice (21, 22), including markers consistent with neuroinflammation (Fig. 2D and fig. S2, C and D), demyelination (Fig. 2E and fig. S2E), neurodegeneration (fig. S2, F and G), and loss of myelin-generating glutathione S-transferase pi (GST-pi) positive mature oligodendrocytes (fig. S2H). Cis mAb also rescued the pathologies in the cortex overlying the corpus callosum or the corpus callosum including morphological changes, demyelination and neuroinflammation (figs. S3 and S4). These pathological changes were corroborated by impaired synaptic plasticity via recording field excitatory postsynaptic potentials (fEPSPs) (Fig. 2F), executive function assessed using the T maze (fig. S2I), and spatial working memory ability quantified by novel object recognition tests (Fig. 2G), all of which were rescued by cis mAb.
Fig. 2. Cis mAb treatment of BCAS mice blocks the development of VCID-like pathology and brain dysfunction at 1 and 6 months after the surgery.
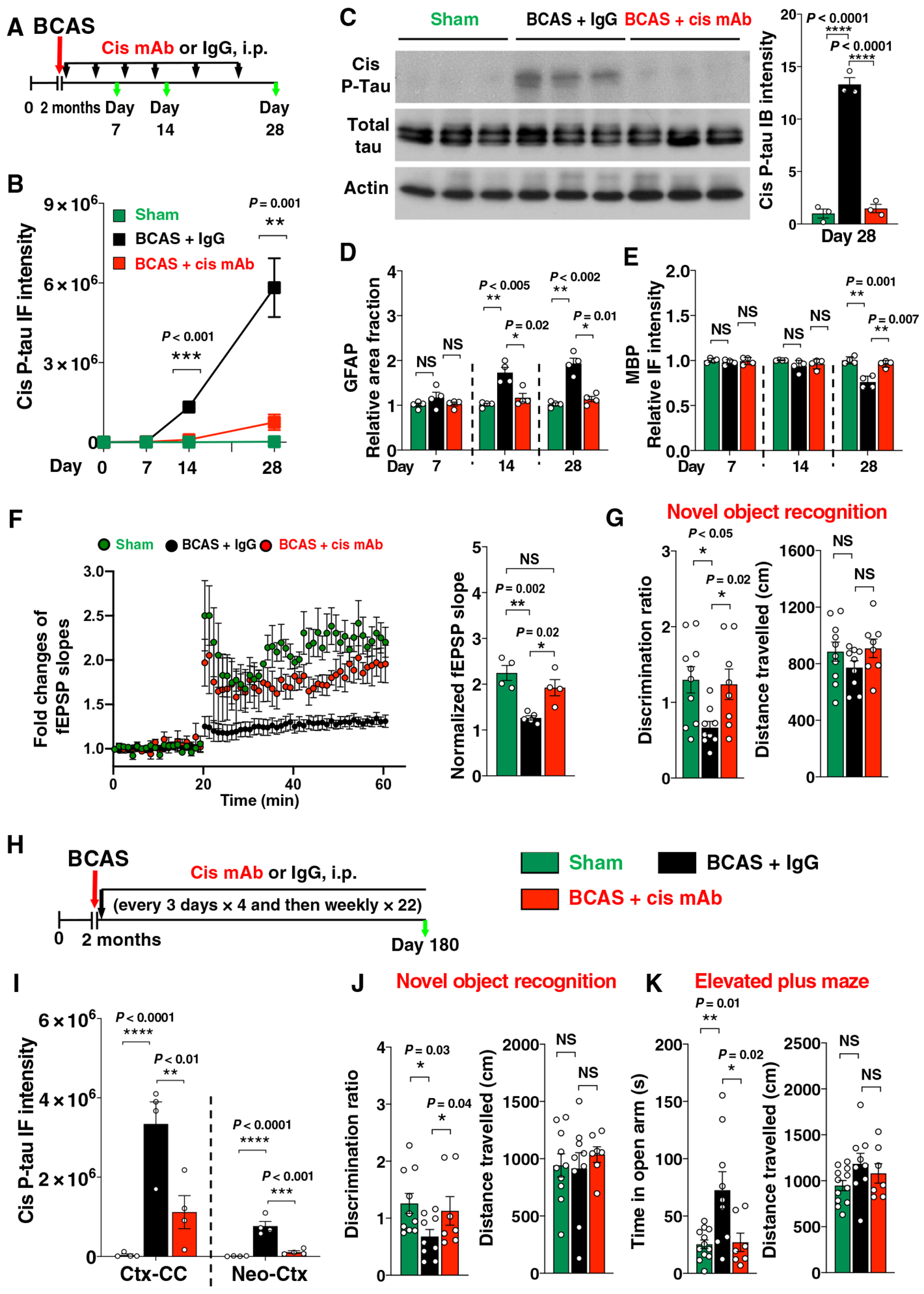
(A) Experimental setup for treating BCAS mice with cis mAb for 28 days. Two-month-old WT mice underwent either sham or BCAS operation, followed by treatment with either cis mAb or IgG isotype control for up to 28 days [300 μg per mouse, intraperitoneally (i.p.), every 3 days for four times, and then 150 μg per mouse every week afterward]. Black arrows, mAb injection; green arrows, functional or pathological assays. (B to E) Ctx-CC from sham, BCAS + IgG, or BCAS + cis mAb mice were examined by immunoblotting for cis P-tau and total tau (C), immunostaining and quantitation for cis P-tau (B), or GFAP (D) or MBP (E). (F) Long-term potentiation (LTP) recording of hippocampal slides. fEPSP slopes over time are shown in dot graph (left), and normalized fEPSP slope for the last 10 min is shown in bar graph (right). (G) Discrimination ratio (left) and distance traveled (right) in novel object recognition assays. (H) Experimental setup for treating BCAS mice with cis mAb for 6 months. Two-month-old WT mice were subjected to either sham or BCAS operation and treated with cis mAb or IgG isotype control for 6 months (300 μg, i.p., every 3 days for four times, and then 200 μg every week afterward). (I) Quantitation of cis P-tau IF intensity from different mouse brain regions. (J) Discrimination ratio (left) and distance traveled (right) in novel object recognition assays. (K) The time mice stay in open arm (left) and distance traveled (right) in elevated plus maze assays. The data were presented as means ± SEM, and the P value was calculated using one-way ANOVA with post hoc Dunnett’s multiple comparison test. NS, not significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To characterize the longer-term effects of cis mAb treatment, we performed BCAS to a new cohort of mice and then treated them with cis mAb or placebo for 6 months (Fig. 2H). Compared with 28 days after BCAS, there was notable progression of pathologies and behavioral deficits at 6 months. None of the BCAS mice had detectable tau tangle epitopes (fig. S5, A to C), consistent with the absence of tangles in patients with VaD. Cis mAb treatment of BCAS mice again eliminated cis P-tau (Fig. 2I and fig. S5D), inhibited markers consistent with neuroinflammation and demyelination (fig. S5, E and F), and partially prevented the loss of mature oligodendrocytes (fig. S5G). These pathological effects were consistent with functional outcomes. Placebo-treated mice displayed impairment not only in spatial memory (Fig. 2J and fig. S5H) but also in motor memory, as assayed by the accelerating rotarod test (fig. S5I). In the accelerating rotarod test, all three groups of the mice had no difference during the day 1 training phase (baseline), indicating comparable basal motor function. However, with repeated testing on days 2 (first trial) and 3 (second trial), sham control mice remained on the accelerating rotarod much longer than on day 1 (baseline), but duration on the rotarod did not increase in placebo-treated BCAS mice, indicating impaired motor learning and memory. Last, because cis P-tau was present in the neocortex at 6 months after BCAS and cortical cis P-tau contributes to risk-taking behavior after TBI (12), we subjected these BCAS mice to the elevated plus maze tests. Most BCAS mice treated with immunoglobulin G (IgG) placebo displayed “risk-taking” behavior, daring to explore the two open or “aversive” arms over significantly more time than sham mice, despite similar distance were traveled among groups (P = 0.01; Fig. 2K). The behavior of cis mAb–treated BCAS mice was comparable to that of the sham mice in all the behavioral tests performed. Thus, cis mAb restored most VCID-like neuropathology and brain dysfunction at 1 and 6 months after BCAS.
Pin1 is inhibited in patients with VaD and mice with VCID, and Pin1 overexpression reduces cis P-tau, preventing VCID-like pathology and dysfunction in BCAS mice
We next examined Pin1 given that it is the only enzyme known to reduce cis P-tau in vitro and in vivo and that the serum is known to induce Pin1 expression in other conditions (9, 10, 14, 23). Active Pin1 was significantly reduced in the subregions with high cis P-tau, especially in the cingulate cortex overlying corpus callosum in human VaD brains and in the cortex overlying corpus callosum in mouse BCAS brains (human, P = 0.001; mouse, P = 0.004; Fig. 3A and figs. S6 and S7A). Moreover, a putative Pin1 enhancer genetic change, E06-21879, which was predicted to affect Pin1 expression (24), is most clearly associated with VaD in a recent genomewide association study (P = 6.2 × 10−41) (Fig. 3B) (25). These results collectively show that Pin1 is reduced in the brains of both patients with VaD and VCID model mice.
Fig. 3. Pin1 is inhibited in BCAS, whereas brain-specific Pin1 overexpression blocks VCID-like pathology and brain dysfunction in BCAS mice.
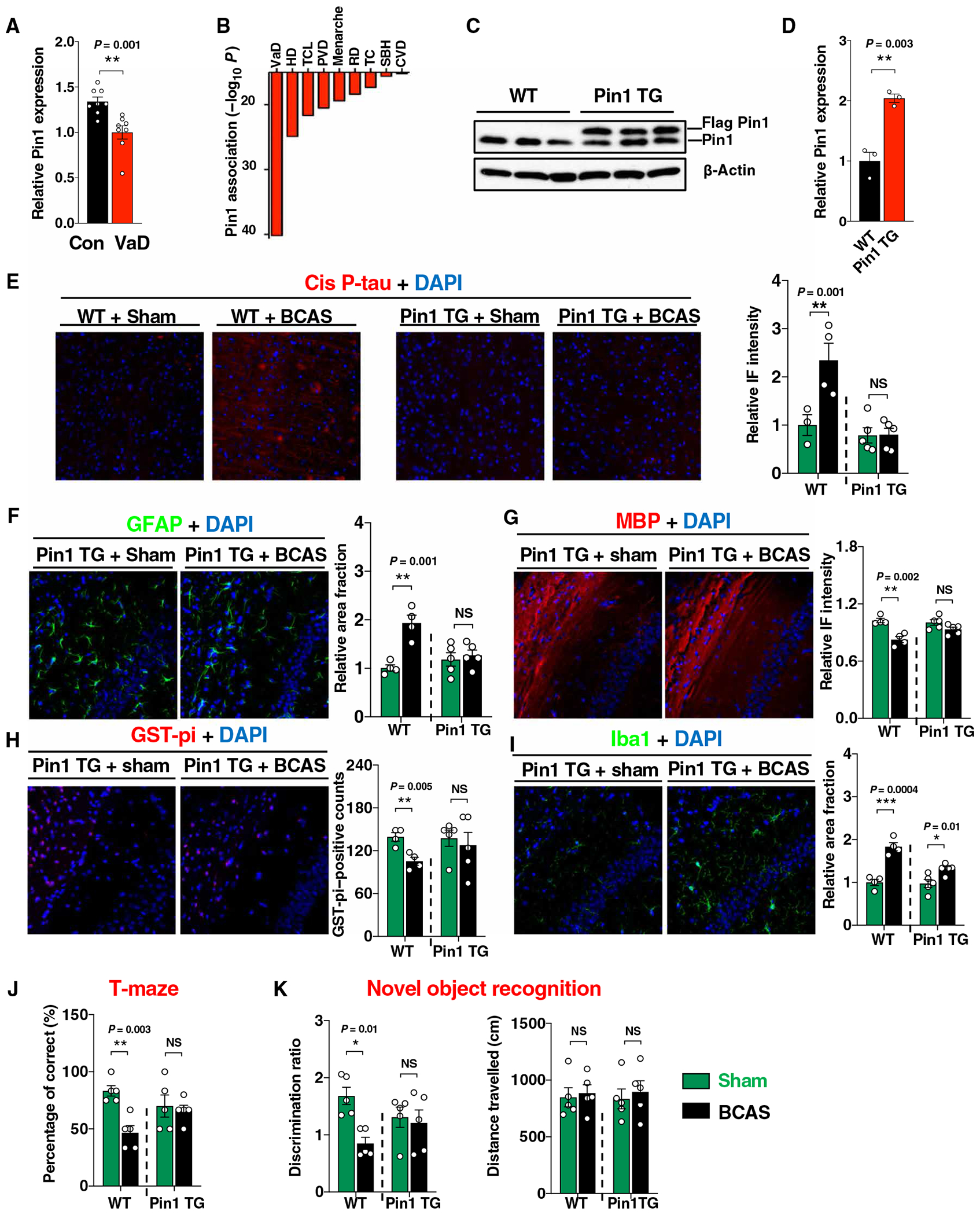
(A) Quantitation of active Pin1 immunoreactivity in the CCTx-CC of human VaD brain and normal control. (B) Top disease association with a putative Pin1 enhancer SNP E06-21879. VaD, vascular dementia; DD, Hodgkin’s disease; TCL, T cell lymphomas; PVD, peripheral vascular disease; RD, respiratory disorders; TC, thyroid cancer; SBH, subarachnoid hemorrhage; CVD, cerebrovascular disease; MSD, multisystem degeneration. (C and D) Immunoblotting of cortical lysate of 2-month-old Pin1 TG mice with Pin1 antibody (C) and quantified (D). (E to I) Indicated mouse brain sections were analyzed by IF cis P-tau (E), GFAP (F), MBP (G), GST-pi (H), and Iba1 (I) antibodies, followed by quantified intensity in the Ctx-CC at 28 days after surgery. (J) Bar graphs showing the percentage of correct choices in T maze. (K) Bar graphs showing discrimination ratio (left) and distance traveled (right) in novel object recognition assays. The data in (D) to (K) were presented as means ± SEM. The P value in (D) was calculated using unpaired two-tailed Student’s t test. The P values in (E) to (K) were calculated using one-way ANOVA with post hoc Dunnett’s multiple comparison test. *P < 0.05, **P < 0.01, and ***P < 0.001.
To examine whether Pin1 plays any role in VCID-like pathologies and brain dysfunction, we used brain-specific Pin1 transgenic (Pin1 TG) mice (Fig. 3, C and D), which overexpress Pin1 under the Thy1.2 promoter and are resistant to cis P-tau induction and neurodegeneration induced by tau overexpression (14, 26). Pin1 TG mice and wild-type (WT) littermates at 2 months of age were subjected to BCAS surgery, followed by the assessment of VCID-like pathologies and executive function changes 28 days after the surgery. In contrast to WT littermates, Pin1 TG mice had much reduced induction of cis P-tau (Fig. 3E), neuroinflammation marker GFAP (Fig. 3F), demyelination (Fig. 3G), and reversed loss of mature oligodendrocytes (Fig. 3H) after BCAS, although Iba1 immunoreactivity was still increased (Fig. 3I). Moreover, BCAS-induced impairment in executive function was also absent in the Pin1 TG mice, as assessed using the T maze and novel object recognition tests (Fig. 3, J and K). Thus, Pin1 was reduced and correlated with cis P-tau in patients and mice with VCID, and Pin1 overexpression prevented VCID-like pathology and memory loss in model mice.
DAPK1 KO reduces Pin1 inhibition and cis P-tau induction and prevents VCID-like pathology and dysfunction in BCAS mice
To further examine upstream regulators leading to Pin1 inhibition after neurovascular insufficiency, we examined death-associated protein kinase 1 (DAPK1), because this kinase is known to be activated after stroke (27) and to phosphorylate Pin1 S71, thereby inhibiting Pin1 isomerase activity in neuronal cells (28). Both DAPK1 and Pin1 S71 phosphorylation were increased in the Ctx-CC in BCAS mice (28 days after surgery) and patients with VaD, despite decreases in active Pin1 expression (figs. S7, B to D, and S8, A to D). To further test whether DAPK1 functions in induction of cis P-tau, VCID-like pathology, and brain dysfunction, we subjected DAPK1 KO mice (28) and WT littermates to BCAS. DAPK1 KO potently eliminated the induction of Pin1 S71 phosphorylation (fig. S8, C and D), cis P-tau (fig. S8, E and F), and markers consistent with neuroinflammation (fig. S8, G to J) and demyelination (fig. S8, K and L) in BCAS mice. Moreover, DAPK1 KO mice did not have impaired executive functions 1 month after BCAS surgery, in contrast to the WT littermate controls, as assessed using the T maze and novel object location recognition tests (fig. S8, M and N). Thus, BCAS leads to cis P-tau induction likely due to Pin1 inhibition by activated DAPK1, whereas reducing cis P-tau by Pin1 overexpression or DAPK1 KO prevented VCID-like pathology and impairment in executive function in BCAS mice.
Cis mAb prevented and ameliorated progression of AD-like neurodegeneration and memory loss in mice
Given well-known vascular contributions to AD and cis P-tau as an early pathogenic tau species in human AD (14), we wondered whether eliminating cis P-tau inhibit progressive neurodegeneration and cognitive decline in AD-like tauopathy mice. To this end, we used htau mice, which express the human tau gene in the place of the mouse one and develop age-dependent tau hyperphosphorylation, NFT-like pathologies, neuronal loss, and cognitive deficits resembling AD (29). In 3-month-old htau mice, cis P-tau was induced and localized to axons without tau tangles (fig. S9, A and B). Cis mAb treatment fully prevented learning and memory deficits, as assessed by the Morris water maze (Fig. 4, A and B; fig. S9, C to E; and movies S1 to S3), which was supported by almost full elimination of cis P-tau induction (Fig. 4, C and D, and fig. S9F) and attenuation of NFT-like pathology (Fig. 4, E and F, and fig. S9, G to J). Thus, cis mAb prevented the development of tangles and cognitive dysfunction in the AD-like tauopathy mice.
Fig. 4. Cis mAb prevents and attenuates the development of NFT-like pathology and functional impairments in htau mice.
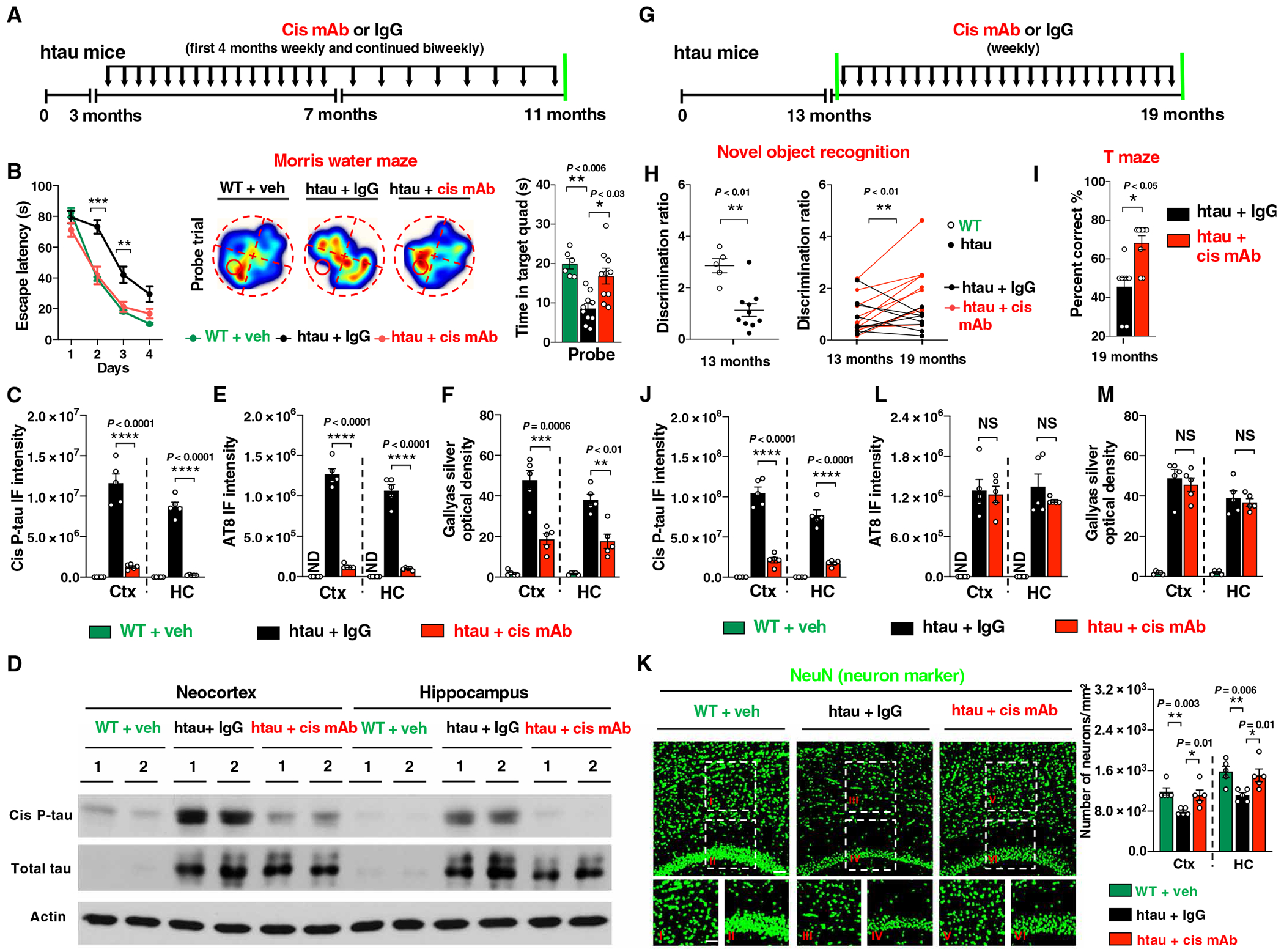
(A) Experimental setup. Three-month-old htau mice underwent treatment of cis mAb or IgG controls for 8 months, followed by functional and pathological examinations. Black arrows, antibody injection; green lines, functional or pathological assays. (B) Effect of cis mAb treatment on behavioral deficits in htau mice, as assayed by the Morris water maze in htau mice escape latency (left) in the acquisition trials and trajectories and time spent in target quadrant in the probe trial (right). (C to F) Effects of cis mAb treatment on the accumulation of cis P-tau and the development of tangle-like pathology in htau mice, as assayed by immunoblotting of cis P-tau and total tau (D); IF staining of cis P-tau (C) in the neocortex and hippocampus; IF with AT8 antibody (E); and Gallyas silver staining (F) to detect tangle-like pathology in neocortex and hippocampus after 8 months of treatment. (G) Experimental setup. Thirteen-month-old htau mice were subjected to treatment of cis mAb or IgG isotype control for 6 months. Black arrows, antibody injection; green lines, functional or pathological assays. (H and I) Effects of cis mAb treatment on the novel object recognition and T maze. Dot graph showing longitudinal assessment of discrimination ratio before and after treatment with either cis mAb (red) or control (black) (H). Bar graph showing the percentage of correct choices in T maze after treatment (I). (J and K) Effects of cis mAb treatment of aged htau mice on the accumulation of cis P-tau, neuronal loss, and tangle-like pathology, as shown by IF for cis P-tau (J), with AT8 mAb (L), Gallyas silver staining (M) to detect tangle-like pathology, and NeuN antibody (K) to detect neuronal loss in neocortex and hippocampus. Inset images are high magnifications of representative areas. ND, not detectable. The data were presented as means ± SEM. The P values in (H) (left) and (I) are computed using unpaired two-tailed Student’s t test. The P value in (H) (right) is computed using paired two-tailed Student’s t test. The P values in (B) to (E) and (J) to (K) were calculated using one-way ANOVA with post hoc Dunnett’s multiple comparison test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To evaluate whether cis mAb treatment had any therapeutic effects in mice with existing AD-like tau pathologies and cognitive symptoms, we treated 13-month-old aged htau mice with cis mAb for 6 months (Fig. 4G). Before treatment, htau mice had cognitive deficits, and most placebo-treated mice further deteriorated over the 6 months of the treatment (Fig. 4H and fig. S10, A to C). In sharp contrast, cognitive deficits in cis mAb–treated mice were ameliorated, as assessed by the novel object recognition test and the T-maze (Fig. 4, H and I). These functional outcomes were correlated with brain pathology. Cis mAb treatment eliminated the cis P-tau in the cortex and hippocampus (Fig. 4J and fig. S10, D and E) and rescued neuronal loss in the neocortex and hippocampal CA1 subfield as well as atrophy of the hippocampal CA1 layer (Fig. 4K). Cis mAb treatment did not reduce NFT-like pathology (Fig. 4, L and M, and fig. S10, F to I), consistent with the findings that NFTs may not be neurotoxic (30). Thus, cis P-tau was required for the progression of neurodegeneration and cognitive impairment in two independent mouse models relevant to VCID and AD.
BCAS in young mice induces diverse cortical cell type–specific transcriptomic changes, and ~85-90% of the global alterations are recovered by cis mAb
The pathological and functional evaluations mainly focus on a limited number of known factors and phenotypes, and they are unlikely to reveal the complicated alterations and interactions among different cell types. Published bulk transcriptomic studies have largely focused on the acute response within 48 hours after vascular insults and do not correlate with chronic phenotypes relevant to dementia (31). Because pathologies and behavioral changes are obvious at 28 days after BCAS, before progressive neurodegeneration over 1 year (21, 22), we reasoned that transcriptomic profiling may be informative at this time for understanding the impact of cis P-tau and for evaluating the efficacy of cis mAb in treating VCID in different cell types. Thus, we used single-nucleus RNA sequencing (RNA-seq) to profile the transcriptomic changes of total ~15,000 cortical cells from BCAS mice treated with cis mAb or IgG control for 28 days, referencing to sham littermates (Fig. 5A). We recovered 25 distinct cell clusters based on their expression profiles and then collapsed similar clusters into five major cell types (excitatory neurons, inhibitory neurons, astrocytes, oligodendrocytes, and endothelia) based on the expression of cell type–specific markers (Fig. 5, B and C, and fig. S11A).
Fig. 5. BCAS induces diverse cortical cell type–specific transcriptomic changes, and the vast majority of the global alterations are recovered by cis mAb.
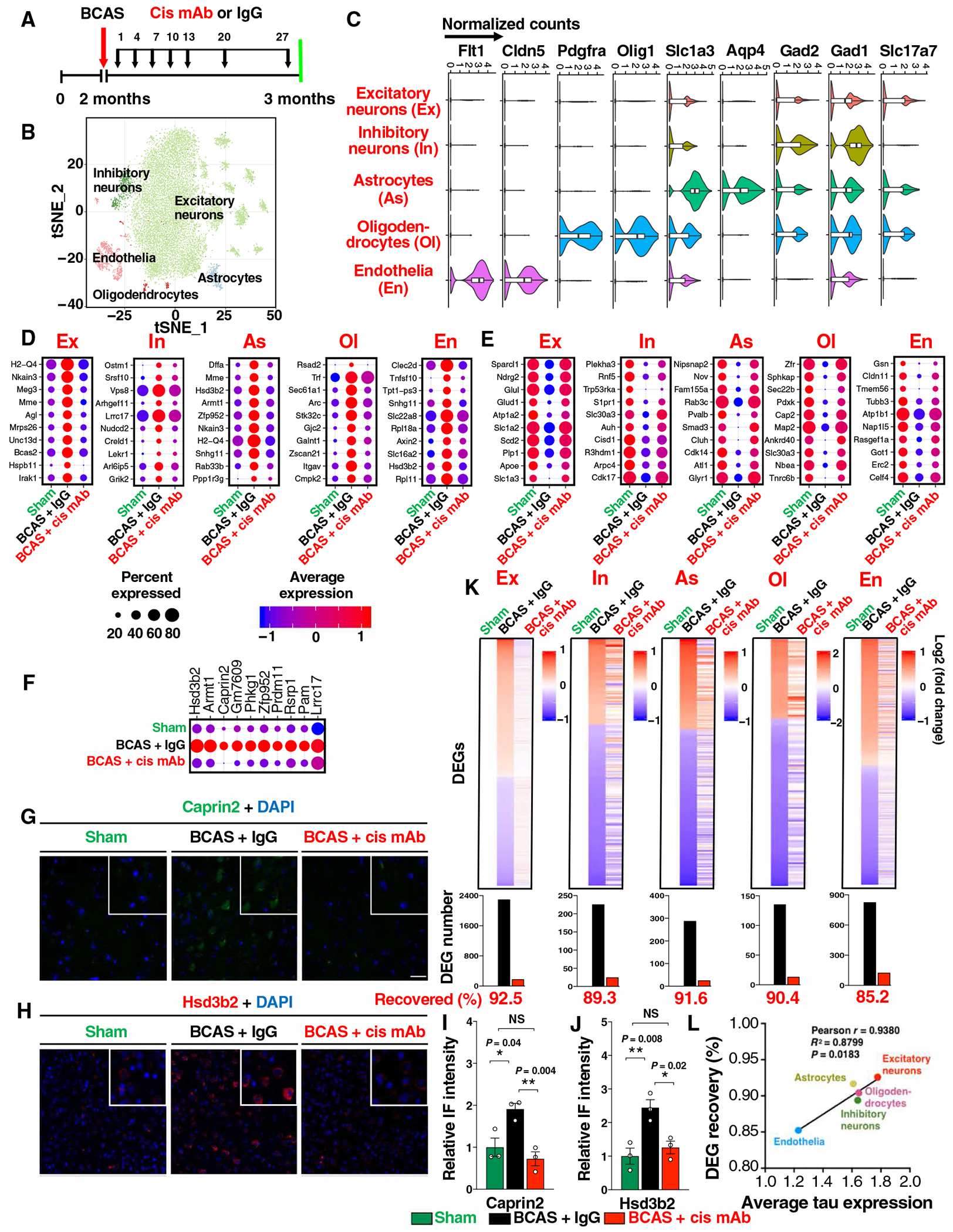
(A) Experimental setup of the single-nucleus RNA-seq for BCAS mice treated with IgG or cis mAb, with sham littermate as controls. (B) tSNE (t-distributed stochastic neighbor embedding) plots of color-coded cortical cell types. (C) Normalized expression of marker genes for different cell types: Slc17a7 (glutamatergic neurons, Ex), Gad1 and Gad2 (GABAergic neurons, In), Aqp4 and Slc1a3 (astrocytes, As), Olig1 and Pdgfra (oligodendrocytes and their progenitor cells, Ol), and Cldn5 and Flt1 (endothelia, En). (D to F) Top up-regulated (D) or down-regulated DEGs (E) in different cell types in BCAS mice with or without cis mAb treatment, and a list of up-regulated excitatory neuronal DEGs that have not been clearly linked to neurodegeneration or stroke (F). Dot plots are color coded with average expression and sized with percentage of cells expressing the gene. (G to J) Validation of two DEGs Caprin2 (G and I) and Hsd3b2 (H and J) with IF staining (G and H), with quantitations (I and H). The data in (I) and (J) were presented as means ± SEM, and the P values were calculated using one-way ANOVA with post hoc Dunnett’s multiple comparison test. (K) Top: Log2-transformed relative normalized expression of each gene compared to sham. Cell types are labeled on the top. Bottom: Number of DEGs are shown in bar graphs. Genes that are differentially expressed in BCAS + IgG but not in BCAS + cis mAb mice (referencing to sham mice) are defined as recovered genes. (L) Xyplot showing correlation between average tau expression and the recovered DEG percentage in five major cell types.
We identified unique differentially expressed genes (DEGs) in the five major cell types by comparing BCAS mice with sham littermate controls (table S2). Many top up-regulated (Fig. 5D) and down-regulated (Fig. 5E) DEGs have been implicated in stroke and/or neurodegeneration, which were effectively restored by cis mAb, both at the average expression and the number of DEG-expressing cells. The up-regulated DEGs included two major histocompatibility complex class I genes implicated in neuroinflammation, Meg3 in ischemic neuronal death, Mme in demyelinating neuropathy, and down-regulated DEGs included Glu1 implicated in VCI and Slc1a2 in AD. DEGs in nonneuronal cell types also include genes that have been linked to neurodegeneration. One such example is endothelia-specific decreased expression of Cldn11, a claudin that regulates tight junction formation and endothelial barrier function, consistent with damage in blood-brain barrier function and a possible link to subsequent demyelination (32). We have also observed oligodendrocyte-specific up-regulation of an interferon-stimulated gene Rsad2 and endothelia-specific up-regulation of Tnfsf10 and Clec2d, two genes that have been linked to promote apoptosis and inflammation in endothelial cells, consistent with increased inflammation (33, 34). Our analyses also revealed many top DEGs that, to our knowledge, have not been linked to neurodegeneration (Fig. 5F), including Lrrc17, a negative nuclear factor κB regulator; Hsd3b2, an enzyme in steroid biosynthesis; and Phkg1, a kinase in glycogenolysis. We validated the differential expression and therapeutic response to cis mAb for both DEGs examined (Fig. 5, G to J).
The recovery of top DEGs by cis mAb treatment led us to systematically evaluate the cis mAb–dependent recovery of DEGs in different cell types. Hundreds to thousands of DEGs were identified after BCAS (table S3), with excitatory neurons being the most affected, as is in human AD (35). The number of DEGs was smaller in other cell types, which could potentially be due to reduced statistical power in the lower abundant cell types. About 85-90% of BCAS- induced DEGs were recovered by cis mAb treatment (Fig. 5K), and the extent of the recovery in different cell types was correlated with their tau expression (Fig. 5L), the source for cis P-tau. These results indicate that BCAS induced diverse cell type–specific transcriptomic changes in mice, and the vast majority of these alterations were recovered by cis mAb, unbiasedly and comprehensively demonstrating the potency of cis-targeted immunotherapy in restoring the global transcriptomic changes in VCID mice at the single-cell resolution.
BCAS-induced transcriptomic changes in young mice are related to myelin and axon function, resembling patients with AD, and largely recoverable by cis mAb
To understand the biology underlying the transcriptomic changes in BCAS mice, we performed Gene Ontology and gene set enrichment analyses. The down-regulated DEGs in excitatory neurons, the most affected cell type (table S2), were most significantly associated with myelin sheath (P = 10−65; Fig. 6A), which was also widely down-regulated in other cell types (fig. S11, B and C). Both the average expression and the number of excitatory neurons that expressed these DEGs were down-regulated, but 83.8% (83 of 99) of these DEGs were significantly recovered by cis mAb (P < 0.0001; Fig. 6B and fig. S11D). These results are consistent with the above findings that BCAS induced prominent demyelination, with the previous findings that demyelination is likely the most prominent pathology in VaD (1, 2) and with the data showing that widespread down-regulation of myelination-related genes in different cell types is substantial in human AD (35). The next most prominent pathways associated with down-regulated DEGs in the excitatory neurons were axon/synapse processes, microtubule/cytoskeleton, and guanosine 5′-triphosphate (GTP)/nucleoside signaling. We validated four of four microtubule-related DEGs examined (Tubb5, Tppp, ApoE, and Fkbp4) by immunostaining (Fig. 6, C to G, and fig. S12, A to D). Furthermore, cis mAb treatment appeared to induce all the four hemoglobin genes in different cortical cell types (fig. S12E). We confirmed with immunostaining that hemoglobin was reduced in the Ctx-CC in BCAS mice and that cis mAb restored hemoglobin to a concentration higher than sham controls (Fig. 6H and fig. S12F). It has been shown that hemoglobin up-regulation extends neuronal activity and reduces hypoxic zones in the brain under hypoxia (36). Thus, cis mAb treatment of BCAS mice may help cortical cells to function better under hypoxic conditions.
Fig. 6. BCAS in young mice induces the global transcriptomic changes resembling those in patients with AD, most of which are recovered by cis mAb.
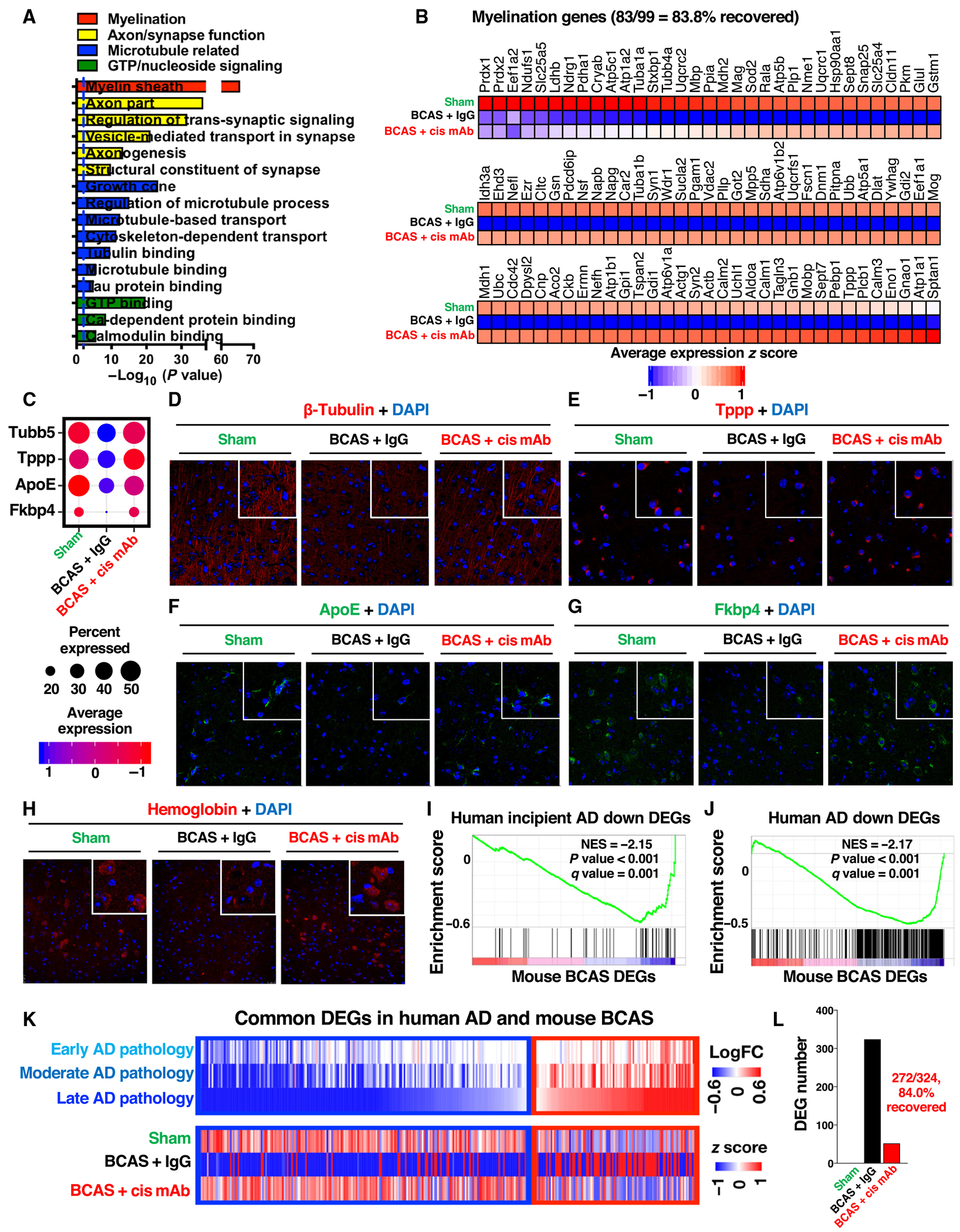
(A) Enriched Gene Ontology (GO) terms with different classes color coded. (B) Heatmap showing average expression z score. (C to G) Dot plots (C) and IF staining validations (D to G) for four microtubule-related down-regulated DEGs. (H) IF staining validations of down-regulated hemoglobin genes in BCAS mouse and recovery in cis mAb–treated mice. (I and J) Gene set enrichment analyses of down-regulated gene sets in patients with incipient AD (I) and AD (J), within BCAS mouse DEGs in excitatory neurons. (K and L) Heatmaps of the shared DEGs in excitatory neurons in young BCAS mice and patients with AD (K) and bar graphs showing the cis mAb recovery percentage of shared DEGs (L).
Gene set enrichment analysis revealed that the only gene set from the curated MSigDb C2 database that was consistently negatively enriched in all the five major cell types was the down-regulated gene sets in human AD brains (P < 0.05; fig. S13) (37). The gene sets that are down-regulated in patients with incipient AD and AD were both highly enriched in our BCAS down-regulated DEGs in the excitatory neurons (P < 0.001) (Fig. 6, I and J), the most abundant and affected cell type from our (table S2) and published analyses (35, 37). We also observed similarity in the direct comparison of enriched down-regulated DEGs (Fig. 6K), 84% (272 of 324) of which were recovered by cis mAb in BCAS mice (Fig. 6L). These results demonstrate that BCAS induced the transcriptomic changes in young mice resembling those in patients with AD, offering transcriptomic evidence for the similarity between VCID and AD. In addition, our data showed that the vast majority of the transcriptomic changes were recovered by cis mAb in BCAS mice, supporting efficacy and potential clinical relevance of cis mAb immunotherapy.
Purified soluble cis P-tau is sufficient to induce progressive neurodegeneration and brain dysfunction by causing cistauosis and axonopathy
Given that cis P-tau is induced early in VCID and its elimination rescued VCID in mice, a major question remains if cis P-tau is sufficient to induce progressive neurodegeneration and brain dysfunction in WT animals. To address this question, we used cis mAb to affinity purify soluble cis P-tau from TBI mice because severe TBI rapidly produces a large quantity of cis P-tau without tau oligomerization, aggregation, or tangle epitopes, which could complicate assays of cis P-tau neurotoxicity. Purified cis P-tau, but not recombinant tau, caused neurotoxicity in SY5Y cells, which was blocked by cis mAb treatment (fig. S14, A and B). Purified cis P-tau also induced death in primary neurons, which was blocked by the pancaspase inhibitor Z-VAD-FMK (fig. S14C). Thus, purified cis P-tau, but not recombinant tau, induced neuronal death and could be blocked by cis mAb.
Cortical injection of brains lysates or purified tau aggregates from tau TG mice or patients with tauopathy induced progressive neurodegeneration in mice (38). To test whether cis P-tau could induce progressive neurodegeneration, we stereotactically injected purified soluble cis P-tau or recombinant tau bilaterally into the upper and lower layers of the neocortex of 3-month-old WT mice, followed by behavioral testing at 1 and 10 months (fig. S14D). At 1 month after injection, cis P-tau caused risk-taking behaviors (fig. S14, E and F), whereas recombinant tau even at four times higher doses had no detectable effects (fig. S14, G to I), consistent with that tau oligomers or pathogenic tau, but not normal tau, are neurotoxic (38). At 10 months after injection, we found not only the persistence of risk-taking behaviors but also the appearance of sensorimotor and cognitive behavioral changes (fig. S14, J and K), suggesting progressive neurodegeneration.
To confirm that the above phenotypes and their progression are due to cis P-tau, not its associated factors, we performed new experiments using cis mAb to eliminate cis P-tau in cis P-tau–injected mice (Fig. 7A). Cis mAb prevented cis P-tau from inducing brain dysfunction at 1 and 10 months (Fig. 7, B to F), as assayed by various behavioral tests. Moreover, the progression of neurodegeneration was further supported by the presence of cis P-tau, cistauosis, and axonopathy in a brain region distant from the injection site at 10 months after injection, which were fully restored by cis mAb (Fig. 7G). Evidence included cis P-tau in various brain subregions (fig. S15), impaired synaptic plasticity (Fig. 7H), ultrastructural pathologies of disrupted axonal microtubules and mitochondria (Fig. 7I), increased cleaved caspase-3 immunoreactivity (fig. S16, A and B), demyelination (fig. S16, C and D), and early tangle-like AT8 epitope (fig. S16, E and F). In contrast, no obvious behavioral deficits were detected when cis P-tau was injected to tau null mice at 1 or 10 months after the injection (fig. S17), indicating a requirement of endogenous tau. Thus, cis P-tau was sufficient to induce progressive neurodegeneration by causing cistauosis and axonopathy dependent on endogenous tau resembling a prion (38).
Fig. 7. Stereotactic cortical injection of purified cis P-tau is sufficient to induce prion-like progressive neurodegeneration and brain dysfunction.
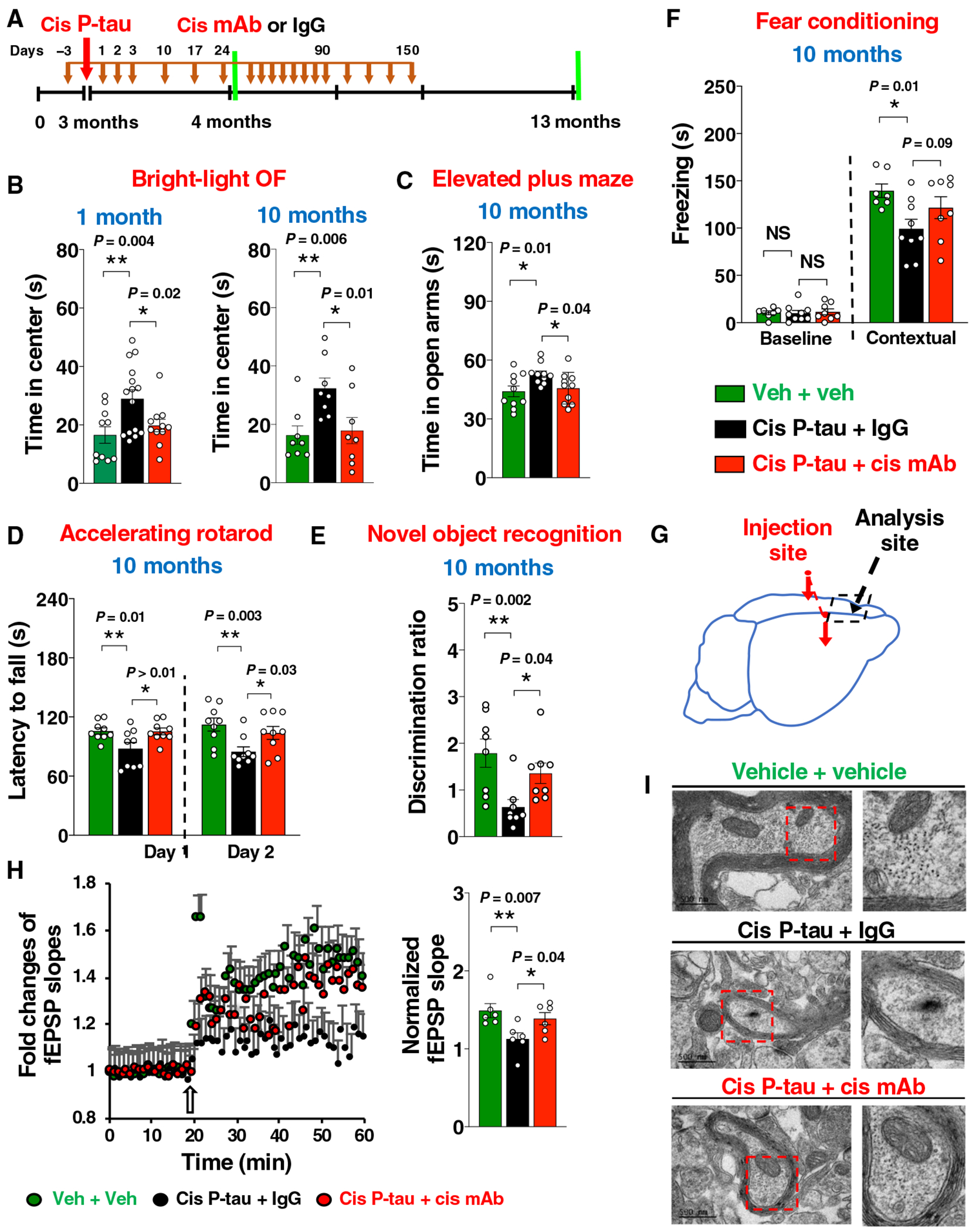
(A) Experimental setup. Red arrows, stereotaxic injections; black arrows, mAb injections; green lines, functional and pathological assays. (B to F) Behavioral assessment of mice at different time (labeled in blue) after cis P-tau injection and mAb treatment. Bar graphs showing time spent in center in bright-light OF (open field) assays (B), time in open arms in elevated plus maze (C), latency to fall in accelerating rotatord (D), discrimination ratio (E) in novel object recognition, and freezing time in fear conditioning (F). (G to I) Cortical LTP (H) and ultrastructural pathologies of axonal microtubules and mitochondria (I) were determined in mPFC away from the injection site (G) at 10 months after the injection. The data were presented as means ± SEM, and the P values were calculated one-way ANOVA with post hoc Dunnett’s multiple comparison test. *P < 0.05 and **P < 0.01.
Injected cis P-tau–induced transcriptomic changes are relevant to cistauosis and axonopathy and are also found in VCID mice and patients with early AD pathology
We next asked whether injected cis P-tau induced transcriptomic changes relevant to axonopathy, VCID and AD. To this end, we profiled 7577 cortical cells for their transcriptomic changes at 10 months after cis P-tau injection in mice, when the progression of pathological and behavioral changes was obvious. We again collapsed distinct cell clusters into the five major cell types (Fig. 8, A to C, and fig. S18A) and identified hundreds of cell type–specific DEGs from cis P-tau–injected mice (table S4). Despite a much smaller number of DEGs in cis-injected mice, the top ontology terms associated with the transcriptomic changes in the excitatory neurons in cis-injected mice were similar to those in the BCAS mice, again highly related to myelin, axon, synapses, and microtubule function (Fig. 8D), with an exception that GTP/nucleoside signaling was not reduced after cis injection. This difference might be expected, given that many serum-containing growth factors known to activate GTP/nucleoside signaling (39) were reduced by BCAS, but not by cis injection.
Fig. 8. Injected cis P-tau induces the conserved transcriptomic changes that are not only highly relevant to cistauosis and axonopathy but are also found in early VCID and AD.
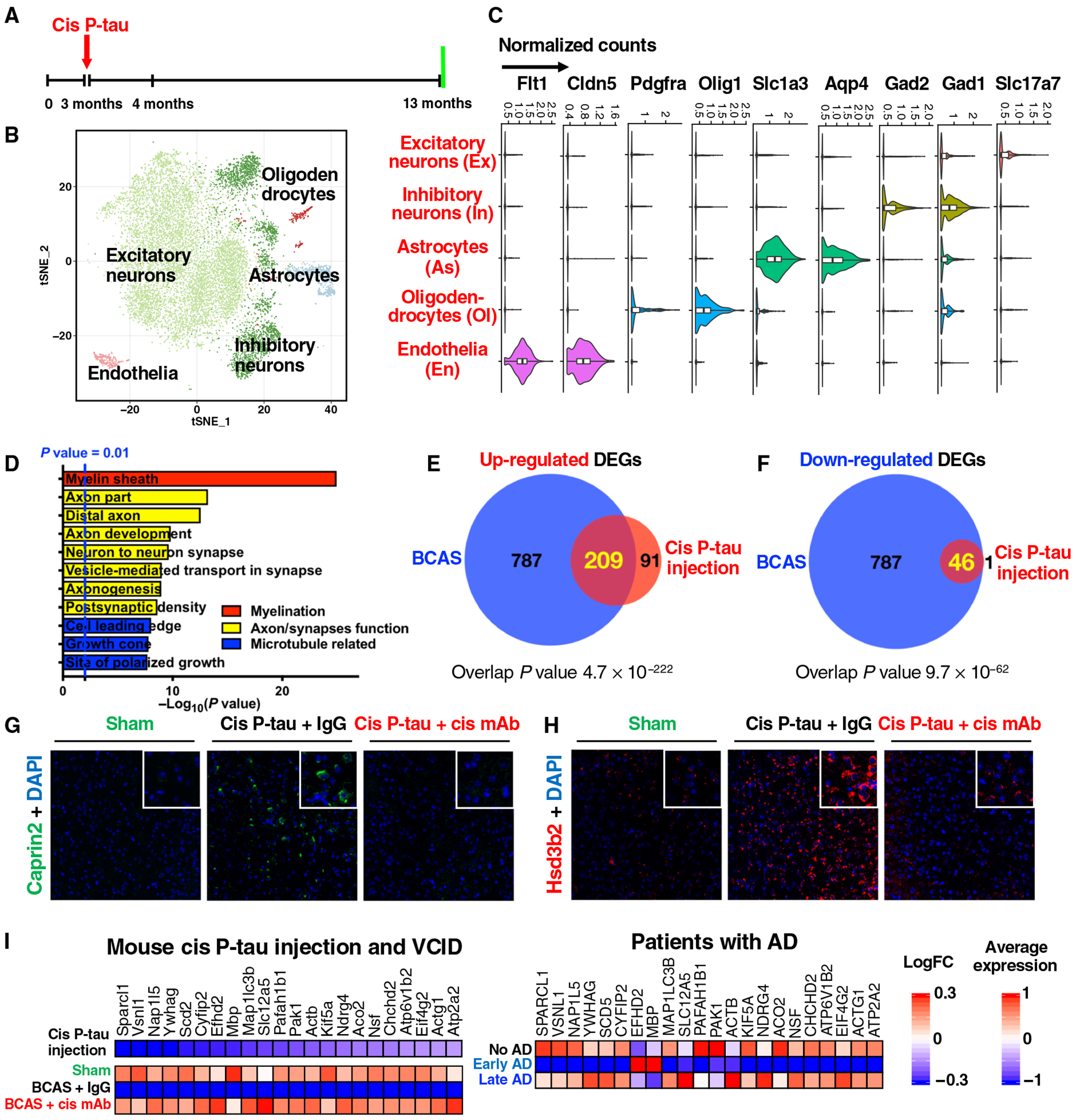
(A) The setup of the single-nucleus RNA-seq experiments for cis P-tau–injected mice, with vehicle controls. (B) tSNE plots of different color-coded cortical cell types from vehicle or cis P-tau–injected mice. (C) Normalized expression of marker genes for different cell types: Slc17a7 (glutamatergic neurons, Ex), Gad1, Gad2 (GABAergic neurons, In), Aqp4, Slc1a3 (astrocytes, As), Olig1, Pdgfra (oligodendrocytes and their progenitor cells, Ol), Cldn5, and Flt1 (endothelia, En). (D) Enriched GO terms with different classes color coded. (E and F) Venn diagram of the shared up-regulated (E) and down-regulated excitatory neuronal DEGs (F) between the cis P-tau—injected mice and BCAS mice. Two-tailed Fisher’s exact test is implemented to compute the P values for the overlap. (G and H) Validation of two up-regulated DEGs Caprin2 (G) and Hsd3b2 (H) and their response to cis mAb by IF staining. (I) Twenty-four conserved genes that are commonly down-regulated in the excitatory neurons of cis P-tau–injected mice, BCAS mice, and human patients only with early, but not late, AD pathology (35).
The similarity in DEG ontology terms between cis P-tau injection and BCAS led us to systematically compare their transcriptomic changes. We found highly significant overlap in the DEGs in the excitatory neurons, the most affected cell type in both models. About 70% (209 of 300) of the cis P-tau up-regulated DEGs were also increased in the BCAS mice (P = 4.7 × 10−222), and ~ 98% (46 of 47) of the cis P-tau down-regulated DEGs were also decreased in the BCAS mice (P = 9.7 × 10−62) (Fig. 8, E and F). We confirmed four of four common DEGs examined (Caprin2, Hsd3b2, Ndrg2, and Mbp) in cis-injected mice (Fig. 8, G and H, and fig. S18, B to E) and BCAS mice (Fig. 5, G to J, and fig. S12, G to J) by immunostaining. The commonly up-regulated DEGs were associated with synapse function (fig. S18F), including Grin2a and Grin2b, two N-methyl-d-aspartate (NMDA) receptors activated in stroke and implicated in neuronal death (27), and EphA7 implicated in neuron injury, inflammation, and axon targeting (40). The commonly down-regulated DEGs (46 of the 47 cis P-tau down-regulated genes) were associated with myelination, axon, and microtubule function (Fig. 8D). Ninety-eight percent of the common DEGs (250 of 255) were recovered by cis mAb (fig. S18, G and H). Thus, we identified the cistauosis transcriptomic signature shared in cis P-tau–injected and BCAS mice.
Given that cis P-tau was induced early in VCID, the cistauosis transcriptomic signature might reflect the conserved transcriptomic changes occurring in patients with early AD pathology. We therefore examined the recently published single-cell transcriptomic analysis of human brains with early and late AD pathology (35). Forty-two of the 46 down-regulated cistauosis DEGs shared between BCAS and cis P-tau–injected mice have well-defined human homologs, and 22 of these 42 genes passed the human gene expression abundance filter for the gene set enrichment analysis. We observed a significant negative enrichment of the cistauosis transcriptomic signature in patients only with early (P = 0.01; fig. S18I,), but not late, AD pathology. Ninety-one percent (20 of 22) of the commonly down-regulated DEGs in cis P-tau–injected mice and BCAS mice were also down-regulated in the cortical excitatory neurons of human patients only with early, but not late, AD pathology (fig. S8I), revealing the presence of the cistauosis transcriptomic signature at early, but not late, stage of AD and VCID. Again, these early down-regulated DEGs were related to myelination (ACTB, ACTG1, ATP6V1B2, NSF, and YWHAG) and axon process (KIF5A, ACTB, ACTG1, PAFAH1B1, and PAK1). Thus, in the most affected excitatory neurons, cis P-tau induces conserved gene expression changes that are found in mouse VCID and human AD with early pathology.
DISCUSSION
Increasing evidence supports that neurovascular dysfunction contributes to cognitive impairment and dementia including AD, but the underlying molecular links remain elusive and there is no effective therapy (1, 2). Here, we found robust cis P-tau in axons without detectable NFTs in patients with VaD and BCAS mice. Reducing cerebral blood flow in mice robustly induced cis P-tau before VCID-like pathological and behavioral changes. Moreover, eliminating cis P-tau using cis mAb rescued most VCID-like neuropathology and brain dysfunction, and the beneficial effects were still present after 6 months even if cerebral blood flow was chronically reduced by ~50%. At this time point, BCAS mice displayed not only cis P-tau and widespread neurodegeneration but also functional impairment in spatial memory, motor memory, and risk-taking behavior. All these pathological and functional changes were largely rescued by cis mAb. Pin1 was inhibited, and its inhibitory kinase DAPK1 is activated in BCAS mice. Eliminating cis P-tau using brain-specific Pin1 overexpression or DAPK1 KO in BCAS mice prevented the development of VCID-like pathology and cognitive impairment. Cis mAb also prevented and ameliorated progression of AD-like neurodegeneration and memory loss in mice. Together, these results support the idea that neurovascular insults induce cis P-tau that, in turn, drives progressive neurodegeneration in VCID including AD, likely due to inhibition of Pin1 by activated DAPK1 (fig. S18J).
This concept has been further supported by the unbiased single-cell transcriptomic analysis. BCAS induced hundreds to thousands of DEGs in diverse cortical cell types, with most being associated with myelin and axon processes. Cis mAb recovered about 85-90% of the transcriptomic changes, with the extent of recovery in different cell types being correlated with their cellular tau expression, the source of cis P-tau. Thus, cis-targeted immunotherapy on VCID might have important therapeutic results. Cortical injection of purified soluble cis P-tau is sufficient to induce progressive neurodegeneration by causing cistauosis, axonopathy, and conserved transcriptomic signature found in early VCID and AD. The cis P-tau injection failed to induce pathogenic phenotypes in tau KO mice, suggesting that endogenous tau is required for cis P-tau toxicity, consistent with the prion-like behavior for other pathogenic tau proteins (41). Purified cis P-tau induced a fraction of the DEGs caused by BCAS at the early stage. These commonly down-regulated DEGs in cis P-tau–injected and BCAS mice were also down-regulated in human patients with early AD pathology (35), with most being related to myelin and axon processes. Thus, our results consistently indicate that cis P-tau underlies neurodegeneration and cognitive decline after neurovascular insufficiency.
Our results potentially offer a highly effective and specific targeted immunotherapy for neurodegeneration in AD and resulting from neurovascular insults and TBI, with some unique features, comparing with other tau immunotherapies (42). First, cis mAb specifically targets only the pathogenic cis P-tau, without affecting the physiologic tau including trans P-tau (12–14), which is critical as tau immunization induces neurodegeneration (43). Second, unlike other tau immunotherapies against tau oligomers, aggregates, or tangles, cis mAb targets a disease driver that appears long before these tau epitopes in TBI/CTE (12, 13), VCID and AD. Third, whereas most other tau antibodies show efficacy in tau TG mice, we have demonstrated the efficacy of cis mAb in non-TG model mice of CTE (12, 13) and VCID. This might be important because most human tauopathies do not arise from tau mutations or overexpression.
In addition, we have leveraged the statistical power of single-nucleus RNA-seq to unbiasedly and comprehensively evaluate the therapeutic response of the mouse cortex to the cis mAb. Proper evaluation of therapeutic response of preclinical drugs, especially for neurodegenerative diseases, has been challenging. One limitation is the inconsistency of therapeutic efficacy between model mice and human patients, given that studies in mice often focus on a limited number of hypothesized pathogenic factors and phenotypes. We show that cis P-tau induces the conserved transcriptomic changes found in mouse VCID and human AD with early pathology and that cis mAb therapy restores ~85-90% of DEGs in VCID mice, including changes found in human early AD brains. Thus, cis mAb might be potent and specific for treating VCID, AD, and TBI/CTE at early stages.
The therapeutic potential of cis mAb might be enhanced by the potential use of P-tau conformations as biomarkers for tauopathies. CSF P-tau (pT231) is a well-known AD biomarker for both diagnosis and tracking of MCI to AD progression (44). In patients with TBI, high P-tau (pT231) concentrations in the CSF (13) or blood (16) correlate with poor clinical outcome. In addition, our current findings that cis P-tau is a major driver of VCID and AD further support the idea of using P-tau conformation-specific mAbs to identify appropriate patients for cis-targeted therapy and to assess their therapeutic response (12–14, 17). Given the efficacy of cis mAb in treating BCAS and htau mice with ongoing or already developed neurodegeneration, one may expect elimination of cis P-tau using cis mAb to be still effective even after symptoms have already developed. A follow-up study with careful control of dose and delayed timing of cis mAb treatment will be interesting to potentially extend the clinical value of this approach. These findings provide a rationale for clinical evaluation of the safety and efficacy of cis mAb for early diagnosis, prevention, and treatment of cognitive impairment and dementia after neurovascular insults and in AD.
Our study has several limitations. First, the droplet-based single-nucleus RNA-seq technology has its limitations. Because of the low transcript capture efficiency, low amount of RNA-input from single-nucleus and the stochastic nature of gene expression among cells, the statistical power to perform DEG analyses is limited by the number of cells. Thus, we were unable to detect sufficient microglia cells to assess the apparent neuroinflammation and instead focused on five major cell types identified, as discussed (45, 46). Second, the mechanism leading to DAPK1 activation remains unclear. It has been shown that cerebral ischemia recruits DAPK1 to the NMDA receptor complex and apparently leads to calcium influx in the cortex, but whether and how cerebral hypoperfusion causes DAPK1 activation remains to be defined. Third, how cis P-tau leads to the cistauosis signature observed in VCID mice and patients with early AD is unknown, although nuclear tau may affect transcription during neuronal stress (47).
Despite the limitations, our histopathological, ultrastructural, electrophysiological, behavioral, and single-cell genomic analyses consistently demonstrated that cis P-tau underlies VCID and AD by inducing the conserved transcriptomic signature and axonopathy and could be effectively targeted by immunotherapy, uncovering previously unrecognized pathogenic mechanisms and offering a promising therapeutic approach for VCID and AD.
MATERIALS AND METHODS
Study design
In this study, we detected robust cis P-tau in patients with VCID and characterized the cis P-tau induction kinetics and pathological roles in mice after BCAS. We subsequently examined and found that both Pin1 overexpression and DAPK1 KO abolished the VCID-induced cis P-tau and rescued most VCID or AD-like pathological and functional outcomes. Given the known vascular contributions to AD, we tested whether cis mAb could ameliorate progressive neurodegeneration and cognitive decline in htau mice, an AD-like mouse model. Furthermore, we profiled the single-cell transcriptome in five major cortical cell types in mice 1 month after BCAS, with or without cis mAb treatment. Last, we investigated whether purified cis P-tau was sufficient to cause brain pathology, behavioral deficits, and the gene expression profile found in BCAS mice and patients with AD. The sample sizes were estimated considering the variation and mean of the samples before the experiments, and the number of biological replicates was reported in the relevant figure legends. Animals were randomly assigned groups for in vivo studies, and group allocation and outcome assessment were also done by different people in a double-blinded manner for mAb treatment and behavioral experiments. No animals or samples were excluded from any analysis except few mice died immediately after BCAS surgery. All histopathological examinations were blinded to injury and treatment status. Data acquisition and analyses were obtained in an unbiased fashion.
Human brain specimens
Discarded fixed human brain tissues with neuropathologically verified VaD and their age-matched controls were obtained from the Northwestern Alzheimer’s Disease Center or a rapid autopsy program of the Netherlands Brain Bank (NBB), Netherlands Institute for Neuroscience, Amsterdam (average postmortem delay, 6 hours) (open access: www.brainbank.nl/) (table S1). All materials have been collected from donors for or from whom a written informed consent for a brain autopsy, and the use of the material and clinical information for research purposes had been obtained by the NBB. Institutional Review Board approval for tissue donation and our studies on discarded human samples was obtained through Beth Israel Deaconess Medical Center and NBB.
Animals
Male C57BL/6 WT, htau, or tau KO mice obtained from the Jackson laboratory, and Pin1 TG mice and DAPK1 KO mice were previously generated in our laboratories (26, 28). Animal housing and experiments were described in Supplementary Materials and Methods.
Statistical analysis
All data are presented as the means ± SEM, followed by the two-tailed Student’s t test for quantitative variables between two groups or one-way analysis of variance (ANOVA) with post hoc Dunnett’s multiple comparison test for three or more groups, as indicated in the legend. P values are shown in the figures or figure legend.
Supplementary Material
Acknowledgments:
We are grateful to M. E. Greenberg and O. Dagliyan for expertise and help with single-nucleus RNA-seq experiments.
Funding:
C.Q. and O.A. are Alzheimer’s Association Research Fellows, C.-Y.T. is a National Science Council Postdoctoral Fellow from Taiwan, and P.A. is supported by an NIH Training Grant 2T32AG000222-26 to C.-Y.T. E.H.B. is supported by NIA P30 AG013854 to E.H.B. This work was supported by NIH grants R01AG055559 to K.P.L., E.H.L. and X.Z.Z. and R01AG046319, U01NS096835, Alzheimer’s Association (DVT-14-322623), Alzheimer’s Research UK and Weston Brain Institute (MCDN-15-368711), Thome Memorial Foundation in Alzheimer’s Disease Drug Discovery Research to K.P.L, and National Football League grant to W. Meehan, K.P.L., and X.Z.Z. and gift donations from the Owens Family Foundation to X.Z.Z. and K.P.L.
Footnotes
Competing interests:
K.P.L. and X.Z.Z. are inventors of many Pin1-related patents, including cis P-tau antibody technology, which was licensed by BIDMC to Pinteon Therapeutics. Both K.P.L. and X.Z.Z. are the scientific founders of and own equity in Pinteon. Their interests were reviewed and are managed by BIDMC in accordance with its conflict of interest policy. The other authors declare that they have no competing interests.
Data and materials availability:
Raw data for single-nucleus RNA sequencing experiments were deposited under NCBI BioProject PRJNA521781, and all other data are available in the main text or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Iadecola C, The pathobiology of vascular dementia. Neuron 80, 844–866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisler K, Nelson AR, Montagne A, Zlokovic BV, Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci 18, 419–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, Spillantini MG, A century of Alzheimer’s disease. Science 314, 777–781 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Mandelkow E, Tau in physiology and pathology. Nat. Rev. Neurosci 17, 5–21 (2016). [DOI] [PubMed] [Google Scholar]
- 5.DeKosky ST, Blennow K, Ikonomovic MD, Gandy S, Acute and chronic traumatic encephalopathies: Pathogenesis and biomarkers. Nat. Rev. Neurol 9, 192–200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pluta R, Ulamek-Koziol M, Januszewski S, Czuczwar SJ, Tau protein dysfunction after brain ischemia. J. Alzheimers Dis. 66, 429–437 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuo Q.-z., Lei P, Jackman KA, Li X-L, Xiong H, Li X-L, Liuyang Z.-y., Roisman L, Zhang S.-t., Ayton S, Wang Q, Crouch PJ, Ganio K, Wang X-C, Pei L, Adlard PA, Lu Y-M, Cappai R, Wang J-Z, Liu R, Bush AI, Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Lu KP, Hanes SD, Hunter T, A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380, 544–547 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP, The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399, 784–788 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP, Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424, 556–561 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Lu KP, Zhou XZ, The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, Wei S, Luo ML, Albayram O, Huang P, Rotenberg A, Ryo A, Goldstein LE, Pascual-Leone A, McKee AC, Meehan W, Zhou XZ, Lu KP, Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albayram O, Kondo A, Mannix R, Smith C, Li C, Tsai CY, Herbert MK, Qiu J, Monuteaux M, Driver JA, Yan S, Gormley W, Puccio AW, Okonkwo DO, Lucke-Wold B, Bailes J, Meehan W, Zeidel M, Lu KP, Zhou XZ, Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun 8, 1000 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP, Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 149, 232–244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McEwan WA, Falcon B, Vaysburd M, Clift D, Oblak AL, Ghetti B, Goedert M, James LC, Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. U.S.A 114, 574–579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubenstein R, Chang B, Yue JK, Chiu A, Winkler EA, Puccio AM, Diaz-Arrastia R, Yuh EL, Mukherjee P, Valadka AB, Gordon WA, Okonkwo DO, Davies P, Agarwal S, Lin F, Sarkis G, Yadikar H, BS ZY, Manley GT, Wang KKW; TRACK-TBI Investigators, Comparing plasma phospho tau, total tau, and phospho tau–total tau ratio as acute and chronic traumatic brain injury biomarkers. JAMA Neurol. 74, 1063–1072 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ, Potential of the antibody againstcis–phosphorylated tau in the early diagnosis, treatment, and prevention of alzheimer disease and brain injury. JAMA Neurol. 73, 1356–1362 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Safety and Tolerability of PNT001 in Healthy Adults (2019); https://clinicaltrials.gov/ct2/show/NCT04096287. [Google Scholar]
- 19.Ferrer I, Aguilo Garcia M, Carmona M, Andres-Benito P, Torrejon-Escribano B, Garcia-Esparcia P, Del Rio JA, Involvement of oligodendrocytes in tau seeding and spreading in tauopathies. Front. Aging Neurosci. 11, 112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balczon R, Morrow KA, Zhou C, Edmonds B, Alexeyev M, Pittet JF, Wagener BM, Moser SA, Leavesley S, Zha X, Frank DW, Stevens T, Pseudomonas aeruginosa infection liberates transmissible, cytotoxic prion amyloids. FASEB J. 31,2785–2796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venkat P, Chopp M, Chen J, Models and mechanisms of vascular dementia. Exp. Neurol 272, 97–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R, Ihara M, Takahashi R, Tomimoto H, Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 38, 2826–2832 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Ryo A, Liou Y-C, Wulf G, Nakamura M, Lee SW, Lu KP, PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol 22, 5281–5295 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao T, He B, Liu S, Zhu H, Tan K, Qian J, EnhancerAtlas: A resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics 32, 3543–3551 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, Marchini J, Smith SM, Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim J, Balastik M, Lee TH, Liou YC, Sun A, Finn G, Pastorino L, Lee VM-Y, Lu KP, Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J. Clin. Invest 118, 1877–1889 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, Balel C, Wang M, Jia N, Zhang W, Lew F, Chan SL, Chen Y, Lu Y, DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140, 222–234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim BM, You MH, Chen CH, Lee S, Hong Y, Hong Y, Kimchi A, Zhou XZ, Lee TH, Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis. 5, e1237 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polydoro M, Acker CM, Duff K, Castillo PE, Davies P, Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci 29, 10741–10749 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polydoro M, Dzhala VI, Pooler AM, Nicholls SB, McKinney AP, Sanchez L, Pitstick R, Carlson GA, Staley KJ, Spires-Jones TL, Hyman BT, Soluble pathological tau in the entorhinal cortex leads to presynaptic deficits in an early Alzheimer’s disease model. Acta Neuropathol. 127, 257–270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarabi AS, Shen H, Wang Y, Hoffer BJ, Backman CM, Gene expression patterns in mouse cortical penumbra after focal ischemic brain injury and reperfusion. J. Neurosci. Res 86, 2912–2924 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Gow A, Southwood CM, Li JS, Pariali M, Riordan GP, Brodie SE, Danias J, Bronstein JM, Kachar B, Lazzarini RA, CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 99, 649–659 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Lai J-J, Cruz FM, Rock KL, Immune sensing of cell death through recognition of histone sequences by C-type lectin-receptor-2d causes inflammation and tissue injury. Immunity 52, 123–135.e6. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li JH, Kirkiles-Smith NC, McNiff JM, Pober JS, TRAIL induces apoptosis and inflammatory gene expression in human endothelial cells. J. Immunol 171, 1526–1533 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraus DW, Colacino JM, Extended oxygen delivery from the nerve hemoglobin of Tellina alternata (Bivalvia). Science 232, 90–92 (1986). [DOI] [PubMed] [Google Scholar]
- 37.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW, Incipient Alzheimer’s disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. U.S.A 101,2173–2178 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clavaguera F, Tolnay M, Goedert M, The prion-like behavior of assembled tau in transgenic mice. Cold Spring Harb. Perspect. Med 7, ea024372 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zemva J, Schubert M, The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer’s disease: Possible therapeutic implications. CNS Neurol. Disord. Drug Targets 13, 322–337 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Coulthard MG, Morgan M, Woodruff TM, Arumugam TV, Taylor SM, Carpenter TC, Lackmann M, Boyd AW, Eph/Ephrin signaling in injury and inflammation. Am. J. Pathol 181,1493–1503 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Wegmann S, Maury EA, Kirk MJ, Saqran L, Roe A, DeVos SL, Nicholls S, Fan Z, Takeda S, Cagsal-Getkin O, William CM, Spires-Jones TL, Pitstick R, Carlson GA, Pooler AM, Hyman BT, Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 34, 3028–3041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Congdon EE, Sigurdsson EM, Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol 14, 399–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O, Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch. Neurol 63, 1459–1467 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT, Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 103, 26–35 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Kalish BT, Cheadle L, Hrvatin S, Nagy MA, Rivera S, Crow M, Gillis J, Kirchner R, Greenberg ME, Single-cell transcriptomics of the developing lateral geniculate nucleus reveals insights into circuit assembly and refinement. Proc. Natl. Acad. Sci. U.S.A 115, E1051–E1060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA, Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, Colin M, Bonnefoy E, Buee L, Galas MC, Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem 286, 4566–4575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW, Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161,1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolock SL, Lopez R, Klein AM, Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. bioRxiv, 357368 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Satija R, Farrell JA, Gennert D, Schier AF, Regev A, Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, Huang D, Xu Y, Huang W, Jiang M, Jiang X, Mao J, Chen Y, Lu C, Xie J, Fang Q, Wang Y, Yue R, Li T, Huang H, Orkin SH, Yuan G-C, Chen M, Guo G, Mapping the mouse cell atlas by Microwell-seq. Cell 172, 1091–1107.e17 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data for single-nucleus RNA sequencing experiments were deposited under NCBI BioProject PRJNA521781, and all other data are available in the main text or the Supplementary Materials.