

Abstract
The serotonin transporter (SERT) is the primary target for the selective serotonin reuptake inhibitors (SSRIs). However, the structural basis for the extraordinarily high binding affinity of the widely-prescribed SSRI, paroxetine, to human SERT (hSERT) has not yet been fully elucidated. Our previous findings unveiled a plausible ambiguity in paroxetine’s binding orientations that may constitute an integral component of this SSRI’s high affinity for hSERT. Herein, we investigate factors contributing to paroxetine’s high affinity by modifying both the ligand and the protein. We generated a series of Bromine (Br)-containing derivatives and found that the one in which the 4-F of paroxetine had been replaced with the chemically-similar but more electron-rich Br atom (13) has the highest affinity. By comparatively characterizing the binding of paroxetine and 13 to both WT and a construct harboring a paroxetine-sensitive mutation in the binding cavity, we identified a mechanistic determinant responsible for the pose ambiguity of paroxetine, which can guide future drug design.
Keywords: Paroxetine, Serotonin Transporter, Selective Serotonin Reuptake Inhibitors, Organocatalysis, Asymmetric Chemistry, Structure-Activity Relationship
INTRODUCTION
Serotonergic signaling mediates many physiological processes in both the central and peripheral nervous systems, such as mood, appetite, sleep, memory, sexual behavior, and neuroendocrine function.1 The serotonin (5-hydroxytryptamine [5-HT]) transporter (SERT) tightly regulates these processes by clearing 5-HT from the synaptic cleft and recycling it into the presynaptic neuron, thereby terminating 5-HT neurotransmission. Given SERT’s crucial role in human physiology, its dysfunction has been implicated in a wide array of diseases, and thus, it has a long history as a target for pharmaceutical development. Furthermore, recreational drugs, such as 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”), bind to SERT2 and may cause long-lasting damage to serotonergic neurons.3, 4 Therefore, structure-activity relationship studies of SERT ligands are critical to improve understanding of ligand-transporter interactions and to illuminate mechanisms underlying these processes.
Human SERT (hSERT) is the primary target for the selective serotonin reuptake inhibitors (SSRIs), such as the FDA-approved and widely-prescribed escitalopram and paroxetine, both of which competitively inhibit 5-HT transport by stabilizing hSERT in an outward-open conformation.5 Of all the SSRIs, paroxetine binds with the highest affinity to hSERT (Ki < 1 nM) and is highly selective.6, 7 It is one of the most therapeutically useful SSRIs, prescribed for the treatment of a wide array of neuropsychiatric disorders such as depression, panic disorder, and obsessive-compulsive disorder.8 Despite its therapeutic value, the molecular determinants of paroxetine binding to hSERT have been disputed.9–11
A crystal structure of a thermostable but transport-deficient hSERT construct, in complex with a Fab fragment was solved with paroxetine bound in the central primary binding (S1) site (PDB ID 5I6X);10 however, one of the thermostabilizing mutations (T439S) was located within this site (Figure 1), which could conceivably distort both the ligand binding pocket as well as the orientation of bound ligand.12 Although a different construct in which the Ser439 had been mutated back to Thr was subsequently employed to determine the structure of two additional hSERT-paroxetine complexes, one via X-ray diffraction (PDB ID 6AWN)9 and the second via cryo-electron microscopy (PDB ID 6DZW),13 this construct is still inactive, and the data are of considerably lower resolution.
Figure 1.
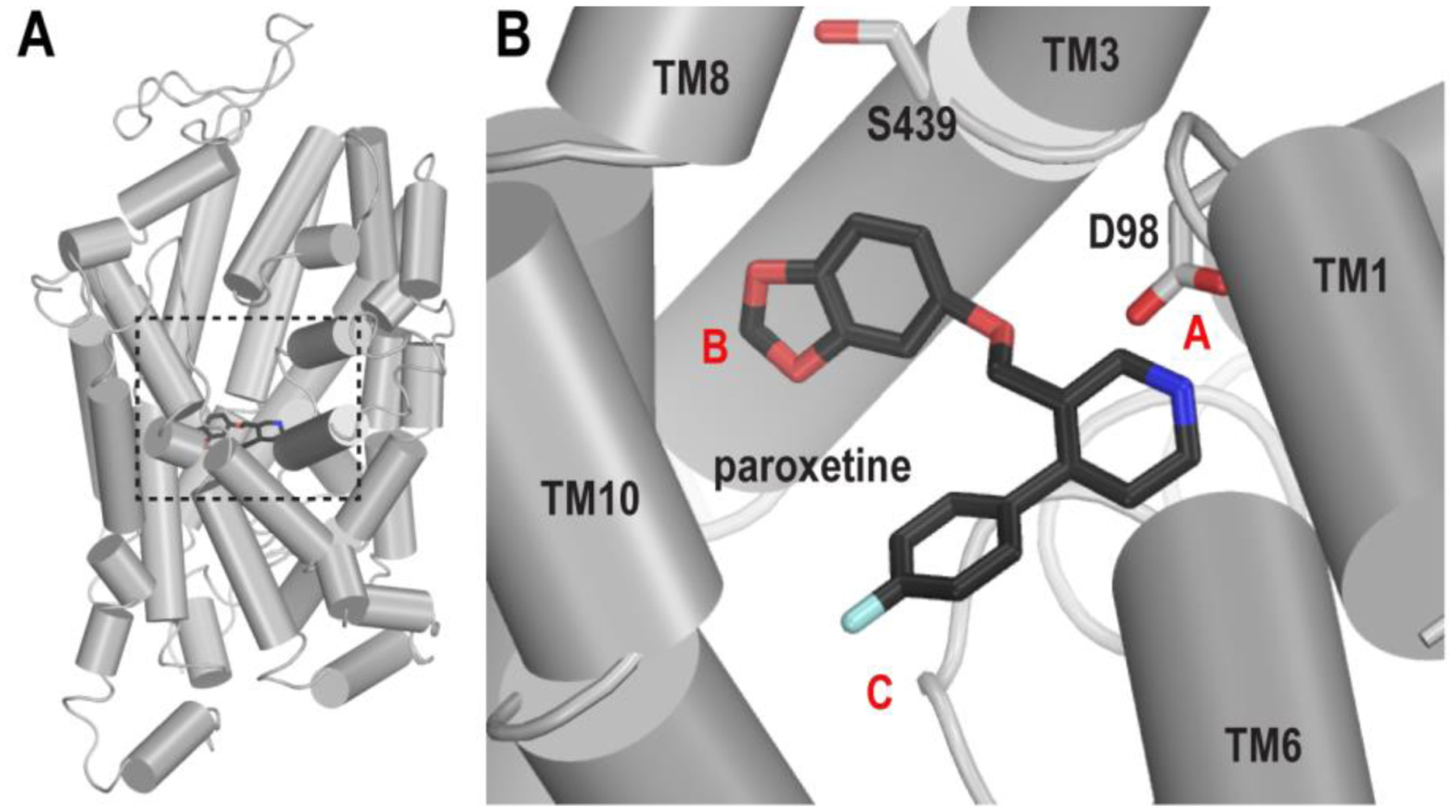
The binding pose of paroxetine in the S1 site of hSERT. (A) The crystal structure of hSERT in complex with paroxetine (PDB ID 5I6X) is viewed parallel to the membrane normal in a cylindrical representation. The S1 site is enclosed by a black dotted box. (B) The paroxetine pose in the S1 site of the structure. Asp98 and Ser439 (see text) are shown in white sticks. The locations of subsites A, B, and C are labeled with red letters.
The S1 site can be divided into three subsites A, B, and C (Figure 1B).14 In all three hSERT/paroxetine structures, the paroxetine piperidine nitrogen interacts with the carboxylate of Asp98 within subsite A, while the benzodioxole and F-phenyl moieties reside in subsites B and C, respectively.9, 10, 13 As defined before,12 this binding pose is designated ABC. In a previous study using a combined in silico and in vitro approach, the “flipped” paroxetine orientation in which the benzodioxole and the F-phenyl occupy subsites C and B, respectively, was proposed and validated11; it is dubbed pose ACB in the current study. We have previously characterized these two potential paroxetine binding poses in the S1 site of hSERT using computational approaches.12 The results of our molecular dynamics (MD) simulations and free energy calculations indicated that both poses are feasible, and that the entropic component, including both pose ambiguity and oscillation of the Phe341 side chain in the S1 site, contributes to paroxetine’s remarkably high affinity.
To investigate the mechanism of this pose ambiguity, here we design and synthesize a series of paroxetine derivatives, evaluating their respective affinities for hSERT-WT. By further examining the effect of the most potent Br-derivative on a single-residue, paroxetine-sensitive hSERT mutant, i.e., A169D11, with both in vitro and in silico approaches, we reveal a mechanistic basis for paroxetine’s pose ambiguity.
RESULTS
Synthesis and characterization of Br-substituted paroxetine analogs.
We hypothesized that by identifying a position on paroxetine that would tolerate both the steric bulk and electronic properties of a Br atom, we could further investigate paroxetine binding interactions in the S1 site of hSERT-WT. In addition, it is well known that “heavy” or electron-rich atoms such as Br scatter X-rays at unique absorption edges (λ = 0.92 in the case of Br) to create an anomalous signal in anomalous difference Fourier electron density maps, allowing for unambiguous assignment of the atom’s position.15, 16 Hence, we reasoned that, with Br acting as a “beacon”, a high-affinity Br-substituted paroxetine analog would provide a critical tool for X-ray crystallographic analyses.
To this end, we designed N-substituted paroxetine derivatives to probe the bulk tolerated at the piperidine nitrogen and to identify a suitable handle with which to append a heavy Br atom, i.e., 4’-Br-phenyl. We decided to synthesize both tertiary amines as well as an analogous amide because, while most monoamine transporter ligands require a free amine for optimal binding interactions17 (with Asp98 in hSERT), a photoactive paroxetine amide has been reported.18 Finally, we replaced the 4-F in paroxetine to generate 4-bromoparoxetine (13) for the purpose of scrutinizing its potential as a structurally similar but heavy-atom-bearing tool.
Synthetic schemes are shown in Figure 2 and experimental detail is described in Supporting Information. In brief, commercially available and enantiopure (-)-trans-paroxetine (Figure 2A) was N-alkylated using either 1-bromohexane or 1-bromopropane in a suspension of K2CO3 in acetonitrile to provide 1 and 2, respectively. Synthesis of N-phenylalkyl derivatives 3-5 (Figure 2A) was achieved by the reductive amination of paroxetine using triacetoxyborohydride and the appropriate aldehyde in THF. An X-ray crystal structure of 4’-bromobenzyl 3 confirmed that the 3C and 4C substituents are in a trans-diequatorial conformation (Figure S1), consistent with previous paroxetine derivatives.19 Finally, amide 6 (Figure 2B) was synthesized via an EDC [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide] coupling of (-)-trans-paroxetine with 4-bromophenylacetic acid and catalytic DMAP (4-dimethylaminopyridine).
Figure 2.
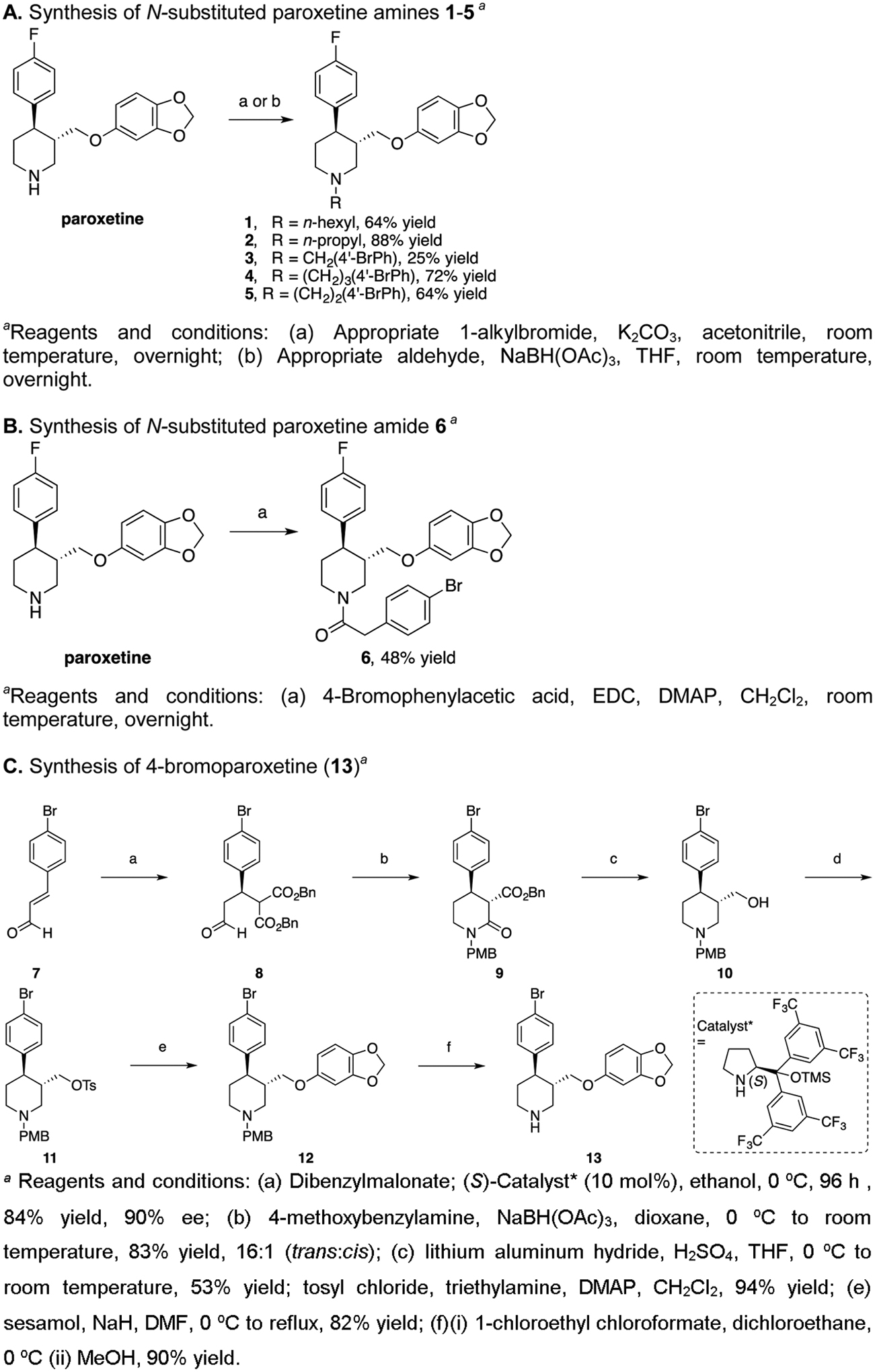
Synthetic schemes for paroxetine analogs
Jørgensen’s asymmetric organocatalytic Michael addition of malonates to α,β-unsaturated aldehydes20 was applied toward the synthesis of 4-bromoparoxetine (13, Figure 2C). 4-Bromocinnamaldehyde (7)21 was subjected to a conjugate addition of dibenzyl malonate using 10 mol% of the commercially available L-proline-derived (S)-Catalyst* [(S)-2-[bis(3,5-bistrifluoromethylphenyl) trimethylsilanyloxymethyl]-pyrrolidine] to provide the β-malonylaldehyde 8 in 84% yield and 90% enantiomeric excess (ee).20 Reductive amination with commercially available 4-methoxybenzylamine and in situ cyclization gave lactam 9. A global reduction using alane, generated from lithium aluminum hydride and sulfuric acid,22 provided aminoalcohol 10, which was tosylated at the OH to give 11, and displaced with sesamol, giving 4-methoxybenzyl (PMB)-protected 4-bromoparoxetine 12 in 82% yield. Deprotection of the piperidine nitrogen by first treating with 1-chloroethyl chloroformate and subsequent methanolysis provided (-)-4-bromoparoxetine (13) in 90% yield.
Other reports of paroxetine23, 24 used 1H NMR spectroscopy to assign the stereochemistry around the piperidine ring. (-)-trans-paroxetine C3 (2.60 ppm) and C4 (2.76 ppm) protons both have triplet of doublet splitting patterns, coupled to one another at J = 12 Hz, consistent with protons in a trans-diequatorial stereochemistry. Similarly, 4-bromoparoxetine (13) C3 and C4 protons are at 2.57 and 2.74 ppm, respectively, coupled to each other in a trans-orientation at J = 12 Hz. Additionally, compound 5 has a high specific optical rotation −96.4 {(similar to paroxetine), cf. [α22D] = −89.4 (c = 0.75, CH3OH)},23 and its enantiopurity was determined to be ≥99% ee by comparing (-)-13 with its racemate using chiral HPLC analysis. Finally, presence of the 4-bromo was proven by mass spectrometry, where isotope splitting was observed at 389.1 and 391.1.
Pharmacological characterization of paroxetine analogs.
Subsequent to their syntheses, the analogs were tested for their respective abilities to inhibit both [3H]citalopram binding to rat SERT (rSERT) from rat brain tissue and [3H]5-HT transport by hSERT stably expressed in HeLa cells. Notably, these two SERT homologs share 92% identity, with 100% conservation of all amino acid residues in the S1 pocket.
Compound binding affinities for rSERT are shown in Table S1 and Figure 3A. In this study, (-)-trans-paroxetine bound to rSERT with picomolar affinity (Ki = 0.311 nM). Akin to what was previously reported,19 N-methylation of paroxetine to compound 14 resulted in a 9.5-fold decrease in potency (Ki = 2.95 nM). The detrimental effect of N-methylation of paroxetine or N-substitution with CH2F has also been documented.25 A more pronounced loss in binding affinity was observed when the N-alkyl chain length of paroxetine was extended to an N-propyl (2) and an N-hexyl (1) (Ki = 98.0 and 395 nM, respectively). Despite this trend correlating longer N-alkyl chain lengths with diminished ligand potencies, a curious anomaly emerged when the Br-phenyl was appended to the piperidine nitrogen via varying linker chain lengths. While compounds with one-carbon-linked (3) and three-carbon-linked (4) chain lengths bound to rSERT with relatively low affinities (Ki = 204 and 101 nM, respectively), the one with a two-carbon-linked chain length (5) bound 2.4–4.9 times more tightly (Ki = 41.9 nM). This unexpected outcome suggests that the Br-phenyl moiety may form favorable interactions near the S1 site, depending on the linker length. By contrast, N-carboxylation, which removes the charge and rigidifies the linker, abrogated affinity, as illustrated by comparing amide 6 (Ki = 3,820 nM) with its two-carbon-linked amine counterpart 5. These data support the critical role of the salt bridge between the ligand amine and the side-chain carboxylate of the conserved Asp98,10 and call into question the reported high affinity for the paroxetine-based photolabel.18
Figure 3.
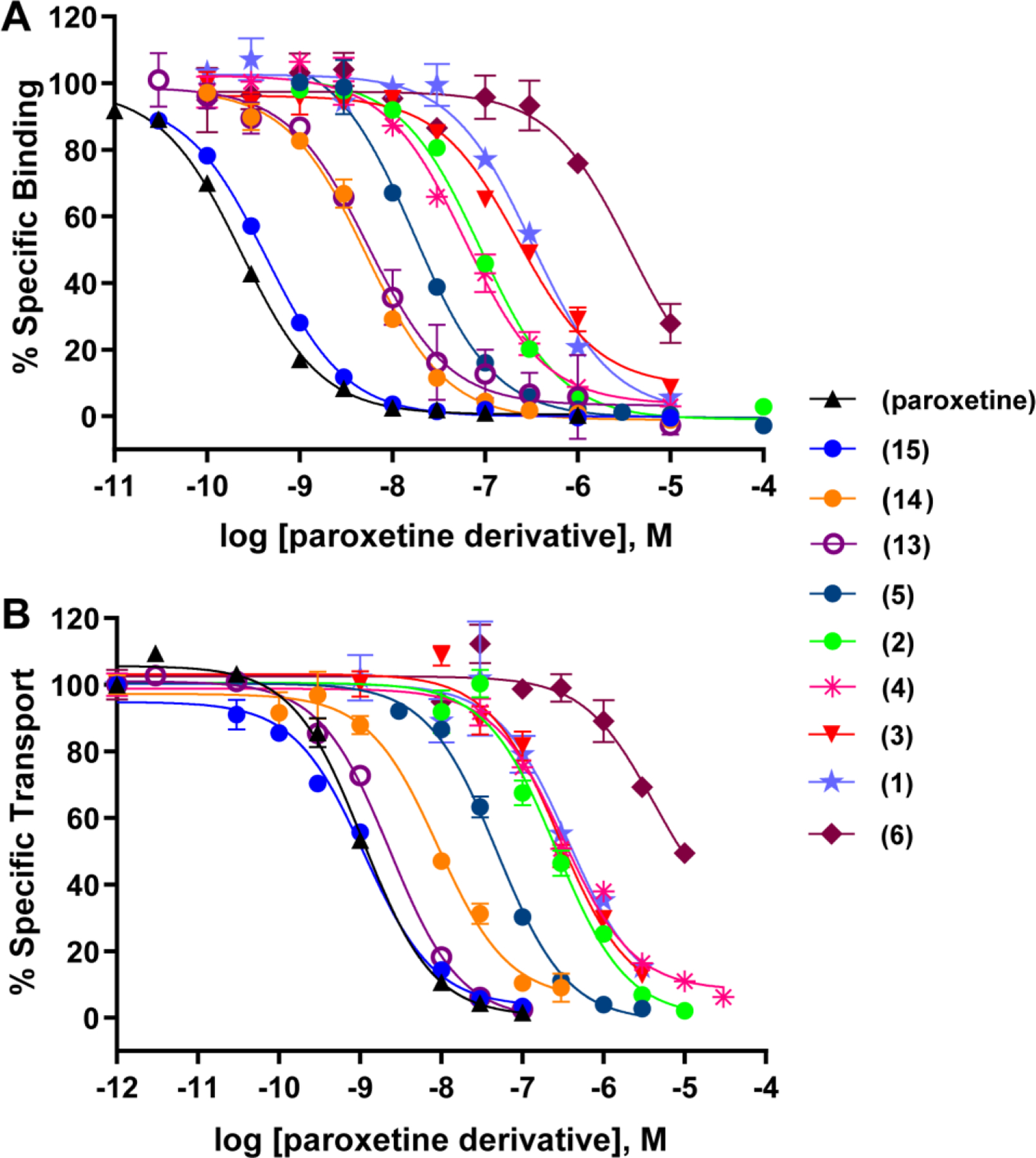
Potencies of paroxetine derivatives on SERT. (A) Inhibition of [3H]citalopram binding to rSERT by the indicated compounds. (B) Inhibition of [3H]5-HT transport by hSERT by the indicated compounds.
The desfluoro analog of paroxetine (15) bound to rSERT similarly as paroxetine (Ki = 0.557 and 0.311 nM, respectively), implying that the 4-F is not required for high affinity binding to SERT but likely imparts metabolic stability to this clinically used drug.26 When the 4-F on paroxetine was replaced with the 4-Br (13, Ki = 4.90 nM), however, binding affinity to rSERT decreased 16-fold. Nevertheless, binding experiments using [3H]paroxetine instead of [3H]citalopram, and using hSERT transfected HEK293T cells, resulted in only a ~4-fold reduction in binding affinity for 13 as compared to paroxetine (Ki=0.073 ± 0.009 nM vs. 0.32 ± 0.028 nM). This relatively modest decrease parallels a similar decrease in binding affinity for a 4-CH2F analog, using [3H]paroxetine as the radioligand.25
All paroxetine derivatives were also evaluated for their respective abilities to inhibit [3H]5-HT transport by hSERT in stably-transfected HeLa cells (Table S1, Figure 3B). In general, these data correlated well with binding affinities at rSERT (Figure S2). For example, paroxetine and desfluoroparoxetine (15) exhibited comparable binding affinities at rSERT (Ki = 0.311 and 0.557 nM, respectively) and uptake inhibition potencies at hSERT (Ki = 0.90 and 0.97 nM, respectively). Likewise, 4-bromoparoxetine (13) inhibited [3H]5-HT uptake at hSERT with comparable potency (Ki = 3.13 nM) to its binding affinity for rSERT (Ki = 4.90 nM).
Characterization of paroxetine and 4-bromoparoxetine (Br-paroxetine) binding modes in the hSERT-A169D mutant.
Our previous functional characterization of three SERT homologues revealed that Ala169 in TM3 located in the S1 site plays a vital role in paroxetine recognition.11 As demonstrated in Davis et al.11 and also in this study (Table 1), mutating this residue to aspartate, the corresponding residue in the SERTs from Drosophila and chicken, significantly diminished the apparent affinity of paroxetine for hSERT-A169D (~30-fold based on inhibition of [125I]RTI-55 binding, and 7-fold based on inhibition of [3H]5-HT uptake). Strikingly, by contrast, Br-paroxetine was insensitive to the A169D mutation in both binding and uptake assays (Table 1). Interestingly, in the presence of the A169D mutation, the affnity of paroxetine was reduced to that of Br-paroxetine. Thus, it appears that the mutation disrupts ligand-protein interactions unique to paroxetine.
Table 1.
Paroxetine and Br-Paroxetine Inhibition of RTI-55 Binding to and 5-HT Transport by hSERT
| Analog | Structure | Ki (nM) RTI-55 Binding | Ki (nM) 5-HT Transport | ||||||
|---|---|---|---|---|---|---|---|---|---|
| WT | A169D | Pa | Sensitivityb | WT | A169D | Pa | Sensitivityb | ||
| Paroxetine |  |
0.08±0.02 | 2.10±0.18 | 0.003 | 26.3 | 1.95±0.27 | 13.62±2.31 | 0.002 | 7.0 |
| Br-Paroxetine (13) |  |
1.07±0.30 | 1.72±0.39 | 0.091 | 1.6 | 2.30±0.27 | 6.37±0.74 | 0.009 | 2.8 |
P-values calculated with the Student’s unpaired T-test. Values less than 0.05 indicate a statistically significant difference between the Ki values of the WT and A169D variants toward paroxetine or Br-paroxetine.
Refers to the “sensitivity” of paroxetine versus Br-paroxetine toward the A169D mutation in hSERT.
To examine the impact of the A169D mutation at atomistic level, we carried out extensive MD simulations of the hSERT-A169D construct in complex with paroxetine or Br-paroxetine in either the ABC or ACB pose, and compared the results to those of the corresponding WT simulations12 (Table S2). Based on the pair-wise ligand RMSD calculations of the most representative frames between the mutant construct and WT (see Methods), we found that, for both paroxetine and Br-paroxetine, the A169D mutation destabilized the ACB pose more than it did the ABC pose (Figure 4 A–E). Specifically, when both ligands were modeled in the ABC pose, the orientation of the benzodioxol substituent, which abuts residue 169, was not affected by the mutation. By contrast, in the ACB pose, with the 4-halophenyl moiety facing the negatively-charged Asp169, the ensuing electrostatic repulsion with the electronegative F- of paroxetine or steric hindrance with the bulky Br- of Br-paroxetine were both disruptive.
Figure 4.
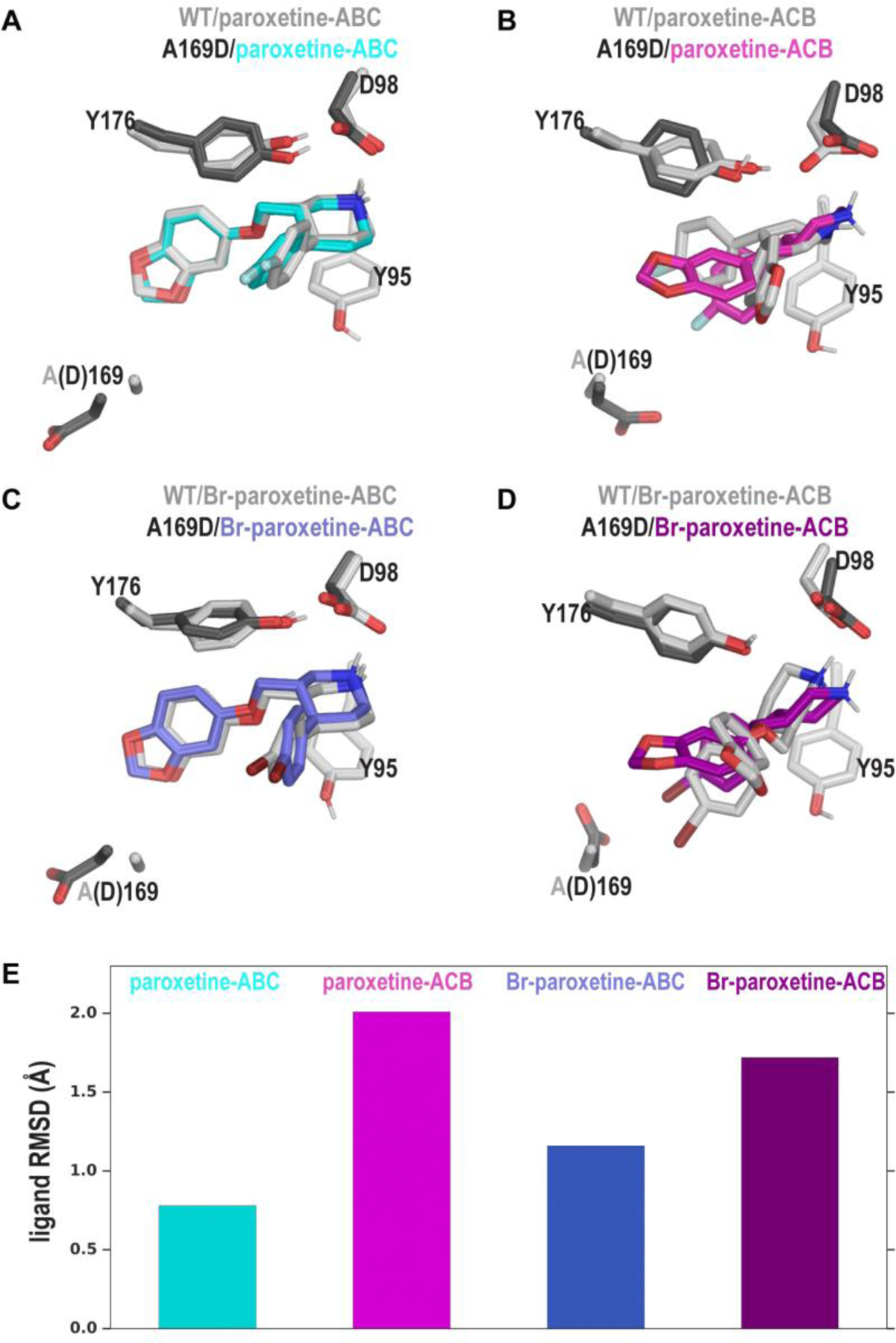
Distinct effects of the A169D mutation on the ABC and ACB poses of paroxetine and Br-paroxetine (13). (A-D) Zoomed-in views of the S1 site showing the ABC and ACB poses of paroxetine (A and B) and Br-paroxetine (C and D) in which the A169D mutant models (colored) are superposed on their respective WT models (gray). In our simulations, the ABC poses of both paroxetine and 13 were not significantly affected by the mutation, while the side chain of Asp169 flips away from the S1 site (A and C). In contrast, for both paroxetine and 13, their ACB poses were obviously distorted by the mutation – while the negatively charged sidechain of Asp electrostatically repelledthe F- of paroxetine (B), it appeared to attract the σ-hole of the Br- of Br-paroxetine (D). However, in the latter case, the sidechain of Asp and the bulky Br-moiety sterically hinder each other. Destabilization of ligand poses by the A169D mutation is quantified by ligand RMSDs of the A169D mutants with respect to their corresponding WT complexes (E).
Thus, for paroxetine, it is likely that the disruption of the ACB pose by the A169D mutation is responsible for the decrease of the experimentally determined affinity. For Br-paroxetine, the near insensitivity of the ligand to the A169D mutation suggests that Br-paroxetine is less likely to take the ACB pose that can be disrupted by the mutation, and prefers the ABC pose (Figure 4 C,E). Combining both the experimental and computational findings, we thus deduced that the mutation may restrict both paroxetine and Br-paroxetine to adopt the ABC pose, resulting in similar binding affinities.
We next investigated why the N-substituted Br-containing derivatives might have significantly reduced affinities, using the highest affinity derivative 5 as an example. Analysis of our MD simulations of hSERT in complex with 5 (Table S2) indicated that, compared to the corresponding paroxetine and Br-paroxetine poses, the additional Br-phenyl moiety on the charged nitrogen of 5 protrudes in between transmembrane segments (TMs) 1, 2, 6, and 7: a hydrophobic cavity formed by residues Val92, Phe133, Leu137, Leu344, and Thr364 (Figure 5A). In addition, the Br-phenyl moiety distorts the orientation of the Tyr95 side chain (Figure 5). Given that Tyr95 is a crucial residue for high-affinity recognition of antidepressants,27 we suggest that such a disruption may account for the diminished affinity of 5 as well as other N-substituted paroxetine derivatives.
Figure 5.
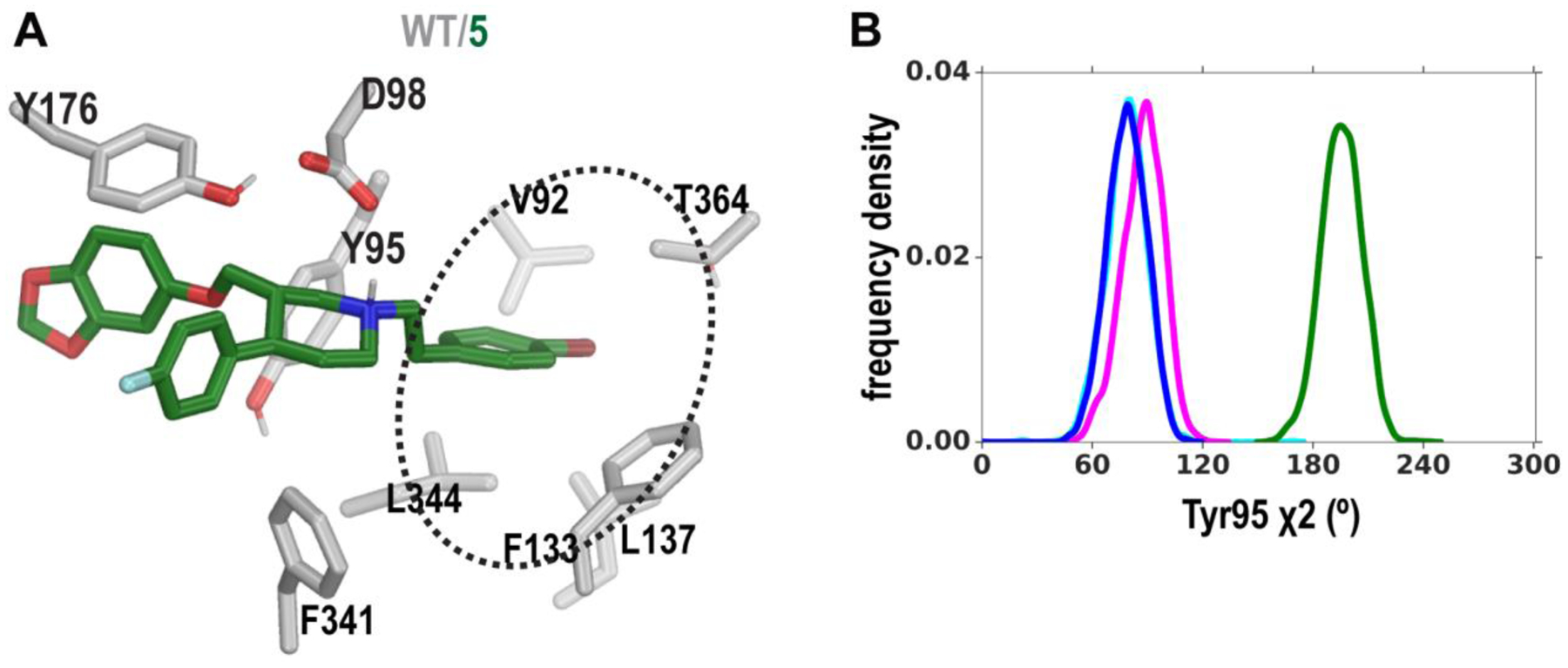
The conformational distortion near the S1 site induced by compound 5. (A) The binding pose of compound 5 in the S1 site viewed from the same angle as those in Figure 4, illustrating the distortion of Tyr95’s side chain. The Br-phenyl moiety of compound 5 protrudes into a hydrophobic cavity (dotted ellipse). (B) Distributions of the Tyr95 χ2 dihedral angle in the MD simulations are compared among the paroxetine- (blue), Br-paroxetine- (magenta) and 5- (green) bound hSERT conformations.
DISCUSSION AND CONCLUSIONS
We previously discovered, using hSERT-WT models, that paroxetine and Br-paroxetine (13) can stably bind in both ABC and ACB poses without disrupting the S1 site. However, our exhaustive computational analysis could not discern the preferred pose for each ligand. Although our combined mutagenesis and computational data presented herein still support the postulate that paroxetine can adopt both poses, we found that replacing the 4-F with 4-Br unexpectedly shifts the preference towards the ABC pose. However, we cannot exclude the possibility that the favored interaction between the σ-hole{Wilcken, 2013 #9164} of 4-Br, which has a partial positive charge, and the negatively charged side chain of Asp169 in the ACB pose may cancel out the disruptive impact of the mutation.
Of the five Br-containing compounds, Br-paroxetine (13) not only is the most structurally similar to paroxetine itself but is also the most potent inhibitor of 5-HT uptake. Together with the other paroxetine derivatives, none of which exhibits improved binding affinity or uptake inhibition potency, paroxetine appears to have achieved optimal affinity for hSERT with such a scaffold, in particular by maximizing a favored entropic contribution to its binding free energy.12
Nevertheless, our findings establish that a certain degree of symmetry exists between subsites B and C of the S1 crevice of hSERT, and such symmetry may be leveraged to improve ligand binding from the entropy perspective. Notably, it opens the door to rationally design or optimize compounds with high(er) binding affinities with other scaffolds not only for hSERT but also for homologous transporters, many of which are crucial therapeutic targets and share similar S1 site architectures, such as the dopamine and norepinephrine transporters.
Supplementary Material
ACKNOWLEDGMENT
We thank Randy Blakely for the pcDNA3.1 plasmid encoding hSERT-WT. We thank L. Keith Henry for the stable hSERT-WT HeLa cell line. The work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Funding Sources
Support for this research was provided by the National Institute on Drug Abuse–Intramural Research Program, Z1A DA000610-02 (A.H.N.), and Z1A DA000606-03 (L.S.); the Goodman-Gilman Yale Scholar Award (S.K.S.); the Alfred P. Sloan Foundation (S.K.S.); the Brain and Behavior Research Foundation (S.K.S.); and the NIH/National Institute of Mental Health Grants R00MH083050 (S.K.S.) and R01MH100688 (S.K.S.).
Footnotes
Supporting Information
Supplementary methods, Supplementary figures S1 and S2, and supplementary tables S1-S10.
REFERENCES
- 1.Lucki I, The spectrum of behaviors influenced by serotonin. Biol Psychiatry 1998, 44 (3), 151–62. [DOI] [PubMed] [Google Scholar]
- 2.Parrott AC; Sisk E; Turner JJ, Psychobiological problems in heavy ‘ecstasy’ (MDMA) polydrug users. Drug Alcohol Depend 2000, 60 (1), 105–10. [DOI] [PubMed] [Google Scholar]
- 3.Morgan MJ, Ecstasy (MDMA): a review of its possible persistent psychological effects. Psychopharmacology (Berl) 2000, 152 (3), 230–48. [DOI] [PubMed] [Google Scholar]
- 4.Lyles J; Cadet JL, Methylenedioxymethamphetamine (MDMA, Ecstasy) neurotoxicity: cellular and molecular mechanisms. Brain Res Brain Res Rev 2003, 42 (2), 155–68. [DOI] [PubMed] [Google Scholar]
- 5.Soderhielm PC; Andersen J; Munro L; Nielsen AT; Kristensen AS, Substrate and Inhibitor-Specific Conformational Changes in the Human Serotonin Transporter Revealed by Voltage-Clamp Fluorometry. Mol Pharmacol 2015, 88 (4), 676–88. [DOI] [PubMed] [Google Scholar]
- 6.Thomas DR; Nelson DR; Johnson AM, Biochemical effects of the antidepressant paroxetine, a specific 5-hydroxytryptamine uptake inhibitor. Psychopharmacology 1987, 93 (2), 193–200. [DOI] [PubMed] [Google Scholar]
- 7.Cool DR; Leibach FH; Ganapathy V, High-affinity paroxetine binding to the human placental serotonin transporter. Am J Physiol 1990, 259 (2 Pt 1), C196–204. [DOI] [PubMed] [Google Scholar]
- 8.Bourin M; Chue P; Guillon Y, Paroxetine: a review. CNS Drug Rev 2001, 7 (1), 25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman JA; Gouaux E, Structural basis for recognition of diverse antidepressants by the human serotonin transporter. Nat Struct Mol Biol 2018, 25 (2), 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coleman JA; Green EM; Gouaux E, X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532 (7599), 334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis BA; Nagarajan A; Forrest LR; Singh SK, Mechanism of Paroxetine (Paxil) Inhibition of the Serotonin Transporter. Sci Rep 2016, 6, 23789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abramyan AM; Slack RD; Meena S; Davis BA; Newman AH; Singh SK; Shi L, Computation-guided analysis of paroxetine binding to hSERT reveals functionally important structural elements and dynamics. Neuropharmacology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coleman JA; Yang D; Zhao Z; Wen PC; Yoshioka C; Tajkhorshid E; Gouaux E, Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sorensen L; Andersen J; Thomsen M; Hansen SM; Zhao X; Sandelin A; Stromgaard K; Kristensen AS, Interaction of antidepressants with the serotonin and norepinephrine transporters: mutational studies of the S1 substrate binding pocket. J Biol Chem 2012, 287 (52), 43694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tiefenbrunn T; Forli S; Happer M; Gonzalez A; Tsai Y; Soltis M; Elder JH; Olson AJ; Stout CD, Crystallographic fragment-based drug discovery: use of a brominated fragment library targeting HIV protease. Chem Biol Drug Des 2014, 83 (2), 141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendrickson WA, Anomalous diffraction in crystallographic phase evaluation. Q Rev Biophys 2014, 47 (1), 49–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitayama S; Shimada S; Xu H; Markham L; Donovan DM; Uhl GR, Dopamine transporter site-directed mutations differentially alter substrate transport and cocaine binding. Proceedings of the National Academy of Sciences of the United States of America 1992, 89 (16), 7782–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chudzik J; McCarthy D; Bakish D; Ravindran A; Hrdina PD, Synthesis and characterization of an aryl-azidoparoxetine. A novel photo-affinity probe for serotonin-transporter. Biochemical pharmacology 1995, 50 (8), 1211–5. [DOI] [PubMed] [Google Scholar]
- 19.Plenge P; Mellerup ET; Honore T; Honore PL, The activity of 25 paroxetine/femoxetine structure variants in various reactions, assumed to be important for the effect of antidepressants. J Pharm Pharmacol 1987, 39 (11), 877–82. [DOI] [PubMed] [Google Scholar]
- 20.Brandau S; Landa A; Franzen J; Marigo M; Jorgensen KA, Organocatalytic conjugate addition of malonates to alpha,beta-unsaturated aldehydes: asymmetric formal synthesis of (-)-paroxetine, chiral lactams, and lactones. Angew Chem Int Ed Engl 2006, 45 (26), 4305–9. [DOI] [PubMed] [Google Scholar]
- 21.Davis MC; Groshens TJ, Synthesis of p-Quinquephenyl from E,E-1,4-Bis(4-bromophenyl)-1,3-butadiene. Synthetic Communications 2010, 41 (2), 206–218. [Google Scholar]
- 22.Agoston GE; Wu JH; Izenwasser S; George C; Katz J; Kline RH; Newman AH, Novel N-substituted 3 alpha-[bis(4’-fluorophenyl)methoxy]tropane analogues: selective ligands for the dopamine transporter. J Med Chem 1997, 40 (26), 4329–39. [DOI] [PubMed] [Google Scholar]
- 23.Amat M; Bosch J; Hidalgo J; Canto M; Perez M; Llor N; Molins E; Miravitlles C; Orozco M; Luque J, Synthesis of enantiopure trans-3,4-disubstituted piperidines. An enantiodivergent synthesis of (+)- and (-)-paroxetine. J Org Chem 2000, 65 (10), 3074–3084. [DOI] [PubMed] [Google Scholar]
- 24.Germann D; Ma G; Han FX; Tikhomirova A, Paroxetine Hydrochloride. Prof Drug Sub Excip 2013, 38, 367–406. [DOI] [PubMed] [Google Scholar]
- 25.Mathis CA; Gerdes JM; Enas JD; Whitney JM; Taylor SE; Zhang Y; Mckenna DJ; Havlik S; Peroutka SJ, Binding Potency of Paroxetine Analogs for the 5-Hydroxytryptamine Uptake Complex. Journal of Pharmacy and Pharmacology 1992, 44 (10), 801–805. [DOI] [PubMed] [Google Scholar]
- 26.Regalado EL; Zhuang P; Chen YD; Makarov AA; Schafer WA; McGachy N; Welch CJ, Chromatographic Resolution of Closely Related Species in Pharmaceutical Chemistry: Dehalogenation Impurities and Mixtures of Halogen Isomers. Anal Chem 2014, 86 (1), 805–813. [DOI] [PubMed] [Google Scholar]
- 27.Henry LK; Field JR; Adkins EM; Parnas ML; Vaughan RA; Zou MF; Newman AH; Blakely RD, Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J Biol Chem 2006, 281 (4), 2012–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.