

Abstract
The influence of genetic background on driver mutations is well established; however, the mechanisms by which the background interacts with Mendelian loci remain unclear. We performed a systematic secondary-variant burden analysis of two independent cohorts of patients with Bardet–Biedl syndrome (BBS) with known recessive biallelic pathogenic mutations in one of 17 BBS genes for each individual. We observed a significant enrichment of trans-acting rare nonsynonymous secondary variants in patients with BBS compared with either population controls or a cohort of individuals with a non-BBS diagnosis and recessive variants in the same gene set. Strikingly, we found a significant over-representation of secondary alleles in chaperonin-encoding genes—a finding corroborated by the observation of epistatic interactions involving this complex in vivo. These data indicate a complex genetic architecture for BBS that informs the biological properties of disease modules and presents a model for secondary-variant burden analysis in recessive disorders.
A persistent hurdle in interrogating the role of genetic background in human genetic disorders is our limited understanding of the properties and distribution of contributory alleles. The challenge is particularly acute in rare disorders, in which the allele frequency of both causal variants and secondary contributory alleles (that is, alleles in loci other than the primary locus) is often low. As such, population-based studies are hampered by the lack of statistical power. At the same time, transitioning from a single-gene-centric disease architecture to a systems-based one defined by biological modules can inform causality, penetrance and expressivity1.
Bardet–Biedl syndrome (BBS), a model ciliopathy, presents an opportunity to study secondary-variant burden. We and others have shown previously that patients with BBS can carry secondary pathogenic variants in known BBS genes2. In rare examples, such alleles can modify penetrance3, whereas, more commonly, they are thought to modulate expressivity4,5. However, initial population-based studies have failed to detect an enrichment for secondary alleles in trans (that is, alleles in loci other than the primary locus), suggesting that either some of the examples were exceptions, or that the incidence, distribution and frequency of such alleles might be different from a priori assumptions6. Here, we studied two BBS cohorts with unambiguous recessive pathogenic mutations in 17 established BBS genes to measure: (1) whether there is enrichment for secondary variants beyond the driver locus; and (2) if so, whether the excess variation is concentrated within discrete disease modules or whether it is randomly distributed.
As a first step, we used targeted exon capture to sequence 102 families of Northern European7 ancestry (the discovery cohort), all of whom had bona fide pathogenic recessive mutations in one of 17 known BBS genes (Fig. 1a). We also sequenced an ancestry-matched control cohort of 384 individuals (the NEU (Northern European) control cohort) using the same exon-capture technology. After establishing a negligible false-positive rate (Methods and Supplementary Table 1), we then asked whether individuals with a clinical diagnosis of BBS have an increased burden of trans secondary variants that lie beyond the primary locus, in addition to the recessive diagnostic changes. If the recessive event at the primary locus is sufficient to drive the entire spectrum of symptomatology pertaining to disease manifestation (null hypothesis), we would expect no difference in allele burden between patients with BBS and controls; however, if additional secondary variants in trans with the primary locus contribute to disease, we would expect to see an enrichment of such changes in cases (burden hypothesis).
Fig. 1 |. Graphical outline of the study and mutational burden across cases and controls.
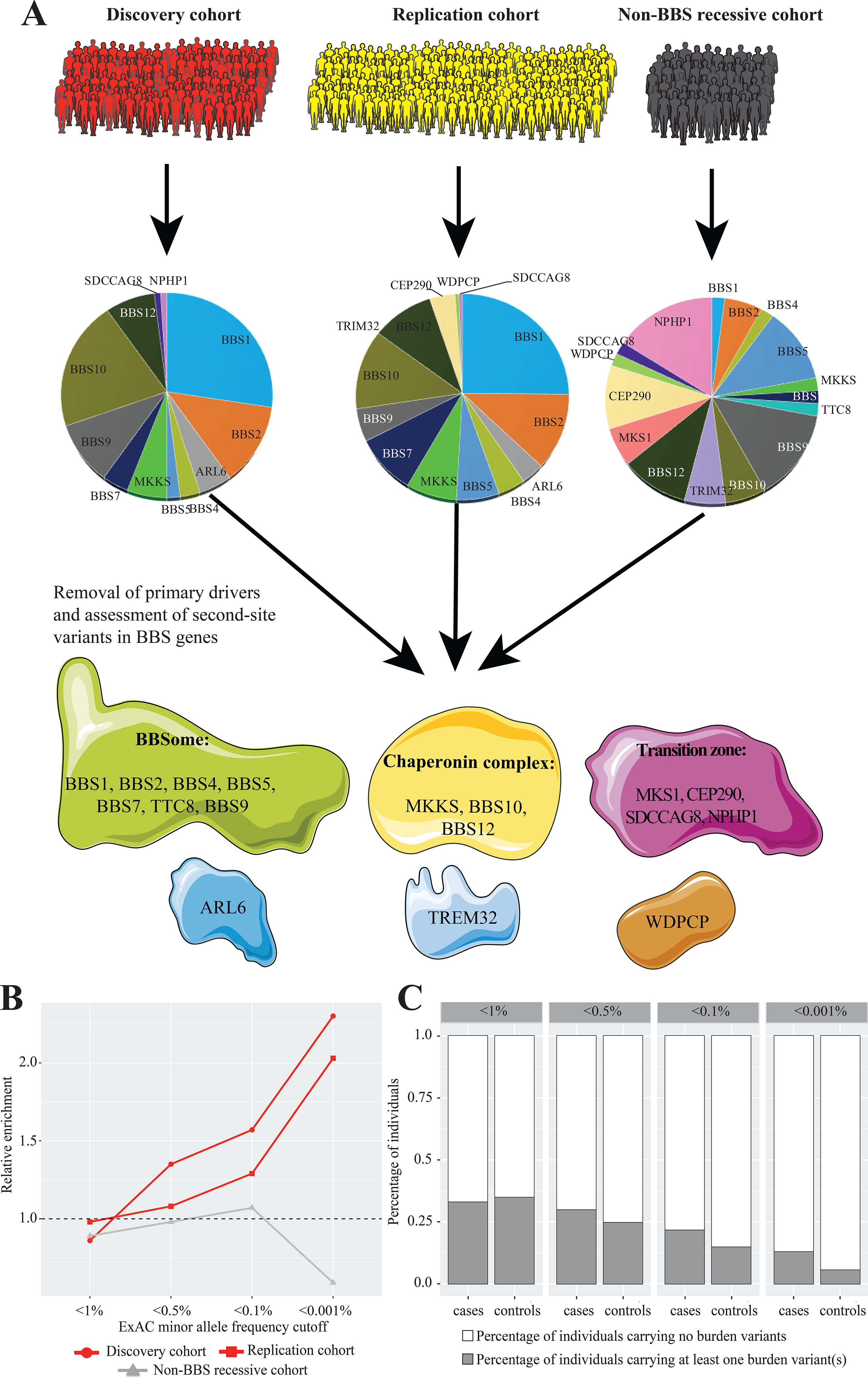
a, Distribution of primary drivers underlying BBS in the cases within the discovery and replication cohorts and across the 50 individuals with recessive BBS mutations but no BBS-compatible diagnosis. b, Mutational burden across the BBS proteins following removal of the primary locus identified for each patient. The graph shows 2.3-fold and twofold enrichment for the discovery and replication cohorts (red lines), respectively, for ultra-rare alleles (MAF < 0.001%). In contrast, the non-BBS cohort (gray line) is depleted for the same alleles. The burden for each cohort has been normalized to the control, which is represented by a dashed horizontal line. c, Distribution of individuals with burden-contributing alleles across the four MAF bins (1, 0.5, 0.1 and 0.001%), showing progressive enrichment for burden alleles in cases versus controls in the rarer allele categories.
To distinguish between the two posits, we used a combined multivariate and collapsing (CMC) test8 of rare variants at varying in-cohort minor allele frequency (MAF) cutoffs, restricting our analysis to nonsynonymous heterozygous secondary variants beyond the primary locus. Within Northern European7 descent individuals (n=84 cases and n=384 controls), we observed a significant enrichment for burden-contributing heterozygous trans variants beyond the primary-driver locus (Table 1) in cases versus controls (one-sided Fisher’s exact test; P=9×10−3; odds ratio (OR)=2.12) at an in-cohort MAF of 0.1% (singletons). This result replicated when evaluating the entire discovery cohort (n=102 cases and n=384 controls) by including 18 additional individuals of mixed European ancestry (one-sided P=2×10−3 and OR=2.29 at an in-cohort MAF of 0.1%).
Table 1:
Burden analysis for variants beyond the primary driver locus
| Discovery cohort | Replication cohort | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cases | Controls | MAF cutoffa | ORb | P-valueb | Cases | Controls | MAF cutoffa | ORb | P-valueb |
| 102 | 384 | AC=1 | 2.29 | 0.002 | 175 | 488 | AC=1 | 1.61 | 0.02 |
| AC=2 | 2.24 | 0.001 | AC=2 | 2.05 | 0.0002 | ||||
| 84c | 384 | AC=1 | 2.12 | 0.009 | |||||
| AC=2 | 2.08 | 0.007 | |||||||
Fixed in-cohort minor allele frequency (MAF) cutoffs of singletons (AC=1) and doubletons (AC=2).
Odds ratio (OR) and one-sided p-value calculated using Fisher’s exact test.
Subset of individuals with Northern European ancestry.
To rule out the possibility that population structure or other technical artifacts were responsible for the observed signal, we next evaluated the distribution of synonymous changes across the case–control cohorts. In the absence of population structure, synonymous variants should be distributed evenly among cases and controls; we found no significant difference in the distribution of singleton synonymous variants in the entire discovery cohort (n=102), suggesting that neither population structure nor another technical artifact were confounders (Supplementary Tables 2 and 3). Similarly, we detected no differences when evaluating the distribution of doubletons and tripletons that were: (1) shared between cases and controls; or (2) specific to either the discovery or control cohorts. This finding further supports no evidence of population substructure (Supplementary Tables 2 and 3).
Next, we classified variants according to their population frequency in the Genome Aggregation Database (gnomAD) dataset, reasoning that alleles that are rare in the population are more likely to disrupt functional nucleotides. Such an analysis was facilitated by the availability of large-scale reference datasets of human genetic variation that lend the opportunity to recognize low-frequency genetic variants (on the order of the disease prevalence), rather than being limited to the in-cohort-level allelic frequency. For each BBS case with a recessive primary locus, we measured the incidence of burden alleles in the other 16 BBS genes at four gnomAD-based MAF cutoffs: 1, 0.5, 0.1 and 0.001%. In agreement with the CMC test, we found a significant enrichment for MAF < 0.001% (ultra-rare alleles) in cases versus controls (2.5-fold enrichment; P=0.02; OR=2.46; Fig. 1b and Extended Data Fig. 1a, b).
To corroborate our findings, we assembled a replication cohort of 641 individuals of mixed ancestry composed of 175 individuals with a secured molecular genetic BBS diagnosis in one of the 17 BBS genes (replication cohort) and 488 control individuals (replication control cohort). Using the CMC test, we observed a significant enrichment for burden alleles in cases versus controls (one-sided P=0.01 and OR=1.65 at an in-cohort MAF of 0.1%; Fig. 1b and Extended Data Fig. 1c), similar to the discovery case–control dataset. Burden analysis of synonymous variants in cases versus controls in the replication cohort was not significant, arguing against the possibility that confounding factors drive the signal (Supplementary Table 4). Using population-based allele frequencies, we observed a significant (2.5-fold) enrichment of ultra-rare alleles (MAF < 0.001%; 25 changes out of 175 cases versus 35 changes out of 488 controls; P=5×10−3; OR=2.19; Fig. 1b), similar to the discovery cohort. Critically, testing for burden in synonymous variants showed no association (P=0.21), while testing for balancing of singletons, doubletons and tripletons was likewise bereft of evidence of population stratification bias (P=0.56 for singletons; P=0.34 for doubletons; P=0.80 for tripletons; Supplementary Table 5). We attribute this observation to the stringent filtering of population-based MAFs (always considering the highest possible MAF) that reduced the possible inflation of population-specific alleles.
Meta-analysis of the two case cohorts (discovery and replication) using a Cochran–Mantel–Haenszel (CMH) procedure taking into account the cohort status as a stratification variable corroborated the enrichment of rare secondary-burden alleles in patients with BBS compared with controls (CMH chi-squared test; P=1.6×10−4; CMH pooled OR=1.89). The enrichment remained when considering population-based allele frequencies in BBS (29 changes out of 191 cases versus 53 changes out of 850 control individuals; P=4×10−4; OR=2.58; Extended Data Fig. 1d). To obtain further evidence of the specific MAF fraction contributing to burden, we counted and normalized the number of individuals with changes in each of the four established MAF bins (1%>MAF>0.5%, 0.5%>MAF>0.1%, 0.1%>MAF>0.001% and 0.001%>MAF>0%). Consistent with our previous observations, the signal for burden was driven exclusively by ultra-rare alleles (0.001%<MAF< 0%; P=3×10−4; OR=2.33; Fig. 1c and Extended Data Fig. 2).
To consider the possibility that the observed signal could be driven by population substructure, ancestry or relatedness, we performed formal testing of relatedness by either PLINK or KING in the discovery cohort, which revealed no evidence of endogamy. In addition, we asked whether any two patients with the same recessive mutation (for example, that encoding p.Met390Arg in BBS1) had the same trans allele in another BBS gene (arguing for a potential population-driven enrichment). Overall, in 46 homozygous recessive cases from the discovery cohort and 99 homozygous cases from the replication cohort, we identified no instances of a primary-driver allele and second-site variant occurring more than once (Supplementary Table 1).
Finally, we tested whether this signal was specific to the BBS genes in our cohort. We extracted all genetic variation from our existing sequencing data for a set of genes mutated in recessive primary ciliary dyskinesia, which were chosen to have a comparable cumulative coding sequence length to the 17 BBS genes (~38 kilobases) and, using the same methods as described earlier, tested for burden. We found no differences in 68 cases from the discovery cohort for which sequencing data were available versus 384 NEU controls, using either population-based (OR=1.18; P=0.72) or in-cohort frequencies (OR=0.64; P=0.13) (Supplementary Table 6).
Next, we turned our attention to the question of topology and nature of the trans-acting variation. We found that this group of variants in BBS genes was more likely to disrupt strongly conserved genomic segments in the combined BBS cases than in combined controls. We focused on synonymous constraint elements (SCEs) in 29 mammalian genomes, which show protein-coding conservation and also strong non-coding constraint in synonymous positions, and thus are likely to contain overlapping functional elements, such as RNA secondary structures or binding sites for regulatory proteins9. We found that secondary variants overlapped SCEs for five out of 76 BBS case variants (6.6%) but only for six out of 371 control variants (1.6%) (hypergeometric P=0.025). Although these numbers are low and as such warrant caution, our data suggest that burden-contributing variants are more likely to disrupt functional elements in BBS cases.
To gain further insight into the properties that distinguish the exomes of individuals diagnosed with BBS from the exomes of individuals who carry chance recessive changes in BBS genes, we parsed ~10,000 exomes from the Baylor Molecular Diagnostics Laboratory. We identified 50 individuals with a clinical genetic disorder other than BBS who carried homozygous rare (MAF < 1%) alleles in one of the BBS genes (non-BBS recessive cohort). In contrast with our 277 bona fide BBS cases, the 50 non-BBS recessive individuals showed no significant burden in ultra-rare BBS alleles compared with the 872 combined controls (P=0.44; Fig. 1b and Extended Data Fig. 1d), consistent with the model that burden alleles contribute to the clinical manifestation of BBS. We further determined that lack of burden in secondary variants was independent of zygosity at the primary locus (Methods and Supplementary Table 7).
In addition to burden alleles, we also studied differences in the primary locus mutations of individuals with BBS and non-BBS recessive individuals. Overall, 45% (125/277) of the patients with BBS harbored two highly disruptive mutations (that is, nonsense, frameshifts, deletions) in the primary locus in contrast with only 2% (1/50) of the non-BBS recessive cases (P=5×10−7). Furthermore, for the individuals harboring at least one missense mutation in the primary locus, we attributed a weight for the impact of these nonsynonymous changes using BLOSUM (score ranges between −3 and 3, with lower scores denoting more deleterious substitutions)10. BLOSUM scores of the BBS case variants were significantly lower and thus probably more disruptive than the scores for the non-BBS recessive individuals, with 76% (90/118) of the bona fide BBS cases having a negative BLOSUM score, compared with only 43% (21/49) of the non-BBS recessive individuals (P=4×10−5; Extended Data Fig. 3). Taken together, our data are consistent with increased mutational load both in the primary locus and in secondary trans-acting variants that may interact epistatically with the primary locus.
Next, we asked whether the observed burden alleles are distributed randomly across BBS genes. For this question, we tested the observed versus expected probability of an individual with a mutation in any given BBS gene to have a secondary variant in a second BBS gene. Although we detected a fivefold increase in the likelihood of a primary BBS5 event co-occurring with a secondary event in BBS2 compared with in other BBS genes (BBS5→BBS2; P=0.04; Fig. 2a), the low-genetic-interaction events detected did not withstand multiple correction testing, therefore not allowing for statistically robust conclusions. Our data further revealed that the frequency of primary drivers was not a predictor of interactions. This was exemplified by BBS1 and BBS10, which despite contributing almost 40% of the recessive drivers in our cohorts were not enriched for secondary trans variants in any other specific BBS gene. Neither our burden observations nor the specific pairings were driven by gene size (for example, CEP290 was the largest of the evaluated BBS loci but showed neither a mutational enrichment nor an interaction enrichment in our analyses).
Fig. 2 |. Genetic and modular interactions in BBS cases and controls.
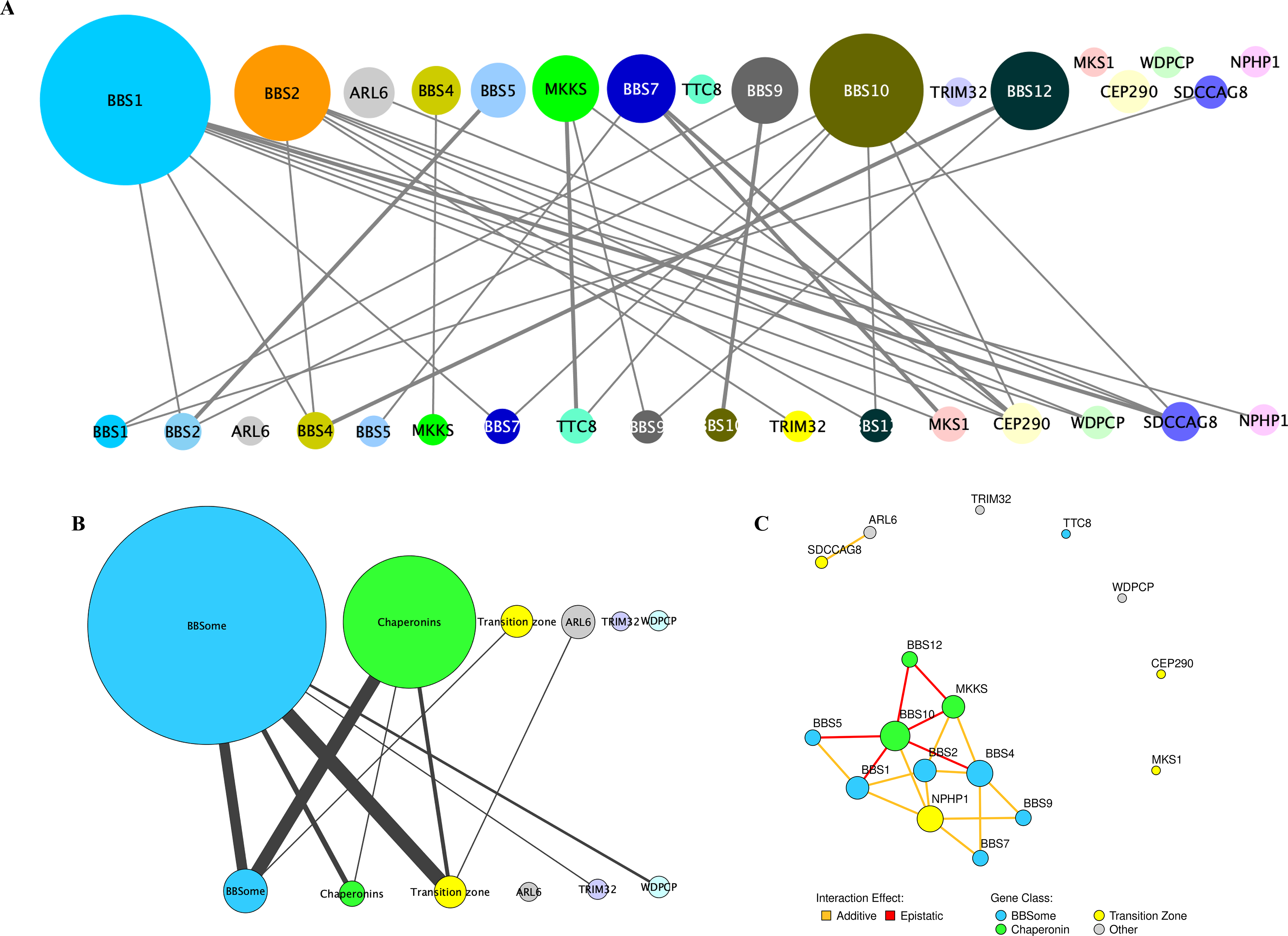
a, In the meta-analysis of BBS cases, BBS1, BBS2, BBS10 and BBS12 harbored recessive driver alleles most frequently (top), but there was an even distribution of burden-contributing alleles (bottom) across the 17 BBS loci. b, Analysis of modules within the BBS proteome (the BBSome (BBS1, BBS2, BBS4, BBS5, BBS7, TTC8 and BBS9); chaperonin complex (MKKS, BBS10 and BBS12); and transition zone (MKS1, CEP290, SDCCAG8 and NPHP1) and three genes that belonged to no yet-defined module (ARL6, TRIM32 and WDPCP)) revealed that the components of the chaperonin complex were driving the majority of modular interactions in BBS. c, Schematic showing the functional outcome (epistatic versus additive) upon suppression of select modular interactions using an in vivo zebrafish system. Epistatic interactions among loci are shown in red and additive genetic interactions are shown in yellow. The gene nodes are color coded according to the functional module they belong to. In each panel, the primary loci are represented as circles in the top half and the loci contributing to mutational load are represented as circles in the bottom half. The size of the circles corresponds to the number of individuals carrying changes in the primary and burden-contributing loci, respectively, and the thickness of the lines connecting primary and burden-contributing loci corresponds to the frequency at which a genetic interaction is observed, with thicker lines representing more common interactions.
To probe deeper into the nature and distribution of interactions, we asked whether this burden was distributed randomly across the set of 17 BBS genes. We noted that primary–secondary locus interactions extended beyond the observed pairwise relationships to macromolecular complexes. We therefore clustered the 17 BBS genes into three previously defined modules: the BBSome complex (BBS1, BBS2, BBS4, BBS5, BBS7, TTC8 and BBS9)11–13; the transition zone complex (MKS1, CEP290, SDCCAG8 and NPHP1)7,14,15; and the chaperonin complex (MKKS, BBS10 and BBS12)16,17. For the remaining three genes (WDPCP, TRIM32 and ARL6), we had no information about the macromolecular complexes to which their transcribed proteins belong. Restricting ourselves to alleles at a MAF of < 0.001% (the bin with the most robust evidence for burden enrichment), we collapsed all alleles per module and plotted all of the observed interactions. We then calculated the number of interactions within and across modules (Fig. 2b). The distribution was non-random: across the combined set of 277 individuals with BSS, most recessive events were contributed by chaperonin-encoding components (Bonferroni-corrected P=4×10−3; Fig. 2b), suggesting that this module potentially has the most physiologically potent interaction capability. We note that the genes belonging to the chaperonin complex also displayed the lowest degree of conservation compared with genes belonging to other BBS-related modules (average conservation of chaperonin complex between human and zebrafish orthologs=34% versus 52% in transition zone proteins and 73% in BBSome complex proteins; Supplementary Table 8), consistent with a potentially dynamic interaction pattern involving this module. Once the biophysical sites of interaction between the BBSome and chaperonin complex are resolved, it will be interesting to ask whether second-site variation maps to regions of biophysical affinity and/or sites of faster evolution within each molecule.
As part of our gene discovery work, we tested pairs of BBS genes for their ability to interact in vivo. In this model, we suppressed each gene (either alone or in pairs in sub-effective doses in zebrafish embryos) and asked whether double suppression affected the severity of the phenotype in an additive or multiplicative fashion18. Post hoc retrieval of interaction data across 19 gene pairs revealed 13 additive and six epistatic interactions (Fig. 2c)17,19–22. Strikingly, all six observed epistatic interactions involved one of the chaperonins, whereas none of the non-chaperonin pairings showed epistasis (P=0.003).
In this study, we provide direct evidence for the enrichment of ultra-rare alleles in patients with BBS with diagnosed recessive mutations. The degree of enrichment (around twofold) was reproducible and only detectable for ultra-rare alleles (MAF < 0.001%), consistent with population-level selection against these mutations, and indicating that they are probably deleterious and occurring at functional positions. We suspect that this occurs because the majority of variants are probably detrimental to protein function at this MAF; the study of larger cohorts should allow for the detection of more common alleles, especially if it is coupled with rigorous functional studies to determine their effect. We also note that the non-random distribution of these variants underscores biological substructure in this module. Although we were cautious to avoid overinterpretation of our data, our observations support the intuitive expectation that genetic interactions within macromolecular complexes are more likely to be additive, but interactions between complexes are more likely to be epistatic. For BBS, understanding the biochemical interplay between the chaperonin module and the BBSome will probably further improve our understanding of the role of the additional variation.
We do not know the contribution of burden to the phenotype. If the discovered variants were necessary for classically defined penetrance, we would anticipate a substantial fraction of asymptomatic individuals with recessive BBS mutations, which is not true. The most parsimonious explanation is that the second-site variants in other components of the BBS-associated modules represent modifiers of expressivity. In such a scenario, the second-site variants, which would be silent under steady-state circumstances, could lead to the expression of novel traits/endophenotypes in the presence of a diseased environment dictated by the primary locus. This concept is consistent with the canalization phenomenon described for molecules such as the chaperone HPS90 (refs. 23,24). However, a definitive indication that this hypothesis is true cannot be achieved without detailed and harmonized clinical data on disease endophenotypes and onset of the spectrum of symptomatology, as well as increased numbers of patients. Our current data indicate that ~22% of our patients carried one or more additional ultra-rare variants at the MAF < 0.1% cutoff. This is likely to be an underestimate of burden since additional trans alleles are likely to: (1) be of higher frequency than we had statistical power to detect in this cohort; (2) lie elsewhere in the known BBS genes; and (3) exist in non-BBS-causing recessive loci that are nonetheless relevant to the function of the BBS biological modules.
Finally, we emphasize the fact that studies of genetic burden in rare genetic disorders can be powered substantially by biological insight. Had we attempted agnostic genome-wide tests, or even tests across the entire set of ciliary genes, the observed burden would have been invisible because of the size of the denominator. Focusing on a comprehensive set of known disease-causing genes allows a rational, functionally driven hypothesis that also avoids the pitfalls of single-gene candidate studies. Indeed, our findings are concordant with recent studies of other genetic disorders, including Charcot– Marie–Tooth disease25 and retinitis pigmentosa26, although the latter example might be confounded by genetic drift. We speculate that the systematic measurement of burden of other genetically heterogeneous disorders in which all of the causal genes are considered as one baseline entity (essentially a functional operon) will reveal similar burden observations, which in turn can be used to understand biological and biochemical substructure.
Methods
Research participants.
We assessed two cohorts of individuals who fulfilled the clinical diagnostic criteria for BBS27. The discovery cohort comprised 102 patients of Northern European descent and the replication cohort comprised 175 unrelated individuals of mixed ancestry (Supplementary Table 1). The control cohorts included 384 Northern European individuals (NEU control cohort) and 488 whole-exome-sequenced and targeted-sequenced unaffected individuals of mixed ancestry (replication control cohort). A cohort of 50 individuals with a non-BBS genetically identified disorder, who were harboring at least two alleles with a MAF of < 1% in one of the 17 BBS loci evaluated (BBS1, BBS2, ARL6/BBS3, BBS4, BBS5, MKKS/BBS6, BBS7, TTC8/BBS8, BBS9, BBS10, TRIM32/BBS11, BBS12, MKS1/ BBS13, CEP290/BBS14, WDPCP/BBS15, SDCCAG8/BBS16 and NPHP1), was also assembled (non-BBS recessive cohort). Informed consent was obtained from all controls, individuals with BBS and their willing family members according to protocols approved by the institutional review boards of the Duke University Medical Center, Université de Strasbourg (Comité Protection des Personnes; EST IV; N°DC20142222) and Baylor College of Medicine. We obtained peripheral whole-blood samples from participants and extracted DNA according to standard methods.
Targeted exome capture and next-generation sequencing.
The patients with BBS and control individuals were sequenced either by regional capture of the exons and intron–exon boundaries of 785 ciliary genes prioritized from the ciliary proteome28, or by direct Sanger sequencing of each exon in a Clinical Laboratory Improvement Amendments–approved laboratory29. We captured regions of interest with a custom NimbleGen targeted liquid capture device (12,000 exons; 1.9 megabases of target) according to the manufacturer’s instructions, pooled 23 samples per pool and subsequently performed next-generation sequencing with an Illumina HiSeq 2000 platform (paired-end 100-base-pair reads with two pools per lane and ten lanes in total). The samples were sequenced with a mean of 110× coverage, with 92% of bases achieving >20× coverage and 99% of targeted regions captured. To minimize biases introduced by the technological methods used, and consequently increase false-positive signals, we employed the same capture library, sequencing platform and base-calling software for the discovery cohort and NEU control cohort and similar ancestry composition for the replication cohort and replication control cohort. Our base-calling method re-detected all 152 previously identified pathogenic recessive mutations in our discovery cohort of cases (50 families with compound heterozygous mutations and 52 with homozygous mutations; Supplementary Table 1), indicating a negligible false-negative rate. To eliminate the possibility of signal being driven by false-positive events, we used Sanger sequencing to test all of the heterozygous burden-contributing secondary alleles at a MAF of < 0.1% in both cases and controls. We confirmed 21 variants in the discovery cohort and 45 in the NEU control cohort. The non-BBS recessive cohort was sequenced by whole-exome capture methodology and underwent the same analysis as previously described30.
Mutation burden analysis.
We performed a unidirectional gene-based association test8 using the mutational target of 17 BBS genes as a single grouping unit (BBS1, BBS2, ARL6/BBS3, BBS4, BBS5, MKKS/BBS6, BBS7, TTC8/BBS8, BBS9, BBS10, TRIM32/BBS11, BBS12, MKS1/BBS13, CEP290/BBS14, WDPCP/BBS15, SDCCAG8/BBS16 and NPHP1). To assess the contribution of burden alleles beyond the primer driver locus, the genotype of each patient with BBS at the respective driver locus was set as a homozygous reference. Missing genotypes in both cases and controls were imputed to reference. Restricting to nonsynonymous single-nucleotide variants with a high impact on protein function (missense and nonsense consequences as annotated by SnpEff version 4.3)31, we performed a CMC test of rare variants at various in-cohort MAF cutoffs, as well as at the optimum in-cohort frequency cutoff selected using a variable threshold approach32, as implemented in the RVTESTS package33. To mitigate potential bias due to imperfect ancestry matching between cases and controls, we repeated the burden analysis excluding 18 individuals with non–Northern European ancestry. We assessed mutational burden in individuals with BBS, controls and the cohort of non-BBS recessive individuals with a known primary recessive driver gene (Supplementary Table 1). For inclusion, variants fulfilled the following criteria: (1) probably disruptive to protein sequence (that is, nonsynonymous, nonsense, frameshifting, bona fide splice sites within three base pairs of exon–intron junctions, and copy number variants described previously19,20); (2) ancestry-matched MAF < 1% (as obtained from the Genome Aggregation Database browser (https://gnomad.broadinstitute.org/) and/or the Greater Middle East Variome (http://igm.ucsd.edu/gme/index.php)); and (3) restricted to the 17 BBS genes sequenced across all three cohorts. The statistical significance between control and affected individuals with variants probably contributing to burden was compared among groups (discovery cohort versus NEU control cohort; replication cohort versus exome control cohort; and combined BBS cohorts (discovery+replication) versus combined control cohorts (NEU control cohort+exome control cohort) using a two-tailed Fisher’s exact probability test.
Synonymous constraint elements.
We searched for SCEs in every coding sequence in consensus coding sequence Homo sapiens release 9 using FRESCo software34. FRESCo analysis was performed using a window size of nine and sequences and distances taken from the 29-mammal alignment35. The resulting list of SCEs is available at https://data.broadinstitute.org/compbio1/SynonymousConstraintTracks/hg19/SCE.hg19.bed.gz. We tested each burden single-nucleotide variant in each individual and found the following changes to be contained in an SCE for cases: p.Ser753Phe (AR201–06; BBS9); p.Pro13Ser (AR831–03; BBS4); p.Glu532Lys (KK011–04; SDCCAG8); p.Arg295Gln (Fam93_ NNMR18; BBS4) and p.Gln505Glu (Fam14_AKX44; SDCCAG8), along with the following changes for controls: p.Ser29Phe (37 − 10101398_C31NDACXX7-IDMB39; NPHP1); p.Ser788Phe (246 − 10104594_C30WYACXX-4-IDMB66; BBS9); p.Met616Ile (DM414–1000; NPHP1); p.Ile45Leu (DM773–1000; NPHP1); p.Gly72Arg (DM1377–1000; ARL6); p.Ser788Phe (DM1377–1001; BBS9).
Zygosity analysis in the non-BBS recessive cohort.
To evaluate whether the lack of clinical symptomatology in the patients who were non-BBS recessive was due to the zygosity state of alleles with different directions of effect (that is, compound heterozygosity for a damaging allele and a benign or hypomorphic allele would be expected to have a more modest impact than homozygosity for the damaging allele alone), we conducted our burden analysis of nonsynonymous variants in the non-BBS recessive cohort, distinguishing between carriers with homozygous recessive alleles and individuals with compound heterozygous alleles at the primary locus. We found no significant burden of rare damaging variants in either patient group compared with control cohorts (Supplementary Table 7), arguing against an effect of zygosity at the primary locus on the manifestation of the clinical phenotype.
BLOSUM scoring.
BLOSUM scores were calculated using the blosum62 matrix obtained from biopython version 1.58. The BLOSUM score ranged from −3 to 3, with lower numbers indicating biochemically more different pairs of amino acids and higher scores indicating more radical amino acid changes. For BBS cases with two missense changes at the recessive locus, the change with the higher BLOSUM score was taken into consideration.
Data availability
A summary of the genotypes presented in the present study is given in Supplementary Table 1. However, restrictions apply to the availability of whole-exome data, patient consent forms and Institutional Review Board instructions. Selected data can be made available from the corresponding author upon reasonable request and inclusion of appropriate Institutional Review Board documentation.
Extended Data
Extended Data Fig. 1 |. Distribution of burden-contributing variation across case and control cohorts.

a, Burden-contributing variants across cases of the Discovery cohort (n= 102), Replication cohort (n= 175), and Meta-analysis of all BBS cases (n= 277) across four population-based minor allele frequency (MAF) cutoffs (1%, 0.5%, 0.1%, and 0.001%). b, Values of burden-contributing variation between BBS cases of the Discovery cohort (n= 102) and the cohort of NEU controls (n= 384) showing a 2.5-fold enrichment for ultra-rare (MAF < 0.001%) alleles in cases compared to controls. c, BBS cases of the Replication cohort (n= 175) and the Replication control cohort (n= 488) showing a 2.5-fold enrichment for ultra-rare (MAF < 0.001%) alleles in cases compared to controls. d, Collapsed case, control and “non-BBS recessive” cohorts. e, f, Distribution of individuals with burden-contributing alleles with MAF < 1% (e) and with MAF < 0.001% (f) in control individuals (blue bars) and BBS cases (orange bars).
Extended Data Fig. 2 |. Distribution of burden-contributing variation across case and control cohorts in each of four discrete MAF bins.

a, The Discovery case cohort shows a 2.5-fold enrichment for ultra-rare (0.001% > MAF > 0%) alleles compared to controls. b, The Replication cohort shows a 2-fold enrichment of such alleles compared to the exome control cohort. c, The BBS case meta-analysis shows a 2.2-fold enrichment compared to the combined control cohorts. d, Collapsed cohorts. a–d show plots across four MAF bins (1% > MAF > 0.5%, 0.5% > MAF > 0.1%, 0.1% > MAF > 0.001%, and 0.001% > MAF > 0%).
Extended Data Fig. 3 |. Estimate of protein impact for the least disruptive of the diagnostic variants for BBS cases and non-BBS recessive individuals, with at least one missense change in the primary locus.

With high BLOSUM62 scores denoting biochemically similar amino acid changes and lower scores marking radical amino acid changes, the graph shows evidence for bona fide BBS cases harboring more disruptive variants.
Supplementary Material
Acknowledgements
We thank the patients and their families for participating in this study, as well as their caring physicians who provided clinical information and referred the patients for diagnostic analyses. We are grateful to C. Glodosky and A. Krentz from PreventionGenetics and R. Haws from Marshfield Clinic for providing information on clinical BBS exome data. We thank A.-S. Jaeger and M. Antin for technical assistance. This study was supported by NIH grants GM121317-13, HD042601 and DK072301 (to N.K.), grants R35GM127131, R01MH101244 and U01HG00908 (to S.R.S.) and R01 HG004037 (to I.J.), as well as GENCODE Wellcome Trust grant U41 HG007234 (to I.J.). R.A.L. is a senior scientific investigator of research to prevent blindness. N.K. is a distinguished Valerie and George D. Kennedy professor.
Footnotes
Competing interests
N.K. is a founder of, and holds significant stock in, Rescindo Therapeutics.
References
- 1.Skafdas E et al. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol. Psychiatry 19, 504–510 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beales PL et al. Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet–Biedl syndrome. Am. J. Hum. Genet 72, 1187–1199 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katsanis N et al. Triallelic inheritance in Bardet–Biedl syndrome, a Mendelian recessive disorder. Science 293, 2256–2259 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Badano JL et al. Dissection of epistasis in oligogenic Bardet–Biedl syndrome. Nature 439, 326–330 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Cardenas-Rodriguez M et al. The Bardet–Biedl syndrome-related protein CCDC28B modulates mTORC2 function and interacts with SIN1 to control cilia length independently of the mTOR complex. Hum. Mol. Genet 22, 4031–4042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaheen R et al. Characterizing the morbid genome of ciliopathies. Genome Biol. 17, 242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto EA et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet 42, 840–850 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li B & Leal SM Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet 83, 311–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin MF et al. Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes. Genome Res. 21, 1916–1928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henikof S & Henikof JG Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA 89, 10915–10919 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loktev AV et al. A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell 15, 854–865 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Nachury MV et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Wei Q et al. The BBSome controls IFT assembly and turnaround in cilia. Nat. Cell Biol 14, 950–957 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Gonzalo FR et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet 43, 776–784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seeger-Nukpezah T et al. The centrosomal kinase Plk1 localizes to the transition zone of primary cilia and induces phosphorylation of nephrocystin-1. PLoS ONE 7, e38838 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JC et al. MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet–Biedl syndrome, is a novel centrosomal component required for cytokinesis. J. Cell Sci 118, 1007–1020 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Stoetzel C et al. Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet–Biedl syndrome. Am. J. Hum. Genet 80, 1–11 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leitch CC et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet–Biedl syndrome. Nat. Genet 40, 443–448 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Lindstrand A et al. Recurrent CNVs and SNVs at the NPHP1 locus contribute pathogenic alleles to Bardet–Biedl syndrome. Am. J. Hum. Genet 94, 745–754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindstrand A et al. Copy-number variation contributes to the mutational load of Bardet–Biedl syndrome. Am. J. Hum. Genet 99, 318–336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoetzel C et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet 38, 521–524 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Zaghloul NA et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet–Biedl syndrome. Proc. Natl Acad. Sci. USA 107, 10602–10607 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutherford SL & Lindquist S Hsp90 as a capacitor for morphological evolution. Nature 396, 336–342 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Karras GI et al. HSP90 shapes the consequences of human genetic variation. Cell 168, 856–866.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzaga-Jauregui C et al. Exome sequence analysis suggests that genetic burden contributes to phenotypic variability and complex neuropathy. Cell Rep. 12, 1169–1183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikopoulos K et al. A frequent variant in the Japanese population determines quasi-Mendelian inheritance of rare retinal ciliopathy. Nat. Commun 10, 2884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beales PL, Elcioglu N, Woolf AS, Parker D & Flinter FA New criteria for improved diagnosis of Bardet–Biedl syndrome: results of a population survey. J. Med. Genet 36, 437–446 (1999). [PMC free article] [PubMed] [Google Scholar]
- 28.Hjeij R et al. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet 93, 357–367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Redin C et al. Targeted high-throughput sequencing for diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet–Biedl and Alström syndromes. J. Med. Genet 49, 502–512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N. Engl. J. Med 369, 1502–1511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cingolani P et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price AL et al. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet 86, 832–838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhan X, Hu Y, Li B, Abecasis GR & Liu DJ RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–1426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sealfon RS et al. FRESCo: finding regions of excess synonymous constraint in diverse viruses. Genome Biol. 16, 38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindblad-Toh K et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A summary of the genotypes presented in the present study is given in Supplementary Table 1. However, restrictions apply to the availability of whole-exome data, patient consent forms and Institutional Review Board instructions. Selected data can be made available from the corresponding author upon reasonable request and inclusion of appropriate Institutional Review Board documentation.