

Abstract
Background
TRAF3IP2 (Act1) is an adapter protein that interacts with IL-17R via its SEF/IL-17R (SEFIR) domain and coordinates two separate pro-inflammatory pathways following IL-17 cytokine stimulation.
Objective
To elucidate the immunological consequences of TRAF3IP2 homozygous mutations to improve treatments for immunodeficiency patients with chronic mucocutaneous candidiasis.
Methods
We describe two patients presenting with chronic mucocutaneous candidiasis that harbor biallelic nonsense mutations in TRAF3IP2. The cellular and molecular features of this genetic defect were assessed using in vitro cytokine assays and protein analysis.
Results
We show the homozygous mutation causes complete loss of protein expression. We also show that the absence of TRAF3IP2 was associated with a defective response to combined IL-2/IL-25 (IL-17E) stimulation.
Conclusion
Failure to initiate normal signaling downstream of IL-17R engagement likely contributes to the patients’ recurrent fungal infections. These findings add to our molecular understanding of genetic defects affecting this critical pathway of anti-fungal immunity.
Keywords: TRAF3IP2, Act1, IL-17, signaling, Chronic mucocutaneous candidiasis, anti-fungal immunity, recurrent fungal infections
Introduction:
Interleukin (IL)-17-mediated responses are necessary for protective mucosal immunity against fungal pathogens such as Candida albicans. Chronic mucocutaneous candidiasis (CMC) is a noninvasive and persistent Candida infection of the skin and oral surfaces that can occur when IL-17 signaling is deficient [1]. The TRAF3IP2 gene is expressed ubiquitously in human tissues and encodes the 574 amino acid long TRAF3 interacting protein 2 (TRAF3IP2, previously known as CIKS or Act1) [2]. TRAF3IP2 is an adapter protein that interacts with IL-17R via its“similar expression to fibroblast growth factor genes and IL-17R (SEFIR) domain and coordinates two distinct pro-inflammatory pathways following IL-17 stimulation. The first pathway induces nuclear factor-kappaB (NF-κB) activity, while the second leads to activation of mitogen-activated protein kinases (MAPKs) p38 and ERK which stabilizes the CXCL1 and CXCL2 transcripts [2]. Given its critical role in multiple pro-inflammatory pathways, there is growing evidence that genetic changes in TRAF3IP2 underlie human immune-mediated diseases. An early study showed that deleterious missense mutations in the SEFIR domain of TRAF3IP2 lead to CMC in humans [3]. A more recent study reported biallelic nonsense TRAF3IP2 variants in a patient with CMC but did not conclusively show their effects at the mRNA or protein level [4]. Here we describe two unrelated patients who presented with CMC and who harbor biallelic nonsense mutations in TRAF3IP2, enabling us to evaluate the physiological effects of complete loss of TRAF3IP2 protein expression.
Results and Discussion:
Patient (Pt. 1) is a 15-year-old Turkish male born to consanguineous parents who presented at 2 months of age with severe thrush and treated with a one-month course of intravenous antifungal treatment (Fig. 1a). At the time of active disease, direct microscopic examination of a smear from the oral monilial lesions demonstrated fungal hyphae. He was diagnosed with CMC and pityriasis lichenoides chronica. Recurrent episodes of candidiasis primarily affected the mouth and tongue and often affected the fingers, neck, or intertriginous areas (Fig. 1b). Over time, he was placed on prophylactic antifungal treatment with oral fluconazole, which led to amelioration of his symptoms. In addition to fungal infections, he had other cutaneous manifestations, including scanty psoriasis-like plaques and desquamation over the scalp, knee, and inguinal areas; erythematous lesions at axillary and retroauricular locations; and livedo reticularis on the thighs (Fig. 2a). His hair was mildly discolored in some regions. From an unrelated family, Pt. 2 is a 10-year-old Turkish girl born to consanguineous parents who presented at 1 year of age with treatment-resistant oral thrush, angular cheilitis, and aphthous stomatitis (Fig. 1a,b). Pt. 2 also suffered from chronic intertrigo and folliculitis on her trunk but had no scalp, nail, or tooth abnormalities (Fig. 2b). Fluconazole treatment and prophylaxis was effective and did not lead to resistance. For both patients, moniliasis recurs upon interruption of prophylaxis. Neither patient exhibited increased susceptibility to bacterial or viral infections and immunophenotyping of peripheral blood mononuclear cells (PBMCs) revealed normal lymphocyte subsets and minor immune abnormalities in the patients (Table 1). Serum immunoglobulin levels were normal, and both patients are within normal height and weight ranges for their ages.
Figure 1.
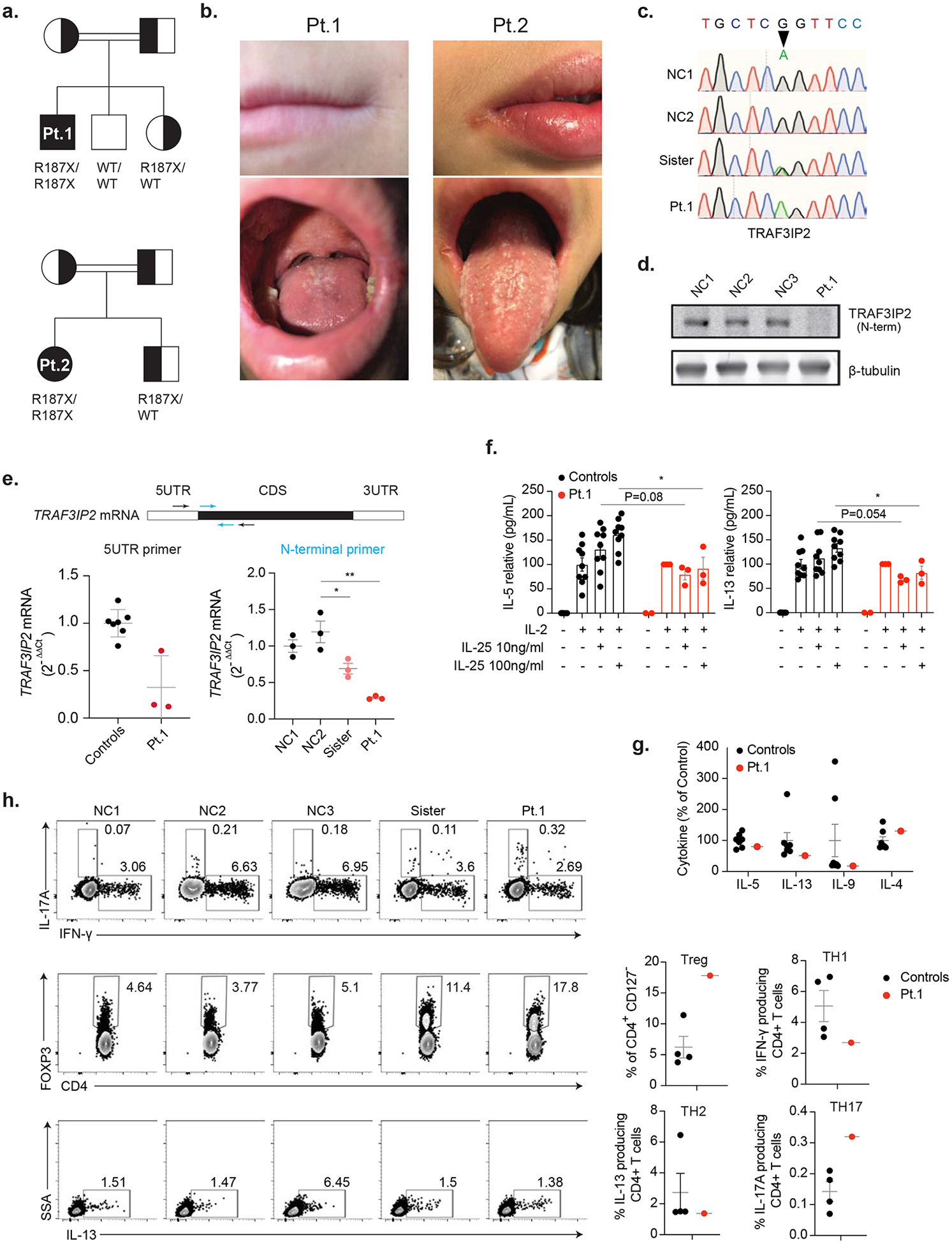
(a) Pedigrees for Patient 1 (Pt. 1) and Pt. 2 showing TRAF3IP2 allele inheritance. Double lines indicate consanguinity. X indicates stop codon; WT is wild-type sequence. (b) Angular cheilitis (top row) and oral thrush (bottom row) in Pt. 1 and Pt. 2. (c) TRAF3IP2 genomic DNA sequence electropherograms, for two normal controls (NC), sister and patient. Arrow shows the missense mutation. (d) Immunoblot showing TRAF3IP2 protein expression in T cell blasts with β-tubulin loading control. (e) The N-terminal primer set to show the TRAF3IP2 mRNA levels in control and Pt. 1 T cell blasts. Each control dot represents an individual donor while each patient dot represents a repeat measurement from an independent experiment. (f) Peripheral blood mononuclear cells (PBMC) were cultured for 7 days with exogenous IL-2 and IL-25 at the indicated concentrations and were measured using a multiplex cytokine assay. Cytokine concentrations were normalized to 100 pg/ml with IL-2 only stimulation. (g) The indicated cytokines from controls and patient were measured using a multiplex cytokine assay. (h) Flow cytometric analysis of Treg cells, IL-17A, IFN-γ and IL-13 producing CD4+ T cells. Statistical comparison is shown on the right. n.s.: not significant, *p < 0.05, **p < 0.01.
Figure 2.
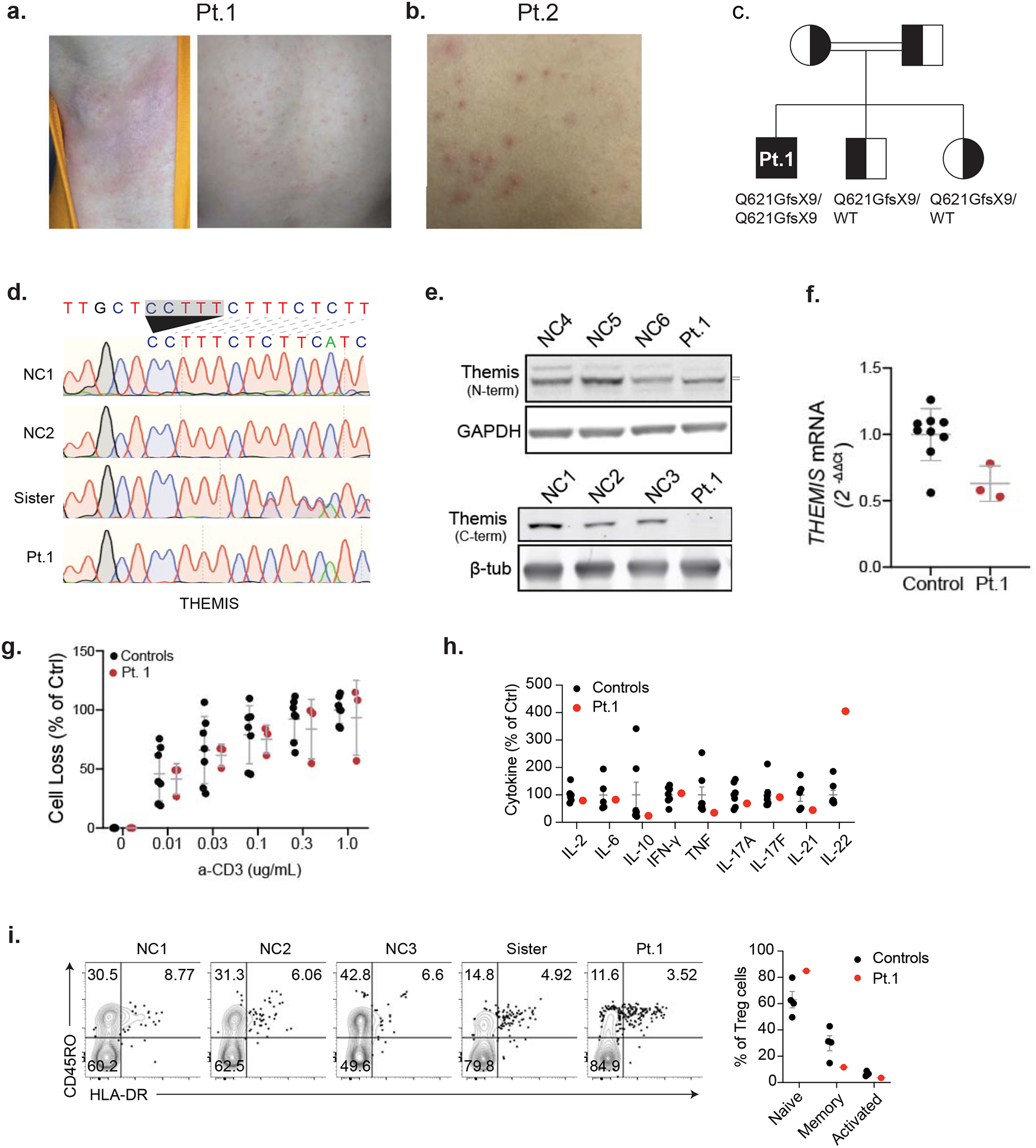
(a,b) Erythematous lesions on axillary skin (left) and pityriasis lichenoides (right) in Patient 1 (Pt. 1). Folliculitis on the trunk of Pt. 2. (c) Family pedigree for Pt. 1 showing THEMIS allele inheritance. Double lines indicate consanguinity. (d) THEMIS genomic DNA sequence electropherograms for normal controls (NC), sister and patient. Arrow shows the deletion. (e) Immunoblot of lysates derived from T cell blasts using an N-terminal THEMIS antibody protein with GAPDH loading control (Top). Immunoblot using C-terminal directed antibody with β-tubulin loading control (bottom). (f) THEMIS mRNA levels in control and Pt. 1 T cell blasts. Each control dot represents an individual donor while each patient dot represents a repeat measurement from an independent experiment. (g) Cell death of T cell blasts proliferating in IL-2 following restimulation with the indicated dose of soluble cross-linked anti-CD3 for 18 hours. Each control dot represents an individual donor while each patient dot represents a repeat measurement from an independent experiment. (h) The indicated cytokines in serum from NC, healthy sister, and patient were measured using a multiplex cytokine assay. (i) Isolated PBMCs were stained with FOXP3 and surface maker CD45RO and HLA-DR. Statistical comparison is shown on the right.
Table 1.
Key features of patients’ clinical disease and immunological workup.
| Demographics | Pt. 1 | Pt. 2 |
|---|---|---|
| Y.O.A. (as of 10/2019) | 15 | 10 |
| Sex | male | female |
| Ethnic background of parents | caucasian | caucasian |
| Primary Diagnosis | Chronic mucocutaneous candidiasis, recurrent skin rash, pityriasis lichenoides | Chronic mucocutaneous candidiasis |
| Age at onset/presentation | 2 months | 1 year |
| Secondary diagnoses | Moniliasis episodes recur as soon as prophylaxis is interrupted. Experiences cutaneous rash at extremities, neck and intertriginous skin. | Chronic intertrigo, folliculitis |
| Lymphadenenopathy | no | no |
| Splenomegaly | no | no |
| Hematologic abnormalities | no | no |
| Malignancy/lymphoma | no | ^ no |
| Autoimmunity or autoinflammatory disease | no | no |
| Immunodeficiency or susceptibility to infection | yes, chronic mucocutaneous candidiasis, recurrent/destructive Otitis Media | yes, chronic mucocutaneous candidiasis |
| Liver abnormalities | no | no |
| Antibody Abnormalities | no | no |
| Lung involvement | no | no |
| Height (percentile) | 10–25th | 25–50th |
| Weight (percentile) | 50th | 25–50th |
| Other significant symptoms | no | no |
| Immunological Workup | ||
| Quantitative Immunoglobulins (mg/dl) | IgG: 1260 (876–2197) IgA: 73 (108–447) IgM: 75 (75–448) IgG1: 758 (547–1474) IgG2: 363 (77–706) IgG3: 35 (36–238) IgE: 686 ng/ml | IgG: 1490 (431–1803) IgM: 87 (126–449) IgA: 138 (45–164) |
| CBC with differential | WBC: 10800 (4500–13000) ANC: 6000 (1500–8000) ALC: 2800 (1400–4500) Eo: 1000 (44– 400) Hb: 11.4 (11– 16) Plt: 338000 (150000– 400000) | WBC: 8500 (4500–13800), ANC: 2700 (1500–8600), ALC 4900 (1800–5600), Eo: 100, Hb 9.6 (11–14), Plt 335000 (182000–564000) |
| Flow Cytometry | CD3: 50%(47–76) CD3+4: 33% (23–48) CD3+8: 27% (25–50) CD19: 30% (14–44) CD16+56: 22% (4–23) | CD3+: 50% (55–88), CD3 +4+: 43% (24–50), CD3+8+:16% (17–42), CD19 +:14% (7–22), CD3−16/56+:14% (4–29) |
To investigate genetic contributions to CMC in these patients, we performed whole genome sequencing of DNA isolated from Pt.1 PBMCs, as well as DNA from his unaffected parents and brother. For Pt. 2, we used next-generation sequencing to target CMC-associated genes (IL17F, IL17RA, IL17RC, and TRAF3IP2) in DNA from patient PBMCs and carried out DNA sequencing by the Sanger method of the TRAF3IP2 gene in DNA from her unaffected sister and health donors (Fig.1c). Both patients harbored rare, homozygous nonsense mutations in TRAF3IP2 (NM_147686.3:c.559C>T,p.Arg187*) that segregated with disease in both kindreds that were heterozygous in the parents (Table 2). Homozygosity for this variant is not observed in healthy individuals reported in gnomAD, and this variant is not observed in a healthy Turkish population in the Greater Middle Eastern Variome database [5, 6]. It is worth noting that Pt. 1 also carried homozygous frameshift mutations in the second-to-last exon of the gene encoding THEMIS, a molecule that regulates TCR signal strength and is critical for thymocyte development and survival (Fig. 2c) [7, 8]. We confirmed the homozygous and heterozygous frameshift mutations in patient and sister, respectively (Fig. 2d). The levels of THEMIS transcripts were moderately reduced in Pt. 1 PBMCs, and lysates contained a slightly truncated protein product that could not be detected with a C-terminal targeted antibody (Fig. 2e,f). However, the patient did not display abnormal T cell subsets or increased sensitivity to restimulation-induced cell death (RICD) after TCR re-engagement, as would be expected for loss of THEMIS protein function, suggesting that this mutation may not account for the disease (Fig. 2g and Table 1).
Table 2.
Rare homozygous variants in Pt. 1, identified by WGS using a recessive model of disease inheritance from a consanguineous family.
| Worst Annotation | |||||||
|---|---|---|---|---|---|---|---|
| RNF223 | 1:1007306 | Exon 2/2 | G | A | c.641 C>T | p.Pro214Leu | missense |
| RNF223 | 1:1007730 | Exon 2/2 | C | T | c.217G>A | p.Val73Ile | missense |
| ATPSCKMT (FAM173B) | 5:10239295 | Exon 2/5 | G | A | c.190C>T | p.Arg64Ter | nonsense |
| TRAF3IP2 | 6:111912731 | Exon 2/9 | G | A | c.559C>T | p.Arg187Ter | nonsense |
| TBC1D32 | 6:121602737 | Exon 14/32 | T | C | c.1561A>G | p.Ile521 Val | missense |
| THEMIS | 6:128040861 | Exon 6/7 | CCTTT | C | c.1979_1982delAAAG | p.Glu621GlyfsTer9 | frameshift/stop |
| USPL1 | 13:31195297 | Exon 2/9 | T | C | c.20T>C | p.Ile7Thr | missense |
| HHIPL1 | 14:100118886 | Exon 2/8 | G | A | c.581 G>A | p.Arg194His | missense |
| GPR132 | 14:105517528 | Exon 3/3 | G | A | c.946C>T | p.Arg316Cys | missense |
| MYBPC2 | 19:50939323 | Exon 4/28 | C | T | c.251 C>T | p.Pro84Leu | missense |
| MYBPC2 | 19:50946971 | Exon 11/28 | C | T | c.1031 C>T | p.Pro344Leu | missense |
| POLA1 | X:24757653 | Exon 20/37 | A | G | c.2184A>G | p.Ile728Met | missense |
| WNK3 | X:54278026 | Exon 14/24 | T | A | c.2462A>T | p.His821 Leu | missense |
| G | A | c.4481 C>T | p.Pro1494Leu | missense |
| OMIM | |||||
|---|---|---|---|---|---|
| 0.00186776 | G/A | G/A > | G/G | G/G | none |
| 0 | C/T | C/C | C/T | C/T | none |
| 0.00026499 | A/A | G/A | G/A | G/A | none |
| 8.28E-06 | A/A | G/A | G/A | G/G | 615527, familial Candidiasis |
| 0.00018284 | C/C | T/C | T/C | T/T | none |
| C/C CC | TTT/C | CCTTT/C | CCTTT/C | none | |
| 0.00094032 | C/C | T/C | T/C | T/C | none |
| 0.00062418 | A/A | G/A | G/A | G/A | none |
| 0.00010728 | A/A | G/A | G/A | G/G | none |
| 0.00011153 | C/T | C/C | C/T | C/T | none |
| 7.46E-05 | C/T | C/T | C/C | C/C | none |
| 0 | G/G | A/A | A/G | A/A | 301220,301030 Van Esch-O’Driscoll syndrome |
| 0 | A/A | T/T | T/A | T/T | none |
| 0 | A/A | G/G | G/A |
We predicted that premature termination in the second exon of TRAF3IP2 transcripts would lead to loss of TRAF3IP2 at the protein level. Indeed, immunoblot of lysates derived from Pt. 1 T cells revealed a complete loss of protein, and reduced mRNA levels suggesting nonsense-mediated decay (Fig. 1d,e). Due to sample unavailability, we were unable to perform direct tests on Pt. 2 cells, but we infer that since it was the same variant, there would be identical defects. We therefore investigated the effects of protein loss on IL-17 signaling in Pt. 1 lymphocytes. The p.T536I substitution in TRAF3IP2, reported by Boisson et al., was shown to impair IL-5 and IL-13 production after co-stimulation with IL-2 and IL-25 [3]. We observed this same defect in PBMCs from Pt. 1, which showed a good cytokine response to IL-2 stimulation alone but no synergistically enhanced response following combined IL-2/IL-25 stimulation (Fig. 1f). Consistent with the cell stimulation, IL-5 and IL-13 were present at a lower level in patient’s serum (Fig. 1g) with relatively normal levels of other T helper (TH) cell cytokines (Fig. 2h). We noticed the amount of IL-22 in patient was much higher in patient (Fig. 2h), this is may because TRAF3IP2 patient carried a higher IL-22 producing T cells [3]. We also observed a higher proportion of IL-17A producing TH17 cells in patients (Fig. 1h), which is consistant with previous TRAF3IP2 patient [3]. But we detected a lower proportion of IFN-γ producing TH1 cells in our patients, which may reflect a TH1/TH17 counterbalance in vivo. To further clarify the proportion of TH cells in our TRAF3IP2 patient, we also measured Treg cells and IL-13-producing TH2 cells. We found that the patient presented a higher proportion of Treg cells and a lower proportion of TH2 cells (Fig. 1h and Fig. 2i). Failure to initiate signaling downstream of IL-17R engagement likely underlies this defective IL-25 response, which could contribute to the patients’ CMC. IL-25 signals though the IL-17 receptor which requires TRAF3IP2 to transmit a signal. This defect will be present for all IL-17R-mediated signaling. Defective IL-17 signaling in epithelial cells likely contributes importantly to susceptibility to candidiasis.
TRAF3IP2 has also been associated with immune-mediated diseases other than CMC (Table 3). One GWAS study associated a single nucleotide polymorphism (SNP) (rs33980500, p.D10N) in the TRAF6 binding site with increased risk of psoriasis and psoriatic arthritis [9]. A recent report of patients with biallelic missense mutations (p.T438N) in the SEFIR domain demonstrated a causal relationship between TRAF3IP2 loss-of-function and development of scalp discoid lupus erythematosus and folliculitis [10]. Interestingly, TRAF3IP2 knockout mice do not completely recapitulate phenotypes observed in human TRAF3IP2 deficiency (Table 3). For example, one of the earliest mouse models indicated that complete knockout of TRAF3IP2 or B cell-specific deletion resulted in B cell abnormalities and severe autoimmunity, explained by hyperactive NF-κB activation upon CD40/BAFF-R stimulation [11]. More recent models differ in that the mice manifested either with no disease or with hyper IgE, lymphadenopathy and splenomegaly, and atopic dermatitis in the absence of overt B cell anomalies [12, 13].
Table 3.
Summary of literature related to TRAF3IP2 deficiency, including mouse models and previously reported cases of humans harboring deleterious mutations. KO: knockout, Hypergam: hypergammaglobulinemia, LAD/SM: lymphadenopathy and splenomegaly, CMC: chronic mucocutaneous candidiasis, PNA: pneumonia.
| Cited | Organism Studied | TRAF3IP2 Protein Alteration | Variant effect on TRAF3IP2 protein | Increased B cells | Autoimmunity | Hypergam | Hyper IgE | LAD/SM | Atopic dermatitis | CMC | Bronchiecta sis / PNA | Scalp lesions +/− alopecia | Folliculitis | Psoriasis (or psoriasis-like skin disease) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Qian, 2004 | mouse | full KO and B-cell specific KO | no protein (not shown) | + | + | + | + | |||||||
| Matsushima, 2010 | mouse | C-term truncation | no protein | + | + | + | ||||||||
| Claudio, 2009 | mouse | full KO | no protein (not shown) | |||||||||||
| Huffmeier, 2010 | human | D10N missense in TRAF6 binding | full-length protein (not shown) | + | ||||||||||
| Boisson, 2013 | human | T583I missense in SEFIR domain | full-length protein | + | + | |||||||||
| Bhattad, 2019 | human | R283X | truncated protein (not shown) | + (from infections) | + | + | ||||||||
| Nemer, 2020 | human | T438N missense in SEFIR domain | full-length protein | + | + | |||||||||
| Pt. 1 (this report) | human | R187X | no protein | + | + | + | ||||||||
| Pt. 2 (this report) | human | R187X | no protein (not shown) | + | + |
In contrast to the first TRAF3IP2 knockout mouse, our patients and those reported previously did not manifest B cell abnormalities or autoimmune features indicative of dysregulated CD40/BAFF-R signaling. Boisson et al. reasoned that the T536I point mutation in TRAF3IP2 specifically disrupts interaction with IL-17R, leaving CD40 and BAFF-R interactions intact. Bhattad et al. report premature stop mutations leading to CMC without B cell abnormalities but were unable to definitively show complete loss of TRAF3IP2 protein in patient cells. Our work reveals that even complete loss of TRAF3IP2 protein in humans does not impact B cell survival but manifests as an inborn error of IL-17 immunity leading to CMC due to insufficient antifungal immunity.
MATERIALS AND METHODS
Human subjects
Written informed consent was provided by all human subjects (or their legal guardians) in accordance with Helsinki principles for enrollment in research protocols that were approved by the Institutional Review Board of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. Patient and healthy control blood was obtained at the National Institutes of Health under approved protocols. Mutations will be automatically archived by Online Mendelian Inheritance in Man (OMIM) at time of publication and whole-genome data will be submitted in dbGaP.
Genetic analysis methods
For Patient 1 (Pt. 1) and family members, genomic DNA was obtained from probands and family members by isolation and purification from peripheral blood mononuclear cells (PBMCs) using Qiagen’s DNeasy Blood and Tissue Kit. DNA was then submitted for whole-genome sequencing (WGS) through a collaboration with MERCK Pharmaceuticals. WGS was performed by massively parallel sequencing by Illumina HiSeq sequencing system on the collected DNA. For individual samples, WGS produced 30× to 60× average sequence coverage. All sequenced DNA reads were mapped to the hg19 human genome reference by Burrows-Wheeler Aligner with default parameters. Single nucleotide variant and indel calling were performed using the Genome Analysis Toolkit version 3.4 (the Broad Institute). Variants were then annotated by variant effect predictor and prioritized by GEMINI (GEnome MINIng) on the basis of population allele frequency, functional prediction, and potential genetic models (autosomal-recessive or de novo models). These methods were previously described.[14]
Primary Cells and Human Serum
Patient or control blood was subjected to a Ficoll density gradient centrifugation, after which PBMCs were collected from the interface and washed extensively in PBS. PBMCs were then used directly or pan T cells were expanded by the addition of 1 ug/mL anti-CD3 and 1 ug/mL anti-CD28 to PBMCs at 1×106 cells/mL in complete RPMI. After 72 hours, stimulatory antibodies were washed out and cells were placed in and maintained in fresh complete RPMI supplemented with 100 I.U./mL IL-2 resulting in activated, cycling T cells that we term, “T cell blasts.” Human serum was collected from healthy donors at the NIH and pooled (at least three donors) and heat shocked (56°C for 30 minutes) prior to use.
FOXP3 and Intracellular cytokine staining
Isolated PBMCs were treated with PMA (100ng/ml) plus Ionomycin (500ng/ml) with the presence of monensin (BioLegend Cat. No. 420701) in the 5-6 hours of cell culture activation. The cells were suspended and stained with surface marker first for 30 minutes at room temperature. Intracellular cytokine staining was carried out by using Fixation/Permeabilization Solution Kit (BD Biosciences, 554714). FOXP3 staining was done by using Foxp3 / Transcription Factor Staining Buffer Set (Thermo Fisher, 00-5523-00).
Media
Human T cell blasts were grown in RPMI 1640 (Gibco/Thermo Fisher, Waltham, Mass) supplemented with 10% heat-inactivated FBS (Gibco), 1% penicillin/streptomycin (Gibco), 1% l-glutamine (Gibco), 50 μM 2-mercaptoethanol (Sigma-Aldrich, St Louis, Mo), and either with or without 100 I.U./mL IL-2. Human PBMCs were stimulated with IL-2 and IL-25 in X-Vivo15 media supplemented with 1% penicillin/streptomycin (Gibco), 1% l-glutamine (Gibco), 50 μM 2-mercaptoethanol (Sigma-Aldrich, St Louis, Mo), and 5% human serum.
Antibodies and reagents
Anti-CD3 (clone: HIT3a) and anti-CD28 (clone: CD28.2) antibodies for T cell stimulation were purchased from Biolegend. Anti-TRAF3IP2 antibody was purchased from Ebioscience (Cat #14-4040-80), anti-THEMIS was from Thermo Fisher (Cat #PA5-56740), anti-β-tubulin was from Millipore Sigma (Cat #05-661), and anti-GAPDH was from Cell Signaling Technology (Cat #2118S). AlexaFluor-conjugated secondary antibodies were purchased from Thermo Fisher Scientific (Cat #32735 and Cat #32729).
Restimulation-induced cell death assays
Patient or control T cell blasts, between 2 and 3 weeks after initial activation, were collected and resuspended in fresh IL-2-containing media. Cells were then added to 96-well plates in triplicate and stimulated with the indicated dose of anti-CD3 (clone: HIT3a, BioLegend) in the presence of 1 ug/mL Protein A for 18 hours, as previously described.[15] Cells were then collected by repeat pipetting and stained with AnnexinV-APC and propidium iodide and analyzed by flow cytometry. The percentage of live cells was determined by AnnexinV and propidium iodide staining and expressed as a percentage of cells lost in the control condition due to TCR restimulation.
IL-25-induction of Th2 cytokines
IL-25-induced IL-5 and IL-13 production was measured as previously described.[3, 16] Briefly, PBMCs from the patient or healthy controls were resuspended to 1×106 cells/mL in complete X-VIVO15 media and 1×105 cells were transferred in triplicate to 96-well round bottom plates. Cells were then left untreated or treated at a final concentration of 0.5×105 cells/mL with 50 I.U./mL IL-2 with or without recombinant human IL-25 (IL-17E) (R&D systems, 8134-IL) at the indicated final concentrations for 7 days at 37°C. Supernatants were then analyzed using the LEGENDplex™ Human Th Panel (13-plex) to assess IL-5 and IL-13 levels. Values are normalized to the average control level after IL-2 stimulation.
Quantitative RT-PCR
Quantitative RT-PCR was performed as described previously.[14] Briefly, total RNA was isolated with the RNeasy kit (Qiagen, Gaithersburg, MD). cDNA was synthesized from 1 μg of total RNA using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). cDNA was brought to a total volume of 100 μL in double-distilled H2O and either immediately used or stored at −80°C for future use. Quantitative RT-PCR for TRAF3IP2, THEMIS, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was performed using the Power SYBR Green (Thermo Fisher) method on a 7900HT machine (ABI) using 2.5 μL of cDNA reaction product. Results were analyzed by using the ΔΔCt method and normalized to the first healthy normal donor values.
| GAPDH Forward | GATGACATCAAGAAGGTGGTG |
| GAPDH Reverse | ACCACCTGGTGCTCAGTGTAG |
| Themis Forward | CAGGCATCTATCTTGAAGGCTCT |
| Themis Reverse | GAAGCAAGGATGCCCTAGTCT |
| TRAF3IP2 Forward | TGGATCTAAGCCTCATCTGTATCC |
| TRAF3IP2 Reverse | AACCACAGACTCAGACGCAG |
| TRAF3IP2 N-terminal Forward | GAACCGAAGCATTCCTGTGGAGG |
| TRAF3IP2 N-terminal Reverse | CTGTTGGGTGCCATGTTCCTTATATTTGG |
Immunoblotting
To analyze total TRAF3IP2 and THEMIS protein levels, T cell blasts were washed in PBS and lysed in 1× Triton lysis buffer with complete protease inhibitor cocktail (Roche, Basel, Switzerland) on ice for 20 minutes. The lysates were then cleared at 14,000 × g at 4°C for 15 minutes. Lysates were then diluted with 2X SDS sample buffer (Quality Biologicals, Gaithersburg, MD) supplemented with 10% BME. Approximately 2 × 106 cell equivalents were separated by SDS-PAGE on 4% to 20% precast tris-glycine gels (Invitrogen) and transferred to a nitrocellulose membrane (Invitrogen). Membranes were blocked with 3% BSA in Tris-buffered saline with 0.01% Tween-20 (TBST) for 30 minutes at room temperature before incubating with primary antibody overnight at 4°C. After 3× 5-minute washes with TBST at room temperature with rocking, fluorescent secondary antibody was added for 2 hours at room temperature. After 5× 5-minute washes in TBST and 1 wash in PBS, membranes were analyzed on the LI-COR Odyssey imaging system or the Azure c300 imaging system.
Statistical Analysis
Graphs are generated by averaging at least 3 different experiments. Comparative statistics were not performed between patients and controls because patient samples represent biological repeats, not independent biological samples. To compare cytokine production between control samples a paired non-parametric Wilcoxon matched-pairs signed rank test was used to compare treated and untreated samples. *p < 0.05, **p < 0.01.
Capsule Summary.
We show that complete loss of TRAF3IP2 protein in humans does not impact B cell survival but manifests as an inborn error of IL-17 immunity leading to chronic mucocutaneous.
Clinical Implications.
Our aim is to help with the diagnosis and treatment of immunodeficiency patients by elucidating the molecular mechanisms leading to chronic fungal infections.
ACKNOWLEDGMENTS
The authors would like to thank Andrew Oler, Ke Huang, Sandhya Xirasagar, Darrell Hurt, and other members of the Bioinformatics and Computational Biosciences Branch (BCBB), NIAID for variant assessment and bioinformatics support. We thank MERCK Pharmaceuticals for performing whole genone sequencing and allowing us to access to their patient information. Finally, we thank the patient, family members, and healthy controls for participating in this study, and without whom it would not be possible to conduct this research.
FUNDING
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. This work was also funded by a fellowship grant (1-16-PDF-025, to Dr. Comrie) from the American Diabetes Association, a F12 postdoctoral fellowship (1FI2GM119979-01, to Dr. Comrie) from the National Institute of General Medical Sciences, NIGMS, NIH, The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- NF-κB
Nuclear Factor-kappaB
- TRAF3IP2
TRAF3 interacting protein 2
- SEFIR
“similar expression to fibroblast growth factor genes and IL-17R”
- CMC
Chronic Mucocutaneous Candidiasis
- PBMCs
Peripheral Blood Mononuclear Cells
- RICD
Restimulation-Induced Cell Death
- SNP
Single Nucleotide Polymorphism
- IL-25
IL-17E
- PMA
Phorbol myristate acetate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Okada S, Puel A, Casanova JL, Kobayashi M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL-17 immunity. Clin Transl Immunology. 2016;5(12):e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doyle MS, Collins ES, FitzGerald OM, Pennington SR. New insight into the functions of the interleukin-17 receptor adaptor protein Act1 in psoriatic arthritis. Arthritis Res Ther. 2012;14(5):226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 2013;39(4):676–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattad S, Dinakar C, Pinnamaraju H, Ganapathy A, Mannan A. Chronic Mucocutaneous Candidiasis in an Adolescent Boy Due to a Novel Mutation in TRAF3IP2. J Clin Immunol. 2019;39(6):596–9. [DOI] [PubMed] [Google Scholar]
- 5.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48(9):1071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi S, Cornall R, Lesourne R, Love PE. THEMIS: Two Models, Different Thresholds. Trends Immunol. 2017;38(9):622–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lesourne R, Uehara S, Lee J, Song KD, Li L, Pinkhasov J, et al. Themis, a T cell-specific protein important for late thymocyte development. Nat Immunol. 2009;10(8):840–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E, et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet. 2010;42(11):996–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nemer G, El-Hachem N, Eid E, Hamie L, Bardawil T, Khalil S, et al. A novel TRAF3IP2 variant causing familial scarring alopecia with mixed features of discoid lupus erythematosus and folliculitis decalvans. Clin Genet. 2020. [DOI] [PubMed] [Google Scholar]
- 11.Qian Y, Qin J, Cui G, Naramura M, Snow EC, Ware CF, et al. Act1, a negative regulator in CD40-and BAFF-mediated B cell survival. Immunity. 2004;21(4):575–87. [DOI] [PubMed] [Google Scholar]
- 12.Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182(3):1617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsushima Y, Kikkawa Y, Takada T, Matsuoka K, Seki Y, Yoshida H, et al. An atopic dermatitis-like skin disease with hyper-IgE-emia develops in mice carrying a spontaneous recessive point mutation in the Traf3ip2 (Act1/CIKS) gene. J Immunol. 2010;185(4):2340–9. [DOI] [PubMed] [Google Scholar]
- 14.Comrie WA, Faruqi AJ, Price S, Zhang Y, Rao VK, Su HC, et al. RELA haploinsufficiency in CD4 lymphoproliferative disease with autoimmune cytopenias. J Allergy Clin Immunol. 2018;141(4):1507–10 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snow AL, Marsh RA, Krummey SM, Roehrs P, Young LR, Zhang K, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest. 2009;119(10):2976–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, et al. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. 2008;181(6):4299–310. [DOI] [PubMed] [Google Scholar]