

Abstract
Rationale:
Increased endothelial permeability and defective repair are the hallmarks of several vascular diseases including acute lung injury (ALI). However, little is known about the intrinsic pathways activating the endothelial cell (EC) regenerative programs.
Objective:
Studies have invoked a crucial role of sphingosine-1-phosphate (S1P) in resolving endothelial hyperpermeability through the activation of the G-protein coupled receptor, sphingosine-1-phosphate receptor 1 (S1PR1). Here we addressed mechanisms of generation of a population of S1PR1+ EC and their pivotal role in restoring endothelial integrity.
Methods and Results:
Studies were made using inducible EC-S1PR1−/− (iEC-S1PR1−/−) mice and S1PR1-GFP reporter mice to trace the generation of S1PR1+ EC. We observed in a mouse model of endotoxemia that S1P generation induced the programming of S1PR1lo to S1PR1+ EC, which eventually comprised 80% of the lung EC. The cell transition was required for reestablishing the endothelial junctional barrier. We observed that conditional deletion of S1PR1 in EC increased endothelial permeability. RNA-seq analysis of S1PR1+ EC showed enrichment of genes regulating S1P synthesis and transport, specifically sphingosine kinase 1 (SPHK1) and SPNS2. Activation of transcription factors EGR1 and STAT3 was required for transcribing SPHK1 and SPNS2, respectively and both served to increase S1P production and amplify S1PR1+ EC transition. Furthermore, transplantation of S1PR1+ EC population into injured lung vasculature restored endothelial integrity.
Conclusion:
Our findings show that generation of the S1PR1+ EC population activates the endothelial regenerative program to mediate endothelial repair. Results raise the possibility of harnessing this pathway to restore vascular homeostasis in inflammatory vascular injury states.
Keywords: Endothelial permeability, endothelial regeneration, proliferation, S1PR1, EGR1, STAT3, SPNS2, SPHK1, endothelial cell, endovascular repair, pulmonary edema, Mechanisms, Pulmonary Biology, Translational Studies, Vascular Biology
Graphical Abstract
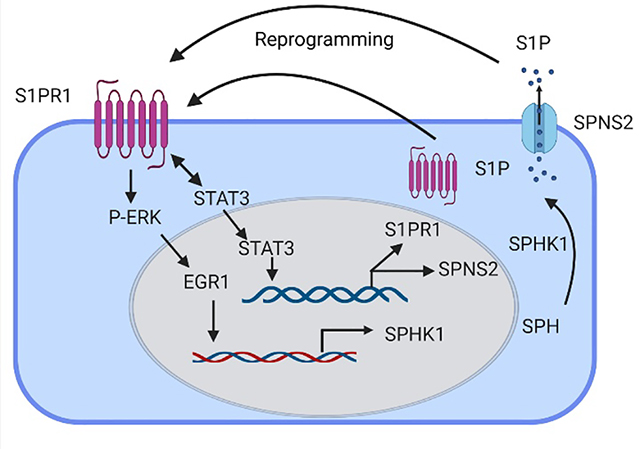
INTRODUCTION
The vascular endothelium regulates the transport of nutrients, protein, water, and leukocytes across the vessel wall that is essential for maintaining tissue and fluid homeostasis and robust immune responses.1 Thus, injury of the endothelial barrier is itself considered as an underlying mechanism of inflammation and edema formation, the hallmarks of several diseases including acute lung injury (ALI).2–4 Vascular endothelial injury induced by inflammatory factors such as, LPS, activates signaling cascades leading to breakdown of adherens junctions (AJs).5–7 However, the intrinsic repair pathways responsible for restoring endothelial barrier integrity and tissue homeostasis remain unclear. Our previous studies showed that early developmental signals involving reactivation of transcriptional factors FoxM1 and Sox17 in injured adult microvessel EC promoted endothelial regeneration.5,8 These transcription factors functioned in a multifaceted complex manner involving expression of cyclin genes, and thus may be linked to the pathogenesis of cancer.9,10
To identify a more effective solution, we focused on the well-known endothelial barrier reparative property of sphingosine-1-phosphate (S1P) receptor (S1PR1) expressed in EC.11,12 S1PR1 belonging to the family of seven transmembrane domain G-protein coupled receptors (GPCRs)1,13 is expressed in the developing vasculature and adult tissue including lungs, brain, and immune organs.14,15 S1PR1 plays a key role in the early phase of angiogenesis.16–18 EC specific deletion of S1PR1 was embryonically lethal due to defective vasculature formation.15 In vascular injury models, the S1PR1 agonist, S1P, reduced lung injury induced by ALI.12,18–21 While these studies focused on the importance of S1P generation and activation of S1PR1 in repairing the endothelium6,21, a crucial unanswered question is whether there is a population of EC expressing S1PR1 constitutively that can be mobilized or programmed after injury to repair the damaged endothelium. Here, using EC-S1PR1 knockout mice and S1PR1-GFP reporter mice, we identify a central role of an S1PR1+ EC population in driving vascular repair. We demonstrated that the S1PR1+ EC population was generated preceding endothelial repair and reestablished endothelial barrier integrity. Our genetic data showed that conditional EC-specific deletion of S1PR1 increased vascular endothelial permeability. Adoptive transfer of S1PR1+ EC integrated into the injured intima to restore endothelial integrity. We demonstrated that transcription factors EGR1 and STAT3 activated during EC injury, transcribed SPHK1 and S1P transporter SPNS2, respectively to promote the generation of S1P and efflux, and thus mediated vascular repair. These studies identified previously unknown population of programmed S1PR1+ EC with the capacity to restore endothelial integrity in inflammatory disease states such as ALI.
METHODS
Data Availability.
Details about materials and methods used to conduct this study as well as statistical analysis are provided in online Data Supplement and the Major Resources Table. The authors declare that all supporting data are available within the article and its online supplementary files.
RESULTS
Tracing the generation of S1PR1+ EC.
We first studied the effects of conditionally deleting S1PR1 on vascular endothelial permeability using inducible EC-S1PR1−/− (iEC-S1PR1−/−) mice (Figure 1A).26 Tamoxifen deleted 80% of S1PR1 in lung EC consistent with its predominant expression in endothelium1,13,40,41 (Figure 1B through 1D). EC-S1PR1 deletion had no significant effect on the expression of S1PR2 or S1PR3 in these studies (Figure 1D). EC-specific deletion of S1PR1 in mice induced lung edema and endothelial injury as evident by increased wet-dry weight ratio (Figure 1E) and lung transvascular albumin flux (Figure 1F).
Figure 1: Conditional deletion of S1PR1 in endothelial cell increases vascular permeability.

A, iEC-S1PR1−/− mice were generated by crossing S1PR1fl/fl mice with mice expressing Cre under the control of tamoxifen-inducible 5’ enhancer stem cell leukemia (Scl-CreERT) promoter. Five weeks old iEC-S1PR1−/− and S1PR1fl/fl mice were exposed to 80 mg/kg tamoxifen i.p. for indicated time points, followed by one-week rest. B-C, Lung S1PR1 protein expression at 11th day post-tamoxifen injection. B, a representative blot from three independent experiments. C, densitometry of S1PR1 expression calculated as fold increase over actin. D, qPCR of indicated genes in the lungs of iEC-S1PR1−/− and S1PR1fl/fl mice taking GAPDH as internal control and expressed as fold change relative to S1PR1fl/fl. The plot shows individual scatter with mean ±SD (n=3). E-F, lung vascular injury was determined post LPS administration (10 mg/kg i.p.) by measuring wet-to-dry weight ratio (E) and Evans blue albumin extravasation in lung parenchyma (F). The plots show individual values ±SD (n=7 mice/group). Statistical significance was assessed by unpaired t test for C, D, E and F (See also Online Table II). C, p=0.0001; D, p=4.64E-06; E, p=5.32E-05 and F, p=0.0001 indicate significance relative to S1PR1fl/fl. ns= not significant.
We used S1PR1-GFP reporter mice generated by crossing H2B-GFP mice with S1PR1knockin mice to study generation and expansion of S1PR1-expressing EC (Online Figure IA).22,42 We tracked GFP+ EC in vivo in LPS model of lung vascular endothelial injury based on the sublethal dosage of LPS (10mg/kg i.p.) coupled to a discrete phase of endothelial repair.6,7 LPS induced lung endothelial injury occurring with the same kinetics and degree as control mice (H2B-GFP mice) (data not shown). Using FACS analysis to determine the time course of S1PR1 activity (recorded by the increase in GFP+ cells), we found that LPS significantly increased S1PR1 activity 8h post LPS exposure; the activity reached maximum at 16h when 23% of lung EC were GFP+(Figure 2A and 2B). At 24h, GFP+ EC number fell to baseline (Figure 2A and 2B). GFP+ EC were not detected in control mice (Figure 2A and 2B). LPS also induced lung edema at 4h and 8h consistent with increase endothelial permeability whereas the edema declined towards basal levels at 24h (Figure 2C).6,7 Imaging showed significantly increased S1PR1 activity at 16h post LPS induced injury as compared to untreated mice or H2B-GFP mice (Figure 2D and 2E).
Figure 2: Kinetics of S1PR1 activity following LPS-induced lung vascular injury.

A and B, FACS analysis of lung cell suspension obtained from S1PR1-GFP reporter mice or H2B-GFP (control) mice following LPS (10 mg/kg, i.p.) administration. A, scatter dot plot of GFP gated cells. B, quantification of GFP+ cells as percent of total lung cells at each time (n=7 mice/group). C, LPS-induced (10 mg/kg i.p.) vascular injury in S1PR1-GFP reporter mice was determined by measuring lung wet-dry weight ratio (n=7 mice/group). D, a representative micrograph of GFP+ cells in the lung sections from S1PR1-GFP reporter or H2B-GFP mice 16h post LPS challenge. E, quantification of GFP+ cells from multiple lung sections. Scale bar 50 μm (n=4 mice/group). B, C and E show individual scatter with mean ±SD. Statistical significance was assessed by one-way ANOVA followed by Post hoc Tukey’s multiple comparisons test for B, C and E (See also Online Table II). B, p=5E-14, p=6.1E-14; C, p=5.1E-14, p=5E-14; E, p=3.72E-09 and p=0.0006 indicate significance relative to 0h LPS. ns=not significant.
On measuring S1P generation, we observed that it also occurred in a delayed manner after lung endothelial injury by LPS.6 To address the possibility that increased S1PR1 activity was the result of S1P generation post-injury, we activated S1PR1 by directly injecting S1P43,44 or the S1PR1 agonist, CYM-544245,46 i.v. in S1PR1-GFP reporter mice. S1PR1 at baseline was expressed in only 2.5–6% of lung cells whereas S1P increased S1PR1 activity by ~2-fold within 2h, which remained at this level upto 4h (Online Figure IB and ID). Compared to S1P, CYM-5442 increased S1PR1 activity faster45,46 i.e., within 1h which increased further by ~3-fold at 4h (Online Figure IC and 1D), indicating the ability to pharmacologically increase the population of S1PR1+ cells. Thus, in contrast to S1P and S1PR1 agonist, the slow rise in S1PR1 activity in S1PR1-GFP reporter mice after LPS challenge likely reflected the observed delayed generation of S1P.6
We next studied the generation of S1PR1+ EC population in response to LPS using FACS analysis. We immunostained cells with anti-CD45 and CD31 antibodies (Online Figure IIA) to identify S1PR1+ EC population. We found that EC (which were CD31 positive, CD45 negative) accounted for ~70–80% of GFP+ cells (termed S1PR1+ EC) in lungs of S1PR1-GFP reporter mice after 8h to16h of LPS challenge (Figure 3A and 3B). Immunostaining with an endothelial specific marker, vWF confirmed that S1PR1+ EC were also vWF+ (Figure 3C and 3D; and Figure 3C inset). We observed far lower S1PR1 activity in lung hematopoietic cells (CD31+CD45+GFP+) (Online Figure IIB), alveolar epithelial cells (GFP+ EpCAM+), and fibroblasts (Online Figure IIC and IID) as compared to EC (Figure 3A and 3B). Thus, LPS activated signaling induced the generation of S1PR1+ EC population.
Figure 3: Lung vascular injury expands the proliferative S1PR1+ endothelial cell population.

A and B, GFP-gated EC (GFP+CD31+CD45−) from LPS (10 mg/kg, i.p) exposed lungs of S1PR1-GFP reporter mice. A, representative FACS plot at indicated time points. B, quantification of GFP+CD31+CD45− (S1PR1+ EC) as a percentage of total lung EC (CD31+CD45−) post injury (n=7 mice/group). C and D, lung sections were immunostained with endothelial-specific marker, von-Willebrand Factor (vWF) and S1PR1+ EC were determined using confocal analysis. C, a representative micrograph. The inset shows x5 magnified vessel. Scale bar 50 μm. D, number of S1PR1+ EC over vessel area in lungs of S1PR1-GFP reporter mice (n=4 mice/group). E, a representative micrograph showing BrdU+S1PR1+ EC in unexposed or LPS-exposed S1PR1-GFP reporter mice lungs. Mice received LPS (10 mg/kg, i.p) followed by BrdU (80mg/kg, i.p.) injection 4h before harvesting lungs. Lungs sections were immunostained with anti-BrdU antibody (n=3 mice/group). Scale bar 50 μm. F, a representative scatter plot of showing S1PR1+ EC proliferation using CD31+CD45−Ki-67+ antibodies after without or with LPS challenge (10 mg/kg, i.p). B and D individual data with mean ±SD. Data in B were analyzed using one-way ANOVA followed by Post hoc Tukey’s multiple comparisons test, while in D unpaired t test was used (See also Online Table II). B, p=0.0211, p=2.56E-15, p=4.23E-15, p=5.48E-14; and D, p=3.14E-05 indicate significance relative to time “0h LPS or No LPS”. ns=not significant.
Bone marrow progenitor EC also have the capacity to generate S1P47, suggesting that these EC may also be responsible for driving S1PR1+ EC generation after lung injury in S1PR-GFP reporter mice. We therefore determined GFP+ cells in the bone marrow of S1PR1-GFP reporter mice and found that bone marrow contained only 6.2% GFP+ cells (Online Figure IIIA). We isolated bone marrow from S1PR1-GFP reporter mice and transplanted them into irradiated WT mice. At 5 weeks, we challenged these chimeric mice with LPS as above and harvested their lungs at 16h post injury (Online Figure IIIB). FACS and confocal analyses demonstrated that lungs from chimeric mice contained only 4% S1PR1+ EC, like the observation in S1PR1-GFP mice under basal conditions (Online Figure IIIC and IIID). Thus, these results show that the majority of S1PR1+ EC were derived from lung resident EC.
Proliferative expansion of S1PR1+ EC promotes resolution of endothelial injury.
As the key question is the source of S1PR1+ EC, we speculated, that the surviving EC post-injury expanded through proliferation to restore vascular integrity.5,8 In S1PR1-GFP reporter mice, GFP expression was only evident in the non-proliferating cells. We injected 5-bromo-2′-deoxyuridine (BrdU), 4h before sacrificing the reporter mice after LPS challenge to determine whether S1PR1+ EC showed proliferative capacity. Immunostaining showed that S1PR1+ EC expressed BrdU at 16h in the S1PR1-GFP reporter mice lungs as compared to baseline (Figure 3E). FACS analysis with anti-Ki-67 antibody, a proliferation marker48, also showed that 4–8% of S1PR1+ EC were Ki-67+ (Figure 3F and Online Figure IVA and IVB). TUNEL assay ruled out that S1PR1+ EC became apoptotic (Online Figure IVC). Thus, these results show the key role of S1PR1+ EC proliferation during the repair phase of lung EC injury.
Transcription regulation of S1PR1+ EC generation.
We performed RNA-seq analysis to assess the transcriptional machinery responsible for programming S1PR1+ EC using flow sorted S1PR1+ EC. We found that at 8h and 16h post LPS challenge, 860 genes were differentially expressed in S1PR1+ EC as compared to unchallenged EC (Online Figure IVD). Ingenuity Pathway analysis showed enrichment of proliferation and cell cycle pathways in S1PR1+ EC (Online Figure VA and VB). On screening the top 100 upregulated genes at 8h or 16h in S1PR1+ EC, we found increases in the expression of S1P generating genes41,49, SPHK1 and S1P plasma membrane transporter, SPNS2 (Figure 4A and Online Figure VC). We also found increase in the expression of early growth response 1 (EGR1), the transcription factor belonging to the immediate early genes (IEGs) family in the S1PR1+ EC.50 These findings were confirmed by qPCR using non-GFP and S1PR1+ EC flow sorted from lungs at baseline or from S1PR1-GFP reporter mice at 8h and 16h post LPS challenge. In contrast to non GFP-EC, S1PR1+ EC showed up to 5-fold increase in SPHK1 mRNA, but no significant difference was observed in SPHK2 mRNA expression (Figure 4B). We also observed 1.5-fold increase in SPNS2 mRNA expression at 8h, which further increased to 3-fold at 16h (Figure 4B). In addition, there was a time dependent increase in EGR1 mRNA in S1PR1+ EC (Figure 4B). S1PR1+ EC also consistently showed increased expression of SPHK1, SPNS2 and EGR1 proteins (Figure 4C and 4D) and 2-fold greater S1P generation than non-GFP EC (Figure 4E).
Figure 4: RNA-seq analysis of S1PR1+ endothelial cells.

A, S1PR1+ EC were flow sorted from unexposed or LPS administered (10 mg/kg, i.p) S1PR1-GFP reporter mice at indicated time points and RNA-seq analysis was performed. A, volcano plot of genes in S1PR1+ EC at 16h post LPS challenge versus 0h. B, validation of indicated gene expression in S1PR1+ EC taking GAPDH as an internal control (n=3). Gene expression is shown as fold change relative to respective genes at 0h LPS. C-D, A Representative immunoblot shows SPHK1, SPNS2 and EGR1 protein expression in flow sorted S1PR1+ EC post 16h LPS stimulation, while plot shows densitometric analysis of indicated proteins expressed as fold increase over actin (n=3). E, S1P concentration was quantified using liquid chromatography-mass spectrometry in S1PR1+ EC versus non-GFP EC flow sorted at 16h post LPS exposure (n=3 mice/group). F, qPCR analysis of indicated genes in HLMVEC after with or without 1μg/ml LPS stimulation from three different experiments. GAPDH was used as an internal control. Gene expression is shown as fold change relative to 0h LPS. G-H, A representative immunoblot shows SPHK1, SPNS2 and EGR1 expression in HLMVEC after stimulation with LPS (1μg/ml) and corresponding densitometry of indicated proteins expressed as fold increase over actin (n=3). B, D, E, F and H show individual data along with mean ±SD. Data in B, F, and H were analyzed using one-way ANOVA followed by Post hoc Tukey’s multiple comparisons test. Unpaired t test was used in D and E (See also Online Table II). C, p=0.0001, p=1.44E-12, p=8.38E-09, p=2.06E-05 indicate significance relative to 0h LPS; D, p=0.0004, p=0.0013, p=5.2E-05 indicate significance relative to No LPS; E, p=0.0009 indicate significance relative to EC; F, p=1.36E-11, p=5.58E-13, p=1E-11 (SPHK1); p=8.83E-07, p=1.36E-11 (SPNS2); p=4.2E-07, p=9.14E-13, p=9.43E-08 (EGR1) indicate significance relative to 0h LPS; H, p=1.23E-12, p=5.36E-12, p=2.61E-14 (SPHK1); p=3.25E-8, p=6.52E-11, p=2.8E-12 (SPNS2); p=3.21E-6, p=5.63E-8 and p=3.98E-10 (EGR1) indicate significance relative to 0h LPS. ns=not significant.
To assess whether LPS induced the expression of these genes in human lung EC, we stimulated human lung microvascular endothelial cells (HLMVEC) with LPS. LPS increased SPHK1 mRNA expression by 4-fold at 8h, which remained at this level till 16h (Figure 4F). However, LPS did not increase SPHK2 mRNA expression (Figure 4F). A gradual increase in SPNS2 and EGR1 mRNA expression was also observed post LPS challenge (Figure 4F). Like, mouse S1PR1+ EC, LPS-stimulated HLMVEC also showed increased SPHK1, SPNS2 and EGR1 protein expression (Figure 4G and 4H).
EGR1 induces SPHK1 expression in S1PR1+ EC.
We next addressed the possible role of EGR1 in mediating SPHK1 expression in EC and increasing their potential to generate S1P and transition to the S1PR1+ EC population. We first depleted EGR1 in HLMVEC using siRNA, which decreased SPHK1 mRNA and protein expression (Figure 5A through 5C and online figure VD through VE). Depletion of S1PR1 similarly depleted EGR1 and SPHK1 (Figure 5D).
Figure 5: EGR1 induces transcription and expression downstream of S1PR1.

A–C, SPHK1 mRNA (A) or protein expression (B–C) following EGR1 depletion in HLMVEC quantified as described in Figure 4B and 4D (n=3). GAPDH was used to normalize RNA while actin was used as loading control for quantifying protein expression. mRNA level quantified as fold change relative to cells transfected with scrambled siRNA. D, mRNA expression of S1PR1, EGR1 and SPHK1 in control versus S1PR1-depleted HLMVEC was quantified taking GAPDH as internal control (n=3). mRNA level quantified as fold change relative to cells transfected with scrambled siRNA. E, human SPHK1 promoter region with three EGR1 binding sites. F, HLMVEC were stimulated with LPS (1μg/ml) for indicated times and chromatin immunoprecipitation (ChIP) followed by qPCR was performed to amplify EGR1 binding sites in SPHK1 promoter (n=3). G, a representative gel shows qPCR-amplified product. H, HLMVEC transducing wild type (WT) or mutated (MT) SPHK1 luciferase promoter constructs were stimulated with LPS (1μg/ml) for indicated times. SPHK1 promoter activity was determined as described in online methods (n=3). I–J, a representative immunoblot shows ERK phosphorylation in flow sorted S1PR1+ EC versus non-GFP EC 16h post LPS stimulation of S1PR1-GFP reporter mice (n=3), whereas plot shows densitometry of phosphorylated ERK expressed as fold increase over total ERK. K, mRNA level of EGR1 and SPHK1 in HLMVEC stimulated with LPS (1μg/ml) and with/without specific ERK inhibitor (5μM) (n=3). mRNA level quantified as fold change relative to unstimulated cells. A, C, D, F, H, J and K show individual value along with mean ±SD. Data in plots F, H, and K were analyzed using one-way ANOVA followed by Post hoc Tukey’s multiple comparisons test, whereas unpaired t test was used for A, C, D, and J (See also Online Table II). A, p=0.0002; C, p=0.0012, p=0.0007; D, p=0.0012, p=0.0006, p=0.0003 indicate significance relative to siSC; F, p=0.0012, p=2.06E-06; H, p=3.27E-08 indicate significance relative to 0h LPS; J, p=0.0003 indicate significance relative to EC; K, p=3.20E-08, p=2.43E-07 (EGR1); p=1.17E-07 and p=1.16E-07 (SPHK1) indicate significance relative to No LPS. ns=not significant.
The SPHK1 promoter contains three EGR1 binding sites (Figure 5E). We, therefore, performed chromatin immunoprecipitation (ChIP) with quantitative real-time PCR to assess if S1PR1 increased EGR1 binding to the SPHK1 promoter regions. Relative to unstimulated cells, LPS significantly increased the binding of EGR1 to SPHK1 promoter (Figure 5F and 5G). To establish the causal role of EGR1 in inducing SPHK1 expression, we mutated EGR1 binding sites on the SPHK1 promoter and transfected WT or mutated SPHK1 promoter in HLMVEC, and found that LPS failed to increase SPHK1 promoter activity in EC transducing the mutated SPHK1 promoter (Figure 5H).
To establish whether EGR1-induced SPHK1 expression was required to mediate endothelial integrity, we determined transendothelial electrical resistance (TEER) in EGR1 depleted HLMVEC and found that depletion of EGR1 significantly reduced endothelial barrier function (Online Figure VIA). However, addition of S1P restored barrier function to basal levels (Online Figure VIA) corroborating the above findings that EGR1-mediated expression of SPHK1 was required to generate S1P that in turn ligated S1PR1 and normalized the EC barrier function.
As ERK induces EGR1 transcriptional activity51,52, we next addressed the role of ERK-EGR1 signaling in the mechanism of SPHK1 expression. We found that ERK phosphorylation, a measure of ERK activity,53 was enhanced in S1PR1+ EC that were flow-sorted post LPS challenge as compared to baseline S1PR1+ EC (Figure 5I and 5J). LPS similarly increased ERK activity in HLMVEC within 4h and the activity remained greater than baseline for up to 16h post-LPS stimulation (Online Figure VIB and VIC). Inhibition of ERK phosphorylation with SCH77298454, a specific ERK inhibitor, reduced both basal and LPS-induced EGR1 and SPHK1 expression (Figure 5K) demonstrating the critical role of ERK-induction of EGR1 in upregulating SPHK1 expression and thereby the S1P production required for generation of the S1PR1+ EC population.
STAT3 induces SPNS2 expression downstream of S1PR1.
Because EGR1 depletion did not produce any significant effect on SPNS2 expression (data not shown), we next performed in-silico analysis and found that STAT3 bound both S1PR1 and SPNS2 promoters. Thus, we tested that LPS increased STAT phosphorylation, a measure of STAT activity55, in S1PR1+ EC. LPS increased STAT3 phosphorylation in HLMVEC at 4h, which remained elevated for up to 16h post-LPS (Online Figure VIB and VID). S1PR1+ EC also showed increased STAT3 phosphorylation compared to other STAT proteins in which phosphorylation remained essentially unaltered (Figure 6A and 6B, and Online Figure VIE). Inhibition of ERK had no significant effect on STAT3 phosphorylation post-LPS stimulation (Online Figure VIF and VIG), indicating S1PR1 induced STAT3 activation independent of the ERK pathway. Depletion of S1PR1 in EC prevented ERK and STAT3 phosphorylation further indicating that LPS stimulated ERK and STAT3 via S1PR1 (Online Figure VIH through VIJ).
Figure 6: STAT3 induces transcription and expression of SPNS2.

A–B, phosphorylation of STAT3 (P-STAT3) from flow sorted EC or S1PR1+ EC (n=3). A, a representative immunoblot. B, densitometry of P-STAT3 expressed as fold increase over total STAT3. C, expression of indicated genes in STAT3 depleted HLMVEC was determined after LPS exposure (1μg/ml) using GAPDH as an internal standard (n=3). mRNA level was quantified as fold change relative to cells transfected with scrambled siRNA. D, human SPNS2 promoter region with two STAT3 binding sites. E–F, binding enrichment of STAT3 on SPNS2 promoter was determined by ChIP assay following without or with LPS stimulation (1μg/ml) as described in Figure 5F and 5G (n=3). G, SPNS2 promoter activity in HLMVEC transfected with wild type (WT) or mutated (MT) SPNS2 promoter constructs after with or without LPS (1μg/ml) stimulation (n=3). B, C, E and G show individual values along with mean ±SD. Data in E and G were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test, while unpaired t test was used for B and C (See also Online Table II). B, p=0.0009 indicates significance relative to EC; C, p=0.0002, p=0.0002 and p=4.43E-05 indicate significance relative to siSC; E, p=0.0123, p=2.32E-05 and G, p=2.09E-09 indicate significance relative to 0h LPS. ns=not significant.
Next, we depleted STAT3 and found that STAT3 depletion prevented SPNS2 and S1PR1 expression without altering EGR1 and SPHK1 expression (Figure 6C). The human SPNS2 promoter contains two STAT3 binding sites (Figure 6D) which was confirmed using chromatin immunoprecipitation assay (Figure 6E and 6F). We thus mutated STAT3 binding sites on SPNS2 promoter and transfected WT or mutated SPNS2 promoter in HLMVEC to establish the role of STAT3 in inducing SPNS2 expression. LPS failed to increase SPNS2 promoter activity in EC transduced with the mutated SPNS2 promoter (Figure 6G).
Depletion of STAT3 or SPNS2 significantly reduced basal endothelial barrier function (Online Figure VIK and VIL). Furthermore, S1P addition did not produce any significant effect on the barrier function (Online Figure VIK and VIL), confirming that STAT3 induced expression of S1PR1 and SPNS2 was required for S1P-mediated enhancement of EC barrier function. These results together demonstrate that S1PR1 expression induced EGR1 and STAT3 transcriptional activation and thereby induced S1P generation and transport by SPHK1 and SPNS2, respectively.
S1PR1+ EC mediate endothelial repair.
To examine whether the S1PR1+ EC population could repair the injured endothelium, we used EC-S1PR1 null mice in which S1PR1 was conditionally deleted in EC using tamoxifen (Figure 1A). We transplanted ~1.0×106 S1PR1+ EC or control EC (non-GFP EC) flow sorted under same conditions from 16h LPS exposed S1PR1-GFP reporter mice into iEC-S1PR1−/− mice and determined the endothelial injury response 24h and 48h post-transplantation. We observed that S1PR1+ EC repaired the endothelium of EC-S1PR1−/− mice (Figure 7A). Immunostaining of lung sections from EC-S1PR1−/− mice with anti-vWF antibody showed that S1PR1+ EC became integrated in the microvessels (Figure 7B and 7C).
Figure 7: S1PR1+ endothelial cells promote resolution of lung endothelial injury.

A, S1PR1+ EC (GFP+CD31+CD45−) or non-GFP EC (GFP−CD31+CD45−) were flow sorted at 16h post LPS challenge (10 mg/kg i.p.) of S1PR1-GFP reporter mice. S1PR1+ EC or EC (~1.0×106) were then injected i.v. into S1PRfl/fl or EC-S1PR1 null mice. Subsequently, lung vascular injury was determined by measuring lung wet-dry weight ratio after 24h or 48h post transplantation (n=6 mice/group). B, a micrograph from EC-S1PR1 null lungs receiving S1PR1+ EC or non-GFP EC were stained with anti-GFP and anti-vWF antibodies to assess integration of S1PR1+ EC into intima of the vessel. Micrograph of experiments were repeated multiple times. Scale bar 50 μm. C, respective quantification of S1PR1+ EC quantified as number of S1PR1+ EC over vessel area (n=4). A and C show individual data along with mean ±SD. Data in A were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons test, whereas unpaired t test was used for C (See also Online Table II). A, p=9.92E-09, p= 3.62E-10 indicate significance relative to mice receiving EC and C, p=0.0086 indicate significance relative to EC.
SPHK1 and STAT3 inhibition prevents S1PR1+ EC generation and augments injury.
To address the reparative role of generated S1PR1+ EC in vivo, we determined the effects of inhibiting SPHK1 or STAT3 in S1PR1-GFP reporter mice on S1PR1+ EC generation. Here, PF-543, an inhibitor of SPHK1 or S3I-201, a STAT3 inhibitor, was injected i.v. into mice 1h post i.p. LPS challenge. LPS failed to generate the S1PR1+ EC population in S1PR1-GFP reporter mice receiving the inhibitors of SPHK1 (Figure 8A and 8B) or STAT3 (Figure 8C and 8D). However, inhibition of SPHK2 activity had no significant effect on S1PR1+ EC generation (Online Figure VIIA and VIIB). Furthermore, lung vascular injury persisted in mice receiving SPHK1 or STAT3 inhibitors (Figure 8E). In parallel, we determined the effects of inhibiting STAT3 and SPHK1 activities on LPS-induced EC barrier injury. We found that SPHK1 and STAT3 inhibitors augmented LPS-induced barrier loss in HLMVEC (Online Figure VIIC). Also, LPS increased endothelial permeability in SPNS2-depleted EC as compared to control EC (Online Figure VIID). These findings together demonstrate the crucial role of SPHK1 and STAT3◊SPNS2 pathway in restoring endothelial barrier following LPS challenge.
Figure 8: SPHK1 and STAT3 activities are required for S1PR1+ endothelial cell generation in vivo and resolution of lung vascular injury.

SPHK1 inhibitor, PF-543 (5 mg/kg, i.v) (A, B and E) or STAT3 inhibitor, S3I-201 (5 mg/kg, i.v) (C, D and E) were injected in S1PR1-GFP reporter mice. FACS analysis or lung injury was determined as described in Figure 2. A and C show FACS dot plots of lungs from these mice after with and without inhibitors whereas, B and D depict corresponding quantification (n=7 mice/group). E, lung vascular injury response following treatment with indicated inhibitors as described in Figure 1E (n=5 mice/group). B, D and E show individual data with mean ±SD. Data were analyzed using one-way ANOVA followed by Post hoc Tukey’s multiple comparisons test (See also Online Table II). B, p=0.0003, p=1.77E-10, p=1.99E-11, p=1.99E-11 indicate significance relative to -PF 543 (No SPHK1 inhibitor); D, p=6.38E-14, p=5.78E-14 indicate significance relative to -S3I-201 (No STAT3 inhibitor); E, p=0.0007, p=2.94E-09, p=7.71E-10 indicate significance relative to No LPS; p=0.0002 and p=2.22E-06 indicate significance relative to LPS exposed mice. ns=not significant.
DISCUSSION
In the present study, we demonstrated the generation of heretofore unknown population of S1PR1+ EC showing marked regenerative capacity that restored lung vascular integrity in LPS challenged mice. We showed that the S1PR1+ EC acquired their reparative property through augmenting S1P generation and S1P transport into the extracellular milieu. Furthermore, i.v. injected population of S1PR1+ EC integrated within injured endothelium of EC-S1PR1 null mice. This transition from S1PR1lo EC to reparative S1PR1+ EC was mediated by the transcription factors EGR1 and STAT3, which upregulated the expression of SPHK1 and SPNS2, respectively. SPHK1 expression enhanced S1P generation whereas SPNS2 induced the efflux of S1P in the injury milieu which enabled S1P to function in a paracrine manner. Thus, our data demonstrate the fundamental endothelial barrier-protective role of the induced S1PR1+ EC population.
The loss of vascular endothelial barrier function over time through apoptosis is a key determinant of ALI and other acute inflammatory diseases.4 Vascular repair in lungs and other tissue is required for restoration of normal organ function and tissue homeostasis.15 By tracing the generation of S1PR1+ EC using S1PR1-GFP reporter mice following endotoxemia, we were able to show the importance of S1PR1+ EC in mediating EC repair. This process involved the expansion of S1PR1+ EC due to activation of the transcription factors EGR1 and STAT3.
The S1PR1+ EC population was generated within 8h coinciding with the peak of LPS-induced injury response. About 76% of EC became S1PR1+ at 16h indicating a shift in EC phenotype that preceded the endothelial repair phase. Injection of S1PR1+ EC, i.v. into EC-S1PR1 null mice with endothelial injury resulted in integration of cells in the damaged intima of lungs restored the endothelial barrier. The mechanisms of homing and integration of these cells are not clear. One possibility is that S1PR1+ EC adhered to the exposed extracellular matrix following denudation of EC as in the LPS injury model.2 Another possibility is that transition to S1PR1+ EC may promote barrier re-annealing by activating Rac1 signaling at the adherens junctions, a signaling essential for EC repair.18,43
The S1PR1+ EC population mediating endothelial repair program post-LPS required 8–16h to maximize the generation of S1P. While S1P and S1PR1 agonist, CYM-5442 increased S1PR1+ EC within 2h. LPS is known to induce SPHK1 expression in a delayed manner6,56, a possible explanation for the 8–16h lag in the repair response. LPS is also known to activate S1P lyase and phosphatase that may interfere with ability to rapidly generate S1P.57 We observed that SPNS2, a S1P-specific transporter41, was also induced in a parallel manner with SPHK1 in EC within the same time frame. Upregulation of both SPHK1 and SPNS2 appeared to contribute to endothelial repair by activating S1P transport from EC that may augment S1PR1 signaling in an autocrine/paracrine manner.41,58,59 Thus, our findings show a novel mechanism of generation of S1PR1+ EC required for endothelial repair.
An important question is whether the mechanism of S1PR1+ EC generation can be accelerated to facilitate endothelial repair. We showed that S1P and S1PR1 agonists induced the generation of S1PR1+ EC within hours as opposed to the endogenous generation requiring 8–16h, suggesting a means of enhancing kinetics of S1PR1+ EC generation and the repair process. Because endothelial repair is essential for tissue survival and restoring homeostasis, multiple mechanisms have likely evolved to repair the endothelium in vivo.60–64 These relied on activating transcriptional regenerative programs in EC, transcription factors FoxM1, FoxC2 and Sox17 that induced vascular repair through proliferation of EC.5,8,65 The generation of S1PR1+ EC as shown in the present study also required the coordinated activation of both EGR1 and STAT3 transcription factors. EGR1 transcribed SPHK1 whereas STAT3 transcribed SPNS2. Inhibition of ERK prevented S1P induction of EGR1 and SPHK1 expression indicating it acted upstream of EGR1. Our findings are also consistent with the requirement of EGR1 activation in inducing SPHK1 expression of ERK.52 Inhibition of either SPHK1 or STAT3 prevented the generation of S1PR1+ EC population and thus endothelial repair.
In summary, using S1PR1-GFP reporter mice, we showed that the generation of a S1PR1+ EC population induced endothelial repair following endotoxemia. The production of S1P and its transport by SPHK1 and SPNS2, respectively, maximized the transition of S1PR1lo EC into the S1PR1+ EC population. Transcription factors EGR1 and STAT3 were required to transcribe SPHK1 and SPNS2, respectively. The generation of S1PR1+ EC population activated the endothelial regenerative program that mediated vascular repair, thereby raising the possibility of activating this intrinsic reparative S1PR1+ EC to restore vascular homeostasis and tissue function.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The vascular endothelium (EC) plays a crucial role in maintaining blood vessel functions, such as maintenance of tissue-fluid homeostasis.
An increase in endothelial permeability accumulates plasma proteins and leukocytes in the interstitium, the hallmark of acute lung injury (ALI).
Sphingosine-1-phosphate receptor 1 (S1PR1), belonging to the family of seven transmembrane domain G-protein-coupled receptors, mediates vascular repair.
In studies in vascular injury models, the S1PR1 activation by its agonist, S1P, reduced lung injury secondary to ALI.
What New Information Does This Article Contribute?
We identified an unknown population of S1PR1 active endothelial cells (S1PR1+ EC) in the lung during injury.
S1PR1+ EC generation preceded lung endothelial repair, and these cells vital for reestablishing the endothelial barrier.
Transplantation of S1PR1+ EC into the leaky vasculature of EC-specific S1PR1 null mice induced intimal integration of the cells and repaired barrier.
The activation of the transcription factors, EGR1 and STAT3 by S1PR1 during EC injury transcribed sphingosine kinase 1 (SPHK1) and the S1P transporter SPNS2 to amplify generation of S1P and its efflux, to mediate the vascular repair.
Endothelial barrier dysfunction leads to protein-rich edema formation and neutrophilic inflammation, the hallmarks of lethal disorders such as acute respiratory distress syndrome (ARDS). Present therapeutic measures are unsuccessful in reviving lung vascular endothelial barrier function in ARDS. S1P generation and S1PR1 activation have emerged as a predominant endothelial barrier repair mechanism. A crucial question is whether a population of EC expressing S1PR1 exists or produced after an injury capable of repairing the damaged endothelium remains unanswered. Here, we show the generation of S1PR1+ EC population during injury that reestablished endothelial barrier integrity. Adoptive transfer of S1PR1+ EC population induced integration of the cells in damaged lung vessels to rescue integrity. Further, S1PR1 activated the transcription factors EGR1 and STAT3 during EC injury, which transcribed SPHK1 and S1P transporter, SPNS2, to promote S1P generation and efflux, committing vascular repair. The present studies described a hitherto unexplored population of S1PR1+ EC with the capacity to repair endothelial integrity, which may prevent inflammatory lung vascular injury.
Acknowledgments
SOURCES OF FUNDING
This work was supported by National Institutes of Health, USA grants (HL060678, HL137169 and HL084153). MZA is supported by American Heart Association Postdoctoral Fellowship (AHA Award-19POST34450241). Bioinformatics was performed by the University of Illinois Research Informatics Core, supported in part by the National Center for Advancing Translational Sciences, NIH grant UL1TR002003.
Nonstandard Abbreviations and Acronyms:
- ABC294640
3-(4-chlorophenyl)-adamantane-1-carboxylic acid (pyridin-4-ylmethyl)amide
- EC
Endothelial cells
- HLMVEC
Human lung microvascular endothelial cells
- H2B-GFP
Histone 2B-green fluorescent protein
- OCT
Optimal Cutting Temperature
- PFA
Paraformaldehyde
- PF-543
1-[[4-[[3-methyl-5-[(phenylsulfonyl)methyl]phenoxy]methyl]phenyl]methyl]-2R-pyrrolidinemethanol
- SCH772984
(3R)-1-[2-oxo-2-[4-[4-(2-pyrimidinyl)phenyl]-1-piperazinyl]ethyl]-N-[3-(4-pyridinyl)-1H-indazol-5-yl]-3-pyrrolidinecarboxamide
- siRNA
Small interfering RNA
- S3I-201
2-Hydroxy-4-[[2-(4-methylphenyl)sulfonyloxyacetyl]amino]benzoic acid
- S1PR1+ EC
S1PR1 active endothelial cells
- TEER
Transendothelial electrical resistance
- WT
Wild type
Footnotes
DISCLOSURE
None.
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Mehta D and Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. [DOI] [PubMed] [Google Scholar]
- 2.Thompson BT, Chambers RC and Liu KD. Acute Respiratory Distress Syndrome. N Engl J Med. 2017;377:562–572. [DOI] [PubMed] [Google Scholar]
- 3.Levitt JE, Gould MK, Ware LB and Matthay MA. The pathogenetic and prognostic value of biologic markers in acute lung injury. J Intensive Care Med. 2009;24:151–67. [DOI] [PubMed] [Google Scholar]
- 4.Matthay MA and Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao YY, Gao XP, Zhao YD, Mirza MK, Frey RS, Kalinichenko VV, Wang IC, Costa RH and Malik AB. Endothelial cell-restricted disruption of FoxM1 impairs endothelial repair following LPS-induced vascular injury. J Clin Invest. 2006;116:2333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB and Mehta D. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. 2008;103:1164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, Miao EA, Rehman J and Malik AB. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127:4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu M, Zhang L, Marsboom G, Jambusaria A, Xiong S, Toth PT, Benevolenskaya EV, Rehman J and Malik AB. Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat Commun. 2019;10:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Lu L, Tu J, Zhang J, Xiong T, Fan W, Wang J, Li M, Chen Y, Steggerda J, Peng H, Chen Y, Li TWH, Zhou ZG, Mato JM, Seki E, Liu T, Yang H and Lu SC. Reciprocal Regulation Between Forkhead Box M1/NF-kB and Methionine Adenosyltransferase 1A Drives Liver Cancer. Hepatology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Lee S, Lee S, Kim K, Yang Y, Kim JH, Adams RH, Wells JM, Morrison SJ, Koh GY and Kim I. Sox17 promotes tumor angiogenesis and destabilizes tumor vessels in mice. J Clin Invest. 2013;123:418–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JF, Gordon S, Estrada R, Wang L, Siow DL, Wattenberg BW, Lominadze D and Lee MJ. Balance of S1P1 and S1P2 signaling regulates peripheral microvascular permeability in rat cremaster muscle vasculature. Am J Physiol Heart Circ Physiol. 2009;296:H33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McVerry BJ and Garcia JG. Endothelial cell barrier regulation by sphingosine 1-phosphate. J Cell Biochem. 2004;92:1075–85. [DOI] [PubMed] [Google Scholar]
- 13.Mendelson K, Evans T and Hla T. Sphingosine 1-phosphate signalling. Development. 2014;141:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cahalan SM, Gonzalez-Cabrera PJ, Sarkisyan G, Nguyen N, Schaeffer MT, Huang L, Yeager A, Clemons B, Scott F and Rosen H. Actions of a picomolar short-acting S1P(1) agonist in S1P(1)-eGFP knock-in mice. Nat Chem Biol. 2011;7:254–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel Sand Proia RL. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106:951–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MJ, Evans M and Hla T. The inducible G protein-coupled receptor edg-1 signals via the G(i)/mitogen-activated protein kinase pathway. J Biol Chem. 1996;271:11272–9. [DOI] [PubMed] [Google Scholar]
- 17.Spiegel S and Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11:403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehta D, Konstantoulaki M, Ahmmed GU and Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–8. [DOI] [PubMed] [Google Scholar]
- 19.Hubner K, Cabochette P, Dieguez-Hurtado R, Wiesner C, Wakayama Y, Grassme KS, Hubert M, Guenther S, Belting HG, Affolter M, Adams RH, Vanhollebeke B and Herzog W. Wnt/beta-catenin signaling regulates VE-cadherin-mediated anastomosis of brain capillaries by counteracting S1pr1 signaling. Nat Commun. 2018;9:4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR and English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wadgaonkar R, Patel V, Grinkina N, Romano C, Liu J, Zhao Y, Sammani S, Garcia JG and Natarajan V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296:L603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kono M, Tucker AE, Tran J, Bergner JB, Turner EM and Proia RL. Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. J Clin Invest. 2014;124:2076–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gothert JR, Gustin SE, van Eekelen JA, Schmidt U, Hall MA, Jane SM, Green AR, Gottgens B, Izon DJ and Begley CG. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104:1769–77. [DOI] [PubMed] [Google Scholar]
- 24.Anderson H, Patch TC, Reddy PN, Hagedorn EJ, Kim PG, Soltis KA, Chen MJ, Tamplin OJ, Frye M, MacLean GA, Hubner K, Bauer DE, Kanki JP, Vogin G, Huston NC, Nguyen M, Fujiwara Y, Paw BH, Vestweber D, Zon LI, Orkin SH, Daley GQ and Shah DI. Hematopoietic stem cells develop in the absence of endothelial cadherin 5 expression. Blood. 2015;126:2811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt TT, Tauseef M, Yue L, Bonini MG, Gothert J, Shen TL, Guan JL, Predescu S, Sadikot R and Mehta D. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am J Physiol Lung Cell Mol Physiol. 2013;305:L291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balaji Ragunathrao VA, Anwar M, Akhter MZ, Chavez A, Mao Y, Natarajan V, Lakshmikanthan S, Chrzanowska-Wodnicka M, Dudek AZ, Claesson-Welsh L, Kitajewski JK, Wary KK, Malik AB and Mehta D. Sphingosine-1-Phosphate Receptor 1 Activity Promotes Tumor Growth by Amplifying VEGF-VEGFR2 Angiogenic Signaling. Cell Rep 2019;29:3472–3487 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faul F, Erdfelder E, Lang AG and Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–91. [DOI] [PubMed] [Google Scholar]
- 28.Yazbeck P, Tauseef M, Kruse K, Amin MR, Sheikh R, Feske S, Komarova Y and Mehta D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca(2+) Entry. Sci Rep. 2017;7:42758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chavez A, Schmidt TT, Yazbeck P, Rajput C, Desai B, Sukriti S, Giantsos-Adams K, Knezevic N, Malik AB and Mehta D. S1PR1 Tyr143 phosphorylation downregulates endothelial cell surface S1PR1 expression and responsiveness. J Cell Sci. 2015;128:878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonhoure E, Lauret A, Barnes DJ, Martin C, Malavaud B, Kohama T, Melo JV and Cuvillier O. Sphingosine kinase-1 is a downstream regulator of imatinib-induced apoptosis in chronic myeloid leukemia cells. Leukemia. 2008;22:971–9. [DOI] [PubMed] [Google Scholar]
- 31.Tauseef M, Knezevic N, Chava KR, Smith M, Sukriti S, Gianaris N, Obukhov AG, Vogel SM, Schraufnagel DE, Dietrich A, Birnbaumer L, Malik AB and Mehta D. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J Exp Med. 2012;209:1953–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joshi JC, Joshi B, Rochford I, Rayees S, Akhter MZ, Baweja S, Chava KR, Tauseef M, Abdelkarim H, Natarajan V, Gaponenko V and Mehta D. SPHK2-Generated S1P in CD11b(+) Macrophages Blocks STING to Suppress the Inflammatory Function of Alveolar Macrophages. Cell Rep. 2020;30:4096–4109 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao Y, Smyth GK and Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- 35.McCarthy DJ, Chen Y and Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiner A, Yekutieli D and Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–75. [DOI] [PubMed] [Google Scholar]
- 37.Dreos R, Ambrosini G, Perier RC and Bucher P. The Eukaryotic Promoter Database: expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res. 2015;43:D92–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chava KR, Tauseef M, Sharma T and Mehta D. Cyclic AMP response element-binding protein prevents endothelial permeability increase through transcriptional controlling p190RhoGAP expression. Blood. 2012;119:308–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rayees S, Joshi JC, Tauseef M, Anwar M, Baweja S, Rochford I, Joshi B, Hollenberg MD, Reddy SP and Mehta D. PAR2-Mediated cAMP Generation Suppresses TRPV4-Dependent Ca(2+) Signaling in Alveolar Macrophages to Resolve TLR4-Induced Inflammation. Cell Rep. 2019;27:793–805 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabula Muris C, Overall c, Logistical c, Organ c, processing, Library p, sequencing, Computational data a, Cell type a, Writing g, Supplemental text writing g and Principal i. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons S, Sasaki N, Umemoto E, Uchida Y, Fukuhara S, Kitazawa Y, Okudaira M, Inoue A, Tohya K, Aoi K, Aoki J, Mochizuki N, Matsuno K, Takeda K, Miyasaka M and Ishii M. High-endothelial cell-derived S1P regulates dendritic cell localization and vascular integrity in the lymph node. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inagaki HK, Ben-Tabou de-Leon S, Wong AM, Jagadish S, Ishimoto H, Barnea G, Kitamoto T, Axel R and Anderson DJ. Visualizing neuromodulation in vivo: TANGO-mapping of dopamine signaling reveals appetite control of sugar sensing. Cell. 2012;148:583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG and Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosen H, Stevens RC, Hanson M, Roberts E and Oldstone MB. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem. 2013;82:637–62. [DOI] [PubMed] [Google Scholar]
- 45.Marro BS, Ware BC, Zak J, de la Torre JC, Rosen H and Oldstone MB. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-alpha signaling. Proc Natl Acad Sci U S A. 2017;114:3708–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burg N, Swendeman S, Worgall S, Hla T and Salmon JE. Sphingosine 1-Phosphate Receptor 1 Signaling Maintains Endothelial Cell Barrier Function and Protects Against Immune Complex-Induced Vascular Injury. Arthritis Rheumatol. 2018;70:1879–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao YD, Ohkawara H, Rehman J, Wary KK, Vogel SM, Minshall RD, Zhao YY and Malik AB. Bone marrow progenitor cells induce endothelial adherens junction integrity by sphingosine-1-phosphate-mediated Rac1 and Cdc42 signaling. Circ Res. 2009;105:696–704, 8 p following 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finn J, Sottoriva K, Pajcini KV, Kitajewski JK, Chen C, Zhang W, Malik AB and Liu Y. Dlk1-Mediated Temporal Regulation of Notch Signaling Is Required for Differentiation of Alveolar Type II to Type I Cells during Repair. Cell Rep. 2019;26:2942–2954 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawahara A, Nishi T, Hisano Y, Fukui H, Yamaguchi A and Mochizuki N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science. 2009;323:524–7. [DOI] [PubMed] [Google Scholar]
- 50.Liu L, Tsai JC and Aird WC. Egr-1 gene is induced by the systemic administration of the vascular endothelial growth factor and the epidermal growth factor. Blood. 2000;96:1772–81. [PubMed] [Google Scholar]
- 51.Hasan RN and Schafer AI. Hemin upregulates Egr-1 expression in vascular smooth muscle cells via reactive oxygen species ERK-1/2-Elk-1 and NF-kappaB. Circ Res. 2008;102:42–50. [DOI] [PubMed] [Google Scholar]
- 52.Sysol JR, Natarajan V and Machado RF. PDGF induces SphK1 expression via Egr-1 to promote pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2016;310:C983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lavoie H, Gagnon J and Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol. 2020;21:607–632. [DOI] [PubMed] [Google Scholar]
- 54.Kong Y, Wang H, Lin T and Wang S. Sphingosine-1-phosphate/S1P receptors signaling modulates cell migration in human bone marrow-derived mesenchymal stem cells. Mediators Inflamm. 2014;2014:565369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kano A, Wolfgang MJ, Gao Q, Jacoby J, Chai GX, Hansen W, Iwamoto Y, Pober JS, Flavell RA and Fu XY. Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J Exp Med. 2003;198:1517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suryadevara V, Fu P, Ebenezer DL, Berdyshev E, Bronova IA, Huang LS, Harijith A and Natarajan V. Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang LS, Berdyshev EV, Tran JT, Xie L, Chen J, Ebenezer DL, Mathew B, Gorshkova I, Zhang W, Reddy SP, Harijith A, Wang G, Feghali-Bostwick C, Noth I, Ma SF, Zhou T, Ma W, Garcia JG and Natarajan V. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: role of S1P signalling and autophagy. Thorax. 2015;70:1138–48. [DOI] [PubMed] [Google Scholar]
- 58.Fu P, Ebenezer DL, Berdyshev EV, Bronova IA, Shaaya M, Harijith A and Natarajan V. Role of Sphingosine Kinase 1 and S1P Transporter Spns2 in HGF-mediated Lamellipodia Formation in Lung Endothelium. J Biol Chem. 2016;291:27187–27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spiegel S, Maczis MA, Maceyka M and Milstien S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J Lipid Res. 2019;60:484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. 2012;2:a006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Q, Huang X, Zhang H, Tian X, He L, Yang R, Yan Y, Wang QD, Gillich A and Zhou B. c-kit(+) cells adopt vascular endothelial but not epithelial cell fates during lung maintenance and repair. Nat Med. 2015;21:866–8. [DOI] [PubMed] [Google Scholar]
- 62.Ubil E, Duan J, Pillai IC, Rosa-Garrido M, Wu Y, Bargiacchi F, Lu Y, Stanbouly S, Huang J, Rojas M, Vondriska TM, Stefani E and Deb A. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514:585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao YD, Huang X, Yi F, Dai Z, Qian Z, Tiruppathi C, Tran K and Zhao YY. Endothelial FoxM1 mediates bone marrow progenitor cell-induced vascular repair and resolution of inflammation following inflammatory lung injury. Stem Cells. 2014;32:1855–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu Y, Sun P, Wang JY, Li ZZ, Gao RL, Wang XZ, Phillips WD and Liang SX. Differentiation of CD45/CD31+ lung side population cells into endothelial and smooth muscle cells in vitro. Int J Mol Med. 2019;43:1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xia S, Menden HL, Korfhagen TR, Kume T and Sampath V. Endothelial immune activation programmes cell-fate decisions and angiogenesis by inducing angiogenesis regulator DLL4 through TLR4-ERK-FOXC2 signalling. J Physiol. 2018;596:1397–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Details about materials and methods used to conduct this study as well as statistical analysis are provided in online Data Supplement and the Major Resources Table. The authors declare that all supporting data are available within the article and its online supplementary files.