

Abstract
Traumatic brain injury (TBI) alters brain function and is a crucial public health concern worldwide. TBI triggers the release of inflammatory mediators (cytokines) that aggravate cerebral damage, thereby affecting clinical prognosis. The renin angiotensin system (RAS) plays a critical role in TBI pathophysiology. RAS is widely expressed in many organs including the brain. Modulation of the RAS in the brain via angiotensin type 1 (AT1) and type 2 (AT2) receptor signaling affects many pathophysiological processes, including TBI. AT1R is highly expressed in neurons and astrocytes. The upregulation of AT1R mediates the effects of angiotensin II (ANG II) including release of proinflammatory cytokines, cell death, oxidative stress, and vasoconstriction. The AT2R, mainly expressed in the fetal brain during development, is also related to cognitive function. Activation of this receptor pathway decreases neuroinflammation and oxidative stress and improves overall cell survival. Numerous studies have illustrated the therapeutic potential of inhibiting AT1R and activating AT2R for treatment of TBI with variable outcomes. In this review, we summarize studies that describe the role of brain RAS signaling, through AT1R and AT2R in TBI, and its modulation with pharmacological approaches.
Keywords: Traumatic brain injury, Angiotensin II receptor type 1, Angiotensin II receptor type 2, Neuroprotection, Renin angiotensin system, ANG II receptor antagonists
1. Introduction
Traumatic brain injury (TBI) is a serious cause of injury-related death and disability in the United States, with approximately 2.8 million cases reported each year (Nelson et al. 2019; Taylor et al. 2017). TBI increases the long-term risk of developing neurodegenerative disorders such as Alzheimer's Disease (AD), Parkinson's disease (PD), chronic traumatic encephalopathy (CTE), and epilepsy (Acosta et al. 2015; Annegers and Coan 2000; Mayeux et al. 1995). Although technological and therapeutic advancements in preceding decades have improved, the quality and length of life for those suffering from TBIs, there are no existing FDA-approved pharmacological therapies to improve functional outcomes. TBI is associated with posttraumatic and progressive neuroinflammation, characterized by a complex cascade of inflammatory responses (Finnie 2013). TBI can range from mild to severe and can be classified into two stages, primary and secondary (Chiu et al. 2016; Nguyen et al. 2016). Primary damage is a direct result of the initial injury or insult (Prasetyo 2020). This disrupts cerebral blood flow (CBF) and metabolism, resulting in a shift to anaerobic glycolysis, lactic acid accumulation and increased blood brain barrier (BBB) permeability, and subsequently brain edema. Eventually, inefficient anaerobic metabolism leads to a failure of energy-dependent membrane ion pumps and reduction of ATP stores (Werner and Engelhard 2007). Secondary damage occurs after the primary insult. The release of cellular inflammatory mediators and pro-inflammatory factors, including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin1 ß (IL-1ß), leads to neuroinflammation, followed by cell death, and ischemia (Prasetyo 2020; Werhane et al. 2017). This phase includes depolarization of neuronal membranes, resulting in an increased release of stimulatory neurotransmitters and an influx of sodium and calcium ions. Accumulation of calcium ions activates proteases and lipid peroxidase enzymes, which increase the intracellular concentrations of amino acids, fatty acids and free radicals. Finally, upregulation of caspases, Interleukin 1ß converting enzyme (ICE-like proteins), translocases, and endonucleases leads to alteration of biological membranes and inhibition of deoxyribonucleic acid (DNA) repair (Werner and Engelhard 2007). The renin angiotensin system (RAS) plays a fundamental role in controlling blood pressure, electrolyte balance, and metabolic hemostasis (Sigmund and Grobe 2020). Existence of RAS in the brain indicates that several brain functions are regulated through RAS receptors (Sigmund and Grobe 2020). The aim of this review is to describe the role of RAS alterations in TBI.
2. The Renin angiotensin system (RAS) in the brain
The brain RAS is involved in the regulation of a vast majority of cardiovascular functions, including blood pressure, cardiac output, vascular tone and peripheral circulation (Volpe et al. 2002). However, the presence of an autonomous RAS in the adult brain serves to underline its unique role in the central nervous system (Ganten et al. 1971; Jackson et al. 2018). Modulation of the brain RAS has been implicated in the pathogenesis of several brain disorders including TBI, multiple sclerosis (MS), Alzheimer’s disease (AD) and other dementias, Parkinson’s disease (PD) and stroke (Abiodun and Ola 2020). There are two main pathways for RAS signaling in the brain: the peripheral or forebrain pathway, that incorporates the circumventricular organs (CVOs), and the central pathway for local production of angiotensin, that links the medulla oblongata and the hypothalamus (Jackson et al. 2018). Brain RAS dysregulation was shown to be associated with neurodegeneration, neuroinflammation, and oxidative stress (Abiodun and Ola 2020).
2.1. Angiotensinogen and Renin
Angiotensinogen (AGT) is made in the liver and secreted into the circulatory system, is the precursor of other bioactive angiotensin peptides. This precursor protein is converted to angiotensin I (ANG I) by the renal enzyme renin, a rate limiting RAS enzyme synthesized in the kidneys and secreted into the blood (Fountain and Lappin 2020). Renin catalyzes the cleavage of AGT into ANG I, which is rapidly converted to ANG II by angiotensin converting enzyme (ACE) (Paz Ocaranza et al. 2020). In the brain, renin and AGT are synthesized in astrocytes (Intebi et al. 1990; Jackson et al. 2018; McKinley et al. 2003). Renin is secreted as the inactive molecule prorenin, which can be activated by proteolysis or non-catalytically by binding to a prorenin receptor (PRR). Hyperactivation of renin/prorenin signaling impairs cognitive function due to activation of the Ang II/AT1R axis (Jackson et al. 2018).
2.2. Angiotensin-converting enzymes (ACE, ACE2)
In the canonical RAS pathway, ACE converts ANG I into ANG II, while in the non-canonical or counter-regulatory RAS axis, ACE2 cleaves ANG I and ANG II to yield the peptides ANG-(1–9) and ANG-(1–7) respectively (Paz Ocaranza et al. 2020). ACE is expressed in the central nervous system (CNS) and can be found at high concentrations in the subfornical organ, organum vasculosum laminae terminalis (OVLT), area postrema, and median eminence (McKinley et al. 2003). Upregulation of ACE leads to aggravated inflammation, cell death and impairs cognitive function by increased activation of AT1R signaling (Jackson et al. 2018).
2.3. Angiotensin (Ang) II
Ang II is a vasoactive peptide and stimulates production of reactive oxygen species (ROS), neuroinflammation, neurodegeneration, and neurotoxicity via AT1R signaling. It has been implicated in several diseases including coronary artery disease (CAD), stroke, and hypertension (Forrester et al. 2018; Hirooka et al. 2006; Ito et al. 2001; Landmesser et al. 2002; Rueckschloss et al. 2002). In the brain, Ang II regulates cerebral blood flow (CBF), hormone release and neuroinflammation. It is also highly involved in processes relating to complex cognitive functions (Benicky et al. 2011; Jackson et al. 2018; Saavedra et al. 2011; Villapol et al. 2012).
2.4. Angiotensin II receptor type 1(AT1R)
The angiotensin II receptor type 1 (AT1R) is a G-protein coupled receptor (GPCR) that is localized to astrocytes, neurons, vascular endothelial cells, and oligodendrocytes (Jackson et al. 2018; Saavedra 2005). Although brain activation of the AT1R is critical for various cerebrovascular functions, its hyperactivation results in endothelial dysfunction and excessive inflammation, with progression to disorders like hypertension, brain ischemia and cognitive impairment (Phillips and De Oliveira 2008; Saavedra 1992; Saavedra 2012; Saavedra et al. 2011). From a mechanistic standpoint, ANG II binding and activation of AT1R signaling mediates endothelial dysfunction oxidative stress and inflammation by increasing transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). The activation of NF-κB regulates production of pro-inflammatory cytokines, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Rodriguez-Perez et al. 2015; Villapol et al. 2015). A class of medications known as AT1 receptor blockers (ARBs), approved for management of various cardiovascular conditions, were also shown to have positive effects in brain pathologies. These agents counteract many of the detrimental effects brought about by excessive AT1R stimulation and have shown potential benefits in conditions like stroke, vascular cognitive impairment and Alzheimer’s disease (Torika et al. 2018; Ozacmak et al. 2007; Villapol et al. 2012).
2.5. Angiotensin II receptor type 2 (AT2R)
The angiotensin II receptor type 2, AT2R, is a GPCR found in neurons, astrocytes, oligodendrocytes and microglia (Dupont and Légat 2020; Jackson et al. 2018). It is expressed in parts of the brain related to cognition and behavior, including the cerebral cortex, basal ganglia, and the hippocampus, (Jackson et al. 2018; Umschweif et al. 2014a). In fetal brains, it is involved in neuronal differentiation and synaptogenesis (Timaru-Kast et al. 2012). AT2R stimulation improves cognitive function and cell survival.It reduces oxidative stress and inflammation by multiple pathways including release of nitric oxide (NO) and bradykinin as well as activation of peroxisome proliferator-activated receptorγ (PPARγ), and the serine-threonine phosphatase PP2A (Jackson et al. 2018; Mogi and Horiuchi 2013; Umschweif et al. 2014a). AT2R activation also improves neuronal regeneration and reduces vascular damage (Pelisch et al. 2010).
2.6. Angiotensin II receptor type 4 (AT4R)
Although little information exists regarding the angiotensin II receptor type 4 (AT4R), it is localized to astrocytes (Holownia and Braszko 2007; Jackson et al. 2018). AT4R upregulation contributes to cognitive improvement, cell signal transmission, and anti-inflammation (Jackson et al. 2018; Wright and Harding 2004). Dysregulation of AT4R signaling impairs cognitive function through synaptic dysfunction and neurotransmitter release (Jackson et al. 2018).
2.7. Non-canonical or counter-regulatory RAS axis: ACE-2 / Angiotensin (1–7) / Mas receptor
The non-canonical axis of brain RAS is composed of ACE-2 / Angiotensin (1–7) / Mas receptor, together with angiotensins (1–5), (1–12), (2–8; also known as ANG III), and (3–8; also known as ANG IV) and almandine shares all but its N-terminal amino acid with angiotensin (1,7) (Lu et al. 2013; Paz Ocaranza et al. 2020). ACE-2 cleaves Ang II to yield the angiotensin-related peptide Ang (1–7) which decreases inflammation and oxidative stress and enhances vasodilation (Jiang et al. 2012; Zhang et al. 2008; Zheng et al. 2014). Angiotensin (1–7) is present in many parts of the brain, including the hypothalamus, amygdala, and medulla oblongata (Chappell et al. 1989). Ang (1–7) binds to the Mas receptor (MasR), which initiates a novel signaling cascade resulting in reduced inflammation (Janatpour et al. 2019; Liu et al. 2016). The non-canonical RAS axis ACE-2/Angiotensin (1–7) / MasR is counter regulatory to the canonical Ang II/AT1R axis. This axis therefore contributes to neuroprotection (Feng et al. 2010; Jiang et al. 2013; Mecca et al. 2011).
3. The brain RAS and TBI
Modulation of the RAS is associated with neuroprotection following TBI (Fouda et al. 2016), as shown schematically in Figure 1. The pathophysiology of TBI can be overcome by blocking of AT1R, which serves to prevent vasoconstriction, apoptosis, and neuroinflammation, and activation of AT2R signaling, which leads to vasodilatation, anti-inflammation, and neuro-regeneration (Li et al. 2005; Timaru-Kast et al. 2012; Vadhan and Speth 2020). Together, these reduce secondary ischemia, one of the detrimental post-TBI outcomes (Leker and Shohami 2002). AT1R signaling has implications in neuroinflammation and microglial activation via several different pathways, including the extracellular signal-regulated kinase (ERK1/2) and mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase (JNK) pathways (Timaru-Kast et al. 2019). Upregulation of AT2R expression improves microcirculation, cognitive performance, motor function, and neuroprotection following TBI (Kiprianova et al. 1999; Umschweif et al. 2014a; Yaka et al. 2007). Inhibition of AT1R using a low dose (0.1 mg/kg) of the ARB candesartan after insult in the controlled cortical impact (CCI) mouse model improved neurological function and decreased neutrophil granulocyte infiltration and microglial activation without affecting blood pressure (Timaru-Kast et al. 2019). Although blocking AT1R using candesartan increased polarization of the anti-inflammatory neuroregenerative M2a microglial phenotype, it did not affect the expression levels of the inducible (iNOS) and endothelial (eNOS) nitric oxide synthases following TBI (Timaru-Kast et al. 2019). M2 microglial expression of regenerative markers, chitinase-3-like protein 3 (Ym1) and arginase 1 (Arg1),were shown to vary with age (Timaru-Kast et al. 2019). Young mice release these markers sooner than aged mice after injury, and correlates with the delayed response of aged to injury (Timaru-Kast et al. 2019). A similar age-related difference in expression levels is observed for the mRNAs encoding AT1R and AT2R. Due to a gene duplication, mice express two subtypes of the AT1R (AT1a and AT1b) that share 95% similarity at the amino acid level, while only a single gene exists in humans (Dasgupta and Zhang 2011; Timaru-Kast et al. 2019). When the expression of mRNAs encoding AT1a, AT1b, and AT2R was examined in young (2 months old) and aged (21 months old) mice before TBI, no age-related changes in AT1a mRNA levels were observed but AT1b and AT2R mRNA levels were higher in the naïve aged mice. One to three days after TBI, expression of AT1a mRNA was unchanged in young mice, but decreased to nearly half level in injured aged mice. Young mice also exhibited an increase in AT1b, and AT2R mRNA levels at 24 h and returned to the naïve level by 72 h after TBI. In elderly mice, the levels of AT1b, and AT2R mRNAs did not change within 72 h of TBI. Moreover, candesartan did not significantly reduce the expression of proinflammatory cytokines such as IL-6, IL-1β, and TNF-α after injury in either age group. In aged and young mice mRNA expression of AT1a was not affected 5 days after injury, although brain damage was reduced. Furthermore, candesartan reduced posttraumatic mortality in aged mice (Timaru-Kast et al. 2019). Candesartan treatment decreased intracranial pressure (ICP) and improved BBB permeability in male Wistar rats without impacting brain water content (Khaksari et al. 2018). Another study showed that while CCI resulted in microgliosis, neuronal and capillary loss in young mice, administration of the ANG I derivative peptide Ang (1–7) after TBI contributed to physiological and functional recovery by reducing activation of microglia and astrocytes, decreasing lesion volume, and increasing microvascular density (Janatpour et al. 2019). Although blood pressure was not monitored and only young mice were used, it is unlikely that Ang (1–7) would not have impacted blood pressure in normotensive animals, particularly in light of the observation that Ang (1–7) decreases the microglial inflammatory response (Janatpour et al. 2019). The limitation of this study was that they did not use aged mice, however, others have found that Ang (1–7) reduced microglial inflammatory response in senescence-accelerated mouse-prone 8 (SAMP8) 8-month-old mice, suggesting that some responses to Ang (1–7) treatment might be similar in young and aged mice (Jiang et al. 2018). Further research, in mice of different age groups, is needed to detail the effect of Ang (1–7) in TBI. With stretch injury, ANG II activates proinflammatory cytokines IL-1β, TNF-α, and caspase-3, resulting in BBB impairment and increased brain damage and initiates neuroinflammation and cell death by stimulating transforming growth factor beta 1 (TGF-β1), matrix metalloproteinases (MMPs), and oxidative stress (Abdul-Muneer et al. 2018). These were reversed by the AT1R antagonist losartan. Administration of losartan (100 μg/mL) to cultured rat cortical neurons prior to stretch injury decreased the expression of oxidative and nitric oxide-mediated stress markers such as 4-hydroxynonenal (4HNE) and 3-nitrotyrosine (3NT) (Abdul-Muneer et al. 2018). Treatment of wild type (WT) and AT1R knock out (KO) mice with the AT1R blockers candesartan and telmisartan improved cognitive and motor function (Villapol et al. 2015). AT1R KO mice are less prone to CCI injury than WT, suggesting that AT1R activation has a deleterious effect post-TBI (Villapol et al. 2015). Both sartans reduced lesion volume and improved both short-term and long-term cognitive performance, but candesartan showed more promise with long-term treatment (Villapol et al. 2015). Although a low-dose of candesartan reduced brain injury, medium and high doses (0.5 –1 mg/kg) caused impairments in cerebral perfusion due to reductions in systolic blood pressure (Timaru-Kast et al. 2012 ).Candesartan treatment administered post- injury in mice reduced the expression of nitric oxide (NO) as well as inflammatory cytokines IL-1β, IL-6, and TNF-α (Timaru-Kast et al. 2012). Administration of candesartan post injury also reduced lesion volume and activated PPARγ, thereby reducing neuronal cell death and microglial activation, while the PPARγ antagonist T0070907 reduced this effect. Candesartan decreased expression of TGFβ1 and increased TGFβ3 in cortical and hippocampal astrocytes by unknown mechanisms, leading to reduced neuroinflammation (Villapol et al. 2012). PPARγ activation contributed to the neuroprotective effects of telmisartan and candesartan (Villapol et al. 2015). In a heat-acclimated (HA) mouse model of TBI, HA increased the expression of AT2R, resulting in neuroprotection and neurogenesis (Umschweif et al. 2014a; Umschweif et al. 2014b). This was associated with increased levels of brain-derived neurotrophic factor (BDNF), tropomyosin receptor kinase A (TrkA), tropomyosin receptor kinase B (TrkB), hypoxia-inducible factor 1a (HIF-1a), and nerve growth factor (NGF). Post-injury treatment with the AT2R antagonist PD123319 inhibited these beneficial AT2R mediated pathways and offset neuroprotection. In another study, the AT2R agonist CGP42112A improved cognitive function after injury and increased expression of BDNF, NGF, extracellular-regulated kinase 1/2 (ERK1/2), and protein kinase B (AKT), whereas PD123319 decreased expression of these neurotrophins (Umschweif et al. 2014a). CGP42112A also improved the migration pathway of neuronal progenitor cells (NPCs) in the subventricular zone (SVZ) after TBI (Umschweif et al. 2014a). Although effective in the pre-clinical setting, CGP42112A has several limitations that may preclude its use in the clinic. As a peptide that is rapidly degraded in vivo, it must be administered by continuous intracerebroventricular infusion (Umschweif et al. 2014a). Moreover, its high binding affinity but low potency makes it difficult to work with, as much higher doses are needed for receptor stimulation than for receptor occupancy. Such high doses can result in loss of AT2R selectivity and can counteract its efficacy and result in adverse effects (Iwanami et al. 2015; Unger and Dahlöf 2010).
Figure 1. RAS modulation through AT1R, AT2R, and Angiotensin (1–7)/MasR affect the brain after TBI via different pathways.
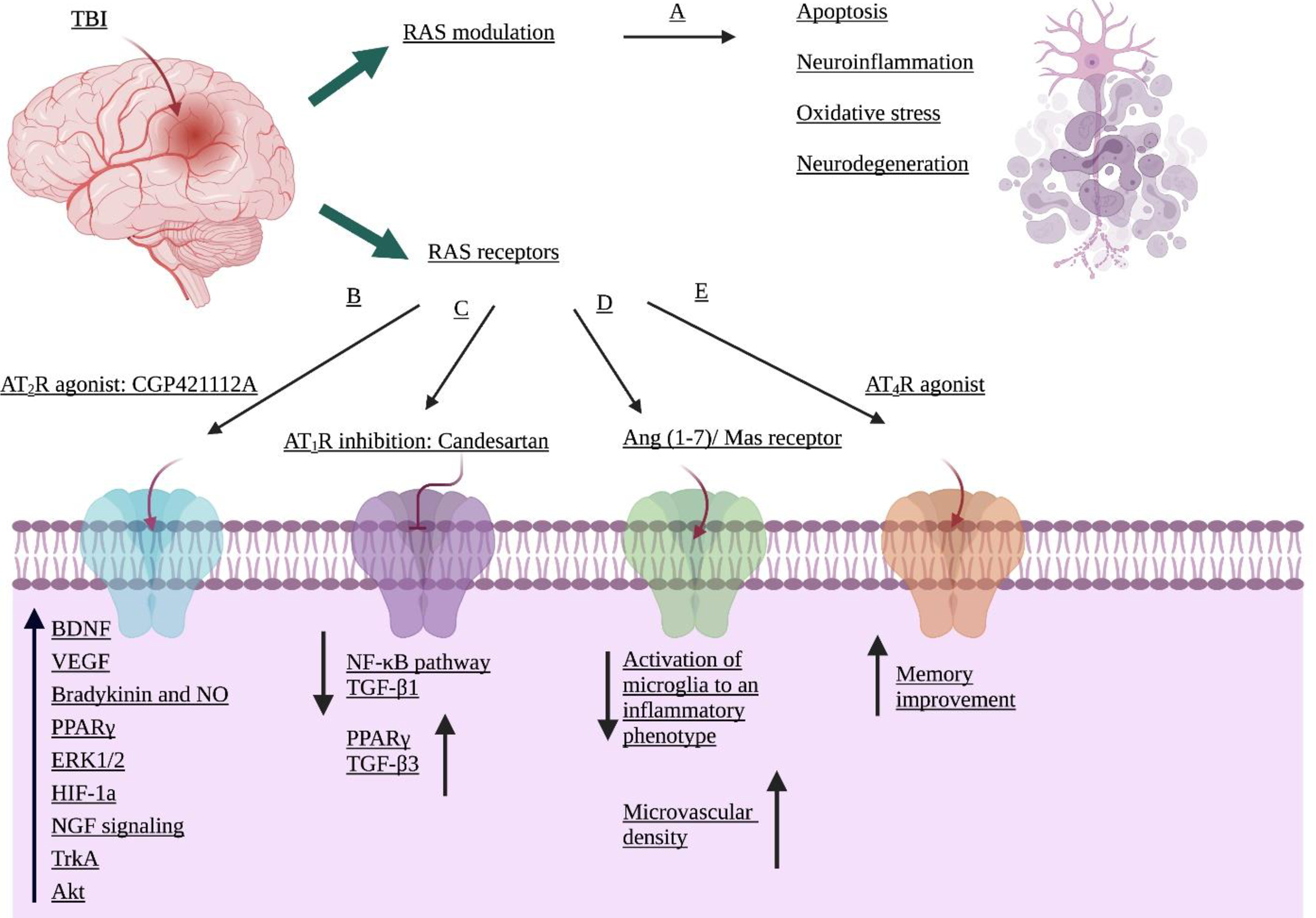
A, RAS alteration following insult. B and C, AT2R activation and AT1R suppression contributes to neuro-regeneration and anti-inflammation by activating various mechanisms. D, Stimulation of Angiotensin (1–7) / MasR reduces microglial activation to an inflammatory phenotype and increases neuroprotection. E, Upregulation of AT4R improves memory function but has not been implicated in TBI.
4. Conclusions
TBI is a critical public health concern and no effective medications are available. Preclinical studies show that RAS modulationis likely to play a significant role in the treatment of TBI. Local RAS in the brain is mediated by signaling through angiotensin II receptors type 1 and 2 (AT1R and AT2Rs). Suppressing AT1R decreases secondary brain damage and neurological impairment following TBI, due to the reduction in neuroinflammation and microglial activation. AT2R upregulation contributes to improved cognitive and motor function in rodent models of TBI. In the brain, AT2R stimulation activates many protective pathways such as BDNF, PPARγ, Akt, and ERK1/2. Inhibition of AT1R with ARBs such as candesartan and telmisartan is limited by their effects on systolic blood pressure. Low dose candesartan therapy shows promise in rodent models of TBI models. In conclusion, more studies using varying times and dosages will be required to assess the impact of AT1R and AT2R signaling in the brains of rodent TBI models.
Table 1.
Summary of experiments on RAS alteration in TBI
| Study (Ref.) | Animal Model | Treatment | Dose and Duration | Result |
|---|---|---|---|---|
| Timaru-Kast, R (Timaru-Kast et al. 2019) | CCI - C57B16N Young, 2 months. Old, 21 months. male mice | Candesartan 10μg/ml | 0.1 mg/kg/ SC/ 30 min after injury | Reduction in brain damage and neurological impairment through microglial response reduction and neutrophil granulocyte infiltration. |
| Janatpour, ZC (Janatpour et al. 2019) | CCI - C57BL/6NCr 8–10-week-old male mice | Ang (1–7) 10.0 mg/mL (micro-osmotic pump); 0.1mg/mL (injections) | 1.0 mg/kg/day 2.5 mg/kg/day |
Ang-(1–7) improved functional recovery and decreases glial activation. |
| Khaksari, M (Khaksari et al. 2018) | Wistar Male rats Diffuse TBI induced by Marmarou method | Candesartan | 0.3 mg/kg IP, 30 min post-TBI | Reduction in brain edema, BBB disruption, oxidant activity, and ICP level using AT1R inhibition. |
| Abdul-Muneer, PM (Abdul-Muneer et al. 2018) | E17 Sprague-Dawley rat embryos Stretch injury | Losartan | (100 μg/mL) 30 min prior to the stretch injury | Losartan downregulated oxidative markers, neuroinflammation and neurodegeneration |
| Villapol, S (Villapol et al. 2015) | CCI - C57BL/6NCr 9-week-old male mice | Candesartan | 0.1, 0.5 or 1 mg/kg IP (after injury) | Neuroprotective. Candesartan is better than telmisartan in chronic condition and low dose of candesartan does not affect blood pressure. |
| C57BL/6J WT control mice AT1R knockout mice | Telmisartan | 1 or 10 mg/kg once per day oral gavage | ||
| PPARγ antagonist (T0070907) | 2 mg/kg (after insult) | |||
| Umschweif, G (Umschweif et al. 2014b) | CHI - Sabra (Weight Drop Injury) 9–10-week-old male mice | AT2 antagonist PD123319 | 10 mg/kg/day for 3 days (after injury) | HA stimulated AT2R and increased neuroprotection and neurogenesis. AT2R inhibition reduced these effects. |
| Timaru-Kast, R (Timaru-Kast et al. 2012) | CCI - C57B16N male mice | Candesartan | low (0.1 mg/kg) med. (0.5 mg/kg) high (1 mg/kg,) SC (30 min after insult) |
AT1R repression declined secondary brain damage and inflammation |
| Umschweif, G (Umschweif et al. 2014a) | CHI - Sabra 9–10-week-old mice | PD 123319 | 10 mg/kg/day | AT2R activation improved neuroprotection and neurogenesis, through different pathways. |
| CGP42112A | 0.1, 1.0, or 10.0 ng/kg/min, 3 days post-CHI | |||
| Villapol, S (Villapol et al. 2012) | CCI - C57BL/6 9-week-old male mice | Candesartan | 1 mg/kg/day | Reduction in neuronal cell death and microglial activation |
| PPARγ antagonist T0070907 | 1.5 mg/kg | |||
Funding
This work was supported by startup funds: Department of Anatomy Neurobiology. UTHSC Memphis TN (TI) and National Institute of Health: R01-NS097800 (TI).
Abbreviations
- AD
Alzheimer's disease
- ACE
Angiotensin converting enzyme
- AGT
Angiotensinogen
- AKT
Protein kinase B
- ANG II
Angiotensin II
- ARB
Angiotensin receptor blockers
- ARG 1
Arginase 1
- BBB
Blood brain barrier
- BDNF
Brain-derived neurotrophic factor
- CBF
Cerebral blood flow
- CCI
Cortical Impact Injury
- CAD
Coronary artery disease
- CHI
Closed head injury
- CNS
Central nervous system
- CTE
Chronic traumatic encephalopathy
- CVOs
Circumventricular organs
- ERK1/2
Extracellular signal-regulated protein kinases 1 and 2
- HA
Heat acclimation
- HIF-la
Hypoxia-inducible factor - 1 alpha
- ICE
Interleukin 1β converting enzyme
- ICP
Intracranial pressure
- iNOS
inducible nitric oxide synthase
- IL-1β
Interleukin 1-β
- IL-6
Interleukin-6
- IP
Intraperitoneal
- JAK/STAT
Janus Kinase and Signal Transducer and Activator of Transcription
- JNKs
c-Jun N-terminal kinases
- KO
Knock out
- MAPK
Mitogen-activated protein kinase
- MMP
Matrix Metalloproteinases
- mRNA
Messenger ribonucleic acid
- MS
Multiple sclerosis
- NF-kB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NGF
Nerve growth factor
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NO
Nitric oxide
- eNOS
Endothelial nitric oxide synthase
- NPC
neural progenitor cell
- OVLT
Organum vasculosum laminae terminalis
- PD
Parkinson’s disease
- PPARγ
Peroxisome proliferator-activated receptor gamma
- PP2A
serine-threonine phosphatase
- PRR
Prorenin receptor
- RAS
Renin-angiotensin system
- ROS
Reactive oxygen species
- SAMP8
senescence-accelerated mouse-prone 8
- SC
Subcutaneous
- SVZ
Subventricular zone
- TGFβ1
Transforming growth factor beta 1
- TGFβ3
Transforming growth factor beta 3
- TNF-α
Tumor necrosis factor alpha
- TrkA
Tropomyosin receptor kinase A
- TrkB
Tropomyosin receptor kinase B
- VCI
Vascular cognitive impairment
- WT
Wild type
- Yml
Chitinase 3- like 3
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Compliance with Ethical Standards
Not applicable.
Data availability
Not applicable.
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
Not applicable.
Informed consent
Not applicable.
References
- Abdul-Muneer P, Bhowmick S, Briski N (2018) Angiotensin II Causes Neuronal Damage in Stretch-Injured Neurons: Protective Effects of Losartan, an Angiotensin T 1 Receptor Blocker. Mol Neurobiol 55:5901–5912 [DOI] [PubMed] [Google Scholar]
- Abiodun OA, Ola MS (2020) Role of brain renin angiotensin system in neurodegeneration: An update. Saudi J Biol Sci 27:905–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV (2015) Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J Cell Physiol 230:1024–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed HA et al. (2019) Angiotensin receptor (AT2R) agonist C21 prevents cognitive decline after permanent stroke in aged animals—A randomized double-blind pre-clinical study Behav Brain Res 359:560–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed HA et al. (2018) RAS modulation prevents progressive cognitive impairment after experimental stroke: a randomized, blinded preclinical trial J Neuroinflammation 15:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annegers JF, Coan SP (2000) The risks of epilepsy after traumatic brain injury Seizure 9:453–457 [DOI] [PubMed] [Google Scholar]
- Benicky J et al. (2011) Angiotensin II AT 1 receptor blockade ameliorates brain inflammation Neuropsychopharmacology 36:857–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell MC, Brosnihan KB, Diz DI, Ferrario CM (1989) Identification of angiotensin-(1–7) in rat brain: evidence for differential processing of angiotensin peptides J Biol Chem 264:16518–16523 [PubMed] [Google Scholar]
- Chiu C-C et al. (2016) Neuroinflammation in animal models of traumatic brain injury J Neurosci Methods 272:38–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta C, Zhang L (2011) Angiotensin II receptors and drug discovery in cardiovascular disease Drug Discov Today 16:22–34 doi: 10.1016/j.drudis.2010.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont AG, Légat L (2020) GABA is a mediator of brain AT 1 and AT 2 receptor-mediated blood pressure responses Hypertens Res:1–11 [DOI] [PubMed] [Google Scholar]
- Feng Y et al. (2010) Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension Circ Res 106:373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnie J (2013) Neuroinflammation: beneficial and detrimental effects after traumatic brain injury Inflammopharmacology 21:309–320 [DOI] [PubMed] [Google Scholar]
- Forrester SJ et al. (2018) Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology Physiol Rev 98:1627–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouda AY, Artham S, El-Remessy AB, Fagan SC (2016) Renin–angiotensin system as a potential therapeutic target in stroke and retinopathy: experimental and clinical evidence Clin Sci 130:221–238 [DOI] [PubMed] [Google Scholar]
- Fountain JH, Lappin SL (2020) Physiology, Renin Angiotensin System. StatPearls Publishing. https://www.ncbi.nlm.nih.gov/books/NBK470410/. [PubMed] [Google Scholar]
- Ganten D et al. (1971) Angiotensin-forming enzyme in brain tissue Science 173:64–65 [DOI] [PubMed] [Google Scholar]
- Hirooka Y, Kimura Y, Nozoe M, Sagara Y, Ito K, Sunagawa K (2006) Amlodipine-induced reduction of oxidative stress in the brain is associated with sympatho-inhibitory effects in stroke-prone spontaneously hypertensive rats Hypertens Res 29:49–56 [DOI] [PubMed] [Google Scholar]
- Holownia A, Braszko JJ (2007) The effect of angiotensin II and IV on ERK1/2 and CREB signalling in cultured rat astroglial cells Naunyn-Schmiedeberg's Arch Pharmacol 376:157–163 [DOI] [PubMed] [Google Scholar]
- Intebi AD, Flaxman MS, Ganong WF, Deschepper CF (1990) Angiotensinogen production by rat astroglial cells in vitro and in vivo Neuroscience 34:545–554 doi: 10.1016/0306-4522(90)90163-X [DOI] [PubMed] [Google Scholar]
- Ito H, Takemori K, Suzuki T (2001) Role of angiotensin II type 1 receptor in the leucocytes and endothelial cells of brain microvessels in the pathogenesis of hypertensive cerebral injury J Hypertens 19:591–597 [DOI] [PubMed] [Google Scholar]
- Iwanami J et al. (2015) Direct angiotensin II type 2 receptor stimulation by compound 21 prevents vascular dementia J Am Soc Hypertens 9:250–256 [DOI] [PubMed] [Google Scholar]
- Jackson L, Eldahshan W, Fagan SC, Ergul A (2018) Within the brain: the renin angiotensin system Int J Mol Sci 19:876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janatpour ZC, Korotcov A, Bosomtwi A, Dardzinski BJ, Symes AJ (2019) Subcutaneous administration of angiotensin-(1–7) improves recovery after traumatic brain injury in mice J Neurotrauma 36:3115–3131 [DOI] [PubMed] [Google Scholar]
- Jiang T, Gao L, Guo J, Lu J, Wang Y, Zhang Y (2012) Suppressing inflammation by inhibiting the NF-κB pathway contributes to the neuroprotective effect of angiotensin-(1–7) in rats with permanent cerebral ischaemia Br J Pharmacol 167:1520–1532 doi: 10.1111/j.1476-5381.2012.02105.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Gao L, Shi J, Lu J, Wang Y, Zhang Y (2013) Angiotensin-(1–7) modulates renin–angiotensin system associated with reducing oxidative stress and attenuating neuronal apoptosis in the brain of hypertensive rats Pharmacol Res 67:84–93 [DOI] [PubMed] [Google Scholar]
- Jiang T et al. (2018) AVE0991, a nonpeptide analogue of Ang-(1–7), attenuates aging-related neuroinflammation Aging (Albany NY) 10:645–657 doi: 10.18632/aging.101419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaksari M et al. (2018) Does inhibition of angiotensin function cause neuroprotection in diffuse traumatic brain injury? Iran J Basic Med Sci 21:615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiprianova I, Sandkühler J, Schwab S, Hoyer S, Spranger M (1999) Brain-derived neurotrophic factor improves long-term potentiation and cognitive functions after transient forebrain ischemia in the rat Exp Neurol 159:511–519 [DOI] [PubMed] [Google Scholar]
- Landmesser U et al. (2002) Role of p47 phox in vascular oxidative stress and hypertension caused by angiotensin II Hypertension 40:511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leker RR, Shohami E (2002) Cerebral ischemia and trauma—different etiologies yet similar mechanisms: neuroprotective opportunities Brain Res Rev 39:55–73 [DOI] [PubMed] [Google Scholar]
- Li J et al. (2005) Angiotensin AT2 receptor protects against cerebral ischemia-induced neuronal injury FASEB J 19:1–25 [DOI] [PubMed] [Google Scholar]
- Liu M, Shi P, Sumners C (2016) Direct anti-inflammatory effects of angiotensin-(1–7) on microglia J Neurochem 136:163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J et al. (2013) The expression of angiotensin-converting enzyme 2-angiotensin-(1–7)-Mas receptor axis are upregulated after acute cerebral ischemic stroke in rats Neuropeptides 47:289–295 doi: 10.1016/j.npep.2013.09.002 [DOI] [PubMed] [Google Scholar]
- Mayeux R et al. (1995) Synergistic effects of traumatic head injury and apolipoprotein-epsilon4 in patients with Alzheimer's disease Neurology 45:555–557 [DOI] [PubMed] [Google Scholar]
- McKinley MJ et al. (2003) The brain renin–angiotensin system: location and physiological roles Int J Biochem Cell Biol 35:901–918 [DOI] [PubMed] [Google Scholar]
- Mecca AP, Regenhardt RW, O’Connor TE, Joseph JP, Raizada MK, Katovich MJ, Sumners C (2011) Cerebroprotection by angiotensin-(1–7) in endothelin-1-induced ischaemic stroke Exp Physiol 96:1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Horiuchi M (2013) Effect of angiotensin II type 2 receptor on stroke, cognitive impairment and neurodegenerative diseases Geriatr Gerontol Int 13:13–18 [DOI] [PubMed] [Google Scholar]
- Nelson LD et al. (2019) Recovery After Mild Traumatic Brain Injury in Patients Presenting to US Level I Trauma Centers: A Transforming Research and Clinical Knowledge in Traumatic Brain Injury (TRACK-TBI) Study JAMA Neurol 76:1049–1059 doi: 10.1001/jamaneurol.2019.1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen R et al. (2016) The international incidence of traumatic brain injury: a systematic review and meta-analysis Can J Neurol Sci 43:774–785 [DOI] [PubMed] [Google Scholar]
- Ozacmak VH, Sayan H, Cetin A, Akyildiz-Igdem A (2007) AT1 receptor blocker candesartan-induced attenuation of brain injury of rats subjected to chronic cerebral hypoperfusion Neurochem Res 32:1314–1321 [DOI] [PubMed] [Google Scholar]
- Paz Ocaranza M, Riquelme JA, García L, Jalil JE, Chiong M, Santos RAS, Lavandero S (2020) Counter-regulatory renin–angiotensin system in cardiovascular disease Nature Reviews Cardiology 17:116–129 doi: 10.1038/s41569-019-0244-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch N et al. (2010) Systemic candesartan reduces brain angiotensin II via downregulation of brain renin–angiotensin system Hypertens Res 33:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MI, De Oliveira EM (2008) Brain renin angiotensin in disease J Mol Med 86:715–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasetyo E (2020) The primary, secondary, and tertiary brain injury Crit Care Shock 23:4–13 [Google Scholar]
- Rodriguez-Perez AI, Borrajo A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL (2015) Interaction between NADPH-oxidase and R ho-kinase in angiotensin II-induced microglial activation Glia 63:466–482 [DOI] [PubMed] [Google Scholar]
- Rueckschloss U, Quinn MT, Holtz J, Morawietz H (2002) Dose-dependent regulation of NAD (P) H oxidase expression by angiotensin II in human endothelial cells: protective effect of angiotensin II type 1 receptor blockade in patients with coronary artery disease Arterioscler Thromb Vasc Biol 22:1845–1851 [DOI] [PubMed] [Google Scholar]
- Saavedra JM (1992) Brain and pituitary angiotensin Endocr Rev 13:329–380 [DOI] [PubMed] [Google Scholar]
- Saavedra JM (2005) Brain angiotensin II: new developments, unanswered questions and therapeutic opportunities Cell Mol Neurobiol 25:485–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra JM (2012) Angiotensin II AT1 receptor blockers as treatments for inflammatory brain disorders Clin Sci 123:567–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra JM, Sánchez-Lemus E, Benicky J (2011) Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: therapeutic implications Psychoneuroendocrinology 36:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigmund CD, Grobe JL (2020) A colorful view of the brain renin–angiotensin system Hypertens Res 43:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckelings UM, Paulis L, Namsolleck P, Unger T (2012) AT2 receptor agonists: hypertension and beyond Curr Opin Nephrol Hypertens 21:142–146 [DOI] [PubMed] [Google Scholar]
- Taylor CA, Bell JM, Breiding MJ, Xu L (2017) Traumatic brain injury–related emergency department visits, hospitalizations, and deaths—United States, 2007 and 2013 MMWR Surveill Summ 66:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timaru-Kast R, Gotthardt P, Luh C, Huang C, Hummel R, Schäfer MK, Thal SC (2019) Angiotensin II receptor 1 blockage limits brain damage and improves functional outcome after brain injury in aged animals despite age-dependent reduction in AT1 expression Front Aging Neurosci 11:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timaru-Kast R et al. (2012) Delayed inhibition of angiotensin II receptor type 1 reduces secondary brain damage and improves functional recovery after experimental brain trauma Crit Care Med 40:935–944 [DOI] [PubMed] [Google Scholar]
- Torika N, Asraf K, Apte RN, Fleisher-Berkovich S (2018) Candesartan ameliorates brain inflammation associated with Alzheimer's disease CNS Neurosci Ther 24:231–242 doi: 10.1111/cns.12802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umschweif G, Liraz-Zaltsman S, Shabashov D, Alexandrovich A, Trembovler V, Horowitz M, Shohami E (2014a) Angiotensin receptor type 2 activation induces neuroprotection and neurogenesis after traumatic brain injury Neurotherapeutics 11:665–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umschweif G, Shabashov D, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E (2014b) Neuroprotection after traumatic brain injury in heat-acclimated mice involves induced neurogenesis and activation of angiotensin receptor type 2 signaling J Cereb Blood Flow Metab 34:1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger T, Dahlöf B (2010) Compound 21, the first orally active, selective agonist of the angiotensin type 2 receptor (AT2): implications for AT2 receptor research and therapeutic potential J Renin Angiotensin Aldosterone Syst 11:75–77 [DOI] [PubMed] [Google Scholar]
- Vadhan JD, Speth RC (2020) The role of the brain renin-angiotensin system (RAS) in mild traumatic brain injury (TBI) Pharmacol Ther:107684. [DOI] [PubMed] [Google Scholar]
- Villapol S, Balarezo MG, Affram K, Saavedra JM, Symes AJ (2015) Neurorestoration after traumatic brain injury through angiotensin II receptor blockage Brain 138:3299–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Yaszemski AK, Logan TT, Sánchez-Lemus E, Saavedra JM, Symes AJ (2012) Candesartan, an angiotensin II AT 1-receptor blocker and PPAR-γ agonist, reduces lesion volume and improves motor and memory function after traumatic brain injury in mice Neuropsychopharmacology 37:2817–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe M, Savoia C, De Paolis P, Ostrowska B, Tarasi D, Rubattu S (2002) The renin-angiotensin system as a risk factor and therapeutic target for cardiovascular and renal disease J Am Soc Nephrol 13:S173–S178 [DOI] [PubMed] [Google Scholar]
- Werhane ML et al. (2017) Pathological vascular and inflammatory biomarkers of acute-and chronic-phase traumatic brain injury Concussion 2:CNC30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Engelhard K (2007) Pathophysiology of traumatic brain injury Br J Anaesth 99:4–9 [DOI] [PubMed] [Google Scholar]
- Wright JW, Harding JW (2004) The brain angiotensin system and extracellular matrix molecules in neural plasticity, learning, and memory Prog Neurobiol 72:263–293 [DOI] [PubMed] [Google Scholar]
- Yaka R et al. (2007) D-cycloserine improves functional recovery and reinstates long-term potentiation (LTP) in a mouse model of closed head injury FASEB J 21:2033–2041 [DOI] [PubMed] [Google Scholar]
- Zhang Y et al. (2008) Central administration of angiotensin-(1–7) stimulates nitric oxide release and upregulates the endothelial nitric oxide synthase expression following focal cerebral ischemia/reperfusion in rats Neuropeptides 42:593–600 doi: 10.1016/j.npep.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Zheng J et al. (2014) Activation of the ACE2/Ang-(1–7)/Mas pathway reduces oxygen-glucose deprivation-induced tissue swelling, ROS production, and cell death in mouse brain with angiotensin II overproduction Neuroscience 273:39–51 doi: 10.1016/j.neuroscience.2014.04.060 [DOI] [PMC free article] [PubMed] [Google Scholar]